

Abstract
A recent study reveals that missense mutations of EWSR1 are associated with neurodegenerative disorders such as amyotrophic lateral sclerosis, but the function of wild-type (WT) EWSR1 in the central nervous system (CNS) is not known yet. Herein, we investigated the neuroanatomical and motor function changes in Ewsr1 knock out (KO) mice. First, we quantified neuronal nucleus size in the motor cortex, dorsal striatum and hippocampus of three different groups: WT, heterozygous Ewsr1 KO (+/−), and homozygous Ewsr1 KO (−/−) mice. The neuronal nucleus size was significantly smaller in the motor cortex and striatum of homozygous Ewsr1 KO (−/−) mice than that of WT. In addition, in the hippocampus, the neuronal nucleus size was significantly smaller in both heterozygous Ewsr1 KO (+/−) and homozygous Ewsr1 KO (−/−) mice. We then assessed motor function of Ewsr1 KO (−/−) and WT mice by a tail suspension test. Both forelimb and hindlimb movements were significantly increased in Ewsr1 KO (−/−) mice. Lastly, we performed immunohistochemistry to examine the expression of TH, DARPP-32, and phosphorylated (p)-DARPP-32 (Thr75) in the striatum and substantia nigra, which are associated with dopaminergic signaling. The immunoreactivity of TH and DARPP-32 was decreased in Ewsr1 KO (−/−) mice. Together, our results suggest that EWSR1 plays a significant role in neuronal morphology, dopaminergic signaling pathways, and motor function in the CNS of mice.
Keywords: EWSR1, central nervous system (CNS), neuron, dopamine, DARPP-32, motor function
Graphical Abstract

INTRODUCTION
EWS RNA binding protein 1 (EWSR1) belongs to the FET family of DNA and RNA binding proteins and shares functional homology. FUS, EWSR1, and TAF15, constituting the FET family, have a significant role in transcription and alternative splicing by interacting with transcription pre-initiation complex and various splicing factors [1,2,3,4,5,6,7]. In addition, FET proteins modulate post transcriptional modification through their RBD and RGG domain [8]. The discovery of EWSR1/EWS gene was first discovered in Ewing sarcoma, an aggressive tumor first described by James Ewing in 1921 that mainly afflicts young adolescents and children [9,10,11]. A translocation between chromosomes 11 and 22, which fuses EWSR1 gene to FLI1, accounts up to 85% of Ewing's sarcoma [12]. It has been subsequently discovered that EWSR1 gene is involved in a broad spectrum of mesenchymal lesions tumors through formation of aberrant fusion genes such as EWSR1-WT1, EWSR1-KLF17, EWSR1-ATF1, and ESWR1-CREB3L1 [13,14,15,16,17]. All of these fusion proteins share a common characteristic of the N-terminal transcription-activating domain of EWSR1 juxtaposed to the C-terminal DNA-binding domain of the fusion partners, creating oncogenic transcription factors that drive proliferation, survival, and transformation of cells [18].
While the role of EWSR1 fusion protein in oncogenesis is relatively known, the role of wild-type EWSR1 remains largely unclear. A previous study suggested that EWSR1 is crucial in meiosis. Ewsr1 knockout (KO) mice show impaired segregation of chromosomes which leads to massive apoptosis of spermatocytes and arrest in gamete maturation [19]. Moreover, Ewsr1 KO mice display symptoms of premature aging, such as reduced bone density, loss of subcutaneous fat, and kyphosis [19]. Ewsr1 KO mice exhibit severe lymphopenia and impaired development of B lymphocytes [19]. Furthermore, a recent study suggests that Ewsr1 deficiency leads to impaired dermal development through the modulation of Drosha and miRNA activity. Otherwise, Ewsr1 KO mice show downregulation of Gnai1, G protein subunit alpha i1, in the spinal cord [20].
