

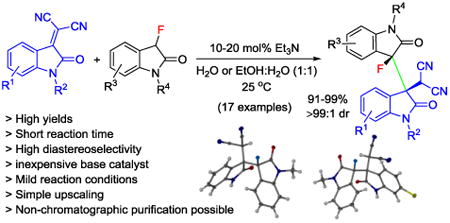
Abstract
A highly diastereoselective organocatalytic reaction for the synthesis of fluorinated 3,3′-bisindolines exhibiting adjacent tetrasubstituted carbon stereocenters is described. A broad variety of heterochiral bisindolines was prepared in 91-99% yield using 3-fluorooxindoles and isatylidene malononitriles in the presence of catalytic amounts of triethylamine in water or aqueous solution. The reaction can be upscaled without compromising yield and diastereoselectivity and the general usefulness of this method was demonstrated with various Michael acceptors and extended to aldol and Mannich reactions.
Keywords: organocatalysis, Michael addition, 3, 3′-bisindolines, organofluorines, green chemistry
Graphical abstract
The high impact of fluoroorganic chemistry in the materials and medicinal sciences continues to spark great interest in synthetic methods that provide practical access to complex fluorinated compounds.[1] The presence of fluorine often affects the lipophilicity, acidity, conformational bias, metabolic stability and other important properties of pharmaceuticals and agrochemicals. Similarly, the reactivity of fluorinated electrophiles and nucleophiles can vary significantly from that of the nonfluorinated analogues. The unique nucleophilicity, propensity to decomposition and unexpected side reactions of fluoroenolates complicate stereoselective carbon-carbon bond formation and this generally requires the development of new methods.[2] The construction of fluorinated tetrasubstituted carbon stereocenters with intermediate fluoroenolates remains particularly difficult.
The 3,3-disubstituted oxindole moiety is a frequently encountered substructure in natural products and drugs and has become a popular synthetic target.[3] To this end, the simultaneous incorporation of contiguous stereocenters has attracted increasing attention.[4] The discovery of the potassium ion channel modulator Maxipost[5] and other biologically active fluorooxindoles has stimulated the development of various fluorination protocols that utilize readily available 3-alkyl and 3-aryloxindoles as starting material.[6] The alternative approach based on C-C bond formation with 3-fluorooxindoles bears relatively unexplored synthetic potential.[7]
Despite the general demand and the progress mentioned above, the synthesis of complex 3-fluorooxindoles exhibiting multiple functional groups and contiguous stereocenters often involves anhydrous conditions, inert atmosphere and low temperatures which increases energy consumption and cost. Encouraged by a report on the “on water” Michael addition of O-silyl difluoroenolates to isatylidene malonitriles,[8] we have developed an environmentally benign method that addresses these issues and produces consistently heterochiral 3,3′-bisindolines from fluorooxindoles using inexpensive Et3N as catalyst (Scheme 1). While the heterogeneous “on water” reaction is limited to preformed difluoroenoxysilanes used in excess and at elevated temperature, our method utilizes stoichiometric amounts of monofluorinated oxindole as the nucleophile precursor at room temperature in water or aqueous ethanol solutions. The reaction affords two adjacent tetrasubstituted carbon stereocenters in almost quantitative yields and with excellent diastereoselectivity. The 3,3′-bisindoline scaffold formed under these conditions is a common structural motif in dimeric alkaloids.[9]
Scheme 1.

Fluoroenolate Michael additions to isatylidene malononitriles.
