

Abstract
Current antivirals effectively target diverse viruses at various stages of their viral lifecycles. Nevertheless, curative therapy has remained elusive for important pathogens (e.g., HIV-1 and herpesviruses), in large part due to viral latency and the evolution of resistance to existing therapies. Here, we review the discovery of viral ‘master’ circuits: virus-encoded auto-regulatory gene networks that can autonomously control viral expression programs (i.e., between active, latent, and abortive fates). These circuits offer a potential new class of antivirals that could lead to intrinsic combination-antiviral therapies within a single molecule—evolutionary escape from such circuit ‘disruptors’ would require simultaneous evolution of both the cis regulatory element (e.g., the DNA-binding site) and the trans element (e.g., the transcription factor) for the circuit’s function to be recapitulated. We review the architectures of these fate-regulating master circuits in HIV-1 and the human herpesvirus cytomegalovirus (CMV) along with potential circuit-disruption strategies that may ultimately enable escape-resistant antiviral therapies.
Keywords: HIV, CMV, transcription, fate regulation, latency, auto-regulatory circuits, feedback, transcriptional fluctuations, stochastic noise
1. VIRAL TRANSCRIPTIONAL REGULATION – THE THERAPEUTIC POTENTIAL
1.1 The Need for New Antivirals
Viral pathogens take an enormous toll on human health. The 1918 “Spanish influenza” outbreak is believed to have killed ~50 million people worldwide while, to date, the HIV-1 pandemic has killed at least 35 million people and infected an additional 36 million people (1). Protective, or neutralizing, vaccines have been developed for a number viruses—leading to worldwide eradication efforts for poliomyelitis virus and smallpox—however, similar successes have not been realized for many other viruses. In the absence of protective vaccines, ~90 antiviral therapeutics have been developed over the past 50 years (for a comprehensive review see (2)). For HIV-1, antiretroviral (ARV) therapeutics approved to target multiple steps in the viral lifecycle have converted HIV-1 from a death sentence in the 1980s and early 1990s to a manageable disease in parts of the world where ARVs are readily accessible. Nevertheless, HIV-1 remains an incurable infection requiring patients to remain on ARVs for their lifetimes and resistance to existing regimens remains a major clinical and epidemiological concern. Likewise, for the human herpesvirus cytomegalovirus (CMV)—the leading known cause of birth defects and transplant failures (3)—a highly effective drug without significant toxicities has yet to be approved and resistance to existing anti-CMV drugs is widespread (4).
1.2 Why Target Viral Transcriptional Regulation?
To control their gene-expression programs and regulate infection outcome (i.e., fate), many diverse viruses encode auto-regulatory gene ‘circuits’. As we review below, the regulatory architectures for the HIV-1 and CMV circuits have been mapped and these circuits are transcriptional feedback circuits—where a viral transactivator protein regulates its own promoter activity—that can autonomously control viral fate (i.e., they are fate-regulating ‘master’ circuits). If these auto-regulatory circuits could be disrupted for antiviral therapy, such circuit ‘disruptors’ would likely be far less susceptible to the evolution of drug-resistant escape mutants (i.e., such therapies would be escape resistant) since evolutionary escape from a molecule that disrupts circuitry would require the virus to simultaneously evolve both the cis regulatory element (e.g., DNA binding site) and the trans elements (e.g., the transcription factor) in order to recapitulate the circuit’s function and associated viral replicative fitness. In other words, circuit disruptors would represent intrinsic combination-antiviral therapy. We review the architectures of these fate-regulating feedback circuits in HIV-1 and the human herpesvirus cytomegalovirus (CMV) along with potential circuit-disruption.
1.3 Unmet Medical Needs
1.3.1 HIV-1
The human immunodeficiency virus type 1 (HIV-1, or “HIV”) infects and depletes CD4+ T lymphocytes, a key white blood cell population that orchestrates the immune response. As a result, HIV-infected individuals cannot mount effective immune responses against common pathogens, leading to acquired immunodeficiency syndrome (AIDS) and death from opportunistic infections. There is no effective vaccine for HIV and a protective vaccine has remained difficult to realize. HIV infection is currently treated using cocktails of ARV drugs. But, ARVs are not a cure. HIV-infected individuals must remain on ARVs for their entire life since HIV persists in long-lived reservoirs of latently infected cells and the virus spontaneously reactivates from these latent reservoirs. When patients interrupt or discontinue ARV therapy, the virus quickly re-establishes pre-treatment levels in the infected individual within one to three weeks (5). Thus, latent reservoirs of virus are considered the greatest obstacle to an HIV cure.
By far, the longest-lived and most-problematic reservoir of latently infected cells is the proviral latent reservoir of CD4+ T lymphocytes (5–7). This population of latently infected cells is small (estimated at 1 cell in 106-107 CD4 T cells in the patient), and difficult to target (8). It is widely believed that ‘purging’ this latent reservoir will be required for an HIV cure (5) and major investments being made on basic and clinical research into initiatives to ‘purge’ the latent reservoir by reactivating latently infected cells using transcriptional activators called latency-reversing agents (LRAs) and then halting viral replication with ARVs (the so called ‘kick-and-kill’ strategies).
Unfortunately, these cure strategies remain largely unrealized and even under best-case in vitro isogenic cell-culture conditions, the most-potent LRAs, delivered at maximal concentrations, only reactivate a fraction of latent HIV (8, 9). This heterogeneous activation profile is ubiquitous for all LRAs tested in culture models (10), primary-cell latency models (11, 12), and even patient cells (13). Hence, to achieve curative therapy or a ‘functional cure’, new approaches that efficiently reactivate latent virus to purge the HIV latent reservoir—or, alternatively, prevent reactivation to stably maintain or ‘lock’ latent HIV within its reservoir—are needed (14, 15).
1.3.2 CMV
CMV is one of eight known human herpesviruses (HHV) and infects a majority of the world’s population. Infection in healthy adults and children is often not overtly symptomatic. However, like all HHVs, CMV can cause life-threatening disease and considerable morbidity in newborns and immune-compromised individuals, including organ and bone marrow transplant patients. In both developed and developing countries CMV remains the leading cause of congenital infection. CMV infection sequelae are often delayed in onset, but in the post-rubella era CMV is the most common cause of childhood hearing loss and a significant cause of neuro-developmental delay. In the United States, CMV is the leading known cause of birth defects—causing more birth defects than Down’s syndrome, fetal alcohol syndrome, or spina bifida (16–19). A decade ago the Institute of medicine assessed the economic impact (medical and educational care) of congenital CMV infection in the US to amount to $1.9 billion per year (20), which is in addition to its role as leading a cause of death in transplant recipients.
CMV-associated disease increased with the AIDS epidemic and continues to increase in conjunction with rises in solid-organ and hematopoietic stem-cell transplantation—the development of tests able to detect CMV antigens in the 1980’s facilitated infection diagnosis in a clinically useful timeframe. However, the current anti-viral drugs given for prevention or treatment of CMV infections are limited in number, have poor bioavailability and have considerable dose-limiting toxicity due to severe adverse side effects and research to develop an effective anti-CMV vaccine has yet to deliver an acceptable candidate. The leading anti-CMV therapy is ganciclovir (GCV), or its pro-drug val-ganciclovir (VGCV), a guanosine analogue that requires activation by a CMV kinase; other nucleic-acid analogues such as cidofovir (a nucleoside monophosphate) or foscarnet (a pyrophosphate analog) do not require prior activation by a viral protein. These treatments are associated with significant systemic toxicity in patients; GCV and VGCV primarily lead to neutropenia while cidofovir and foscaret can cause renal toxicity and electrolyte imbalance. As such, these drugs are not usually given to pregnant women. Resistance has been observed using all of these anti-CMV drugs when mutations affect the virus UL97 kinase or virus DNA polymerase (19). Thus, there is a pressing need for new, more effective, anti-CMV therapies that could be used alone or in combination with existing medications to help prevent or treat CMV infection.