Recently, several studies have discovered mutations of FET family proteins and cytoplasmic aggregates of FET proteins in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) patients, suggesting these genes are associated with neurodegenerative diseases [21,22,23,24,25,26,27]. Mutant forms of FET proteins generate prion-like aggregation and alter their subcellular localization [28]. Importantly, a recent study has found missense mutation of EWSR1 gene in several ALS patients [23]. Similar to other FET proteins, mutant EWSR1 protein is mislocalized in the cytoplasm of the spinal cord tissue [23]. Although the EWSR1 gene has been implicated in ALS patients, no study has yet reported about the roles of WT EWSR1 in the CNS. In the present study, we aimed to determine and characterize the neuroanatomical and motor function difference in conventional Ewsr1 KO mice compared with WT mice. We found that Ewsr1 deficiency leads to impairments of neuronal morphology, dopaminergic signaling pathways, and motor function in mice.
MATERIALS AND METHODS
Generation of Ewsr1 KO mice
Homozygous Ewsr1 KO mice were generated as described previously [19]. Targeting Ewsr1 knockout allele was inserted into TC-1 mouse ES cells. Homologous recombination was screened by PCR and confirmed by Southern blotting. Positive ES cells were injected into C57BL/6J blastocysts, and the resulting chimeras were bred with Black Swiss females (Taconic) to generate F1 heterozygotes. Male heterozygotes were bred with female heterozygotes in order to acquire homozygotes. Genotyping was performed by PCR analysis of tail DNA using two sets of primers: wild-type (WT) (411-bp PCR product): 5'-TGG ATC CTA TGG ATC CTA CAG CCA GGC TCC-3', 5'-TGCTCGCTAGTGCTCTGTGAGCAGGAC-3'; mutant (237-bp PCR product): 5'-TGGATCCTACAGCCAAGCTCC-3', 5'-CCTGTATGAGTCCTGGTGTGGGTC-3'. All animal experiments were carried out in accordance with the Guide for Institutional Animal Care and Use Committee.
Histological evaluation
A total of 16 mice (7 homozygous Ewsr1 KO (−/−) mice, 6 heterozygous Ewsr1 (+/−) mice and 3 wild-type (WT) littermate controls) were transcardially perfused at 3 postnatal weeks with 4% buffered paraformaldehyde. Both female and male mice were used. The brains were removed, post-fixed overnight, and cryoprotected in 30% sucrose (in PBS) after their weight measured. Then the brains were serially cryosectioned at either coronal or sagittal planes. Brain sections were stained with hematoxylin and eosin or cresyl violet (Nissl staining). The staining images were taken under phase contrast microscope (Olympus DP 73, Tokyo, Japan).
Image analysis
Neuronal nucleus size was measured from the motor cortex, striatum, and hippocampus. Then their sizes were analyzed by NIH ImageJ program, followed by the principles of unbiased stereology [29]. Neuronal nucleus size was measured as the cross-sectional area of the nucleus. Measurement of neuronal nucleus size was conducted using freehand selection. A large counting box (399×299 µm) was placed over a slide in a systemic and random manner. Nucleus size was measured from at least 100 neurons per slide, which encompasses the average neuronal nucleus size of the area [30]. For comparison, a corresponding region was examined from each animal: M2 region of motor cortex adjacent to the midsagittal line, a CA3 region of the hippocampus and caudate putamen adjoining lateral ventricle. The counting and measurement were conducted by two individual researchers, and each researcher was blinded to the genetic type.
Tail suspension test
Fore and hind limb movements were assessed as described previously [31]. Using video captures, limb movement was quantified by the number of movements per second (mvmt/sec). Movement of the ventral surface of each mouse was filmed while the mouse was suspended by the tail for 10 seconds, followed by a brief touch down and 10-second suspension, and followed by the second brief touch down and the last suspension for 12 seconds. The total suspension time of each object was not exceeding 32 seconds. Limb movement data was acquired and quantified by blind analysis to the genetic type of mice. Dystonia of both forelimb and hindlimb was recorded at 3 postnatal weeks by counting the number of clasping behavior [31]. In addition, we measured the duration of torso flexion of both WT and Ewsr1 KO mice.