We began our search for a method that yields the targeted 3,3′-bisindoline scaffold under mild homogeneous conditions using commonly used organic solvents and base additives. After initial screening we found that the reaction between isatylidene malononitrile, 1a, and 3-fluorooxindole, 2a, in the presence of 10 mol% of Et3N as catalyst in THF at 25 °C proceeded smoothly and gave exclusively the 3,3′-bisindoline 3 in 99% yield and with >99:1 diastereoselectivity (Table 1, entry 1). It Is noteworthy to mention that the reaction can easily be monitored by the disappearance of the dark red color of the isatylidene malononitrile derivatives as the 3,3′-bisindoline products are colorless. This initial result prompted us to further investigate the role of catalyst and solvent. As expected, formation of product 3 was not observed under strictly heterogeneous “on water” conditions in the absence of base (Table 1, entry 2). Further screening revealed that the reaction occurs in a variety of solvents including acetone, dichloromethane and ethanol (Table 1, entries 3-5). However, when acetone was used as solvent, 23% of 2-(2-oxo-3-(2-oxopropyl)indolin-3-yl)malononitrile along with 76% of 3,3′-bisindoline 3 was obtained due to competition of the Michael addition of acetone to 1a. Having found a remarkable solvent compatibility, we envisioned a green protocol and investigated water and aqueous solutions as solvent in the presence of 10 mol% of Et3N as catalyst. Fortunately, all these reactions yielded 3 in quantitative amounts and excellent diastereoselectivity (Table 1, entries 6-8). The addition of small amounts of the base allows partial dissolution of 1a and 2a in the form of its enolate in water. The reaction then occurs within 3 hours in the water phase rather than “on water”. The absence of any “on water” reaction acceleration is in agreement with the observation that the formation of 3 is even faster under perfectly homogeneous conditions when a water/ethanol mixture is used as solvent (compare entries 7 and 8). It is noteworthy that replacement of Et3N by inorganic bases such as NaHCO3, Na2CO3 or K2CO3 led to lower yield and longer reaction times (Table 1, entries 9-11). Changes in the catalyst loading only affected the reaction rate but did not compromise the stereoselectivity (entries 12 and 13).
Table 1.
Optimization of the Michael addition of 3-fluorooxindole 2a to the isatylidene malononitrile 1a.a
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Catalyst | Solvent | Time (h) | Yield (%)b | drc |
| 1 | Et3N | THF | 2 | 99 | >99 : 1 |
| 2 | No base | H2O | 12 | nr | -- |
| 3 | Et3N | Acetone | 1 | 76 | >99 : 1 |
| 4 | Et3N | CH2Cl2 | 2 | 93 | >99 : 1 |
| 5 | Et3N | EtOH | 1 | 97 | >99 : 1 |
| 6 | Et3N | THF:H2O (1:1) | 2 | 99 | >99 : 1 |
| 7 | Et3N | EtOH:H2O (1:1) | 2 | 98 | >99 : 1 |
| 8 | Et3N | H2O | 3 | 99 | >99 : 1 |
| 9d | NaHCO3 | H2O | 18 | 16 | >99 : 1 |
| 10 | Na2CO3 | H2O | 18 | 45 | >99 : 1 |
| 11 | K2CO3 | H2O | 18 | 78 | >99 : 1 |
| 12e | Et3N | H2O | 2 | 99 | >99 : 1 |
| 13f | Et3N | H2O | 7 | 81 | >99 : 1 |
Reaction conditions: 0.2 mmol of 1a and 0.2 mmol of 2a in 0.5 mL of solvent, 10 mol% of catalyst, 25 °C,
Isolated yield,
Determined by 1H and 19F NMR,
0.5 mL of sat. NaHCO3,
20 mol% of Et3N,
5 mol% of Et3N.
With the optimized reaction protocol in hand, we continued with exploring the substrate scope. 3-Fluorooxindoles containing a methyl or phenyl group at the oxindole nitrogen were treated with a series of isatylidene malononitriles under optimized reaction conditions. The 3,3′-bisindolines 3 and 4 were obtained in 94-99% yield and with >99:1 dr (Scheme 2). As expected, protection of the nitrogen atom in the isatylidene Michael acceptor does not affect the reaction outcome and we obtained 5 and 6 in 93-95%yield and with excellent stereoselectivity (>99:1 dr). More variations around the aryl ring in 1 revealed that electronically diverse acceptors containing 5-Me, 7-CF3, 5-F, 6-Cl, 5-NO2 or 5-OMe substituents are well tolerated. The corresponding 3,3′-bisindolines 7-12 were produced in very good yields (91-99%) and with unchanged diastereoselectivity. Interestingly, complete precipitation of N,N′-protected-3,3′-bisindolines from the homogenous reaction mixture allowed nonchromatographic product isolation which minimizes labor and solvent waste production.[10]
Scheme 2.

Organocatalytic synthesis of 3,3′-bisindolines 3-12 from 3-fluorooxindoles and isatylidene malononitriles. Reaction conditions: 0.2 mmol of isatylidene malononitrile and 0.2 mmol of 3-fluorooxindole in 0.5 mL of solvent, 10-20 mol% of catalyst, 25 °C. See SI for details.
