


Abstract
RNA sequencing (RNA-Seq) has become the experimental standard in transcriptome studies. While most of the bioinformatic pipelines for the analysis of RNA-Seq data and the identification of significant changes in transcript abundance are based on the comparison of two conditions, it is common practice to perform several experiments in parallel (e.g. from different individuals, developmental stages, tissues), for the identification of genes showing a significant variation of expression across all the conditions studied. In this work we present RNentropy, a methodology based on information theory devised for this task, which given expression estimates from any number of RNA-Seq samples and conditions identifies genes or transcripts with a significant variation of expression across all the conditions studied, together with the samples in which they are over- or under-expressed. To show the capabilities offered by our methodology, we applied it to different RNA-Seq datasets: 48 biological replicates of two different yeast conditions; samples extracted from six human tissues of three individuals; seven different mouse brain cell types; human liver samples from six individuals. Results, and their comparison to different state of the art bioinformatic methods, show that RNentropy can provide a quick and in depth analysis of significant changes in gene expression profiles over any number of conditions.
INTRODUCTION
The orchestration of gene expression in appropriate spatio-temporal coordination is the key biological mechanism for development and life in multicellular organisms. Indeed, we can observe a highly regulated specificity of the expression profile of genes in different cell or tissue types, developmental or cell-cycle stages, physiological conditions, in response to external stimuli, normal and pathological conditions, and so on.
In the last few years, RNA sequencing (RNA-Seq) has become de facto the experimental standard for transcriptome investigations (1), producing estimated expression levels computed either by assembling transcripts from sequence reads (2) or by employing reference genome and/or gene annotations (3,4). Given normalized expression estimates in two or more conditions, the next step is to identify those genes or transcripts that change their expression in a significant way, that is, show changes not simply due to experimental noise or normal biological variation. This is currently a very open and thoroughly investigated line of research, with several different methods and statistical approaches introduced to tackle the problem (among many others, see (4–10), and (11) for a more comprehensive overview), that try to incorporate into a unique statistical framework all the different sources of biological or experimental variability. The most widely used protocols and pipelines for the identification of transcripts or genes with significant changes of expression used today are focused on pairwise comparisons (11), even in case studies where a simultaneous comparison of larger numbers of samples and conditions would be more appropriate.
On the other hand, given a study on more than two conditions, there is no general unique definition of ‘condition specific’ (e.g. tissue-specific) genes. For example, one could require a gene to be exclusively expressed in a single condition, or the expression of a gene in a specific condition to be greater than k times its average across all the conditions studied (12,13). Indeed, different tissue specificity metrics have been introduced for the identification of tissue-specific genes (14), that can be adapted to other multi-condition comparisons. However, these measures consider only relative variation of expression, and thus two genes with very different expression levels will be considered to be ‘equally significant’ if they present the same variation with respect to the respective averages across the samples studied. Furthermore, the assessment of the variability of gene expression should also consider the biological or technical replicates available for each condition.
Indeed, recent multi-tissue, or in general, multi sample studies are still mainly based on pairwise comparisons. For example, a recently published large scale study on 1641 samples from 43 different tissues of 175 individuals (GTEx, (15)) resorted to pairwise comparisons to assess tissue and individual specificity (sample against sample, or one sample against the pooling of the others), and provided only general indices summarizing the variability across tissues or individuals of each gene.
Starting from these premises, we propose here a novel methodology called RNentropy, designed to detect genes with significant changes of expression across any number of conditions. Given normalized expression levels of genes and/or their isoforms, calculated through any of the different tools available (see among many others (3,4,7)), RNentropy is able to identify over- or under-expressed genes in each of the conditions studied. The samples, corresponding to different conditions sequenced in any number of replicates, are compared by taking into account the global gene expression level and at the same time the impact of biological variation across replicates. The result of the analysis is a map that, for each gene, reports in which samples it can be considered over- or under-expressed, as well as the corresponding significance levels. Thus, RNentropy is suitable for any analysis performed on multiple samples, where genes of interest should be retrieved by considering simultaneously all the conditions studied without explicitly designing comparisons among pairs or groups of conditions. We provide some examples of analyses of this kind, showing how the method can be employed in the detection of genes with cell-, tissue- or individual-specific expression patterns.
MATERIALS AND METHODS
Our method works on gene or transcript expression levels derived from a series of RNA-Seq experiments, where samples are sequenced with technical or biological replicates. We underline that the method does not compute by itself normalized measures to be used in the comparisons, but rather works downstream of analysis pipelines that estimate transcript levels from the initial raw sequences. It thus can work on any measure considered suitable for the comparison of transcript levels of a gene across different samples and conditions. One example are normalized levels expressed with measures like ‘Fragments (or Reads) per kilobase of exon per million reads’ (FPKM, RPKM, (16)):
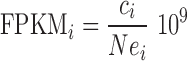 |
where ci is the raw read count associated with gene i, N is the overall number of reads (fragments) used for the quantification, ei is the overall size in bp of the exons of gene i. A variation on this measure are ‘Transcripts per million’ (TPM, (3)), where each gene read count is first divided by exon size, and the results are used to normalize by overall counts. That is, given  for gene i and
for gene i and  over all the genes considered, TPM for each gene are then defined as:
over all the genes considered, TPM for each gene are then defined as:
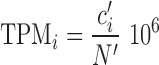 |
While these measures are still widely used, and results based on them still appear on a regular basis, recent literature has pointed out some shortcomings for FPKMs and RPKMs (see (11) and references therein), especially in comparisons of two conditions for the identification of differentially expressed genes. As a consequence, it is currently advised to employ techniques that do not normalize each sample separately, but rather perform the normalization across the conditions and samples that have to be compared (5–7,9). To assess the effect on the results of the measure employed (if any), in our tests we used both TPMs and the counts per million (CPM) output by edgeR after trimmed mean of M-values (TMM) normalization (7).
Finding differentially expressed genes
RNentropy performs statistical tests based on information theory that assess if and how much the expression of a gene across any number of different samples diverges from a given background (e.g. uniform) distribution, computing the probability of obtaining the same test value if the expression pattern observed was actually resulting from the background distribution. After a subset of genes or transcripts has been singled out to have a distribution significantly different from the background, further processing can identify in which of the samples or conditions lie the most significant differences, and thus which are the actual conditions in which each of the genes or transcripts is more ‘specific’, or ‘over-expressed’, or vice versa ‘under-expressed’.
The problem of finding differentially expressed genes across multiple samples has been widely studied since the initial transcriptome studies based on technologies like microarrays. Among many others, an approach that was introduced for the problem (17) comes from information theory and is based on Shannon's entropy (from now on referred to simply as entropy). The idea of entropy is to measure the uncertainty of a random variable according to a series of observations. When a discrete random variable has m possible outcomes, entropy is defined from the frequency with which each outcome is observed as:
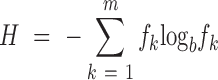 |
where fk is the frequency with which the random variable assumes the k-th value in the observations. If fk = 1 for a single outcome k, then entropy will be zero, and the maximum will be obtained when the variable shows a uniform distribution with fk = 1/m for every outcome. For b the most commonly used values are 2 or e (natural logarithm).
Let T be a gene, and t1…tm its normalized expression levels in m different samples. Let  be the cumulative expression value of T, and
be the cumulative expression value of T, and 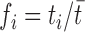 . If we apply the entropy formula to the fi expression values, H(T) will represent the probability with which a RNA assigned to gene T was sequenced in the ith sample studied. The H(T) value will thus be minimum (zero) when all the RNAs assigned to the gene come from a single sample, that is, the gene is expressed only in a single sample. The more unbiased the expression is, the more H(T) will grow, and will be maximum if T has identical expression values across all the samples.