1.4 Existing Antivirals Do Not Target Viral Gene Regulation
Currently approved antiviral therapies target and disrupt multiple stages of the viral lifecycle (Fig. 1A), which can be divided into various stages: (i) attachment and virus entry, (ii) generation of viral gene products using host-cell machinery, (iii) viral genome replication, and (iv) virion assembly, packaging, and release (21). For most stages of the lifecycle, multiple classes of compounds have been approved as antiviral therapeutics (Fig. 1B). By class, the most common therapeutic drugs are nucleic acid analogues (e.g., nucleoside and nucleotide analogues, a.k.a. “nuc” analogoues), which target viral gene replication at various stages including HIV reverse transcription and are used in the treatment of many different viruses (2). Conspicuously, very few approved antiviral therapeutics (2) have targeted viral gene expression (Fig. 1B). The notable example of an approved drug to target viral gene expression was Fomivirsen (22)—an antisense RNA against CMV-related retinitis in AIDS patients, which was approved for use by direct intravitreal injection into the eye in 1998. Fomivirsen has since been withdrawn from the market due to the success of ARV therapy for HIV (23, 24). However, the Fomivirsen example, the first approved antisense therapy, showed that viral gene regulation could be successfully targeted at the level of protein translation and has spurred research on related RNA-based therapeutic approaches, for a review see (25).
Figure 1. Existing antiviral targets and therapies.
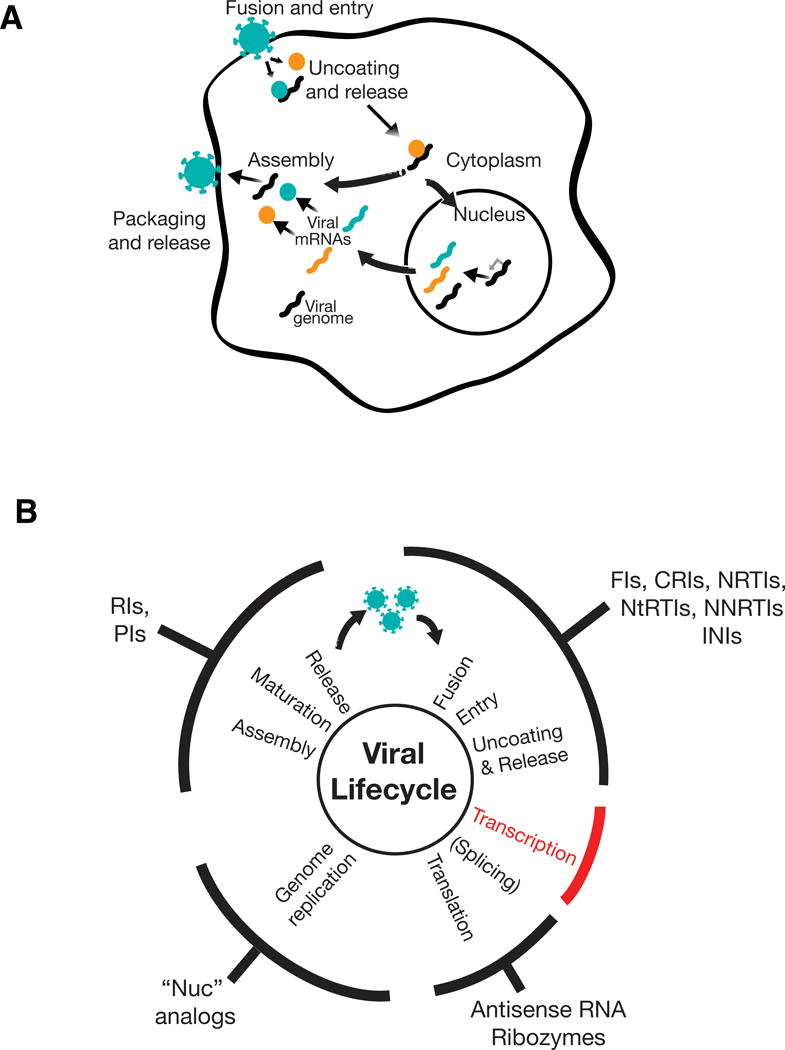
A) General schematic of the viral lifecycle. (i) Viruses recognize specific receptors and associated components on the surface of their target host cells and fuse with the host cell membrane. (ii) This fusion leads to viral entry into the cytoplasm followed by uncoating of the virus and release of components that include the viral proteins (colored circles) and viral RNA or DNA (black squiggle). (iii) The released viral components either induce viral transcription in the nucleus (e.g., DNA viruses other than poxviruses and retroviruses; mRNAs shown in colored squiggles) or directly undergo genome replication in the cytoplasm (e.g., RNA viruses and poxviruses). Genome replication can also be considered to be coincident with transcription (e.g., Influenza or retroviruses, where the genomic RNA is a full-length unspliced mRNA transcript) or follow translation aided by virus-generated products (e.g., Poliovirus). (iv) Viral genomes and products assemble, are packaged into virions, and released from the cell.
B) Targeting stages of the viral lifecyle for antiviral therapies. The generalized viral life cycle (discussed in panel A) is represented by the inner circle with classes of approved antivirals that affect these stages noted on the outer circumference. Antivirals against transcription are notably absent (red). Antivirals abbreviations are as follows (together with representative FDA-approved example): FI—fusion inhibitor (e.g., Enfuvirtide for HIV); CRI—co-receptor inhibitor (e.g., Maraviroc for HIV); NNRTI—non-nucleoside reverse transcriptase inhibitor (e.g., Efavirenz for HIV), NRTI—nucleoside reverse transcriptase inhibitor (e.g., Azidothymidine, AZT, for HIV); NtRTI—nucleotide reverse transcriptase inhibitor (e.g., Tenofivir for HIV); INI—integrase inhibitor (e.g., Raltegravir for HIV); RI—release inhibitor (e.g., Zanamivir for Influenza), PI—protease inhibitor (e.g., Darunavir for HIV or Boceprevir for HCV), RI—release/uncoating inhibitor (e.g., Amantidine for Influenza). Approved RNA-based therapeutics include fomivirsen (antisense RNA) to treat CMV; ribozyme therapies remain experimental. A range of nucleic acid (“nuc”) analogs are FDA-approved to inhibit viral genome replication including Aciclovir for Herpes Simplex 1 (HSV-1), Ganciclovir for CMV, and Sofosbuvir for Hepatitis C Virus (HCV).
In this review, we focus on viral transcriptional regulation as a potential new class of targets for antiviral therapy. In particular, we focus on virus encoded auto-regulatory gene circuits that act as ‘master’ circuits in HIV and CMV, the unique feedback architectures of these circuits, and how these architectures might be targeted and disrupted to manipulate viral fate for antiviral therapy.
2 BACKGROUND: TRANSCRIPTIONAL CIRCUITS AND FATE REGULATION
The selection of a distinct fate, orchestrated by an underlying transcriptional network that processes cell-intrinsic and environmental inputs, is seen throughout biology from embryonic development, where individual cells adopt distinct stereotyped functions, to bacteria (26, 27) and viruses that adopt either active replication or dormant states. These fate decisions are regulated, in large part, by transcriptional programs and viruses represent arguably the simplest examples of fate-selection processes regulated by intrinsic transcriptional circuits. For example, upon infecting an Escherichia coli cell, the bacteriophage λ adopts either the lytic pathway or lysogenic pathway as a result of a core ‘operator’—a mutual-inhibition transcriptional toggle circuit (28, 29)—and, as reviewed below, HIV probabilistically establishes either active replication or viral latency (a quiescent state) based largely on activity of its Tat transcriptional activator—a stochastic positive-feedback circuit (30, 31).