The crystalline nature of the bisindolines greatly facilitates X-ray examination of the relative configuration of the oxindole dimers. Slow evaporation of solutions of compounds 3-6 and 10 in hexanes and ethylacetate gave single crystals suitable for crystallography which proved that all compounds investigated were present in the heterochiral form (Figure 1).[11]
Figure 1.
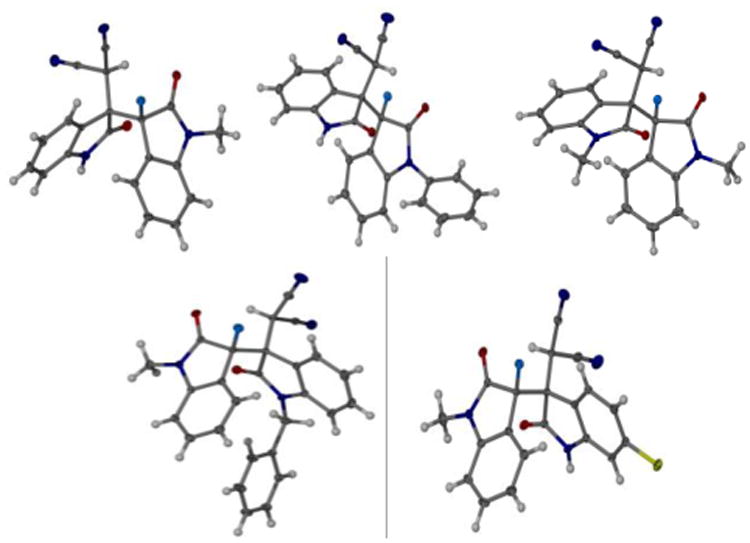
X-ray structures of the 3,3′-bisindolines 3-6 and 10 (top left to right bottom). All products are heterochiral. Only one enantiomer is shown for simplicity.
We then applied 3-fluorooxindoles carrying removable N-protecting groups such as benzyl, p-methoxyphenyl (PMP), p-benzyloxyphenyl (PBP) and diphenylmethyl in essentially the same protocol (Scheme 3). The corresponding products 13-18 were obtained in 91-99% yield and with excellent diastereoselectivity. Altogether, the presence of protecting groups either in the 3-fluorooxindole moiety or in the isatylidene malononitrile Michael acceptor is well tolerated and this can be useful for further selective synthetic modifications of the 3,3′-bisindolines. The reaction however, also proceeds smoothly in the absence of any protecting groups and we obtained 19 in 98% yield with >99:1 dr. Again, crystallographic analysis of single crystals obtained from 18 confirmed that the heterochiral dimer is consistently formed.
Scheme 3.
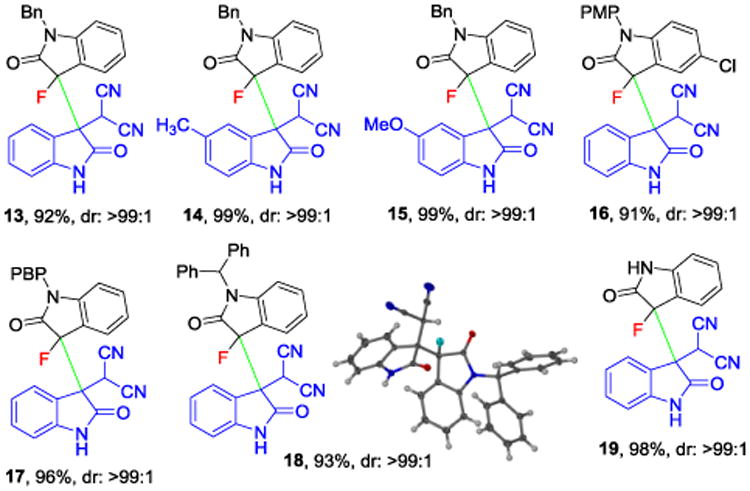
3,3′-Bisindolines carrying a protecting group in the fluorooxindole moiety. See Scheme 2 for reaction conditions.