Immunohistochemistry
Immunohistochemistry was performed in coronal or sagittal sections of WT and Ewsr1 KO mouse brains to detect immunoreactivity of Tyrosine hydroxylase (TH), dopamine and cyclic-AMP regulated phosphoprotein 32 (DARPP-32) and p-DARPP-32 (Thr75). Anti-TH antibody (Cat. No.: AB152, Millipore, MA, USA), anti-DARPP-32 (H3) antibody (Cat. No.: sc-271111, Santa Cruz Biotechnology, CA, USA) and anti-p-DARPP32 (Thr75) antibody (Cat. No.: ab51114, Abcam, MA, USA) were used at 1:200 dilution. The intensity (pixel) of TH, DARPP-32 and p-DARPP-32 (Thr75) was analyzed by Multi-Gauge Software (Fuji photo film Co, Ltd., Tokyo, Japan). Changes of TH, DARPP-32 and p-DARPP-32 (Thr75) immunoreactivity in Ewsr1 KO mice were normalized to the background signal in WT mice.
Western blot analysis
Western blot was conducted as previous describe [20]. Twenty micrograms of protein was electrophoresed on SDS-PAGE (10%) and blotted with anti-TH antibody (Cat. No.: AB152, Millipore, MA, USA) at 1:1000 dilution or anti-DARPP-32 antibody (Cat. No.: sc-271111, Santa Cruz Biotechnology, CA, USA) at 1:500 dilution. Anti-β-Actin antibody (Cat. No.: a1978, Sigma, MO, USA) at 1: 5000 dilution was used as protein loading control.
Quantitative real-time PCR (qPCR)
Total RNA from WT and Ewsr1 KO mice was extracted by using a commercial extraction system (Macherey-Nagel). 500 ng RNA was prepared for cDNA synthesis, using a First strands cDNA Synthesis Kit (Toyobo, Osaka, Japan). The amplification of cDNA from each sample was performed by RT-PCR, using SYBR Green Supermix (Toyobo, Osaka, Japan). PCR cycling conditions were as the following: denaturation for 3 min at 95℃, then 40 cycles of amplification for 15 s at 95℃, 15 s at 60℃, 20 s at 70℃, followed with 30 s at 72℃. The PCR primers were: mouse Th: forward, F: 5'-GATTGCAGAGATTGCCTTCC-3' and reverse, R: 5'-GAAGTGAGACACATCCTCCA-3'; mouse DARPP-32 : forward, 5'-CCCAGCCTTAACCCAGTACTGTTC-3' and reverse, 5'-TGGGCAAGTGGACTGTTCAGAT-3'; mouse Ddc: forward, 5'-TACCCAGCTATGCTTGCAGAC-3' and reverse, 5'-GCGGATAACTTTAGTCCGAGC-3'; mouse Gapdh: forward, 5'-ACC ACA GTC CAT GCC ATC AC-3' and reverse, 5'-TCC ACC ACC CTG TTG CTG T-3'. Gapdh mRNA was used as a control. The mRNA of each sample was normalized to Gapdh mRNA.
Statistical analysis
The data are expressed as the mean±standard error of the mean (SEM). Comparison among WT and Ewsr1 KO mouse group was performed by one-way ANOVA and Tukey's post-hoc tests (SPSS). Differences were considered significant when p value was below 0.05.
RESULTS
Ewsr1 KO mice show reduced neuronal nucleus sizes
At the first series of experiment, we characterized gross anatomical difference of the brain and neuronal nucleus sizes between WT and Ewsr1 KO mice. The whole brain sections of both WT and Ewsr1 KO mice are shown in Fig. 1. Both sagittal and coronal sections did not show any noticeable gross anatomical difference. We then measured and compared the neuronal nucleus sizes in the motor cortex, striatum, and hippocampus between WT and Ewsr1 KO mice (Fig. 2). The neuronal nucleus size was significantly smaller in all three brain regions in Ewsr1 KO mice than in WT mice (Fig. 2; p<0.0001, one-way ANOVA with Turkey). In motor cortex, the neuronal nucleus size of Ewsr1 KO (−/−) mice was significantly smaller than WT mice, while that of heterozygous Ewsr1 (+/−) mice was not significantly different (Fig. 2A, p<0.0001, one-way ANOVA with Turkey). Likewise, the striatal neuronal nucleus sizes of Ewsr1 KO mice were significantly smaller than WT mice (Fig. 2B; p<0.0001, one-way ANOVA with Turkey). In the hippocampus, the neuronal nucleus sizes in both Ewsr1 (+/−) and Ewsr1 (−/−) mice were significantly smaller than WT mice. (Fig. 2C, p<0.0001, one-way ANOVA with Turkey).