2.1 What is a Transcriptional Circuit?
Transcriptional or gene-regulatory circuits are biological analogues of electrical circuits (32, 33) and are comprised of minimal sets of genes, their promoters, the resultant gene products, and the obligate interactions between these components that generate outputs in response to signaling inputs. As the examples below demonstrate, modern-day appreciation of the central roles of these minimal circuits stems from classical studies in bacteriophage (34), which revealed that just a handful of interconnected genes can represent a sufficient module to enable switching between distinct fates.
Historically, the presence of alternate biological or developmental fates (phenotypes) was attributed to cell-extrinsic factors such as environmental signaling cues and/or precise spatial signals. HIV serves a prominent example where several lines of evidence suggested precisely this—that establishment of proviral latency was controlled by the host cell (30, 35)—but it is now clear that the intrinsic transcriptional dynamics of HIV’s circuitry play a pivotal role in regulating latency independent of the host cell environment (discussed below). Below, we describe several well-studied genetic circuits and the fates they control, including the classical bacteriophage λ lysis-lysogeny decision and the synthetic toggle switch in bacteria (both of which were elucidated prior to quantitative mapping of the HIV Tat circuit) as well as B. subtillis competence, stem-cell differentiation, and adipocyte differentiation (which were elucidated after mapping of the HIV Tat circuit). Together, these examples serve to emphasize two aspects: (i) the capabilities of genetic circuits to orchestrate fate regulation by themselves independent of host-cell-mediated effects and (ii) the common mechanisms such as feedback that typically form the core of fate-regulating circuits.
2.2 Examples of Fate-Regulating Transcriptional Circuits
2.2.1 Early examples: Bacteriophage λ lysis-lysogeny circuitry and the synthetic toggle switch
Perhaps the first viral system where fate-regulation was studied is bacteriophage λ (36, 37). After phage λ infects a bacterium, it either replicates and lyses the host cell or integrates into the host chromosome as a prophage (28). In response to cell damage, the prophage can reactivate to induce the lytic pathway. Phage λ fate regulation is governed by a genetic circuit that includes the CI (a.k.a λ repressor) and Cro (a.k.a lytic activator) viral proteins expressed from the PRM and PR promoters, respectively. The proteins and promoters of this circuit are interconnected through operator sites and form a mutual-repression circuit. The operation of this circuit, particularly the per-cell fluctuations in crucial circuit controlling molecules (often reaching very low numbers per cell), was elucidated by a stochastic model from Arkin et al. in 1998 (38) (reviewed by (39)). However, precisely how random or how deterministic the lysis-lysogeny decision is remains actively debated (28, 40, 41).
Biological fate-regulation need not be as complex as in phage λ. This point was established by the development of a synthetic toggle switch in Escherichia coli (29) where individual cells in the population adopted (or toggled) between two alternative fates, a state where reporter protein was expressed and a non-expressing state (incidentally, the toggle switch used the λ -repressor protein as a component). The successful construction of the toggle switch demonstrates that a biological system can be built to function in a predictable manner using first principles from electrical circuit design. Moreover, the ‘toggle switch’ showed that simple genetic circuits are sufficient to generate alternate and reversible transcriptional fates where circuit dynamics can intrinsically control entry and exit independent of cell state.
2.2.3 Contemporary examples: Stochastic regulatory circuits in HIV, bacteria and cellular reprogramming and differentiation
Following the initial finding that the HIV Tat positive-feedback circuit stochastically controlled HIV fate (42), for a review see (43), there were numerous examples of genetic circuits probabilistically controlling cell fate. Among the most elegant examples was work from Elowitz and colleagues showing that bacteria encode an excitable positive-feedback circuit that enables them to probabilistically enter competence states allow uptake of exogenous DNA (44). Similarly elegant work showed that a minimal set of transcription factors responsible for cellular reprogramming, including Oct4, Sox2, and Nanog (45) form a stochastic positive-feedback circuit (46) that can stochastically control cellular reprogramming (47). Another, more recent example of a fate circuit has been described in adipocyte differentiation. Adipocytes arise from a large pool of precursor cells (48) and pre-adipocytes differentiate through a switch-like mechanism with a threshold for activation that involves positive feedback between two key transcription factors, CCAAT/enhancer binding protein α (CEBPA) and peroxisome proliferator-activated receptor γ (PPARG) (49). As with the outcomes of bacteriophage λ, the variability in the numbers of transcription factors at any point in time in a cell has a significant role in the rates of differentiation and the steady-state number of pre-adipocytes and adipocytes.
2.3 Design Principles of Circuits that Regulate Fate
Gene expression is an intrinsically stochastic process characterized by large fluctuations in the reactant biomolecules within individual cells, leading to significant cell-to-cell variability, or noise, even in isogenic cell populations (50). The stochastic fluctuations often provide the necessary impulse for a switch from one state to another and the role of stochastic noise in promoting switching between alternate expression states and cell fates has been demonstrated in multiple systems from viruses, to bacteria, to stem cells (42, 44, 51) indicating that fate-regulating gene circuits function in a noisy biological environment of fluctuating numbers of molecules. Regulatory circuits that act as fate-selection switches must function in these noisy intracellular environments and process this cellular noise (52–54) to generate alternate fate outcomes (i.e., phenotypes) in response to intra- or extra-cellular signaling. In mathematical terms, these circuit-driven phenotypes result from thresholds or bi-stability in the underlying regulatory circuitry—where the bi-stability and multi-stability can be either deterministic or stochastic.
While the regulatory architectures that generate the requisite thresholds and bi-stability are diverse (55–57), transcriptional feedback appears to be a common motif. When feedback regulation is self-cooperative in nature—as measured by a sigmoidal dose-response curve with a Hill coefficient H ≥ 2 (e.g., transcription factors that must homo-multimerize to function or multiple transcription factor binding sites that must be occupied for functioning)—the resulting circuit exhibits signaling thresholds and memory-like responses (i.e., hysteresis), resulting in deterministic bi-stability, for review see (58). Moderately self-cooperative feedback circuits (H ~ 2–3) are not uncommon. However, the CMV circuit, reviewed below, utilizes exceptionally high self-cooperativity (H ≈ 7), from protein homo-multimerization, to set a threshold for activation of its negative-feedback circuit that maintains active infection (59). In contrast, other fate-selection switches need not be self-cooperative or deterministically bistable. For example, the HIV circuit, also reviewed below, regulates viral fate by a non-cooperative positive-feedback circuit (H ≈ 1) where the circuit is stochastically bistable, relying on noise, rather than deterministically bistable (43, 60).
3 THE HIV TAT MASTER CIRCUIT: REGULATION OF PROVIRAL LATENCY
3.1 Fate Selection in HIV: Active Replication Versus Proviral Latency
Upon infection of target CD4+ T cells, HIV typically integrates into the host genome in a biased-random fashion with a strong propensity for actively transcribed loci (i.e., presumably regions of ‘open’ chromatin) (61, 62). The integrated HIV provirus can then enter either an active-replication state—marked by robust transcription from the HIV Long Terminal Repeat (LTR) promoter, expression of viral proteins, production of infectious progeny virions within 24 hours (63), and ultimately cell death within ~40 hours (64)—or he provirus can enter a long-lived proviral latent state where viral transcription is largely quiescent (Fig. 2) (30). Current combination ARV therapy (cART) effectively blocks active replication. However, the virus can spontaneously reactivate from the latent state, leading to active viral replication. This switching ‘on’ from the latent state, combined with the long-lived nature of the latently infected cells (latent reservoir), requires that HIV-infected patients remain on ARV therapy for life (65, 66). Any interruption in ARV therapy rapidly leads to re-emergence of the virus and re-establishment of the pre-treatment ‘set point’ viremia within a few weeks (67, 68).