Since bisindolines are key builing blocks for the synthesis of several alkaloids, many radical dimerization procedures for the synthesis of 3,3′-bridged bisoxindoles have been reported.[12] Because radical dimerization of oxindoles often suffers from low stereoselectivity, the introductions of complementary approaches that overcome this problem has remained important. To the best of our knowledge the 3,3′-bisoxindoline 3-19 have not been reported to date and our organocatalytic method generates a single diastereomer in one step under environmentally benign condition. In addition, this synthetic protocol can easily be upscaled without compromising results. The formation of 3 using 4.0 mmol of 1a and 4.0 mmol of 2a in 10 mL of water gave 1.38 g of 3 with 96% yield and >99:1 diastereoselectivity.
To broaden the general utility of our green chemistry protocol, we employed N-methyl and N-phenyl 3-fluorooxindoles in other Michael additions, an aldol type reaction and a Mannich reaction (Scheme 4). The addition of N-phenyl-3-fluorooxindole (2b) to acrylonitrile gave 20 in 92% yield. Compound 21 was obtained from N-methyl-3-fluorooxindole (2a) and methyl vinyl ketone in 96% yield. We noticed that the reactions catalyzed by a weak organic base (20 mol% of Et3N) takes 2 to 2.5 days whereas the use of stronger base (20 mol% DBU) under identical reaction conditions decreased the reaction time to 2 hours. We were pleased to find that the aldol reaction with diethyl ketomalonate gave 22 in 97% yield and aminomethylation of 3-fluorooxindole using Eschenmoser's salt gave 23 in 99% yield, which altogether underscore the versatility and practicality of C-C bond formation with fluoroenolates in aqueous solution.
Scheme 4.
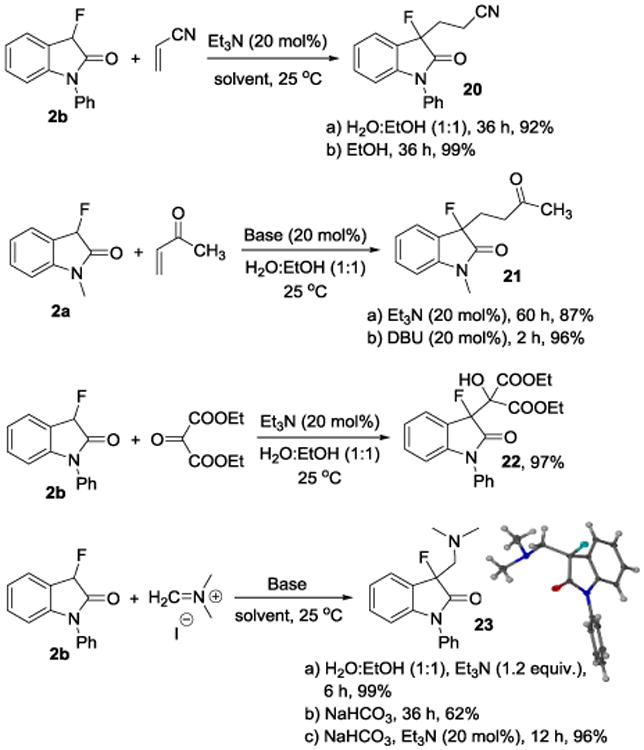
Organocatalytic 1,2- and 1,4-additions with fluorooxindole enolates in aqueous solutions.
In summary, we have developed a highly diastereoselective organocatalytic method for the synthesis of 3,3′-bisindolines containing two adjacent quarternary chiral centers. Excellent yields and stereoselectivities were achieved using inexpensive Et3N as catalyst in water or aqueous solutions at room temperature. This organocatalytic protocol favorably compares to previously reported “on water” Michael additions to isatylidene malonitriles. Furthermore, analysis of a series of single crystal structures of six products revealed that this reaction consistently favors formation of the heterochiral dimer representing an important scaffold observed in cyclotryptamine alkaloids. The general usefulness of this reaction procedure goes beyond the synthesis of fluorinated 3,3′-bisindolines which was demonstrated with the addition of fluorooxindoles to methyl vinyl ketone, acrylonitrile, diethyl ketomalonate and Eschenmoser's salt.