Fig. 1. Whole brain sections of WT and Ewsr1 KO (−/−) mice at 3 weeks of age. (A) Sagittal (upper) and coronal (lower) brain sections of WT mice. (B) Sagittal (upper) and coronal (lower) brain sections of Ewsr1 KO (−/−) mice. Sagittal sections were stained with hematoxylin and eosin. Coronal sections were stained with cresyl violet.

Fig. 2. Neuronal nucleus sizes are altered in the cortex, striatum and hippocampus of Ewsr1 KO (−/−) mice. (A) The neuronal nucleus sizes in the motor cortex were reduced in Ewsr1 KO (−/−) mice. (B) The neuronal nucleus sizes in the striatum were reduced in Ewsr1 KO (−/−) mice. (C) The neuronal nucleus sizes in the hippocampus were reduced in Ewsr1 KO (−/−) mice. The tissues were stained with hematoxylin and eosin staining. Scale bar (black): 20 µm. Data are presented as the mean±SEM. Significantly different at *p<0.05, **p<0.001, ***p<0.0001.

Ewsr1 deficiency leads to motor dysfunction
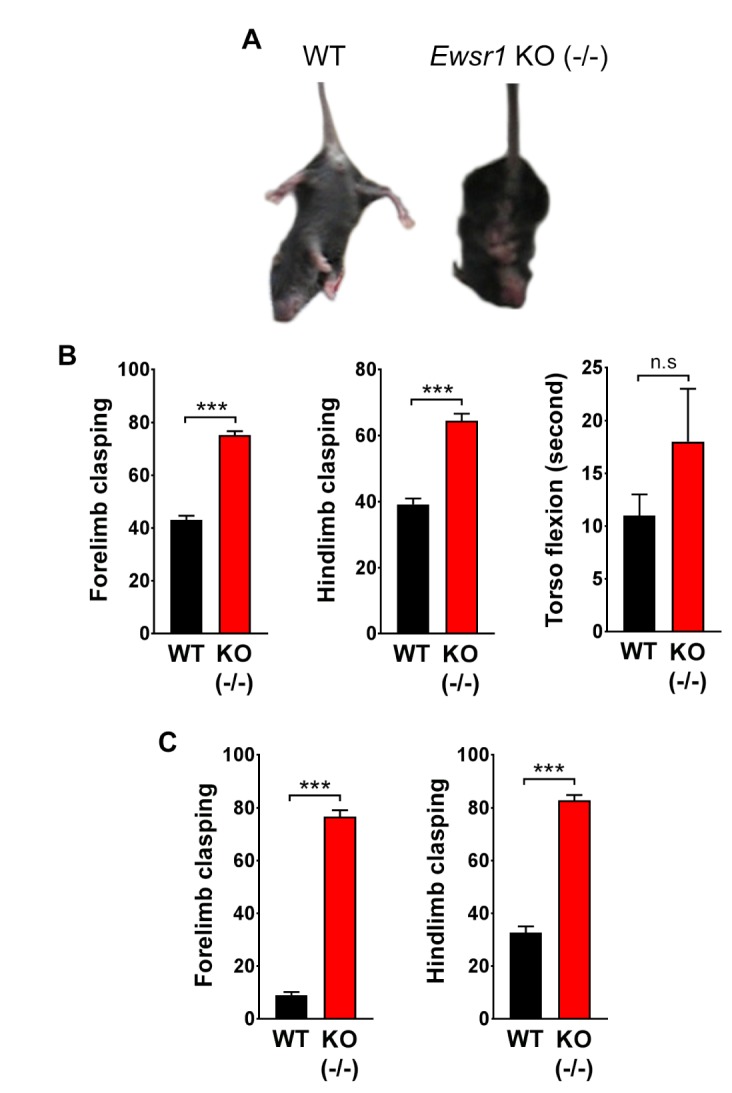
In order to determine whether Ewsr1 deficiency affects motor function, we performed the tail suspension test. When WT mice were suspended by its tail, hindlimb initially extended tangentially from its body, like helicopter propellers. Then its torso flexed laterally to keep the balance. At the same time, hindlimb splayed and forelimb clasped together (Fig. 3A). In contrast, several differences in motor function were observed in Ewsr1 KO mice. The frequency of forelimb and hindlimb movements were much higher (hyperkinesia) in Ewsr1 KO mice compared to WT littermates. Moreover, the coordinative motions among forelimb, hindlimb, and torso were not properly exhibited in Ewsr1 KO mice. Hindlimb and torso also showed flexion-dominant movements in Ewsr1 KO mice. Due to the frequent flexion, an increase in distinct splay movement of hindlimb was observed in Ewsr1 KO mice compared to WT mice at 3 weeks of age and 10 weeks of age (Fig. 3B and C). The frequency of forelimb and hindlimb clasping behaviors in Ewsr1 KO mice was significantly higher compared to WT mice both at 3 weeks of age and 10 weeks of age (Fig. 3B and C, p<0.0001, unpaired Student's t test). Similarly, Ewsr1 KO littermates also showed higher frequency of torso flexion than WT mice at 3 weeks of age but it was not statistically significant (Fig. 3B, p>0.5, unpaired Student's t test).