. If we apply the entropy formula to the fi expression values, H(T) will represent the probability with which a RNA assigned to gene T was sequenced in the ith sample studied. The H(T) value will thus be minimum (zero) when all the RNAs assigned to the gene come from a single sample, that is, the gene is expressed only in a single sample. The more unbiased the expression is, the more H(T) will grow, and will be maximum if T has identical expression values across all the samples.
As it is, entropy considers only relative variation of expression and not absolute expression levels. Also, the distribution of the fi values across m samples is implicitly compared to background expected values of bi = 1/m. To consider cases for which the distribution of the background values is not uniform, relative entropy (or, the Kullback–Leibler divergence between the empirical distribution of the fi values and theoretical one of bi, such that 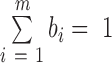 ) can be employed:
) can be employed:
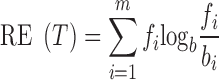 |
The difference is that relative entropy is zero when fi = bi for every i, is higher as the more the observed distribution diverges from the background one, and it is maximum where fi = 1 for some i. To take expression levels into account, we introduce a weighted version of the above formula, where each term is multiplied, instead of for the frequency, for the corresponding expression value ti:
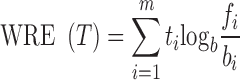 |
This value corresponds to one half of the value of a G-test (goodness of fit test, or log-likelihood ratio test) (18,19), where the null hypothesis is, given values ti and a random distribution with expected frequencies bi, to obtain the following G(T) value by chance:
 |
The G(T) values are distributed with a chi-square distribution with m – 1 degrees of freedom. This fact allows us to associate a P-value with every WRE(T) value, by multiplying it by two and using b = e as the base of the logarithm. The resulting P-value will thus represent the probability of obtaining the ti observed values by chance assuming a random background distribution with frequencies bi. We call this test the global sample specificity test.
To assess the global sample specificity of a single gene we can assume a uniform background distribution with bi = 1/m, but the same framework can be used with any other assumption on the theoretical distribution of the expression values. Resulting P-values have to be corrected for multiple testing, for which we employed the Benjamini–Hochberg procedure, ranking the genes according to increasing P-values, and given a significance P-value threshold α RNentropy considers as significant all genes for which  , where N is the overall number of genes and k is the rank position of the gene.
, where N is the overall number of genes and k is the rank position of the gene.
Once a gene T has been singled out to be significant, another test can be performed for each sample, comparing the expression of T in the sample with its expression in the other m – 1. Hence, the sum will consist of two terms, with expected values  for the sample considered and
for the sample considered and 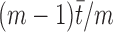 for the others, where
for the others, where 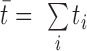 is the cumulative expression of T in all the samples considered. We call this computation the local sample specificity test.
is the cumulative expression of T in all the samples considered. We call this computation the local sample specificity test.
All in all, all genes that passed the global sample specificity test can be reported to be over-expressed in the conditions with expression value higher than the average and that in the local specificity test yield a P-value lower than a threshold 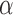 . This test will also consider as significant those genes with expression values significantly lower than the average in some samples, in which they can be then considered to be ‘under-expressed’ or ‘repressed’. The output, thus, summarizes for each gene if it passed the global specificity test, and, if so, if the gene is significantly over- or under-expressed in each of the samples compared.
. This test will also consider as significant those genes with expression values significantly lower than the average in some samples, in which they can be then considered to be ‘under-expressed’ or ‘repressed’. The output, thus, summarizes for each gene if it passed the global specificity test, and, if so, if the gene is significantly over- or under-expressed in each of the samples compared.
Another interesting feature of the tests we just introduced is that they do not necessarily have to be applied to a single gene. So, given any subset of genes belonging to a given pathway or selected according to some criterion, they can be applied to the average expression values of the genes across the samples, in order to assess whether, as a whole, they present a significant variation of expression. Also, it can be applied in a straightforward way to cases where the expression of a gene is split into single transcript or splicing isoform contributions: the result will be that RNentropy will return transcripts and isoforms with a significant variation of expression.
Using replicate experiments
In RNA-Seq analyses, it is essential to work with biological replicates, that is, sequence more than one sample for each of the conditions studied, in order to estimate the amount of experimental or biological variability obtained in the measurements. Hence, methods for the detection of differentially expressed genes assess whether the difference between the mean expression values in two conditions is significant, by comparing it to the variability of the expression estimated from the replicates themselves. The latter is due to two main factors: on one hand experimental noise, derived from all the experimental and computational steps that lead to the final expression values, and on the other the actual biological variation across the samples studied. Indeed, the usage of the term ‘replicates’ in literature is usually employed quite liberally, from the resequencing of the same RNA sample (also referred to as ‘technical’ replicates since all the variation is due to technical reasons), to sequencing of different RNA samples extracted from the same cell line, to sequencing of RNA samples of the same condition (tissue, tumor, etc.) from different individuals. In this latter case, biological variation becomes dominant. As a consequence, raw gene read counts across multiple replicates follow a Poisson distribution in technical replicates (20,21), while they have been shown to be ‘over-dispersed’ in case of biological variation, and best approximated by a negative binomial distribution whose variance depends on a dispersion parameter that can be estimated from the data themselves (6,7,21,22).
Our method does not take explicitly into account the variance of the expression values, but performs a separate test on each replicate by itself. If some samples are defined as replicates of the same condition, a gene will be over- (or under-) expressed in the condition if the local sample specificity test considers it to be significantly over- (under-) expressed in all the replicates. The only difference is that in the local test the expected value against which each replicate of a condition is compared is computed without taking into account the expression values of the other replicates of the same condition. Requiring each single replicate to pass the test is a very stringent criterion, since all replicates have to be significantly above the average expression – while the most widely used methods and tissue-specificity indices simply consider only the mean expression and/or its variance across the replicates. For this reason, in case samples that could be considered ‘replicates’ show high variability of expression (e.g. they come from different individuals), our advice is to process them separately instead of grouping them into replicates, and highlight over-expressed genes shared by more than one sample by post-processing the results.
Assessing similarities among conditions
After differentially expressed genes have been identified in each of the conditions studied, they can be grouped or clustered according to their over- and/or under-expression signature. Also, useful information can be extracted by defining correlations and similarities among the conditions, on the basis of which genes were found to be over-expressed in each, and their overlap. For this task, we included in RNentropy a module to compute point-wise mutual information (PMI, (23)) between every pair of conditions. First, a matrix with one row per gene and one column per sample is built from the output of RNentropy according to the chosen thresholds of global and local P-values. Cell (x, y) of the matrix has value 1 if gene x is found to be over-expressed in condition y, –1 if under-expressed, and zero otherwise. Then, the PMI on over-expressed genes between two columns (conditions) i and j is computed, defined as
 |
where f(i,j) is the fraction of genes passing the global test with 1 in both columns, divided by the product of the fraction of genes passing the global test with 1 in column i by the fraction of genes with 1 in column j. Positive PMI values thus indicate that the number of over-expressed genes shared by the two conditions compared is higher than expected, showing co-association between the two and pointing to relevant correlations between the respective transcriptomes. On the other hand, negative PMI values vice versa point to anti-correlation between the two conditions.
Comparison with other methods
DESeq2 (6) and edgeR (7) are two of the most widely used and best performing tools (26) for the identification of differentially expressed genes between two conditions. Both tools can anyway process any number of conditions split in any number of replicates, and the normalization of read counts and computation of dispersion parameters is performed across all samples. Both methods assess differential expression by selecting two subsets of samples to be compared, each one defining a condition whose samples are considered replicates. They also include the possibility of evaluating the significance of the variation of the expression across all samples considered with ANOVA-like tests. Thus, a framework similar to our method can be designed with both, by comparing the results of the ANOVA-like tests to those of our global significance test. A series of contrasts in which each condition or sample is compared with all the others will be the equivalent of our local significance test.
