

Abstract
Asymmetric thiourea and squaramide catalysis provides access to synthetically versatile α-oxetanyl- and α-azetidinyl alkyl halides exhibiting a tetrasubstituted chiral carbon center with high yields and enantioselectivities. The products are readily transformed with negligible erosion of enantiopurity and excellent diastereoselectivity to a diverse group of multifunctional compounds including fluorooxindoles with two contiguous chirality centers, fluorinated heterocyclic spiranes and polyspiro compounds.
Graphical abstract
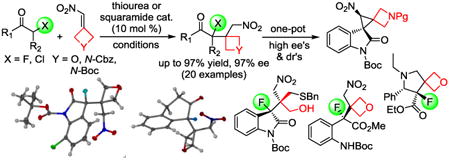
The widespread use of organofluorines and organochlorines in the health sciences continues to stimulate the search for methods that incorporate an oxindole moiety and other privileged pharmacophores into increasingly complex scaffolds. Chiral halogenated compounds displaying a tetrasubstituted stereogenic carbon center, a spiro motif or a proximate oxetane or azetidine ring are among the most challenging synthetic targets (Figure 1).1 The importance of nonracemizing α-fluorinated ketones, esters and amides has been highlighted with the introduction of fluorothalidomide2 and several potassium channel inhibitors, antibiotics and antimalarials carrying a fluorinated chiral carbon center surrounded by a carbonyl and other functionalities.3 In this regard, the placement of an oxetane ring adjacent to a C-F bond is of particular interest. The oxetane ring mimics the dipole and lone electron pair arrangement of a stretched carbonyl group but it excludes the metabolic vulnerability and propensity for α-deprotonation of aliphatic carbonyl compounds. As a result, 3,3-disubstituted oxetanes have become popular nonenolizable ketone surrogates in drug discovery and diversity oriented synthesis.4 Especially the favorable solubility properties, the high metabolic stability, and the remarkable hydrogen bond acceptor capabilities exceeding that of aliphatic ketones and esters have received attention.5 In analogy to oxetanes, the azetidine ring has emerged as a synthetically and medicinally invaluable building block in recent years.6
Figure 1.

Examples of bioactive organofluorines, oxindoles, spiranes and oxetanes.
Stereoselective synthesis with fluoroenolates is challenging and often requires careful enolate formation strategies to control side reactions.7 The medicinal utility of 3-substituted 3-fluorooxindoles,8 has nurtured the development of direct 3-alkyl and 3-aryloxindole fluorination protocols.9 The formation of 3-fluorooxindoles with two adjacent chirality centers, however, remains difficult and catalytic asymmetric C-C bond formations with 3-fluorooxindoles are relatively rare.10 Herein we describe a practical organocatalytic method that accomplishes these tasks with simultaneous incorporation of oxetane and azetidine rings into the 3-fluoro- and 3-chlorooxindole scaffold. This does not only provide unprecedented access to highly enantio- and diastereoenriched halogenated oxindoles exhibiting two contiguous centers of chirality, it also provides a diverse pool of multifunctional chiral compounds including azetidine and oxetane derived spiranes.
Because the 3-substituted oxindole ring is widely present in bioactive compounds and an intensively pursued structural motif,11 we began our search for an organocatalytic method that generates a chiral tertiary alkyl fluoride or chloride moiety adjacent to an oxetane or azetidine ring using N-Boc-3-fluorooxindole, 1a, and the strained nitroalkenes 2a-b as starting materials.12 After initial screening, we achieved quantitative conversion of 1a and 2a to the disubstituted oxetane 3a with 80% ee using 10 mol % of commercially available thiourea I and methyl tert-butyl ether as solvent (Table 1, entry 1). The introduction of the thioureas and squaramides II-IV, which were prepared in 4-5 steps,13 under the same conditions did not improve the enantioselectivity until we employed V14 which gave 3a in 91% ee albeit in lower yield (entries 2-5). Finally, we realized that 3a is produced in excellent yield and ee when the thiourea VI, which was available through a 5-step literature procedure,15 is used as catalyst (entry 6). Only minor modifications of our original protocol were necessary to quantitatively afford the azetidine 3b in 93% ee (entries 7-9). We then turned our attention to N-Boc-3-chlorooxindole, 4. This reaction appeared to be relatively slow and the 3-chloro-3-oxetanyloxindole 5a was initially formed with moderate ee's (entries 10-12). The yield and enantioselectivity improved to 94% and 72% ee, respectively, when the squaramide V was used under cryogenic conditions (entry 13). We therefore prepared VII, VIII and IX in three steps16 for further catalyst optimization and we were pleased to find that IX furnishes 5a in 90% ee at -15 °C (entries 14-16).