Figure 2. Mapping and manipulating HIV’s master circuit.
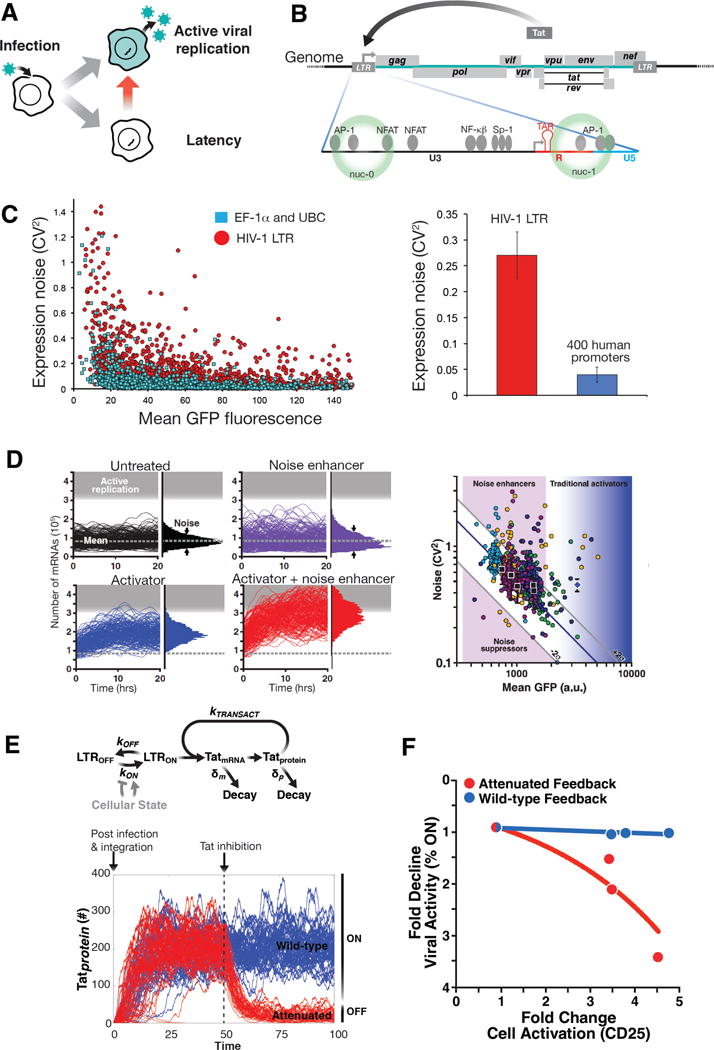
A) HIV fate selection. Post-infection of CD4+ T lymphocytes, HIV enters either a lytic (active replication) state leading to cell death and production of viral progeny or enters a latent (transcriptionally quiescent) state that can be long-lived (years) and subsequently reactivate to active replication (red arrow).
B) Schematic of the HIV-1 genome and the HIV LTR promoter.
C) The HIV LTR is an exceptionally noisy promoter that generates large stochastic fluctuations in expression. Left: Single-cell expression profiles (from quantitative time-lapse microscopy imaging) of ~8,000 cells carrying single integrations of either the HIV LTR promoter or house-keeping promoters (elongation factor 1a or Ubiquitin C promoter) driving GFP. Shown is the measured coefficient of variation squared for each 12-hr single-cell trajectory plotted against that cell’s mean-expression level during the 12-hr period; data quantified from (86). Each dot indicates a unique cell with a unique genomic integration site of the promoter. Right: HIV LTR expression noise (red bar), compared to average expression noise across 400 different human promoters (blue bar, data obtained from supplementary figure 15 in reference (116)). The error bars show 95% confidence intervals. Single-molecule mRNA FISH measurements corroborate these results for the HIV LTR at the mRNA level (117).
D) Mathematical modeling of stochastic gene expression from the LTR predicted that expression noise can be therapeutically exploited for latency reversal. Left: Stochastic simulations of the two-state LTR model of transcription cells (black trajectories, top left). Adding an activator increases mean expression (blue) while adding a noise enhancer (purple) only changes the noise (width of distribution. Combining the two, generates synergy (red trajectories) with more trajectories entering into active replication. Figure adapted from (15). Right: Screening for compounds that modulate noise in HIV promoter expression. Flow cytometry measurements of LTR-d2GFP isoclonal cells exposed to a small molecule library. Each color represents a measurement cluster of compounds screened on a single day. Untreated controls (black squares) vary along the constant-burst-size line (diagonal blue line); error bars calculated from standard deviation for 28 measurements. TNF, which changes both mean and noise was used as a positive control (blue diamond). Figure adapted from (15)
E) Mathematical model of the integrated LTR-Tat positive-feedback circuit predicted that therapeutic targeting of feedback circuitry could promote latency and ‘lock’ HIV into a latent state. (Top) A simplified model of the LTR-Tat circuit. The LTR promoter can toggle between a state where transcriptional elongation is stalled (LTROFF) and a state where elongation proceeds (LTRON) at rates koff and kon, respectively, and Tat protein transactivates the promoter by enhancing transcriptional elongation at a rate ktransact Tat protein and mRNA decay at rates δm and δp, respectively. The ON–OFF toggling of the LTR is the source of the high-magnitude noise while the Tat positive-feedback circuitry amplifies this LTR noise. (Bottom) Stochastic simulations of Tat protein levels (in arbitrary number of molecules) in individual cells over time (from model in left panel). Each trajectory represents an individual cell; 50 single-cell trajectories are shown. The initial conditions and the simulations capture the dynamics of Tat immediately post integration of viral genome into an activated T cell. To examine the effect of Tat inhibition, the effect of adding a Tat inhibitor (which reduces ktransact 10 fold, red) was simulated relative to the wild-type scenario (blue). In the wildtype case, the strong Tat feedback maintains active proliferation (viral ON state), while attenuation of feedback can ‘lock’ active proliferation into latency (OFF) with no recovery.
F) Experimental validation that attenuation of Tat feedback promotes latency (adapted from (76). Data was obtained through flow cytometry analysis of HIV expression in human donor-derived primary CD4+ T lymphocytes during transition from activated to resting in attenuated Tat feedback or wild-type Tat feedback conditions. Tat feedback strength was attenuated via a reversible proteolysis fusion tag (FKBP) where proteolysis can be protected via a small molecule (Shield-1) and cellular transition from activated to resting was induced by removal of anti-CD3/28 activation beads. Shown is the fold change in the number of actively HIV infected cells (as measured by LTR expression) as cells transition to resting state. When Tat feedback strength is wild-type (blue data points; blue trend line), the fold change in HIV expression activity is uncorrelated with cellular relaxation to resting (as measured by the cellular activation marker CD25). When Tat feedback is attenuated (red data points; red trend line), the percentage of active infections is dependent on cell state.