Fig. 3. Tail suspension test shows an increase of limb clasping behaviors in Ewsr1 KO (−/−) mice. (A) Still images of tail suspended WT and Ewsr1 KO (−/−) mice. (B) The number of forelimb and hindlimb clasping was significantly increased in Ewsr1 KO (−/−) mice at 3 weeks of age. The number of torso flexion was slightly increased Ewsr1 KO (−/−) mice. WT (n=3); KO (−/−) (n=3). (C) The number of forelimb and hindlimb clasping was significantly increased in Ewsr1 KO (−/−) mice at 10 weeks of age. WT (n=6); KO (−/−) (n=6). Data are presented as the mean±SEM. Significantly different at *p<0.05, **p<0.001, ***p<0.0001 and n.s, non-significant at p>0.05.
Ewsr1 deficiency alters dopaminergic signaling pathways
To address whether the motor dysfunction is associated with impaired dopaminergic signaling in Ewsr1 KO mice, we examined the expression of TH and the DARPP-32 proteins in the striatum and the substantia nigra of WT and Ewsr1 KO mice. We performed qPCR analysis and found that TH mRNA was significantly decreased in the striatum of Ewsr1 KO (−/−) mice compared to WT mice (Fig. 4A). But DDC (dopamine decarboxylase) and DARPP-32 mRNAs were not significantly changed (Fig. 4A). In addition, we ran Western blot analysis and confirmed that TH protein level was significantly decreased in Ewsr1 KO (−/−) mice (Fig. 4B). DARPP-32 protein level was also significantly reduced in Ewsr1 KO (−/−) mice (Fig. 4B). On the other hand, we performed immunohistochemistry and found that TH immunoreactivity was significantly decreased in the striatal and substantia nigral neurons of Ewsr1 KO mice compared to WT mice (Fig. 4C). DARPP-32 immunoreactivity was also significantly decreased in the striatum and substantia nigra of Ewsr1 KO mice compared to WT mice (Fig 4D; p<0.0001, unpaired Student's t test). In contrast, the immunoreactivity of p-DARPP-32 (Thr75) was significantly increased in Ewsr1 KO mice (Fig. 4D; p<0.0001 and p<0.001 for striatum and substantia nigra, respectively, unpaired Student's t test).
Fig. 4. Dopaminergic signaling pathways are impaired in Ewsr1 KO (−/−) mice at 3 weeks of age. (A) qPCR analysis showed that Th mRNA was significantly lower in the striatum of Ewsr1 KO (−/−) mice (n=4) compared to WT mice (n=4) while Ddc (dopamine decarboxylase) and DARPP-32 mRNAs were not changed noticeably. (B) Western blot analysis showed that protein levels of TH and DARPP-32 were significantly decreased in Ewsr1 KO (−/−) mice (n=4) compared to WT mice (n=4). (C) The immunoreactivity of TH and DARPP-32 was markedly decreased in the striatum and the substantia nigra of Ewsr1 KO (−/−) mice, whereas the immunoreactivity of p-DARPP-32 (Thr75) was highly increased. (d) Densitometry analysis confirmed that the immunoreactivity of TH and DARPP-32 was significantly reduced in the striatum and substantia nigra of Ewsr1 KO (−/−) mice while the immunoreactivity of p-DARPP-32 (Thr75) was increased. Scale bar: 100 µm (black); 20 µm (red). WT (n=3); KO (−/−) (n=3). Data are presented as the mean±SEM. *p<0.05, **p<0.001, ***p<0.0001, unpaired Student's t test.