Experimental Section
Representative Procedure for the Organocatalytic Michael addition
2-(2-Oxoindolin-3-ylidene)malononitrile (39 mg, 0.2 mmol) and 3-fluoro-1-methylindolin-2-one (33 mg, 0.2 mmol) were added together with 10 mol% Et3N into 0.5 mL of water. The reaction was stirred at room temperature and monitored by TLC. After 3 hours, the mixture was poured onto brine (10 mL) and extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate as mobile phase to give 3,3′-bisindoline 3 as a colorless solid in 99% yield (71 mg, 0.198 mmol). Rf = 0.4 (hexanes/EtOAc, 1:1); 1H NMR (400 MHz, DMSO-d6) δ = 11.17 (s, 1H), 7.86 (dd, J = 7.7, 1.7 Hz, 1H), 7.61 (dd, J = 7.7, 7.7 Hz, 1H), 7.43 (dd, J = 7.8, 7.7 Hz, 1H), 7.38 (dd, J = 7.7, 1.7 Hz, 1H), 7.13 (dd, J = 7.7, 1.8 Hz, 1H), 7.01 (dd, J = 7.8, 1.7 Hz, 1H), 6.82 (dd, J = 7.8, 7.7 Hz, 1H), 6.09 (s, 1H), 5.82 (dd, J = 7.7, 1.8 Hz, 1H), 3.20 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ = 170.7 (d, Jc-f = 9.3 Hz), 168.9 (d, Jc-f = 21.2 Hz), 144.7 (d, Jc-f = 5.1 Hz), 143.7, 133.6 (d, Jc-f = 3.1 Hz), 132.5, 126.4 (d, Jc-f = 2.7 Hz), 124.6, 123.9, 123.5 (d, Jc-f = 2.7 Hz), 122.6, 121.1 (d, Jc-f = 18.2 Hz), 112.7, 112.1, 111.5, 110.6, 91.3 (d, Jc-f = 198.7 Hz), 56.8 (d, Jc-f = 23.7 Hz), 26.9, 25.4; 19F NMR (376 MHz, DMSO-d6) δ = -164.2; Anal. Calcd. For C20H13FN4O2: C, 66.66; H, 3.64; N, 15.55. Found: C, 66.61; H, 3.81; N, 15.78.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from NIH (GM106260) and the Georgetown Environment Initiative (GEI).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.((Please delete if not appropriate))
References
- 1.a) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (b) Zhu W, Wang J, Wang Si, Gu Z, Aceña JL, Izawa K, Liu H, Soloshonok VA. J Fluorine Chem. 2014;167:37–54. [Google Scholar]; (c) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Chem Rev. 2016;116:422–518. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- 2.a) Saidalimu I, Fang X, He XP, Liang J, Yang XY, Wu FH. Angew Chem Int Ed. 2013;52:5566–5570. doi: 10.1002/anie.201301443. [DOI] [PubMed] [Google Scholar]; b) Saidalimu I, Fang X, Lv W, Yang X, He X, Zhang J, Wu FH. Adv Synth Catal. 2013;355:857–863. [Google Scholar]; c) Zhang P, Wolf C. Angew Chem Int Ed. 2013;52:7869–7873. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]; d) Xie C, Wu L, Mei H, Soloshonok VA, Han J, Pan Y. Org Biomol Chem. 2014;12:7836–7843. doi: 10.1039/c4ob01575d. [DOI] [PubMed] [Google Scholar]; e) Wu C, Li G, Sun W, Zhang M, Hong L, Wang R. Org Lett. 2014;16:1960–1963. doi: 10.1021/ol500517d. [DOI] [PubMed] [Google Scholar]; f) Xie C, Wu L, Han J, Soloshonok VA, Pan Y. Angew Chem Int Ed. 2015;54:6019–6023. doi: 10.1002/anie.201500908. [DOI] [PubMed] [Google Scholar]; g) Xie C, Dai Y, Mei H, Han J, Soloshonok VA, Pan Y. Chem Commun. 2015;51:9149–9152. doi: 10.1039/c5cc02256h. [DOI] [PubMed] [Google Scholar]; h) Saadi J, Wennemers H. Nat Chem. 2016;8:276–280. doi: 10.1038/nchem.2437. [DOI] [PubMed] [Google Scholar]; i) Balaraman K, Moskowitz M, Liu Y, Wolf C. Synthesis. 2016;48:2376–2384. doi: 10.1055/s-0035-1561433. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a general review: Mei H, Acena JL, Soloshonok VA, Roeschenthaler GV, Han J. Eur J Org Chem. 2015:6401–6412.