3.2 The HIV Tat Positive-Feedback Circuit Regulates Viral Fate Selection
Once the phenomenon of HIV latency was identified, a major question arose as to the mechanism by which HIV selects between active replication and proviral latency. An early theory that HIV latency was determined by viral integration site has been repeatedly tested but only small correlations have been found to date (69, 70). The conventional theory for latency establishment held that HIV latency was an ‘epiphenomenon’ resulting from HIV infection during transitioning of CD4+ T-cells (the major target of HIV infection) from an activated to a resting state (30, 35). This ‘epiphenomeon’ theory emerged from observations that activated CD4+ T-cells are highly susceptible to HIV infection and favorable environments for HIV transcription while resting-memory T-cells (the anatomical reservoir of latency in HIV infected patients) have multiple blocks to infection, including being poor environments for HIV transcription. Under this theory, when HIV infects an activated T-cell which then relaxes to become a resting cell, the relaxation silences viral transcription and results in a latent provirus. The implication of this theory was that latency is the unintended consequence of viral tropism for an activated T-cell and that, by itself, latency is of no evolutionary benefit to the virus.
However, the ‘epiphenomenon’ theory was dependent on slow processes of resting memory T-cell formation (71) or the associated chromatin silencing of viral expression (72) which occur over weeks. In contrast, HIV latency formation was measured to be rapid and on the order of hours or days in vivo—in Rhesus macaques latency is establishes within 3 days post infection (73)—and in cell-culture models latency is established immediately post infection in actively dividing cells (74, 75). To reconcile this discrepancy, we, Razooky et al., (76), directly tested whether host-cell transitioning from activated to resting was sufficient to silence HIV transcription. This study found that, once Tat feedback was initiated, viral transcription was surprisingly robust to cellular relaxation and largely independent of cellular state. In other words, cellular transitioning from activated to resting was unable to silence HIV transcription (76). In addition, the Razooky et al. study found that latency could be modulated independent of host cell state by modulating relative levels of the viral trans-activator of transcription (Tat). Together, the results showed that, HIV latency appeared to be part of a fate-regulation program ‘hardwired’ into HIV and not an epiphenomenon being driven by the host cell.
The autonomy of HIV Tat feedback cirucitry once in the ‘on’ state does not imply the absence of any role for host-cell state in determining or biasing latency. In fact, using simulations, Razooky et al. showed that the likelihood of viral latency establishment increases somewhat as cells relax (14, 76) and subsequent work (77) verified this by showing an approximately 2-fold change in latency probability as cells transitioned from activated to resting. Thus, while cellular relaxation can bias latency establishment, the critical subtlety is that latency is established immediately post infection (for in vitro results see (74, 77) and in vivo see (73)), whether in activated or resting T-cells, and does not require cellular relaxation.
The HIV-encoded fate-selection switch, and other lines of evidence suppoorting this model, are examined in-depth elsewhere (43). This debate is of consequence in HIV eradication efforts as the epiphenomenon theory places the focus of latency and latency reversal on host-cell modulation (which has proven exceptionally challenging as a kick-and-kill therapeutic strategy), whereas the viral circuitry hypothesis implies that the HIV Tat circuit ultimately controls latency largely autonomous of host-cell state. Next, we examine the consequences of viral autonomous fate-selection, and strategies to disrupt the virus-encoded gene circuitry.
3.3 Core Elements of HIV’s Viral Fate-Control Program
3.3.1 A weak but ‘bursty’ promoter
The enhancer and modulatory regions of the LTR (Fig. 2) contain cis-acting recognition elements for regulatory cellular transcription factors such as NF-κB p65/p50 heterodimers, NFAT, STAT5 and AP-1 (78, 79). These transcription-factor binding sites relate to how host cell activate state (the availability of transcription factors) affects the likelihood of viral latency. Despite the presence of these cis-acting sites, RNA polymerase II stalls (RNAPII) typically stalls near the TAR region post initiation of transcription at the LTR and terminates resulting in short abortive 59bp transcripts. Initially observed in a few stress-response genes and then extensively studied with HIV, RNAPII stalling is now understood to be a general transcription control mechanism in eukaryotes (80) (81) and in the HIV case is attributed to negative elongation factor (NELF) binding to TAR, DRB sensitivity-inducing factor (DSIF)(30), and the repressive nucleosome positioning along the LTR (82). In particular, the nucleosome arrangement seen at the LTR (82) has been linked to super-Poissonian stochastic fluctuations in transcription (83–85) where the expression of mRNAs from any particular locus occurs in infrequent, episodic bursts of transcription due to the inherent ON-OFF toggling of the LTR promoter.
This super-Poissonian transcriptional noise can be modeled by the classical two-state (a.k.a. “random-telegraph”) model where the RNAPII stalling represents an OFF state, elongation represents an ON state, and multiple RNAPII molecules can elongate while the LTR is in the ON state before it toggles to the OFF state. This toggling results in a burst of transcripts (each RNAPII molecule generating one mRNA) leading to large time-dependent variability in expression where cells can be OFF for long periods of time and then exhibit an intense burst of transcription for a short amount of time. The promoter toggling rates define the frequency and size of each transcriptional burst (i.e., number of mRNAs produced per burst) and in comparison to other human promoters, the HIV LTR is an exceptionally noisy promoter (Fig. 2) (86). This high noise is quite remarkable given the evolutionary plasticity of HIV; all things being equal, the virus presumably should select for LTR-promoter variants that generate the highest replicative fitness, which putatively would be expected to be a low noise promoter. However, as we have argued (14), this high expression noise could be a viral adaptation for enabling latency to act as a probabilistic bet-hedging decision that maximizes HIV transmission.
3.3.2 Strong Tat positive feedback
After initiating transcription at the LTR, RNAPII will elongate and then stall ~70 nucleotides downstream of initiation and the transactivation responsive (TAR) RNA region of the LTR forms a hairpin stem-loop structure within the first 59 nucleotides of each viral transcript (Fig. 2) (79). Tat protein recognizes and binds to TAR at its specific ‘bulge’ recognition site but without any apparent self-cooperative binding (60). Tat recruitment of the positive transcription elongation factor complex (pTEFb) complex to TAR results in a reversal of RNAPII stalling, boosting transcriptional elongation by 50–100 fold (79). The mechanisms of this reversal include the recruitment of cyclin T1 and CDK9, which phosphorylates the carboxyl-terminal domain of RNAP II (and NELF-E) leading to efficient elongation of transcription. Tat activity is also associated with recruitment of chromatin modifying and remodeling complexes to the LTR, leading to extensive post-translational modifications of the LTR chromatin (78, 79). Once initiated, Tat-mediated positive feedback, allows robust HIV transcription independent of cellular relaxation from activated to resting (76). Importantly, attenuation of wild-type Tat-feedback strength results in the circuit being unable to withstand the silencing effects of cellular relaxation and leads to establishment of a quiescent LTR transcriptional profile (Fig. 2).
3.3.3 The integrated circuit: feedback amplification of promoter noise generates a probabilistic ON–OFF switch
A well-known feature of positive-feedback circuitry is that it amplifies not only the mean level of expression, but also fluctuations. In HIV, Tat feedback amplifies bursty transcription from the LTR, causing both amplification of the mean and amplification of fluctuations to the point the circuit stochastically switches between an active and a quiescent expression state (42, 43). Critically, the resultant stochastic switching is inherently ‘probabilistic’ rather than ‘deterministic’. That is, every integrated provirus has a specific probability of establishing latency or active replication and in a population of infected cells (even an isogenic population), only a fraction are in either state. The magnitude of this probability, of say the virus choosing latency, depends on the parameters within the host cell at any given time; this includes the activation state of the cell, which reduces the probability of latency. This is fundamentally different from the deterministic case where all integrations at a specific locus would be expected to adopt the same fate.