For edgeR a matrix of contrasts can be created, where each contrast consists of one condition against all the others. A one-way analysis of variance (ANOVA) is then performed on each gene, combining the results of the single contrasts and the respective statistics into a single F-statistic, with the corresponding P-value and FDR.
For DESeq2, the likelihood-ratio test (LRT) can be employed. The LRT examines two models for the counts, a full model with a certain number of terms and a reduced model, in which each of the single conditions is removed. The test determines if the increased likelihood of the data using the extra terms in the full model is more than expected if those extra terms are truly zero. This is conceptually similar to an analysis of variance (ANOVA), except that in the case of the Negative Binomial general linear model (GLM) employed by DESeq2 it is an analysis of deviance (ANODEV), where the deviance captures the difference in likelihood between a full and a reduced model. Once again, a P-value and the corresponding FDR are associated with each gene. Over-expression in each condition can be then evaluated by performing a separate LRT for the condition against the others. Further details on the parameters and commands we employed for both tools in the tests we present are provided as Supplementary Material.
RESULTS
We benchmarked our methodology on different datasets. The first one consisted of yeast gene expression measured on two different conditions, each with 48 biological replicates, taken from (26,27). The second is a study performed on 18 human RNA-Seq samples derived from six tissues (liver, kidney, brain, lung, muscle, heart) in three age- and sex-matched individuals. Each sample was sequenced in three technical replicates, thus yielding in all 54 samples to be compared. Then, we applied RNentropy to a dataset consisting of seven mouse brain cell types taken from (13). Finally, we show the result of the analysis of liver samples from three male and three female individuals, taken from a study that investigated sex-specific expression in human and primates (28). The first dataset was chosen in order to evaluate the correspondence between experimental and estimated false positive rates. The others show how the features of our methodology are suitable in detecting simultaneous tissue- and/or individual-specific expression in a complex experimental setting, with multiple samples from different individuals or different cell types and subtypes.
In all the following examples, we employed RNentropy with the default threshold of 0.01 for both the corrected global P-value and the local P-value. Each of the tests was run on standard personal or laptop computers, requiring a computation time of at most a few minutes with negligible requirements for memory.
Assessing false positive rates on yeast replicate experiments
Several benchmark studies are available, comparing sensitivity and specificity of tools for the identification of differentially expressed genes. Some encompass the impact of sequencing depth and an evaluation of the reliability of the estimation of transcript levels from read counts, as in (21). In others, benchmark data come from different individuals, as for example in (29,30): in this case, the variability of the data is not due only to the experimental protocol in the production and analysis of the sequences, but also to individual variation, which as we show in the following is a key factor in studies of this kind. Synthetic datasets that produce read counts according to a given random distribution (usually Poisson or negative binomial) have also been employed (22), which in turn are often the same background distributions assumed by most of the methods. Thus, for a first assessment of the capabilities of our method, we chose to employ the data presented in (26,27). It is an experiment performed on Saccharomyces cerevisiae where two conditions (wild-type and a Δsnf2 mutant) were each sequenced in 48 replicate samples. Each replicate corresponds to a different wild-type (WT) or Δsnf2 yeast strain. Since these samples have little biological variation across replicates (estimated negative binomial dispersion of read counts was 0.01 (27), very close to the Poisson distribution), and present little or no problems in data processing (e.g. assigning reads to the correct gene or splicing isoform), they were used to design datasets also for assessing false positive rates of tools for the identification of differentially expressed genes across two conditions. Authors highlighted technical problems in five of the WT and four of the Δsnf2 samples, probably due to uneven priming during the PCR amplification step of library preparation, considering the resulting expression values unreliable and excluding them from downstream analyses. As in the original work (26), we excluded these samples from downstream analyses.
We transformed the original read counts (retrieved from ENA archive project PRJEB5348 https://www.ebi.ac.uk/ena/data/view/PRJEB5348) to TPMs following the formula shown in the previous section, employing for gene exon sizes the yeast gene annotation sacCer3 (31). We then generated different datasets, consisting of two or more groups, each composed of two or more replicates sampled at random from the WT samples. The number of conditions we simulated ranged from 2 to 6, each with a number of replicates ranging from 2 to 5. For each condition/replicate number combination we built 1000 different datasets, each one with a different combination of samples selected at random. We ran RNentropy on each dataset, considering a gene to be significantly over-expressed if it passed the global test with a corrected P-value threshold of 0.01, and all the replicates of at least one condition passed the local test with P-value <0.01 with all the expression values greater than the average (over-expressed) or lower (under-expressed).
The results are summarized in Table 1. Since all genes found to be over- or under-expressed are to be considered false positives, the table shows the false positive rate, computed as the number of genes found to be either over- or under-expressed divided by the overall number of local tests performed on genes. The theoretical false positive rate of our method (0.01) is very close to the empirical one for two replicates, and the latter is <0.01 for three or more replicates in any number of conditions. That is, the more replicates are available the more conservative the method is: this is a feature to be expected given the way the replicates are processed.
Table 1. Results on the yeast 48 WT sample dataset.
| Number of conditions | ||||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||
| RNentropy | 2 Reps | 0.0104 | 0.0110 | 0.0103 | 0.0106 | 0.0104 |
| 3 Reps | 0.0055 | 0.0041 | 0.0035 | 0.0033 | 0.0032 | |
| >3 Reps | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| edgeR | ||||||
| 2 Reps | 0.0083 | 0.0099 | 0.0074 | 0.0078 | 0.0077 | |
| 3 Reps | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |
| >3 Reps | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |
| DESeq2 | ||||||
| 2 Reps | 0.0120 | 0.0099 | 0.0092 | 0.0090 | 0.0089 | |
| 3 Reps | 0.0085 | 0.0074 | 0.0035 | 0.0057 | 0.0035 | |
| >3 Reps | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
False positive rates (number of over- and under-expressed genes passing both the global and the local test of RNentropy, divided by the number of tests performed) for each combination of number of conditions (columns) and replicates (rows). False positive rates for each combination of number of conditions (columns) and replicates (rows) derived from edgeR and DESeq2. FDR threshold for all methods was set to 0.01.
We ran edgeR and DESeq2 on the same datasets, starting from the original read counts, both with a corrected P-value (FDR) threshold of 0.01 (see Supplementary Material). The two-condition comparisons with two replicates confirmed the good performance previously reported (26), with an observed false positive rate around 0.008 for edgeR and 0.01 for DESeq2, very close to the ones of RNentropy. On three or more conditions compared once again the respective FPRs were lower than theoretical estimate of 0.01 either by employing the ANOVA-like tests, the ‘local’ comparisons of one condition against the others, or a combination of both. Increasing the number of replicates had the effect of a significant drop of in false positives for both (more visibly so for edgeR), similar to the one observed with RNentropy, for any number of simulated conditions. All in all, thus, the performance of RNentropy is in line with two of the best performing methods on the same benchmark dataset (26), with an empirical false positive rate well below the threshold of 0.01 with three or more replicates. We then repeated the test with RNentropy by replacing TPM expression values with TMM-normalized counts, with no relevant difference in the false positive rates detected (data not shown).
In order to have a more complete picture of the overall variability of this dataset, we also ran RNentropy with the same parameters used before on all the 48 WT samples, and then on the Δsnf2 dataset (thus including in both also the ‘bad’ samples excluded before). Each sample was processed as a separate condition (hence, 48 conditions in both datasets, with no replicates). The number of genes passing the global test and local test was quite high, more of the 5% of the total. However, we observed that the distribution of the number of over-expressed genes across all the samples was not uniform (which would be an indicator of a problem in the statistical testing) but rather, as shown in Figure 1 for TPM normalized values, skewed towards a few samples with a very high number of genes significantly differing from the others (see some examples in Supplementary Figure S1): these samples correspond to those originally identified as unreliable replicates (in red in the histograms). Notice also how the variability seems to be higher in WT samples, with also replicates not previously considered ‘bad’ (like number 17) with a higher fraction of over-expressed genes. This result points to a further application of our method, that is, the identification of one or more samples with a significant deviation from the others in cases where little or no difference should be expected. Point-wise mutual information is also able to highlight problematic samples, since those yielding the highest numbers of over-expressed genes at the same time show to be anti-correlated with respect to all the other samples, as shown in Supplementary Figure S2.