Table 1.
Optimization of the C-C bond formation with fluoro- or chlorooxindoles and strained nitroalkenes.a
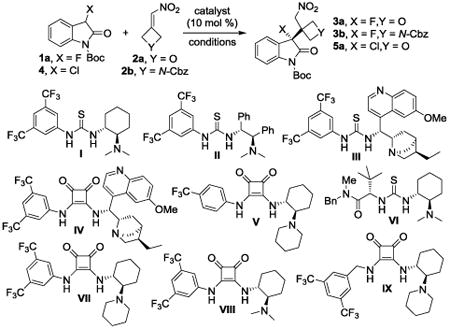
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | catalyst | oxindole | 2 | product | solvent | yieldb (%) | eec (%) |
| 1 | I | 1a | 2a | 3a | MTBE | 100 | 80 |
| 2 | II | 1a | 2a | 3a | MTBE | 100 | 24 |
| 3 | III | 1a | 2a | 3a | MTBE | 100 | 26 |
| 4 | IV | 1a | 2a | 3a | MTBE | 100 | 65 |
| 5 | V | 1a | 2a | 3a | MTBE | 73 | 91 |
| 6 | VI | 1a | 2a | 3a | MTBE | 100 | 94 |
| 7 | VI | 1a | 2b | 3b | MTBE | 64 | 93 |
| 8 | V | 1a | 2b | 3b | EtOAc | 73 | 92 |
| 9d | V | 1a | 2b | 3b | EtOAc | 98 | 93 |
| 10 | I | 4 | 2a | 5a | EtOAc | 81e | 53 |
| 11 | V | 4 | 2a | 5a | EtOAc | 73e | 63 |
| 12 | VI | 4 | 2a | 5a | EtOAc | 48e | 60 |
| 13f | V | 4 | 2a | 5a | EtOAc | 94e | 72 |
| 14g | VII | 4 | 2a | 5a | EtOAc | 52e | 63 |
| 15g | VIII | 4 | 2a | 5a | EtOAc | 45e | 63 |
| 16g | IX | 4 | 2a | 5a | EtOAc | 80e | 90 |
Reaction conditions: alkene 2 (0.15 mmol), organocatalyst I-IX (10 mol %), 1a or 4 (1.1 equiv), solvent (0.45 mL) at 25 °C.
Determined by 1H NMR.
Determined by chiral HPLC.
1.4 equivalent of 1a was used.
Isolated yield.
-10 °C.
-15 °C.
A variety of 3-fluoro and 3-chlorooxindoles were examined to evaluate the scope of this reaction (Table 2). As expected from our optimization efforts, N-Boc 3-fluoro-3-(3-(nitromethyl)oxetan-3-yl)-2-oxindole, 3a, was isolated in 90% yield and 94% ee (entry 1). We then applied the same protocol to several arylhalogenated 3-fluorooxindoles which gave the corresponding oxetanes 3c-f in 89-94% yield and 90-95% ee (entries 2-5). Many other functional groups are tolerated and electron-donating or withdrawing groups in various oxindole positions do not have a noteworthy effect on the yields and enantioselectivities. The products 3g-j carrying an ester, trifluoromethyl, nitro or methoxy group were obtained in very high yields and ee's (entries 6-9). All thiourea VI catalyzed asymmetric reactions with 3-(nitromethylene)oxetane, 2a, were complete within 7 hours. By contrast, fluorooxindole additions to the Cbz and Boc-protected azetidines 2b and c, which were performed in the presence of the squaramide V, required 24 hours. The corresponding azetidinyl fluorooxindoles 3k-m, however, were isolated in 90-97% yield and 92-93% ee (entries 10-13). Excellent results were also achieved with the chlorooxindole 4 and alkenes 2a-c using 10 mol % of IX as catalyst (entries 14-16).17 Slow evaporation of a concentrated solution of 3d in ethanol gave single crystals that were of sufficient quality for crystallographic determination of the absolute configuration. X-ray and chiral HPLC analysis of the same single crystal proved that the major enantiomer of the product formed from 7-chloro-3-fluorooxindole, 1d, and 2a has (R)-configuration when VI is used as catalyst (Scheme 1). During our reaction optimization studies we found that the catalysts V, VI and IX favor the same sense of asymmetric induction and we therefore assume that the reaction generally proceeds via the transition state depicted in Scheme 1.18,19
Table 2.