The probabilistic nature of HIV fate regulation is a particular impediment for curative therapy and the elimination of latency. Even the strongest available LRAs result in only partial latent reactivation, even in idealized cell-culture settings where all cells have the same HIV-integration site (9–12, 87–90). This partial penetrance of reactivation is consistent with a noise-driven probabilistic switch underlying HIV latency—in contrast, for a deterministic system a strong enough activator could theoretically reverse latency in all cells. Next, we discuss strategies for targeting transcriptional fluctuations (noise) and positive feedback that may help overcome current barriers to curative therapy.
3.4 Disrupting the HIV Tat Circuit
3.4.1 Targeting Transcriptional Noise
In a recent study from our group, Dar et al. (15) demonstrated that noise modulation can be used as a form of ‘dithering’ to potentiate HIV reactivation from latency (Fig. 2). Transcriptional activators, such as tumor necrosis factor alpha (TNFα), activate the LTR by increasing the frequency of transcription initiation events (86, 91), essentially increasing the toggling rate from OFF to ON in the two-state random-telegraph model. In contrast, Dar et al. reasoned that increasing the transcriptional burst size—by extending the duration in the ON state while simultaneously decreasing the toggling frequency—would amplify noise and potentiate the effects of transcriptional activators. Such compounds, which modulate the width (variance) of a distribution rather than its mean, would be missed by traditional LRA screens. This noise enhancer effect is the transcriptional equivalent of how Bunsen burners act in physical chemistry by increasing thermal fluctuations to enable crossing of catalyst-lowered activation-energy barriers (i.e., increasing the kT in the Arrenhius equation).
Simulations of the two-state model predicated that noise enhancers, when paired with activators would provide synergistic viral reactivation (Fig. 2). To identify potential noise-modulating compounds, a diverse library of 1600 bioactive small molecules was screened in an isoclonal Jurkat cell line (Fig. 2). This Jurkat T cell line carried one integrated copy of the HIV LTR promoter expressing a short-lived green fluorescent protein (GFP) reporter (d2GFP) —the short GFP half-life focused the screen toward identifying compounds that modulate transcription. The screen identified more than 80 compounds that modulated noise and synergized with conventional transcriptional activators. These noise enhancers reactivated latent cells significantly better than existing best-in-class reactivation drug combinations (and with reduced off-target cytotoxicity). Lastly, just as discussed in other fate-switches where noise acts as an impulse to promote state change, the screen also discovered noise suppressors that stabilized latency (reduced latent to active switching).
3.4.2 Targeting Tat-mediated Positive Feedback
Computational modeling (Fig. 2) and analytical arguments showed that strong (i.e., high gain) Tat positive feedback is critical for HIV to maintain transcriptional autonomy to cellular relaxation (76); to achieve cell-state autonomy, positive feedback must be stronger than the ON à OFF LTR toggling rate (single-cell measurements show Tat feedback meets the high-gain requirement (76)). Consequently, simulations predict that attenuation of Tat feedback would generate spontaneous switching from active proliferation to the latent state and abrogate the HIV circuit’s resilience to cellular silencing (Fig. 2). This prediction was directly tested in donor-derived primary CD4+ T cells using a minimal LTR-Tat circuit. In this system, Tat protein was fused to a fluorescent reporter (Dendra) and a proteolysis domain (FKBP): Dendra fluorescence allowed monitoring of LTR activity while the FKBP fusion allowed for reversible attenuation of Tat feedback strength by altering Tat degradation via a small molecule (Shield-1). When HIV circuits with attenuated Tat positive-feedback strength underwent cellular transitioning from activated to resting-memory, and the associated transcriptional silencing, the buffering seen with wild-type Tat positive-feedback was lost and these attenuated-feedback circuits were silenced in a fashion consistent with host-cell dependence (Fig. 2)
These results highlight the susceptibility of the HIV circuitry to therapies directed at disrupting Tat-mediated positive feedback, either via directly targeting Tat or disrupting the Tat-TAR interaction. As an example of the former, disruption of Tat acetylation-deacetylation pathway can inhibit Tat-mediated transactivation of LTR transcription (92). Similarly, several Tat-TAR interaction disruptors have been discovered (93–95) including most recently the compound Didehydro-Cortistatin A (dCA) (96–98). dCA antiviral abilities are attributed to its binding of Tat’s TAR binding domain. Consistent with our understanding that disruption of Tat positive feedback leads to viral latency, the use of dCA was shown to be effective in suppressing HIV reactivation after stimulation of CD4+ T cells isolated from virally suppressed subjects (97).
4. THE CMV IE2 MASTER CIRCUIT: PROTECTION FROM ABORTIVE INFECTION
In contrast to HIV’s viral latency circuit, another aspect of viral-fate regulation is the operational tradeoff between response speed and amplitude, a constraint common to all signaling circuits (99, 100). This tradeoff—where a faster rate of initial increase is typically obtained at the expense of a higher steady-state level, i.e. a rate-vs.-level tradeoff—creates an evolutionary pressure when quick turn-on of a signaling molecule is essential but the signaling molecule is cytotoxic at high levels. Diverse biological signaling circuits—and even electrical circuits—face this fundamental rate-vs.-level tradeoff (100–102) including inflammatory cytokines (103), the mammalian fever response (104), and of course viral systems (105).
In particular, herpesviruses must quickly express viral genes that modulate the host-cell environment into a replication-favorable state, but these genes often yield cytotoxic products when expressed at high levels and can prematurely damage the cell before an optimal number of viral progeny are produced. It is now clear that herpesviruses, in particular CMV, have evolved specialized transcriptional ‘accelerator’ circuitry to overcome the rate-vs.-level tradeoff (59).
4.1 The IE2 Transcriptional ‘Accelerator’ Circuit circumvents the Rate-vs-Level Tradeoff
CMV’s viral gene-expression program (106) is initiated within the cell by expression of the viral 86-kDa transactivator protein Immediate-Early 2 (IE2). IE2 is a promiscuous transactivator of viral (and human) promoters and is essential for viral replication (107), but also highly cytotoxic (105, 108). Thus, CMV must quickly express IE2 to establish a replication-favorable environment but also limit IE2 levels to avoid prematurely compromising the cell’s ability to produce viral progeny. IE2, along with Immediate-Early 1 (IE1), is encoded by a precursor mRNA expressed from the CMV Major Immediate-Early (MIE) promoter, which directs all subsequent viral gene expression and is considered to be the chief regulator of the lytic cycle (107). The MIE promoter (MIEP) is exceptionally strong and encodes multiple transcription factor–binding sites within its ~500-nucleotide enhancer (109). The MIEP is also auto-repressed by IE2 via direct DNA binding to a 12-nucleotide cis repression sequence (crs) located between positions −13 and +1 relative to the transcriptional start site (110).
Teng et al. (59) mapped the structure and determined the functional consequence of this IE2 auto-repression circuit (Fig. 3A). They demonstrated that CMV infection in different cellular environments and in response to transcriptional activators (e.g. Valproic Acid (VPA), Trichostatin (TSA) or Tumor Necrosis Factor-α (TNF-α)) responds by accelerating IE2 expression without amplifying IE2 protein levels. This accelerator effect was measured using quantitative western blotting and single-cell time-lapse microscopy in cells undergoing infection by a recombinant CMV encoding yellow fluorescent protein (YFP) fused to the IE2 open reading frame (CMV IE2-YFP). Using an integrated approach that couples mathematical modeling with quantitative time-lapse microscopy, the study showed that IE2 negative feedback allows the virus to accelerate IE2 gene expression without any measureable increase in the steady-state expression level. Analysis of virus replication kinetics showed that transcriptional acceleration correlated with increases in virus replication fitness; a 9-fold increase in viral replication was observed relative to controls using a clinical isolate of CMV (strain TB40E).