Figure 1.

False positive rates (fraction of over-expressed genes passing the global and local test of RNentropy in each of the samples) split into the different samples in the comparison of (A) the 48 wild-type yeast replicates and (B) the 48 mutant yeast replicates. Theoretical false positive rate was 0.01.
Human individual samples from six tissues
RNA samples were taken from six different tissues (brain, liver, lung, striated muscle, kidney, heart) from three male individuals. They have already been employed to study RNA editing (24) and the correlation of the expression of mitochondrial genes with mitochondrial DNA abundance (25). The three sample IDs are S7, S12 and S13, and all sample and sequencing details are reported in Supplementary Materials. Each sample was sequenced in three technical replicates, for an overall number of 54 expression profiles. For the quantification of expression levels, each sample was processed using the Rsem software package (3) with default parameters. As reference transcriptome, we employed the UCSC human gene annotation (version 2013–06-14, genome assembly hg19). The annotation comprises 78 829 transcripts assigned to 28 452 different genes. Genes annotated on the mitochondrion or on ‘hap’ chromosomes were not considered, and the respective read counts excluded from downstream normalizations and analyses. We employed both the transcripts per million (TPM) expression values output by Rsem, or the read per million counts (CPM), as output by edgeR after TMM normalization of the original raw counts returned by Rsem. In both cases, we processed the resulting expression values by computing the global and local sample specificity tests on each gene with global (adjusted) and local P-value thresholds of 0.01. The complete results are available as Supplementary Tables S1-S3.
Using TPM values
With TPM values the global sample specificity test returned 14 624 genes of the 28 434 considered as showing an expression pattern with a significant deviation from the uniform distribution, a fraction greater than half of the genes if restricted to the genes showing evidence of transcription in at least one sample. We then deemed a gene that passed this initial test to be over- (under-) expressed in a given individual-specific tissue sample if it passed the local sample specificity test in all the three replicates for the sample, with expression values higher than (or lower than, respectively) the average across all the samples.
The tissue with the highest number genes over-expressed in at least one individual was brain, with more than 6000 genes, followed by heart (∼4000). However, we had only 4578 cases of genes resulting over-expressed in a tissue in all the three individuals, as shown in Table 2a and Figure 2. The trend towards individual specificity changes according to the tissue, with muscle having 40% of the genes over-expressed in all the three individuals, while vice versa heart, kidney and liver have the large majority of genes (∼50%) with expression biased in only one individual. Supplementary Figure S3 confirms this fact, showing how the distribution of gene expression across the three individuals in each of the tissues studied is highly polarized towards one or two of the individuals, with visible differences for each tissue. The different degrees of correlation among the individual transcriptomes are also nicely summarized by the respective point-wise mutual information values, as shown in Supplementary Figure S4.
Table 2. (a) Number of genes in each of the six tissues of the 18 human tissue sample dataset passing the global and local specificity tests, and considered to be over-expressed in one, two and three individuals. Expression values were defined as transcripts per million (TPM). On each tissue, enrichment for over-expressed genes in the UP_TISSUE DAVID analysis (32) is reported on the bottom row, with the tissue yielding the lowest P-value. (b) Percentage of tissue-specific genes, according to the Human Protein Atlas (33), found to be over-expressed by RNentropy, according to the number of individuals. (c) Number of genes over-expressed in each of the six tissues when the nine samples from the different individuals were processed as replicates of the same condition.
| (a) | ||||||
| IND/Tissue | Brain | Heart | Kidney | Liver | Lung | Muscle |
| In 1 | 2487 (41%) | 1921 (48%) | 1358 (51%) | 924 (48%) | 1453 (57%) | 731 (35%) |
| In 2 | 2143 (35%) | 1406 (35%) | 828 (31%) | 332 (17%) | 578 (23%) | 521 (25%) |
| In 3 | 1487 (24%) | 643 (16%) | 460 (17%) | 653 (34%) | 527 (20%) | 808 (39%) |
| Total | 6117 | 3970 | 2646 | 1909 | 2558 | 2060 |
| UP_TISSUE (P-value) | Brain (<10−178) | Heart (<10−73) | Kidney (<10−72) | Liver (<10−225) | Lung (<10−23) | Sk. muscle (<10−131) |
| (b) | ||||||
| Brain | Heart | Kidney | Liver | Lung | Muscle | |
| Genes over-expressed for HPA | 1375 | 195 | 306 | 409 | 174 | 307 |
| Over-expressed for RNentropy: | ||||||
| in any # of individuals (%) | 1256 (91%) | 168 (86%) | 267 (87%) | 392 (96%) | 131 (75%) | 280 (91%) |
| in 1 individual (%) | 141 (10%) | 20 (10%) | 28 (9%) | 22 (5%) | 27 (16%) | 22 (7%) |
| in 2 individuals (%) | 509 (37%) | 54 (28%) | 177 (58%) | 49 (12%) | 40 (23%) | 26 (8%) |
| in 3 individuals (%) | 606 (44%) | 94 (48%) | 62 (20%) | 321 (78%) | 64 (37%) | 232 (76%) |
| Not DE in tissue (%) | 38 (3%) | 17 (9%) | 23 (8%) | 9 (2%) | 21 (12%) | 14 (5%) |
| Not DE in any tissue (%) | 81 (6%) | 10 (5%) | 16 (5%) | 8 (2%) | 22 (13%) | 13 (4%) |
| (c) | ||||||
| Brain | Heart | Kidney | Liver | Lung | Muscle | |
| 2470 | 1050 | 774 | 833 | 896 | 1031 | |
Figure 2.

(A) Number of genes reported as over-expressed by RNentropy in each of the 18 human tissue samples. (B) Number of genes found to be simultaneously over-expressed in a number of samples from 1 to 9. (C) Number of genes over-expressed in each of the six tissues by considering all individual samples from each tissue as replicates.
We confirmed tissue specificity of over-expressed genes, regardless of the number of individuals they were found in, by comparing our results with other resources for the identification or retrieval of tissue-specific genes.
The UP_TISSUE enrichment analysis available at DAVID (32) defines over-expressed genes for more than 100 different human tissues, and permits to analyze sets of genes for enrichment in over-expression for a tissue. Submitting genes reported by RNentropy as over-expressed in each tissue to this analysis returned the corresponding tissue as the most enriched one with very low P-values (Table 2a, bottom).
We then compared our over-expressed genes in each tissue with those defined as having expression ‘tissue-enriched’, ‘tissue-elevated’, or ‘group-enriched’ in the Human Protein Atlas (33), thus selecting genes considered either to be specific for the tissue or for a group of tissues comprising the one studied. As shown in Table 2b, the results of RNentropy are consistent with these definitions of tissue specificity, despite the fact that, also in this case, tissue specificity was defined on the basis of relative enrichment against a much larger sampling of 34 tissues. About 85–95% of the genes defined as tissue-specific in the Human Protein Atlas are also reported by RNentropy, with the sole exception of 75% for lung. Remarkably, as shown in the table, a non negligible fraction of these genes was found to be over-expressed by RNentropy not in every individual. Indeed, it appears that genes are usually considered to be ‘tissue-specific’ show significant changes of expression across different individuals.