Organocatalytic asymmetric synthesis of α-oxetanyl and α-azetidinyl 3-halooxindoles.a
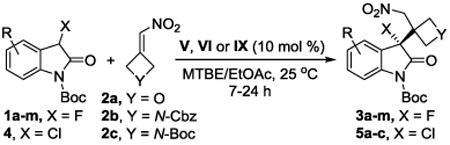
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | X/R | Y | catalyst | product | yielda (%) | eeb (%) |
| 1c | F/H | O | VI | 3a | 90 | 94 |
| 2c | F/6-Cl | O | VI | 3c | 94 | 90 |
| 3c | F/7-Cl | O | VI | 3d | 89 | 94 |
| 4c | F/5-F | O | VI | 3e | 91 | 95 |
| 5c | F/5-Br | O | VI | 3f | 92 | 95 |
| 6c | F/6-CO2Me | O | VI | 3g | 85 | 94 |
| 7c | F/6-CF3 | O | VI | 3h | 97 | 96 |
| 8c | F/5-NO2 | O | VI | 3i | 86 | 93 |
| 9c | F/5-OMe | O | VI | 3j | 93 | 94 |
| 10d | F/H | N-Boc | V | 3k | 97 | 93 |
| 11d | F/5-Br | N-Boc | V | 3l | 93 | 92 |
| 12d | F/H | N-Cbz | V | 3b | 92 | 92 |
| 13d | F/6-CO2Me | N-Cbz | V | 3m | 90 | 92 |
| 14d,e | Cl/H | O | IX | 5a | 80 | 90 |
| 15d,e | Cl/H | N-Boc | IX | 5b | 90 | 97 |
| 16d,e | Cl/H | N-Cbz | IX | 5c | 86 | 96 |
Isolated yield.
Determined by chiral HPLC.
MTBE.
EtOAc.
-15 °C.
The sense of asymmetric induction is based on the crystallographic analysis of 3d.
Scheme 1.
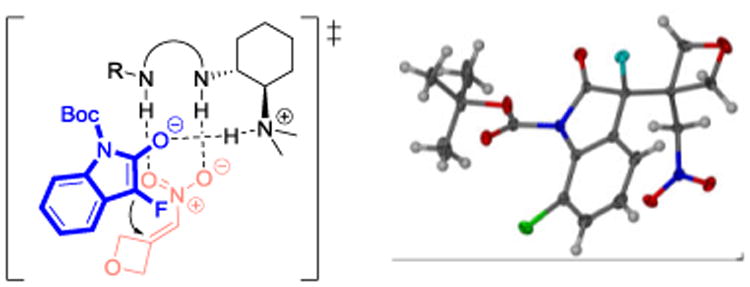
Proposed transition state model and crystal structure of (R)-3d.
We also explored the possibility of organocatalysis using acyclic α-fluoro ketones and the oxetanyl and azetidinyl alkenes 2a-c. Preliminary results with 1-fluoro-2-tetralone, 6, and ethyl 2-fluoro-3-oxo-3-phenylpropanoate, 7, using our standard reaction conditions were promising but additional method development was necessary. We therefore reinvestigated the performance of ten thiourea and squaramide catalysts under otherwise similar conditions (see Supporting Information). As shown in Scheme 2, we were able to produce the oxetane 8a in 84% yield and 87% ee using 10 mol % of squaramide V. The addition of the acyclic fluoro ketoester 7 to 2a-c occurred with excellent yields and only slightly reduced asymmetric induction. These results underscore the general usefulness of our method. The organofluorines 8a-d are versatile building blocks that possess a synthetically challenging fluorinated tetrasubstituted chiral carbon center surrounded by one or more than one carbonyl group and other functionalities.20 Evaporation of a concentrated solution of 8a in CH2Cl2 gave a single crystal suitable for X-ray analysis which confirmed the (R)-configuration.