Figure 3. Mapping and manipulating CMV’s transcriptional ‘accelerator’ circuit.
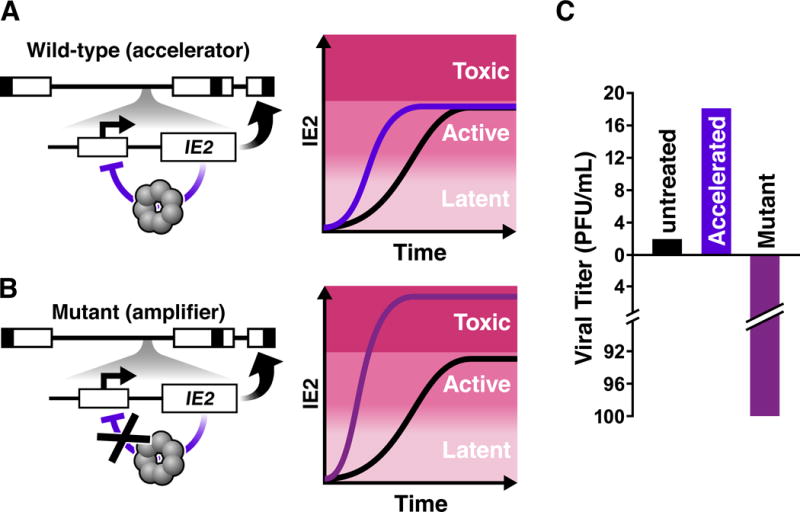
A) (Left) Schematic of the CMV genome (linear form) with the IE2 negative-feedback ‘accelerator’ circuit. As is common, boxes represent terminal and inverted repeat regions of the genome, respectively, while lines represent unique long and unique short genomic regions. The Major Immediate-Early (MIE) locus is encoded within the unique long region of the genome. For simplicity, one the MIE promoter and IE2 are depicted. IE2 is an obligate transactivator of CMV early genes (thick curved arrow). However, IE2 is also cytotoxic at high levels and auto-represses its own MIE promoter as a high-order homo-multimer, forming a highly self-cooperative negative-feedback circuit that limits IE2 expression (purple blunted arrow). (right) Schematic of single-cell imaging trajectories of IE2 protein levels, adapted from (59). This highly cooperative IE2 negative feedback allows accelerated IE2 kinetics but precludes IE2 levels from increasing to levels where IE2 could be cytotoxic (i.e. acts as an ‘accelerator’).
B) (left) Schematic of the CMV recombinant mutant ‘amplifier’ circuit where two nucleotides in the 14 base-pair IE2 DNA-binding site (the ‘cis repression sequence’) within the MIE promoter were mutated. (right) The negative-feedback ‘amplifier’ mutant generates increased IE2 levels, reaching into the cytotoxic regime, upon promoter activation.
C) The IE2 circuit’s effect on viral replicative fitness. Accelerating IE2 accumulation in single-cells by a few hours yields a replicative fitness advantage for the virus (~5–10 fold depending on viral strain). In contrast, disrupting IE2 feedback, by just a two-nucleotide mutation in the MIE promoter, amplifies IE2 levels in individual cells, causing substantial cytotoxicity to the host cell and reducing viral output > 100-fold (Adapted from (59)).
4.2 Core Components of Transcriptional ‘Accelerator’ Circuitry
4.2.1 Highly self-cooperative negative feedback (H≈7)
Based on theoretical arguments originally developed for electrical circuits (100–102) and subsequently applied to synthetic biology (99, 100), it was hypothesized that acceleration of IE2-expression rate without amplification of steady-state IE2 protein levels would likely utilize negative-feedback circuitry. To test this hypothesis, Teng et al. employed a number of quantitative approaches.
First, rate-balance analysis of simplified computational models of the circuit was performed and single-cell IE2-YFP trajectories from infected cells were fit to mathematical model using nonlinear least squares regression to estimate the H value of the IE2 negative feedback. H ≈ 7 generated the best fit to the single-cell time-lapse imaging data. Second, Teng et al. constructed a synthetic reporter cell-line to measure H outside the context of the virus via a dose-response-type approach. Traditionally, ‘open-loop’ systems (i.e., whereby feedback is removed from the system) are used to measure H (60) but, when the dose is cytotoxic, such as IE2, the traditional open-loop dose-response method destroys the cell before the response can be fully measured. To circumvent this cytotoxicity problem, Teng et al. developed a ‘closed-loop’ single-cell analysis method to analyze how a circuit’s output (steady-state protein levels) saturates as a function of increasing promoter activation and varying H values. This method essentially measures the change in steady-state levels as a function of increasing promoter strength. A lentiviral vector encoding the full-length MIEP driving IE2 and GFP (MIEP-IE2-GFP) was compared to a minimal non-feedback lentiviral vector encoding the full-length MIEP driving GFP (MIEP-mCherry-GFP), which acts as the non-feedback control circuit. By increasing the IEP activity using transcriptional activators (e.g. TSA or VPA) the response of each circuit can be measured and these responses can then be compared to theoretically predicted responses for varying H levels. As expected for the non-feedback circuit, a linear increase in activator resulted in a linear increase in GFP steady-state levels. However, for the MIEP-IE2-GFP negative-feedback circuit, the equivalent linear increase in activator input results in a significant saturation in GFP steady state. This saturation in the GFP steady-state values was consistent with the single-cell nonlinear regression analysis and indicated H ≈ 6–8 for IE2 negative feedback.
4.2.2 H≈7 achieved via IE2 protein homo-multimerization
To explore the potential molecular mechanisms whereby H ≈ 6–8 was achieved, Teng et al. (59) examined protein homo-multimerization, a common mechanism for achieving ultra-sensitive or self-cooperative responses (58). IE2 homo-multimerization during CMV infection was examined using polarization anisotropy Förster Resonance Energy transfer (FRET) imaging which can differentiate between monomers and higher-order homo-multimers (111). IE2-YFP exhibited a strong homo-FRET signal at sub-nuclear transcriptional centers, indicating high-order IE2 homo-multimerization, and comparison with theoretical models (112) was used to estimate the number of individual IE2 monomers that might be interacting within an IE2 homo-multimer to generate the measured FRET signal. While the model cannot precisely calculate the number of monomers making up the homo-multimer—since the distance between individual IE2 monomers is not known—a lower limit on the number of IE2 monomers within the homo-multimer can be estimated with confidence, under the most conservative assumption that the distance between each IE2-YFP monomer is the diameter of the YFP molecule (24 Å). Under this maximally conservative assumption, the measured anisotropy shift (r ≥ 0.5 →r ≈ 0.1) is consistent with an IE2 homo-multimer composed of at least five to six IE2 monomers.
These data argued that IE2 homo-multimerization is a core factor in establishing the high Hill coefficient of this transcriptional negative-feedback circuit and that protein homo-multimerization underlies the circuit’s ability to act as an accelerator. Overall, Teng et al. demonstrated that the IE2 circuit encodes a highly self-cooperative negative feedback with an H value sufficient to generate an accelerator that effectively abolishes IE2 amplification under different inputs. IE2 self-cooprativity was verified using three independent measurements: (i) fitting single-cell CMV IE2-YFP trajectories to a mathematical model, (ii) a novel dose-reponse analysis of the IE2 feedback circuit, and (iii) FRET-based imaging of IE2.