Finally, GSEA analysis (34) on genes found over-expressed in each tissue by RNentropy reported enrichment in functional annotations consistent with the respective tissues, as also shown by their overlap with the GSEA results we obtained on tissue-specific genes for the Human Protein Atlas (summarized in Supplementary Table S2). In some tissues the most relevant functional categories remained unchanged if we included in the analysis genes over-expressed in one or two individuals out of three. For example, in brain, that showed the highest degree of individual-specific expression, virtually the same categories, all related to neurons (synapse, axon, etc.), were found to be enriched regardless of the number of individuals genes were over-expressed in. This is the effect of having genes belonging to the same family differentially expressed across the individuals. An example is the expression of genes of the synaptotagmin family, shown in Figure 3A. While the cumulative expression of the members of the family is quite balanced among the three individuals (rightmost column), there are significant biases for most of the genes, that tend to be split between those over-expressed in S13 against those in S7 and S12.
Figure 3.

(A) Expression in brain of genes of the synaptotagmin family, split into the contributions of the three individuals. While most of the genes show remarkable bias for the different individuals, the overall expression of the family is more balanced (rightmost column). (B) Average expression across the samples of genes belonging to the KEGG pathway ‘Cardiac Contraction’. (C) Distribution across the 18 human tissue samples of the expression of genes RPS4Y and FOSL2 (TPM, mean values over the three individual replicates of each sample).
In other tissues, however, enriched tissue-specific functional annotations could be clearly retrieved only for genes over-expressed in all individuals, as for example in muscle or heart. In the latter, the largest number of over-expressed genes was found in individual S7. Functional enrichment analysis on only these genes reported categories like ribosomal proteins, mitochondrion, and respiratory chain (Bonferroni corrected P-value < 10−10), but also cardiac muscle contraction (KEGG pathway enrichment P-value <10−15), heart contraction (<10−7), cardiomyophathy (<10−20), a functional annotation remarkably consistent, like the others, with the cause of death of individual S7, which was acute coronary syndrome (35,36). We then retrieved from the KEGG pathway database (37) the list of genes belonging to the pathway ‘cardiac contraction’, and applied RNentropy to their average expression values across the samples investigated (see Figure 3B). That is, we performed a sample specificity test not on single genes, but on the average expression of all the genes in the pathway, in order to determine whether it changed significantly across the tissues or the individuals. Genes belonging to the pathway resulted to be over-expressed in muscle samples of all the individuals, with similar expression average, but more interestingly over-expressed in heart only for S7.
We also obtained 3884 genes to be considered mainly ‘individual specific’, that is, with expression significantly increased in one or more tissues but always in the same individual. Remarkably, 582 of those genes had individual specificity as a key feature stronger than tissue specificity, that is, resulted to be over-expressed in more than one tissue but always in only one individual. Two examples are shown in Figure 3C. Gene RPS4Y is over-expressed by individual S13, while FOSL2 is over-expressed, in every tissue, in individual S12. As previously discussed this could be due to normal biological variation, but also to specific individual response to the conditions leading to the post-mortem sampling. Other large-scale studies on human gene expression highlighted individual variability (15), also correlating it with eQTLs: RNentropy however permits to study this phenomenon more in depth, with a detailed and simultaneous individual- or tissue-based assessment and classification of the specificity of the expression of each single gene.
Finally, a non negligible total of 1335 genes passing the global test could not be considered to be significantly over-expressed in any sample after the local test. Of these, 529 genes were reported to be under-expressed in at least one sample. Thus, RNentropy identified a small but relevant subset of genes characterized by the fact of being ‘repressed’ in one or more of the samples but not over-expressed in any. Interestingly, about 75% of these genes were found to be under-expressed in liver samples. The most relevant functional annotation for them was ‘cell cycle’ (P-value < 10−6), an observation consistent with the specific expression and/or activation patterns of cell cycle genes in liver cells compared to other cell types (38). Finally, we found most of the remaining genes, passing the global test but not the local, and thus neither over- or under-expressed in any sample, to have variable transcript levels across the replicates, hence not passing the test in all of them as required. Unsurprisingly, this latter group was formed mostly of small/micro/tRNA genes, included in the UCSC annotation we employed, but for which the measures obtained from a total RNA-Seq experiment like the one we performed cannot be considered reliable, with a high variability of the estimated levels across the replicates.
As a further test, we grouped samples by tissue, by considering all the samples of different individuals for the same tissue as biological replicates for the local test (Table 2c). The result of the global test was obviously the same, but since we required all individual samples for a tissue to pass the local test, the number of genes resulting to be over-expressed in at least one tissue was lower, and a larger number of genes was discarded because not passing the test in all the replicates. Overall, we obtained 6603 genes passing the global test and over-expressed in a tissue in all the individual samples. This number is, on the other hand, higher than the number of genes found to be over-expressed in the same tissue in all the three individuals processed separately. This is due to how our method performs the local test on replicates: by processing the individual samples of each tissue separately, a sample has to be over-expressed also considering samples of the same tissue from other individuals; while when samples are grouped by tissue, each sample has to be over-expressed only with respect to other samples from different tissues.
Using TMM normalized counts per million
The same analysis starting from counts per million after TMM normalization yielded very similar results. With respect to the results obtained with TPMs, the number of significant genes passing the global test is slightly lower (14 399), with 13 479 genes passing the local test and over-expressed in at least one sample (Table 3, Supplementary Table S3).
Table 3. Number of genes in each of the six tissues of the 18 human tissue sample dataset passing the global and local specificity tests, and considered to be over-expressed in one, two and three individuals.
| IND/Tissue | Brain | Heart | Kidney | Liver | Lung | Muscle |
|---|---|---|---|---|---|---|
| In 1 | 1428 (33%) | 1495 (58%) | 972 (42%) | 1455 (41%) | 1148 (41%) | 806 (22%) |
| In 2 | 1428 (33%) | 635 (25%) | 836 (36%) | 813 (23%) | 876 (31%) | 916 (26%) |
| In 3 | 1504 (34%) | 419 (16%) | 515 (22%) | 1310 (37%) | 766 (27%) | 1840 (51%) |
| Total | 4360 | 2549 | 2323 | 3578 | 2790 | 3562 |
Expression values analyzed were defined as counts per million, as output by edgeR after TMM normalization.
While in TPM values the original counts are normalized both by gene length and library size, the count per million values are normalized by TMM only with respect to the latter. Indeed, we can observe that genes resulting significant by using TPM are much shorter than those reported by TMM only, with a difference in median length greater than 2000 bp (Supplementary Figure S5). Concerning the specific conditions, the fraction of genes over-expressed in one, two or three individuals in each of the tissues remains remarkably similar for the two measures. That is, the high individual variability of expression can be captured by RNentropy in very similar proportions with both measures (compare percentages of Tables 2 and 3). Moreover, the 12 012 genes passing the global test with both, 35% are considered over-expressed in exactly the same conditions, and for a further 46% there are only one or two samples in which the genes are found over-expressed only with one measure (Supplementary Figures S6 and S7). Here we can observe that, in general, TMM-normalized values tend to yield less genes specific for brain and heart samples, and more for muscle and liver. In other words, the number of genes over-expressed in each of the different tissues tends to be more balanced after TMM normalization than with TPMs, another clear consequence of the normalization technique used to produce the values on which the test is applied. All in all, RNentropy seems to work with consistent results on both measures, and all relevant differences lie solely in the normalization technique employed. We consider further considerations on this aspect outside the scope of this work, referring the reader to specialized literature for comparisons and assessments of different approaches to the normalization of RNA-Seq data, in order to find the one most suitable for analyses aimed at finding differentially expressed genes.