We then sought to evaluate the synthetic value of the multifunctional compounds 3, 5 and 8 (Scheme 3). We observed that 3a is smoothly converted into the α-aryl-α-fluoro-α-oxetanyl ester 9, a relatively small compound with very high functional group density.21 We then turned our attention to the possibility of replacing the oxindole chloride via intramolecular nitronate substitution which would afford a challenging trispiro arrangement having a central cyclopropane ring. Spirooxindoles with an all-carbon quaternary chiral center exhibit a privileged motif frequently encountered in nature.22 In recent years, there has been growing emphasis on the synthesis of optically active spirocyclopropyl oxindoles.23 When we treated the chlorooxindole 5b with base we observed quantitative conversion to the trispiro compound 10. Chiral HPLC analysis confirmed that this cyclopropanation occurs without any sign of racemization and we isolated 10 in 94% yield, 95% ee and more than 99:1 dr. Essentially the same reaction occurred when we employed the CBz-protected azetidine 5c and we obtained diastereomerically pure 11 in 90% yield and 95% ee. The syn-configuration at the cyclopropane ring was determined by COSY and NOESY experiments (see SI).24
Scheme 3.

Synthetic utility of oxetanyl and azetidinyl tertiary alkyl halides.
The presence of carbonyl and nitro groups in the fluoro ketoesters 8 suggested to us that preparation of another type of spiro compounds should be possible. We therefore decided to investigate the possibility of a reductive ring closure with the fluorinated ketoester 8b. After screening various reduction methods we obtained pyrrolidine 12, a spiro compound consisting of two different heterocycles, in 72% yield, with high diastereoselectivity and only slightly diminished ee. The cyclization favors the anti-pyrrolidine ring with 33:1 dr which was confirmed by 19F NMR analysis (see SI). Compound 12 belongs to the class of β-fluoro-β-prolines and fluorinated pyrrolidines which are of medicinal interest.25 Altogether, the cyclizations of 5b, 5c and 8b afford efficient access to a variety of multisubstituted spirocyclic oxetanes and azetidines which have received increasing attention in drug discovery programs targeting rigid substructures of low molecular weight and high functionally density. The oxetane ring provides additional synthetic opportunities.26 Since several groups have discovered oxetane desymmetrizations in recent years,27 we decided to highlight the general utility of our oxetanyl fluorides with a modified Lewis acid promoted ring opening protocol.28 After some reaction optimization using benzylthiol as the nucleophile we obtained (R,S)-13 in 68% yield, 95% ee and with >99:1 dr from (R)-3a. This reaction establishes an all-carbon quaternary chiral center with three N-, O- and S-substituted alkyl moieties next to the tetrasubstituted halogenated stereocenter. The relative stereochemistry was confirmed by NOESY and 1H19F HOESY experiments (SI).
In summary, we have developed a highly enantioselective organocatalytic method that affords a wide range of multifunctional α-oxetanyl- and α-azetidinyl alkyl fluorides and chlorides exhibiting a tetrasubstituted chiral carbon center in high yields. The thiourea or squaramide catalyzed reaction is broad in scope and readily available halogenated oxindoles, acyclic and cyclic ketones or keto esters can be used. This work provides unprecedented access to a diverse pool of complex compounds including fluorooxindoles with two contiguous chirality centers and heterocyclic spiranes.
Supplementary Material
Scheme 2.
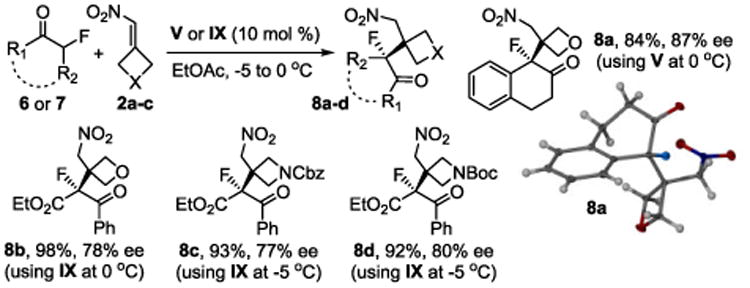
Cyclic and acyclic fluoroenolate additions.