DISCUSSION
In the current study, we measured neuronal nucleus size in three different brain regions such as motor cortex, hippocampus, and striatum of Ewsr1 KO and WT mice. Notably, significant anatomical and neuronal size differences were observed in all three regions of Ewsr1 KO mice compared to WT mice. In the motor cortex and striatum, the neuronal nucleus sizes were significantly smaller in Ewsr1 KO mice compared to WT mice. In the hippocampus, the neuronal nucleus sizes of both heterozygous and homozygous Ewsr1-deficient mice were significantly smaller than that of WT mice. It seems likely that Ewsr1 deficiency leads to neuronal atrophy and consequently alters CNS function in mice. It has been known that hippocampal neuronal atrophy is a typical pathologic event associated with cognitive impairment in neurodegenerative brain disorders such as FTLD. Neuronal atrophy is commonly considered as an intermediate pace of neuronal loss in neurodegenerative disorders [32]. For example, striatal neuronal atrophy has been viewed as an important neuropathological characteristic of Huntington's disease (HD) or spinocerebellar ataxia type 1 (SCA1) [32,33]. We found that Ewsr1 deficient mice showed a remarkable reduction of neuronal nucleus sizes in motor cortex, striatum, and hippocampus, respectively. Our morphological data suggest that the Ewsr1 deficiency may leads to motor impairment through this neuropathological change. Recently, cytoplasmic aggregates of FET proteins are observed in few ALS and FTLD patients, indicating that loss of FUS, EWSR1, and TAF15 function could contribute to pathological etiology of ALS and several FTLD subtypes [23,24,25,26,27,28]. In this context, our finding on the reduction of neuronal nucleus size in the hippocampus of Ewsr1 KO mice supports an idea that the dysfunction of EWSR1 is associated with the pathogenesis of ALS and FTLD. A recent study has found that genetic ablation of Ewsr1 in zebrafish leads to defects in the CNS and higher susceptibility to apoptosis [34]. The zebrafish data suggest that Ewsr1 may regulate migration, survival, and differentiation of neurons. Moreover, Ewsr1 may be responsible for the modulation of neuronal survival and death. In part, understanding the specific function of Ewsr1 in the CNS of mice provides a novel insight on how missense mutations of EWSR1 can be a causative process of neurodegeneration.
Interestingly, we found that Ewsr1 KO mice show hyperkinesia of both forelimb and hindlimb. Ewsr1 KO mice exhibited a significantly higher frequency of clasping and torso flexion compared to WT mice, indicating dysfunction of motor coordination in Ewsr1 KO mice. Based on this finding, we proposed a potential link that Ewsr1 deficiency may affect the motor function via dopaminergic signaling pathways through the striatum and substantia nigra. It is well established that the dopaminergic system is associated with motor function. TH is a rate-limiting enzyme that plays a critical role in synthesizing dopamine. TH catalyzes tyrosine to L-DOPA, which is again converted into dopamine by dopamine decarboxylase (DDC) [35]. Importantly, we found that levels of Th mRNA and TH protein are significantly decreased in the striatum and substantia nigra of Ewsr1 KO mice. It seems likely that Ewsr1 deficiency leads to down regulation of TH expression at the transcription level. DARPP-32 is a signal transduction molecule that is enriched in neurons with dopamine receptors such as medium spiny neurons in the striatum. Dopamine acts upon D1 receptor, which then activates protein kinase A (PKA) and causes subsequent DARPP-32 phosphorylation [36]. The immunoreactivity of DARPP-32 was significantly decreased in Ewsr1 KO mice while the immunoreactivity of phosphorylated (p)-DARPP-32 (Thr75) was increased in Ewsr1 KO mice. The p-DARPP-32 (Thr75) is known to be a negative regulator of dopamine signaling. Thus, our results indicate that the alterations of TH, DARPP-32 and p-DARPP-32 (Thr75) levels by Ewsr1 deficiency deregulate dopaminergic signaling pathways and lead to motor dysfunction. Our previous study showed that EWSR1 possesses multifunctional and undescribed roles in cellular processes, which may lead to the diverse effects in Ewsr1 deficient mice [20]. However, further studies using multiple behavioral and molecular analyses in Ewsr1 WT and KO mice are required to better understand the mechanisms by which EWSR1 regulates motor coordination.