Comparison with other methods
We applied to the original read counts output by Rsem both edgeR and DESeq2. By using the same FDR threshold of 0.01 in the respective ANOVA-like tests, both methods returned as ‘significant’ a much larger number of genes than our method, as shown in Figure 4. Of the 20 094 genes with at least one read mapped in at least one sample given as input to both, 19 632 and 19 391 genes had an associated FDR lower than 0.01 for edgeR and DESeq2, respectively. Thus, these ‘global’ indicators, which are equivalent to the initial selection provided by the global specificity test of RNentropy, provide little or no filtering of the data, since in both cases >96% of the genes showing evidence of transcription are reported as having significant changes of expression across the conditions studied. The results of the two methods after this step are consistent, with a negligible fraction of genes passing the test only for either, and in turn only a small subset of genes resulting to be variable only for RNentropy.
Figure 4.

Performance comparison of RNentropy, DESeq2, and edgeR on the 18 human tissue dataset. Clockwise from top left: (A) Overlap among genes passing the global tests of the three methods. (B) Number of genes found to be over-expressed in each sample by either RNentropy, edgeR, or both. (C) Number of genes found to be over-expressed in each sample by either RNentropy, DESeq2, or both. (D) Distribution of the standard deviation of TPM expression values of all genes found to be over-expressed in at least one tissue by DESeq2 and RNentropy (either or both), by DESeq2 only, by RNentropy only. Wilcoxon rank-sum test on ‘DESeq2 only’ and ‘RNentropy only’ distributions P-value < 2.2e–16.
In single comparisons between one individual tissue sample to the others, reproducing our local test, the two methods had strikingly different behavior (see Figure 4). We again employed for both methods the corrected p-value (FDR) threshold of 0.01, and, as with RNentropy, no explicit expression fold-ratio threshold. Compared to RNentropy, edgeR reported a much higher number of genes as over-expressed in at least one individual-specific tissue sample. We had more than 10 000 genes (half of those considered) over-expressed in at least one individual for brain, liver, lung and kidney, and more than 8000 for heart and muscle.
On the other hand, DESeq2 was much more conservative, with only 10 236 genes found to be over-expressed in at least one condition. We noticed however a sizable number of ‘outliers’ in several of the tests performed, that is, genes whose expression profile could not fit the general linear model and for which the statistical test could not be performed. The more the outliers, the lower the number of over-expressed genes found in a condition, with <200 genes found to be over-expressed in heart and kidney samples.
In general, nearly all genes reported by RNentropy to be over-expressed in at least one condition (using either TPM or TMM normalized CPM) are found to be over-expressed in the same condition(s) also by edgeR. Instead, while for DESeq2 virtually all the over-expressed genes in each condition were found also by edgeR, in some tissues and/or individuals there is a non negligible fraction of genes detected as over-expressed by either DESeq2 or RNentropy, but not by both. The most remarkable difference between the two sets of genes lies in the variance of the expression of the genes. As shown in Figure 4, genes found to be over-expressed only by DESeq2 in at least one condition have an expression variance (measured as TPMs) significantly lower than those found by RNentropy alone. We thus conjecture that when one condition with very little variability (three individual technical replicates of the same tissue sample) is compared to a highly variable set, DESeq2 claims the gene either to be an outlier, or to have a change of expression not significant because its high expression variance leads to an over-estimation of the FDR. Vice versa, where the overall variance is lower, DESeq2 picks as significant genes with little variation in expression (as log-fold ratio of the mean expression values)—not detected as significant by RNentropy.
We then ran both methods on the samples grouped by tissue. Also in this case, more than 19 000 genes passed the global tests for both, in other words, could be considered to be variable enough to be ‘tissue specific’ for some tissue. edgeR reported as over-expressed in a whole tissue about half of the genes that were so in at least one individual, with ∼5000 genes over-expressed in each tissue. Interestingly, DESeq2 in this case returned a much larger number of tissue over-expressed genes than those found by keeping the individual samples separate, also because the testing found just a very few outliers. That is, hundreds of genes that were not considered to be over-expressed in a tissue in any individual were reported to be over-expressed for the same tissue if all the individual samples were merged and considered replicates of the same condition. We conjecture again that in the latter case the method could obtain better and more balanced variance estimates for the statistical tests performed.
Deciding on the basis of comparisons of this kind which method can be considered more reliable clearly depends on what kind of result one expects. Some typical cases of the discrepancy in the results at the individual and tissue level we obtained are shown in Figure 5. Gene ABCC2 is reported to be over-expressed in all three individuals in liver, and in two individuals out of three in kidney by RNentropy, that at the tissue level considers it over-expressed only in liver, since in kidney it does not pass the local test in individual S12. For edgeR at the individual level it is over-expressed in the same five conditions as RNentropy, but at the tissue level edgeR considers it over-expressed also in kidney. Finally, at the individual level DESeq2 quite strikingly does not find it over-expressed in any sample, always reporting corrected P-values greater than 0.01, but considers it over-expressed in liver at the tissue level, that is, on the test performed by comparing the nine liver samples grouped together to the others. Gene AKAP5 is considered to be over-expressed in brain (by combining all nine samples in one condition) by both edgeR and DESeq2, despite the fact that in one individual it has an expression value much lower than other tissues (compare brain_S13 with lung_S13 and kidney_S12), but not by RNentropy for which it passes the test only for individuals S7 and S12.
Figure 5.

Expression values (as log(TPM+1), averaged over replicates of each sample) across the 18 human tissue samples of genes ABCC2 and AKAP5.
Finally, we applied to this dataset two of the most widely used measures introduced for the identification of tissue-specific genes (14), namely the tissue-specificity index (TSI, (39)) and the tau index (40). Given 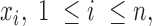 the expression values of a gene over n samples, they are defined as follows:
the expression values of a gene over n samples, they are defined as follows:
 |
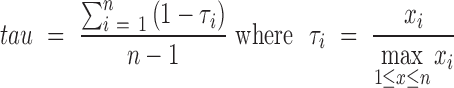 |
As in (14) we considered only genes with TPM > 1 in at least one sample, computed both indices using the average expression value of the three replicates of each sample, and defined a gene to be specific for (over-expressed in) at least one sample if the corresponding index value was greater than 0.85. For both indices, the number of genes found to be over-expressed in at least one sample was significantly lower than RNentropy, with 5627 genes returned by the tau index and, remarkably, only 168 for the TSI.
Indeed, being relative, these two measures were able to capture only those genes with the highest variation in one sample relative to the average, regardless of the estimated transcript levels. That is, the average TPM expression of the 5627 genes relevant for the tau index had median value 20.31. However, the TPM expression median of the 4046 genes found over-expressed also by RNentropy was 43.28, remarkably dropping to TPM 1.62 for the 1581 genes found significant only by the tau index. Also, the ratio between the maximum expression value across the samples and the average was always higher than 5 for the tau index, and had a median ∼7.5 either for genes found significant also by RNentropy or those significant only for the tau index. But, it dropped to 2.68 for those genes found to be significant and over-expressed in at least one sample only by RNentropy. With respect to these indices, thus, RNentropy captures less wide, but still significant, differences in expression. The complete distributions are available as Supplementary Figure S8.
We think that this case study fully shows the capabilities of our method, that provides a quick and comprehensive overview of the variation of gene expression across the tissues and individuals studied. The results are also consistent if the normalization strategy and the measure employed for the quantification of transcript levels are changed. Tools based on pairwise comparisons seem instead to suffer from the high variability of the data, that in turn impacts the estimate of the statistical parameters they are based on, and eventually they yield very different, and somehow inconsistent, results. Tissue specificity indices, finally, are able to capture only those genes with the widest relative variation of expression.