Acknowledgments
We gratefully acknowledge financial support from NIH (GM106260).
Footnotes
Supporting Information: Experimental details, characterization data including NMR spectra and HPLC chromatograms. The Supporting Information is available free of charge on the ACS Publications website.
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.a) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Chem Rev. 2016;116:422. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]; b) Jiang T, Kuhen KL, Wolff K, Yin H, Bieza K, Caldwell J, Bursulaya B, Wu TYH, He Y. Bioorg Med Chem Lett. 2006;16:2105. doi: 10.1016/j.bmcl.2006.01.073. [DOI] [PubMed] [Google Scholar]; c) Fernandes PD, Zardo RS, Figueiredo GS, Silva BV, Pinto AC. Life Sci. 2014;116:16. doi: 10.1016/j.lfs.2014.08.019. [DOI] [PubMed] [Google Scholar]; d) Atkinson B, Beattie D, Culshaw AJ, Dale J, Devereux NJ, Mckenna J. Cyclohexyl amide derivatives as CRF receptor antagonists. 2011;WO2011092293A2 [Google Scholar]; e) Fang QK, Spear KL, Campbell U. Heterocyclic Compounds and Methods of Use Thereof. 2015;US20150336928 [Google Scholar]; f) Burkhard JA, Wuitschik G, Planscher JM, Rogers-Evans M, Carreira EM. Org Lett. 2013;15:4312. doi: 10.1021/ol401705a. [DOI] [PubMed] [Google Scholar]
- 2.a) Takeuchi Y, Shiragami T, Kimura K, Suzuki E, Shibata N. Org Lett. 1999;1:1571. [Google Scholar]; b) Man HW, Corral LG, Stirling DI, Muller GW. Bioorg Med Chem Lett. 2003;13:3415. doi: 10.1016/s0960-894x(03)00778-9. [DOI] [PubMed] [Google Scholar]; c) Tokunaga E, Akiyama H, Soloshonok VA, Inoue Y, Hara H, Shibata N. PLoS ONE. 2017;12:e0182152. doi: 10.1371/journal.pone.0182152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Abad A, Agulló C, Cuñat AC, González-Coloma A, Pardo D. Eur J Org Chem. 2010:2182. [Google Scholar]; b) Yi WB, Huang X, Cai C, Zhang W. Green Chem. 2012;14:3185. [Google Scholar]; c) You Y, Zhang L, Luo S. Chem Sci. 2017;8:621. doi: 10.1039/c6sc03109a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Burkhard JA, Wuitschik G, Rogers-Evans M, Müller K, Carreira EM. Angew Chem Int Ed. 2010;49:9052. doi: 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]; b) Beadle JD, Knuhtsen A, Hoose A, Raubo P, Jamieson AG, Shipman M. Org Lett. 2017;19:3303. doi: 10.1021/acs.orglett.7b01466. [DOI] [PubMed] [Google Scholar]
- 5.Bull JA, Croft RA, Davis OA, Doran R, Morgan KF. Chem Rev. 2016;116:12150. doi: 10.1021/acs.chemrev.6b00274. [DOI] [PubMed] [Google Scholar]
- 6.a) Maetani M, Zoller J, Melillo B, Verho O, Kato N, Pu J, Comer E, Schreiber SL. J Am Chem Soc. 2017;139:11300. doi: 10.1021/jacs.7b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reidl TW, Son J, Wink DJ, Anderson LL. Angew Chem Int Ed. 2017;56:11579. doi: 10.1002/anie.201705681. [DOI] [PubMed] [Google Scholar]