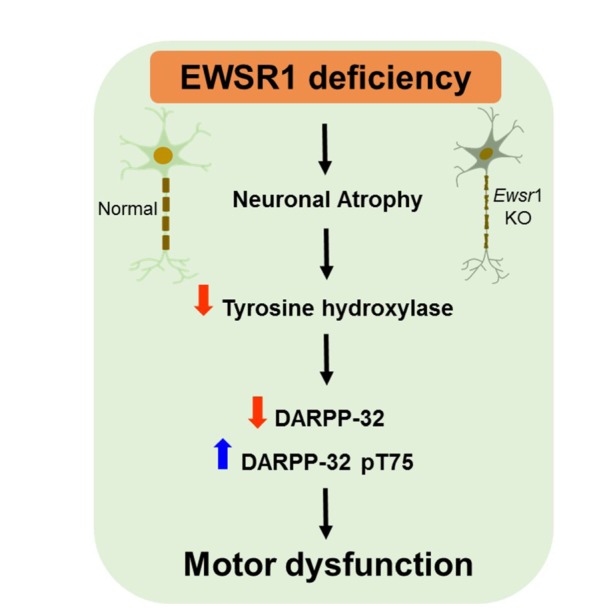
In summary, Ewsr1 deficiency caused the gross neuroanatomical change of the brain, the reduction of neuronal nucleus size, and the deregulation of motor function in mice. Our finding indicates that the motor dysfunction may be closely linked to altered dopaminergic signaling pathways due to the decreased levels of TH and DARPP-32 (Fig. 5). Taken together, our data indicate that EWSR1 plays a significant role in the neuronal morphology and motor function in mice.
Fig. 5. A schematic diagram showing that EWSR1 deficiency reduces neuronal nucleus size (atrophy) and, in parallel, causes the reduction of tyrosine hydroxylase in the substantia nigra. Non-phosphorylated DARPP-32 and phosphorylated (p)-DARPP32 (Thr75) are differentially regulated in the striatum of Ewsr1 mice, indicating that the dopaminergic signaling pathway is abnormaly regulated by EWSR1 deficiency. Consequently, EWSR1 deficiency leads to motor behavioral dysfunction in mice.
ACKNOWLEDGEMENTS
This study was supported by NIH Grant (R01NS067283) (H.R.). This study was also supported by the National Research Foundation of Korea Grant (NRF-2015M3A9A8030034 and NRF-2016M3C7A1904233) from the Ministry of Science, ICT and Future Planning, the National Research Council of Science & Technology (NST) Grant (No. CRC-15-04-KIST) from the Korea government (MSIP), and Grants from Korea Institute of Science and Technology (2E26200 and 2E26663).
References
- 1.Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paronetto MP. Ewing sarcoma protein: a key player in human cancer. Int J Cell Biol. 2013;2013:642853. doi: 10.1155/2013/642853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O, Tora L. EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol. 1998;18:1489–1497. doi: 10.1128/mcb.18.3.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem. 1998;273:27761–27764. doi: 10.1074/jbc.273.43.27761. [DOI] [PubMed] [Google Scholar]
- 5.Chansky HA, Hu M, Hickstein DD, Yang L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res. 2001;61:3586–3590. [PubMed] [Google Scholar]
- 6.Meissner M, Lopato S, Gotzmann J, Sauermann G, Barta A. Proto-oncoprotein TLS/FUS is associated to the nuclear matrix and complexed with splicing factors PTB, SRm160, and SR proteins. Exp Cell Res. 2003;283:184–195. doi: 10.1016/s0014-4827(02)00046-0. [DOI] [PubMed] [Google Scholar]
- 7.Paronetto MP, Bernardis I, Volpe E, Bechara E, Sebestyen E, Eyras E, Valcarcel J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014;7:1211–1226. doi: 10.1016/j.celrep.2014.03.077. [DOI] [PubMed] [Google Scholar]
- 8.Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 9.Meltzer PS. Is Ewing's sarcoma a stem cell tumor? Cell Stem Cell. 2007;1:13–15. doi: 10.1016/j.stem.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Ewing J. Diffuse endothelioma of bone. CA Cancer J Clin. 1972;22:95–98. doi: 10.3322/canjclin.22.2.95. [DOI] [PubMed] [Google Scholar]
- 11.Zucman J, Delattre O, Desmaze C, Plougastel B, Joubert I, Melot T, Peter M, De Jong P, Rouleau G, Aurias A, Thomas G. Cloning and characterization of the Ewing's sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Genes Chromosomes Cancer. 1992;5:271–277. doi: 10.1002/gcc.2870050402. [DOI] [PubMed] [Google Scholar]
- 12.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 13.May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R, Denny CT. The Ewing's sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993;13:7393–7398. doi: 10.1128/mcb.13.12.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology. 2014;64:134–150. doi: 10.1111/his.12269. [DOI] [PubMed] [Google Scholar]