Identification of marker genes in seven mouse brain cell types
A very thorough characterization of the transcriptome of different brain cell types has been presented in (13). In particular, neurons, glia (astrocytes, oligodendrocytes, and microglia) and vascular cells (endothelial cells and pericytes) from mouse brain were investigated by RNA-Seq, and the results, summarizing expression at the gene and alternative splicing level, collected in a user-friendly database. Oligodendrocytes were split into three sub-types, that is, precursor (OPC), newly formed (NFO) and myelinating (MO) oligodendrocyte cells. Pericyte samples were considered by authors to suffer from contamination from other cells: they were thus excluded from downstream analyses for the identification of genes over-expressed in each cell type and the corresponding expression values not included in the database.
RNA-Seq was performed on poly-A RNAs pooled from different individuals, and samples from each cell type sequenced in two replicates. Expression values were defined as FPKMs, and replicates were employed to compute confidence intervals for the resulting values and exclude from downstream analyses genes with unreliable expression estimates. We remark that if replicates are kept separate this step is automatically performed by RNentropy in the computation of local p-values, as also shown in the human individual tissues analysis. Differential expression was calculated as the FPKM of a gene in a given cell type divided by the average FPKM of all other cell types, with the exception of each of the oligodendrocyte sub-types, that were compared only to the other non- oligodendrocyte cells. Thus, expression values and differentially expressed genes were available for seven cell types or sub-types. Differentially expressed genes, potential markers of each cell type, were further confirmed by performing the same analysis with oligonucleotide microarrays. A distinct set of candidate marker genes for each cell was eventually validated by qRT-PCR and in situ hybridization.
We applied RNentropy to the FPKM values of the seven cell types retrieved from the database, with corrected global and local P-value thresholds of 0.01. A sizable number of 6435 genes was reported to be over-expressed in at least one cell type, split among the different samples as summarized in Figure 6 and detailed in Supplementary Table S4. As a reference, we interrogated the database for the top 100 genes with FPKM > 20 specific for each cell type, ranking them by fold enrichment, that is, using the same criteria employed in the original work for the selection of marker genes (Figure 4 in (13)). The overlap between RNentropy over-expressed genes and the marker genes of the database is very high, equaling or close to the 100% of the top marker genes for all cell types (Figure 6, Supplementary Table S4).
Figure 6.

(A) Number of genes found to be over-expressed by RNentropy in each of the seven brain cell types, and number of the top 100 marker genes of each cell type defined according to (13) found to be over-expressed by RNentropy. (B) Heatmap of PMI scores computed on over-expressed genes shared by each pair of samples.
However, striking differences emerge if we consider genes found over-expressed in only one cell type by RNentropy, that is, what should be considered to be real marker genes. In oligodendrocyte cell sub-types (OPC, MO, and NFO cells), the overlap dropped to 50% of the top 100 genes for OPC, 15% for MO, and just 7% for NFO. The reason lies in the fact that for the three oligodendrocyte cell sub-types the ratio was computed only with respect to non- oligodendrocyte cells. Thus, a gene over-expressed in both MO and NFO cells according to this criterion could be reported to be a marker for both (e.g. gene Olig1, Figure 4 from (13)). The similarity between the transcriptome of these cell sub-types is also clear from the PMI heatmap (Figure 6B), with a very high correlation between MO and NFO cells, as well as from the distribution of the expression values of genes over-expressed in the two cell sub-types (Supplementary Figure S9). Further post-processing and manual curation of the results can easily eliminate redundant markers: on the other hand, RNentropy was able to report hundreds of genes exclusively over-expressed in each cell type (minimum 190 for MO cells), thus identifying true cell type specific markers in a more robust and straightforward way, and less dependent on subjective choices of expression thresholds and ratios.
Human liver RNA samples from six individuals
It is very common to have experimental settings in which several samples or conditions are studied together, with little or no a priori knowledge on the degree of similarity among the different expression profiles, nor a clear definition of which samples should be considered as ‘replicates’ and combined together in the comparisons. In this context, where in principle an ‘all against all’ comparison should be performed, it is common practice (and advised, see for example the tutorial for DESeq2 at Bioconductor, ‘Analyzing RNA-seq data with DESeq2’ at DESeq2 page at bioconductor.org. Version May 2017. Section ‘Can I run DESeq2 to contrast the levels of many groups?’) to perform preliminary explorative analyses using methods like clustering or principal component analysis, and from the results define ‘replicates’ and design the most relevant comparisons, usually between conditions that differ most, for finding genes best characterizing the transcriptomes and the respective differences. We show here how RNentropy can encompass both these aspects in a single analysis.
A simple but suitable example is the dataset composed of liver samples from six Nigerian individuals, three males and three females, introduced in (28), as part of a larger study on sex-specific expression in human and its conservation in two other primate species. The original work was focused on finding sex-specific genes, comparing the three male to the three female individual samples both in inter- and intra-species fashion, thus considering as ‘replicates’ samples from the same sex. On the other hand, all the samples could be viewed as ‘biological replicates’ of the same condition (human liver cells). We retrieved from Recount (41) the corresponding read counts for human, and TMM-normalized them edgeR. By performing a male vs female comparison, however, edgeR did not report any gene differentially expressed between males and females, at a FDR threshold of 0.01, neither by employing the glmQLFTest or the glmLRT test, with a few hundreds of genes with un-corrected P-value <0.01.
We processed this dataset by considering each individual as a separate sample and applying RNentropy to the counts per million values output by edgeR after normalization. The results (see Figure 7) reported 2189 genes with significant variation according to the global test and over-expressed in at least one sample, detecting once again relevant biological variation among different individuals. Analysis through the DAVID enrichment tool reported ‘Metabolism’ as the functional annotation better characterizing these genes (P-value < 10−10) that were found to be enriched also in several KEGG pathways related to liver function (metabolic pathways, fatty acid degradation, Bile secretion, and so on). According to the local test, however, RNentropy did find only one gene to be simultaneously over-expressed in all three female samples (and not in any male), and vice versa nine over-expressed in all three male samples and not in females. By limiting sex-specific expression to genes over-expressed in two samples out of three in either sex and none of the other, we detected 98 and 191 genes over-expressed in females and males samples only, respectively. This latter set contained all the genes found to be most variable by edgeR but not passing the FDR threshold. However, samples sharing the larger number of over-expressed genes are mixed, coming from both males and female individuals, with 552 genes found to be over-expressed simultaneously in at least one female and one male sample. Finally, ∼50% of the significant genes showed over-expression in a single individual. All in all, thus, individual variation, regardless of sex, seems to be a feature clearly dominating sex-specific expression. The results of point-wise mutual information analysis are shown in Figure 7C, where it can be clearly seen how one female sample (F3) does not seem to correlate well with any other sample, and how one male sample (M2) has a pattern of over-expressed genes more similar to two females (F1 and F2) rather than the two other males.
Figure 7.

(A) Number of genes over-expressed in each of the six male/female liver samples. ( B) Number of genes simultaneously expressed in x male samples (rows) and y female samples (columns). (C) Heatmap of point-wise mutual information scores computed on over-expressed genes shared between all pairs of samples.
This example shows how RNentropy can combine in a simple and intuitive way in a single run an explorative analysis giving an overview of the similarities among the transcriptomes studied, and at the same time a statistical framework for the identification of differentially expressed genes, without the need to pre-define groups of samples to be considered to be replicates of conditions to be compared. For example, if we wanted to perform an exhaustive analysis detecting genes differentially expressed between any combination of samples, we would have to design 41 pairwise comparisons, a number rising to 162 in case of eight samples, to 637 for ten and so on.
DISCUSSION
We presented here a novel methodology that introduces an entropy-based measure for the identification of biased and/or specific expression across multiple RNA-Seq conditions. The statistical tests based on this measure can provide a quick and effective overview of the variation of the transcriptional landscape in any number of samples from different tissues, cell types, time points, physiological or pathological conditions.