- 7.a) Saidalimu I, Fang X, He XP, Liang J, Yang XY, Wu FH. Angew Chem Int Ed. 2013;52:5566. doi: 10.1002/anie.201301443. [DOI] [PubMed] [Google Scholar]; b) Zhang P, Wolf C. Angew Chem Int Ed. 2013;52:7869. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]; c) Xie C, Wu L, Han J, Soloshonok VA, Pan Y. Angew Chem Int Ed. 2015;54:6019. doi: 10.1002/anie.201500908. [DOI] [PubMed] [Google Scholar]; d) Xie C, Dai Y, Mei H, Han J, Soloshonok VA, Pan Y. Chem Commun. 2015;51:9149. doi: 10.1039/c5cc02256h. [DOI] [PubMed] [Google Scholar]; e) Saadi J, Wennemers H. Nat Chem. 2016;8:276. doi: 10.1038/nchem.2437. [DOI] [PubMed] [Google Scholar]; f) Balaraman K, Moskowitz M, Liu Y, Wolf C. Synthesis. 2016;48:2376. doi: 10.1055/s-0035-1561433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hewawasam P, Gribkoff VK, Pendri Y, Dworetzky SI, Meanwell NA, Martinez E, Boissard CG, Post-Munson DJ, Trojnacki JT, Yeleswaram K, Pajor LM, Knipe J, Gao Q, Perrone R, Starrett JE., Jr Bioorg Med Chem Lett. 2002;12:1023. doi: 10.1016/s0960-894x(02)00101-4. [DOI] [PubMed] [Google Scholar]
- 9.Selected examples: Ishimaru T, Shibata N, Horikawa T, Yasuda N, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2008;47:4157. doi: 10.1002/anie.200800717.; Wu L, Falivene L, Drinkel E, Grant S, Linden A, Cavallo L, Dorta R. Angew Chem Int Ed. 2012;51:2870. doi: 10.1002/anie.201200206. [DOI] [PubMed] [Google Scholar]
- 10.a) Xie C, Zhang L, Sha W, Soloshonok VA, Han J, Pan Y. Org Lett. 2016;18:3270. doi: 10.1021/acs.orglett.6b01516. [DOI] [PubMed] [Google Scholar]; b) Balaraman K, Wolf C. Angew Chem, Int Ed. 2017;56:1390. doi: 10.1002/anie.201608752. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jin Y, Chen M, Ge S, Hartwig JF. Org Lett. 2017;19:1390. doi: 10.1021/acs.orglett.7b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Paladhi S, Park SY, Yang JW, Song CE. Org Lett. 2017;19:5336. doi: 10.1021/acs.orglett.7b02628. [DOI] [PubMed] [Google Scholar]
- 11.a) Biswas P, Paul S, Guin J. Angew Chem Int Ed. 2016;55:7756. doi: 10.1002/anie.201603809. [DOI] [PubMed] [Google Scholar]; b) Sankar MG, Garcia-Castro M, Golz C, Strohmann C, Kumar K. Angew Chem Int Ed. 2016;55:9709. doi: 10.1002/anie.201603936. [DOI] [PubMed] [Google Scholar]; c) Kong W, Wang Q, Zhu J. Angew Chem Int Ed. 2016;55:9714. doi: 10.1002/anie.201603950. [DOI] [PubMed] [Google Scholar]
- 12.For asymmetric cycloadditions, C-S bond formation via 1,4-thioacid addition and pyrazolone additions with strained nitroalkenes see: Phelan JP, Patel EJ, Ellman JA. Angew Chem Int Ed. 2014;53:11329. doi: 10.1002/anie.201406971.; b) Monleon A, Glaus F, Vergura S, Jorgensen KA. Angew Chem Int Ed. 2016;55:2478. doi: 10.1002/anie.201510731. [DOI] [PubMed] [Google Scholar]; c) Phelan JP, Ellman JA. Adv Synth Catal. 2016;358:1713. [Google Scholar]; Asymmetric fluorooxindole addition to unstrained vinyl sulfones and nitroalkenes: Dou X, Lou Y. Org Biomol Chem. 2013;11:5217. doi: 10.1039/c3ob41267a.