- 3.a) Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:8037–8041. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]; b) Antonchick AP, Gerding-Reimers C, Catarinella M, Schürmann M, Preut H, Ziegler S, Rauh D, Waldmann H. Nat Chem. 2010;2:735–740. doi: 10.1038/nchem.730. [DOI] [PubMed] [Google Scholar]; c) Guo C, Song J, Huang JZ, Chen PH, Luo SW, Gong LZ. Angew Chem Int Ed. 2012;51:1046–1050. doi: 10.1002/anie.201107079. [DOI] [PubMed] [Google Scholar]; d) Zhu B, Zhang W, Lee R, Han Z, Yang W, Tan D, Huang KW, Jiang Z. Angew Chem Int Ed. 2013;52:6666–6670. doi: 10.1002/anie.201302274. [DOI] [PubMed] [Google Scholar]; e) Xie W, Jiang G, Liu H, Hu J, Pan X, Zhang H, Wan X, Lai Yi, Mae D. Angew Chem Int Ed. 2013;52:12924–12927. doi: 10.1002/anie.201306774. [DOI] [PubMed] [Google Scholar]; f) Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S. Angew Chem Int Ed. 2012;51:5217–5221. doi: 10.1002/anie.201201132. [DOI] [PubMed] [Google Scholar]; g) Zong L, Du S, Chin KF, Wang C, Tan CH. Angew Chem Int Ed. 2015;54:9390–9393. doi: 10.1002/anie.201503844. [DOI] [PubMed] [Google Scholar]; h) Biswas P, Paul S, Guin J. Angew Chem Int Ed. 2016;55:7756–7760. doi: 10.1002/anie.201603809. [DOI] [PubMed] [Google Scholar]
- 4.a) Ogawa S, Shibata N, Inagaki J, Nakamura S, Toru T, Shiro M. Angew Chem Int Ed. 2007;46:8666–8669. doi: 10.1002/anie.200703317. [DOI] [PubMed] [Google Scholar]; b) Bui T, Syed S, Barbas CF., III J Am Chem Soc. 2009;131:8758–8759. doi: 10.1021/ja903520c. [DOI] [PubMed] [Google Scholar]; c) Wu MY, He WW, Liu XY, Tan B. Angew Chem Int Ed. 2015;54:9409–9413. doi: 10.1002/anie.201504640. [DOI] [PubMed] [Google Scholar]; d) Ohmatsu K, Ando Y, Ooi T. J Am Chem Soc. 2013;135:18706–18709. doi: 10.1021/ja411647x. [DOI] [PubMed] [Google Scholar]; e) Tan B, Candeias NR, Barbas CF. Nat Chem. 2011;3:473–477. doi: 10.1038/nchem.1039. [DOI] [PubMed] [Google Scholar]; f) Guo QX, Liu YW, Li XC, Zhong LZ, Peng YG. J Org Chem. 2012;77:3589, 3594. doi: 10.1021/jo202585w. [DOI] [PubMed] [Google Scholar]; g) Lv H, Tiwari B, Mo J, Xing C, Chi YR. Org Lett. 2012;14:5412–5415. doi: 10.1021/ol302475g. [DOI] [PubMed] [Google Scholar]; h) Zhao J, Fang B, Luo W, Hao X, Liu X, Lin L, Feng X. Angew Chem Int Ed. 2015;54:241–244. doi: 10.1002/anie.201408730. [DOI] [PubMed] [Google Scholar]; i) Engl OD, Fritz SP, Wennemers H. Angew Chem Int Ed. 2015;54:8193–8197. doi: 10.1002/anie.201502976. [DOI] [PubMed] [Google Scholar]; j) Shan J, Cui B, Wang Y, Yang C, Zhou X, Han W, Chen Y. J Org Chem. 2016;81:5270–5277. doi: 10.1021/acs.joc.6b00278. [DOI] [PubMed] [Google Scholar]; k) You Y, Wu ZJ, Chen JF, Wang ZH, Xu XY, Zhang XM, Yuan WC. J Org Chem. 2016;81:5759–5765. doi: 10.1021/acs.joc.6b00896. [DOI] [PubMed] [Google Scholar]
- 5.a) Gribkoff VK, Starrett JE, Jr, Dworetzky SL, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. Nat Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]; b) Hewawasam P, Gribkoff VK, Pendri Y, Dworetzky SI, Meanwell NA, Martinez E, Boissard CG, Post-Munson DJ, Trojnacki JT, Yeleswaram K, Pajor LM, Knipe J, Gao Q, Perrone R, Starrett JE., Jr Bioorg Med Chem Lett. 2002;12:1023–1026. doi: 10.1016/s0960-894x(02)00101-4. [DOI] [PubMed] [Google Scholar]