- 15.Huang SC, Chen HW, Zhang L, Sung YS, Agaram NP, Davis M, Edelman M, Fletcher CD, Antonescu CR. Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer. 2015;54:267–275. doi: 10.1002/gcc.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi S, Szuhai K, Ijszenga M, Tanke HJ, Zanatta L, Sciot R, Fletcher CD, Dei Tos AP, Hogendoorn PC. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res. 2007;13:7322–7328. doi: 10.1158/1078-0432.CCR-07-1744. [DOI] [PubMed] [Google Scholar]
- 17.Lau PP, Lui PC, Lau GT, Yau DT, Cheung ET, Chan JK. EWSR1-CREB3L1 gene fusion: a novel alternative molecular aberration of low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2013;37:734–738. doi: 10.1097/PAS.0b013e31827560f8. [DOI] [PubMed] [Google Scholar]
- 18.Romeo S, Dei Tos AP. Soft tissue tumors associated with EWSR1 translocation. Virchows Arch. 2010;456:219–234. doi: 10.1007/s00428-009-0854-3. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Watford W, Li C, Parmelee A, Bryant MA, Deng C, O'Shea J, Lee SB. Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J Clin Invest. 2007;117:1314–1323. doi: 10.1172/JCI31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KY, Hwang YJ, Jung MK, Choe J, Kim Y, Kim S, Lee CJ, Ahn H, Lee J, Kowall NW, Kim YK, Kim JI, Lee SB, Ryu H. A multifunctional protein EWS regulates the expression of Drosha and microRNAs. Cell Death Differ. 2014;21:136–145. doi: 10.1038/cdd.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 22.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, Kim CE, Frackelton EC, Solski JA, Williams KL, Clay-Falcone D, Elman L, McCluskey L, Greene R, Hakonarson H, Kalb RG, Lee VM, Trojanowski JQ, Nicholson GA, Blair IP, Bonini NM, Van Deerlin VM, Mourelatos Z, Shorter J, Gitler AD. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–371. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]
- 26.Mackenzie IR, Neumann M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012;1462:40–43. doi: 10.1016/j.brainres.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423–434. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svetoni F, Frisone P, Paronetto MP. Role of FET proteins in neurodegenerative disorders. RNA Biol. 2016;13:1089–1102. doi: 10.1080/15476286.2016.1211225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–831. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 30.Meitzen J, Pflepsen KR, Stern CM, Meisel RL, Mermelstein PG. Measurements of neuron soma size and density in rat dorsal striatum, nucleus accumbens core and nucleus accumbens shell: differences between striatal region and brain hemisphere, but not sex. Neurosci Lett. 2011;487:177–181. doi: 10.1016/j.neulet.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- 32.Dell'Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT, Shakkottai VG. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci. 2015;35:11292–11307. doi: 10.1523/JNEUROSCI.1357-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Raamsdonk JM, Pearson J, Rogers DA, Bissada N, Vogl AW, Hayden MR, Leavitt BR. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:1379–1392. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- 34.Azuma M, Embree LJ, Sabaawy H, Hickstein DD. Ewing sarcoma protein ewsr1 maintains mitotic integrity and proneural cell survival in the zebrafish embryo. PLoS One. 2007;2:e979. doi: 10.1371/journal.pone.0000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daubner SC, Le T, Wang S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys. 2011;508:1–12. doi: 10.1016/j.abb.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]