To show the flexibility and the reliability of our methodology, we presented the results of the analysis of different RNA samples. We employed 48 wild type and mutant yeast strains to assess false positive rates; six different human tissues from three individuals, identifying genes with tissue- and/or individual-specific expression patterns; seven mouse brain cell types, defining marker genes for each type; six human liver samples from three male and three female individuals, highlighting patterns of similarities in expression across different individuals.
Several studies comparing the performance of different RNA-Seq analysis tools over different benchmark datasets have appeared in literature in the last few years. Performances change according to the different case studies, and the general consensus is to choose the most suitable tool(s) according to the specific features of the datasets to be analyzed, and more importantly to use more than one tool in order to obtain more consistent results. We think that our method can be in turn very useful in cases like the multi-sample comparisons we presented, providing a quick and comprehensive overview of the transcriptional landscape of the conditions considered, together with the genes characterizing it by showing the most relevant variations of expression. Thus, it can be a very effective addition to the most commonly used pipelines for the comparison of transcriptome landscapes and the identification of differentially expressed genes. The examples we presented also point in a straightforward way to the possibility of applying RNentropy to single cell RNA-Seq experiments, which is indeed the focus of our current research.
DATA AVAILABILITY
RNentropy is available at www.beaconlab.it/RNentropy as a standalone software package (executable files for any LINUX 64 bit platform). C++ source code, and the implementation in the R language are also freely available for download.
All sequencing data produced in the present work are part of a series of sequencing DNA and RNA samples characterizing genomic variability at the individual level, and are available at the dbGaP database (https://www.ncbi.nlm.nih.gov/gap (42)) under accession number phs000870. All normalized read counts used in subsequent analyses are included in the RNentropy software package.
Supplementary Material
ACKNOWLEDGEMENTS
We warmly thank Alessio Valletti and Caterina Manzari for technical support in some experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Italian Ministero dell’Istruzione, Università e Ricerca (MIUR) [PRIN 2009, 2010 and 2012]; Consiglio Nazionale delle Ricerche: Flagship Project EPIGEN, Medicina Personalizzata and Aging Program 2012–2014. Funding for open access charge: Italian National Research Council (CNR) Flagship Project EPIGEN.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013; 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tarazona S., García-Alcalde F., Dopazo J., Ferrer A., Conesa A.. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011; 21:2213–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anders S., McCarthy D.J., Chen Y., Okoniewski M., Smyth G.K., Huber W., Robinson M.D.. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013; 8:1765–1786. [DOI] [PubMed] [Google Scholar]
- 11. Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., Szcześniak M.W., Gaffney D.J., Elo L.L., Zhang X. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dalman M.R., Deeter A., Nimishakavi G., Duan Z.H.. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinformatics. 2012; 13(Suppl. 2):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O’Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N. et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014; 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kryuchkova-Mostacci N., Robinson-Rechavi M.. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017; 18:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mele M., Ferreira P.G., Reverter F., DeLuca D.S., Monlong J., Sammeth M., Young T.R., Goldmann J.M., Pervouchine D.D., Sullivan T.J. et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015; 348:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B.. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628. [DOI] [PubMed] [Google Scholar]
- 17. Wang K., Phillips C.A., Rogers G.L., Barrenas F., Benson M., Langston M.A.. Differential Shannon entropy and differential coefficient of variation: alternatives and augmentations to differential expression in the search for disease-related genes. Int. J. Comput. Biol. Drug Des. 2014; 7:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sokal R.R., Rohlf F.J.. Biometry: The Principles and Practices of Statistics in Biological Research. 2012; 4th ednNY: Macmillan Education. [Google Scholar]
- 19. McDonald J.H. Handbook of Biological Statistics. 2015; 3rd ednBaltimore: Sparky House Publishing. [Google Scholar]
- 20. Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y.. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008; 18:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rapaport F., Khanin R., Liang Y., Pirun M., Krek A., Zumbo P., Mason C.E., Socci N.D., Betel D.. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013; 14:R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soneson C., Delorenzi M.. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013; 14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fano R.M., Hawkins D.. Transmission of information: a statistical theory of communications. Am. J. Phys. 1961; 29:793–794. [Google Scholar]
- 24. Picardi E., Manzari C., Mastropasqua F., Aiello I., D’Erchia A.M., Pesole G.. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci. Rep. 2015; 5:14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. D’Erchia A.M., Atlante A., Gadaleta G., Pavesi G., Chiara M., De Virgilio C., Manzari C., Mastropasqua F., Prazzoli G.M., Picardi E. et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion. 2015; 20:13–21. [DOI] [PubMed] [Google Scholar]
- 26. Schurch N.J., Schofield P., Gierliński M., Cole C., Sherstnev A., Singh V., Wrobel N., Gharbi K., Simpson G.G., Owen-Hughes T. et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?. RNA. 2016; 22:839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gierliński M., Cole C., Schofield P., Schurch N.J., Sherstnev A., Singh V., Wrobel N., Gharbi K., Simpson G., Owen-Hughes T. et al. Statistical models for RNA-seq data derived from a two-condition 48-replicate experiment. Bioinformatics. 2015; 31:3625–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blekhman R., Marioni J.C., Zumbo P., Stephens M., Gilad Y.. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 2010; 20:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo Y., Li C.-I., Ye F., Shyr Y.. Evaluation of read count based RNAseq analysis methods. BMC Genomics. 2013; 14(Suppl. 8):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seyednasrollah F., Laiho A., Elo L.L.. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief. Bioinform. 2015; 16:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Engel S.R., Dietrich F.S., Fisk D.G., Binkley G., Balakrishnan R., Costanzo M.C., Dwight S.S., Hitz B.C., Karra K., Nash R.S. et al. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda). 2014; 4:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dennis G. Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A.. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4:P3. [PubMed] [Google Scholar]
- 33. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. et al. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 34. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buja L.M. The pathobiology of acute coronary syndromes: clinical implications and central role of the mitochondria. Tex Hear. Inst J. 2013; 40:221–228. [PMC free article] [PubMed] [Google Scholar]
- 36. Machado N.G., Alves M.G., Carvalho R.A., Oliveira P.J.. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: can we close the box?. Cardiovasc. Toxicol. 2009; 9:211–227. [DOI] [PubMed] [Google Scholar]
- 37. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M.. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loyer P., Corlu A., Desdouets C.. Regulation of the hepatocyte cell cycle: signaling pathways and protein kinases. Int. J. Hepatol. 2012; 2012:592354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Julien P., Brawand D., Soumillon M., Necsulea A., Liechti A., Schütz F., Daish T., Grützner F., Kaessmann H.. Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS Biol. 2012; 10:e1001328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yanai I., Benjamin H., Shmoish M., Chalifa-Caspi V., Shklar M., Ophir R., Bar-Even A., Horn-Saban S., Safran M., Domany E. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005; 21:650–659. [DOI] [PubMed] [Google Scholar]
- 41. Frazee A.C., Langmead B., Leek J.T.. ReCount: A multi-experiment resource of analysis-ready RNA-seq gene count datasets. BMC Bioinformatics. 2011; 12:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tryka K.A., Hao L., Sturcke A., Jin Y., Wang Z.Y., Ziyabari L., Lee M., Popova N., Sharopova N., Kimura M. et al. NCBI’s database of genotypes and phenotypes: DbGaP. Nucleic Acids Res. 2014; 42:D975–D979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNentropy is available at www.beaconlab.it/RNentropy as a standalone software package (executable files for any LINUX 64 bit platform). C++ source code, and the implementation in the R language are also freely available for download.
All sequencing data produced in the present work are part of a series of sequencing DNA and RNA samples characterizing genomic variability at the individual level, and are available at the dbGaP database (https://www.ncbi.nlm.nih.gov/gap (42)) under accession number phs000870. All normalized read counts used in subsequent analyses are included in the RNentropy software package.