- 6.Selected examples: Shibata N, Suzuki E, Asahi T, Shiro M. J Am Chem Soc. 2001;123:7001–7009. doi: 10.1021/ja010789t.; b) Hamashima Y, Suzuki T, Takano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164–10165. doi: 10.1021/ja0513077. [DOI] [PubMed] [Google Scholar]; c) Shibata N, Kohno J, Takai K, Ishimaru T, Nakamura S, Toru T, Kanemasa S. Angew Chem, Int Ed. 2005;44:4204–4207. doi: 10.1002/anie.200501041. [DOI] [PubMed] [Google Scholar]; d) Ishimaru T, Shibata N, Horikawa T, Yasuda N, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2008;47:4157–4161. doi: 10.1002/anie.200800717. [DOI] [PubMed] [Google Scholar]; e) Deng QH, Wadepohl H, Gade LH. Chem Eur J. 2011;17:14922–14928. doi: 10.1002/chem.201102375. [DOI] [PubMed] [Google Scholar]; f) Shen K, Liu XH, Lin LL, Feng XM. Chem Sci. 2012;3:327–334. [Google Scholar]; g) Li J, Cai Y, Chen W, Liu X, Lin L, Feng X. J Org Chem. 2012;77:9148–9155. doi: 10.1021/jo301705t. [DOI] [PubMed] [Google Scholar]; h) Gu X, Zhang Y, Xu ZJ, Che CM. Chem Commun. 2014;50:7870–7873. doi: 10.1039/c4cc01631a. [DOI] [PubMed] [Google Scholar]; For an orthogonal approach based on asymmetric ring construction, see Wu L, Falivene L, Drinkel E, Grant S, Linden A, Cavallo L, Dorta R. Angew Chem Int Ed. 2012;51:2870–2873. doi: 10.1002/anie.201200206.
- 7.a) Dou X, Lu Y. Org Biomol Chem. 2013;11:5217–5221. doi: 10.1039/c3ob41267a. [DOI] [PubMed] [Google Scholar]; b) Wang T, Hoon DL, Lu Y. Chem Commun. 2015;51:10186–10189. doi: 10.1039/c5cc03289j. [DOI] [PubMed] [Google Scholar]; c) Xie C, Zhang L, Sha W, Soloshonok VA, Han J, Pan Y. Org Lett. 2016;18:3270–3273. doi: 10.1021/acs.orglett.6b01516. [DOI] [PubMed] [Google Scholar]; d) Balaraman K, Wolf C. Angew Chem, Int Ed. 2017;56:1390–1395. doi: 10.1002/anie.201608752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu JS, Liu YL, Tang J, Wang X, Zhou J. Angew Chem, Int Ed. 2014;53:9512–9516. doi: 10.1002/anie.201404432. [DOI] [PubMed] [Google Scholar]
- 9.Steven A, Overman LE. Angew Chem, Int Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]
- 10.For example, 3,3′-bisindoline 5 was quantitatively isolated after reaction of 2-(1-methyl-2-oxoindolin-3-ylidene)malononitrile and 3-fluoro-1-methylindolin-2-one in EtOH:H2O (1:1) in the presence of 10 mol% of Et3N after 3 hours. The precipitate was washed twice with small portions of ethanol to give pure product.
- 11.The CCDC numbers 1569588, 1569590, 1569589,1569587, 1569585, 1569586 and 1569584 contain the the supplementary crystallographic data for compounds 3, 4, 5, 6, 10, 18 and 23, respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 12.a) Hendrickson JB, Göschke R, Rees R. Tetrahedron. 1964;20:565–579. [Google Scholar]; b) Fang CL, Horne S, Taylor N, Rodrigo R. J Am Chem Soc. 1994;116:9480–9486. [Google Scholar]; c) Ghosh S, Chaudhuri S, Bisai A. Org Lett. 2015;17:1373–1376. doi: 10.1021/acs.orglett.5b00032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.