- 13.a) Okino T, Hoashi Y, Furukawa T, Xu X, Takemoto Y. J Am Chem Soc. 2005;127:119. doi: 10.1021/ja044370p. [DOI] [PubMed] [Google Scholar]; b) Bae HY, Some S, Lee Jae H, Kim JY, Song MJ, Lee S, Zhang YJ, Song CE. Adv Synth Catal. 2011;353:3196. [Google Scholar]
- 14.Zhu Y, Malerich JP, Rawal VH. Angew Chem Int Ed. 2010;49:153. doi: 10.1002/anie.200904779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puglisi A, Benaglia M, Annunziata R, Rossi D. Tetrahedron: Asymmetry. 2008;19:2258. [Google Scholar]
- 16.a) Qian Y, Ma G, Lv A, Zhu HL, Zhao J, Rawal VH. Chem Commun. 2010;46:3004. doi: 10.1039/b922120d. [DOI] [PubMed] [Google Scholar]; b) Baran R, Veverková E, Škvorcova A, Šebesta R. Org Biomol Chem. 2013;11:7705. doi: 10.1039/c3ob41709c. [DOI] [PubMed] [Google Scholar]
- 17.For asymmetric oxindole chlorination, see: Zheng W, Zhang Z, Kaplan MJ, Antilla JC. J Am Chem Soc. 2011;133:3339. doi: 10.1021/ja109824x.
- 18.CCDC numbers 1584426 [(R)-3d] and 1584425 [(R)-8] contain the supplementary X-ray data. These can be obtained free of charge from the Cambridge Crystallographic Data Centre.
- 19.Reiter C, López-Molina S, Schmid B, Neiss C, Görling A, Tsogoeva SB. ChemCatChem. 2014;6:1324. [Google Scholar]
- 20.For a related tandem Michael addition/fluorination sequence, see: Wang L, Meng W, Zhu CL, Zheng Y, Nie J, Ma JA. Angew Chem Int Ed. 2011;50:9442. doi: 10.1002/anie.201104565.
- 21.Hamashima Y, Suzuki T, Taskano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164. doi: 10.1021/ja0513077. [DOI] [PubMed] [Google Scholar]
- 22.a) Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; b) Antonchick AP, Gerding-Reimers C, Catarinella M, Schürmann M, Preut H, Ziegler S, Rauh D, Waldmann H. Nature Chem. 2010;2:735. doi: 10.1038/nchem.730. [DOI] [PubMed] [Google Scholar]; c) Cheng D, Ishihara Y, Tan B, Barbas CF., III ACS Catal. 2014;4:743. [Google Scholar]
- 23.a) Dou X, Yao W, Zhou B, Lu Y. Chem Commun. 2013;49:9224. doi: 10.1039/c3cc45369c. [DOI] [PubMed] [Google Scholar]; b) Oseka M, Noole A, Zari S, Oeren M, Jarving I, Lopp M, Kangera T. Eur J Org Chem. 2014:3599. [Google Scholar]; c) Cao ZY, Zhou J. Org Chem Front. 2015;2:849. [Google Scholar]
- 24.Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Angew Chem. 2009;48:8037. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]
- 25.a) Yarmolchuk VS, Mykhailiuk PK, Komarov IV. Tet Lett. 2011;52:1300. [Google Scholar]; b) Hu XG, Hunter L. Beilstein J Org Chem. 2013;9:2696. doi: 10.3762/bjoc.9.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Ruider SA, Muller S, Carreira EM. Angew Chem Int Ed. 2013;52:11908. doi: 10.1002/anie.201306563. [DOI] [PubMed] [Google Scholar]; b) Yang W, Wang Z, Sun J. Angew Chem Int Ed. 2016;55:6954. doi: 10.1002/anie.201601844. [DOI] [PubMed] [Google Scholar]; c) White AR, Kozlowski RA, Tsai SC, Vanderwal CD. Angew Chem Int Ed. 2017;56:10525. doi: 10.1002/anie.201704119. [DOI] [PubMed] [Google Scholar]
- 27.a) Loy RN, Jacobsen EN. J Am Chem Soc. 2009;131:2786. doi: 10.1021/ja809176m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Z, Chen Z, Sun J. Angew Chem Int Ed. 2013;52:6685. doi: 10.1002/anie.201300188. [DOI] [PubMed] [Google Scholar]; c) Maji R, Champagne PA, Houk KN, Wheeler SE. ACS Catal. 2017;7:7332. [Google Scholar]
- 28.Xianming H, Kellog RM. Tetrahedron Asymm. 1995;6:1399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.