4.3 Disrupting the CMV IE2 circuit
Teng et al. (59) where able to examine a recombinant CMV mutant where negative-feedback was ablated via genetic mutation of the crs binding for IE2 on the MIEP. They examined both the full-length virus mutant and a minimal circuit version of this mutant. In the minimal lentiviral vector setting this Δcrs mutant circuit acted as an ‘amplifier’ and produced a ~1.5-fold amplification of IE2 expression levels. In the context of the full-length virus, the Δcrs mutant exhibited significantly decelerated IE2 expression kinetics and when MIEP expression was activated with VPA, or similar perturbation, IE2 levels were amplified rather than accelerated (Fig. 3B). In other words, the Δcrs mutation resulted in the accelerator phenotype being converted into an ‘amplifier’ phenotype. Strikingly, this effect also generated a severe two-log fitness defect for the virus (Fig. 3C).
These results indicate that disruption of the IE2 transcriptional accelerator circuit, either through disruption of IE2 protein-protein interactions or through disruption of IE2 protein-DNA interactions, may inhibit viral replication. More recent work used the synthetic MIEP-IE2-GFP minimal circuit as a platform for cell-based screening to identify small-molecule chemicals that could inhibit the IE2-crs interaction thereby disrupting the negative-feedback circuit (113) and mimicking the genetic disruption of the Δcrs mutant. A number of small-molecule chemicals were identified in this screen of the synthetic IE2-feedback circuit. From the perspectives of evolution and drug-resistance, the IE2-acceletrator circuit presents an intriguing antiviral target in part because viral escape mutants (i.e., amplifier mutants) were inherently transcriptionally decelerated and exhibited significantly diminished replicative fitness compared to wild-type virus (59).
7 OUTLOOK
Given the rapid nature of viral evolution, which generates drug-resistant escape mutants, new antiviral therapies—ideally, targeting novel molecular targets—are a pressing public-health need. The discovery of virus-encoded transcriptional ‘master’ circuits that can autonomously control viral expression programs in diverse virus families such as CMV (Baltimore virus classification Group I) and HIV (Baltimore virus classification group VI), offers a potential new class of antiviral targets. We and others have shown that these transcriptional circuits can be disrupted using small-molecule chemicals (15, 59, 97, 113, 114). Transcriptional circuit disruption could represent intrinsic combination-antiviral therapy where evolutionary escape from circuit ‘disruptors’ requires simultaneous evolution of both the cis regulatory element (e.g., the DNA-binding site) and the trans element (e.g., the transcription factor) for the circuit’s function to be recapitulated. Hence, despite the conventional view that such circuits are challenging therapeutic targets—protein-protein and protein-DNA interactions have conventionally been considered “undruggable” (115)—disruption of viral transcriptional circuits appears possible and carries the potential to be escape-resistant antiviral therapy.
References
- 1.UNAIDS. Global AIDS Response Progress Reporting 2016. World Health Organization Press: Geneva, Switzerland; 2016. [Google Scholar]
- 2.De Clercq E, Li G. Approved Antiviral Drugs over the Past 50 Years. Clin Microbiol Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57(Suppl 4):S178–81. doi: 10.1093/cid/cit629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev. 2010;23:689–712. doi: 10.1128/CMR.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–7. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 6.Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol. 2007;5:95–106. doi: 10.1038/nrmicro1580. [DOI] [PubMed] [Google Scholar]
- 7.Pierson T, McArthur J, Siliciano RF. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. 2000;18:665–708. doi: 10.1146/annurev.immunol.18.1.665. [DOI] [PubMed] [Google Scholar]
- 8.Weinberger AD, Weinberger LS. Stochastic Fate Selection in HIV-Infected Patients. Cell. 2013;155:497–99. doi: 10.1016/j.cell.2013.09.039. [DOI] [PubMed] [Google Scholar]
- 9.Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet. 2008;40:466–70. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- 10.Burnett JC, Lim KI, Calafi A, Rossi JJ, Schaffer DV, Arkin AP. Combinatorial latency reactivation for HIV-1 subtypes and variants. J Virol. 2010;84:5958–74. doi: 10.1128/JVI.00161-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods. 2011;53:54–61. doi: 10.1016/j.ymeth.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–51. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rouzine IM, Weinberger AD, Weinberger LS. An Evolutionary Role for HIV Latency in Enhancing Viral Transmission. Cell. 2015;160:1002–12. doi: 10.1016/j.cell.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dar RD, Hosmane NN, Arkin MR, Siliciano RF, Weinberger LS. Screening for noise in gene expression identifies drug synergies. Science. 2014;344:1392–6. doi: 10.1126/science.1250220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cannon MJ. Congenital cytomegalovirus (CMV) epidemiology and awareness. J Clin Virol. 2009;46(Suppl 4):S6–10. doi: 10.1016/j.jcv.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Barry PA, Lockridge KM, Salamat S, Tinling SP, Yue Y, et al. Nonhuman primate models of intrauterine cytomegalovirus infection. ILAR J. 2006;47:49–64. doi: 10.1093/ilar.47.1.49. [DOI] [PubMed] [Google Scholar]
- 18.Hotez PJ. Neglected infections of poverty in the United States of America. PLoS Negl Trop Dis. 2008;2:e256. doi: 10.1371/journal.pntd.0000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prichard MN, Kern ER. The search for new therapies for human cytomegalovirus infections. Virus Res. 2011;157:212–21. doi: 10.1016/j.virusres.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. 2013;26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2:a007161. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roush W. Antisense aims for a renaissance. Science. 1997;276:1192–3. doi: 10.1126/science.276.5316.1192. [DOI] [PubMed] [Google Scholar]
- 23.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–40. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulamba GB, Hu A, Azad RF, Anderson KP, Coen DM. Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922) Antimicrob Agents Chemother. 1998;42:971–3. doi: 10.1128/aac.42.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–9. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balaban NQ. Persistence: mechanisms for triggering and enhancing phenotypic variability. Curr Opin Genet Dev. 2011;21:768–75. doi: 10.1016/j.gde.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Golding I. Single-Cell Studies of Phage lambda: Hidden Treasures Under Occam’s Rug. Annu Rev Virol. 2016;3:453–72. doi: 10.1146/annurev-virology-110615-042127. [DOI] [PubMed] [Google Scholar]
- 29.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–42. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 30.Siliciano RF, Greene WC. HIV latency. Cold Spring Harbor perspectives in medicine. 2011;1 doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinberger LS. A minimal fate-selection switch. Curr Opin Cell Biol. 2015;37:111–8. doi: 10.1016/j.ceb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Hasty J, McMillen D, Collins JJ. Engineered gene circuits. Nature. 2002;420:224–30. doi: 10.1038/nature01257. [DOI] [PubMed] [Google Scholar]
- 33.Nandagopal N, Elowitz MB. Synthetic biology: integrated gene circuits. Science. 2011;333:1244–8. doi: 10.1126/science.1207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ptashne M, Johnson AD, Pabo CO. A genetic switch in a bacterial virus. Sci Am. 1982;247:128–30. 32, 34–40. doi: 10.1038/scientificamerican1182-128. [DOI] [PubMed] [Google Scholar]
- 35.Coffin J, Swanstrom R. HIV pathogenesis: dynamics and genetics of viral populations and infected cells. Cold Spring Harb Perspect Med. 2013;3:a012526. doi: 10.1101/cshperspect.a012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delbruck M. The Growth of Bacteriophage and Lysis of the Host. J Gen Physiol. 1940;23:643–60. doi: 10.1085/jgp.23.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis EL, Delbruck M. The Growth of Bacteriophage. J Gen Physiol. 1939;22:365–84. doi: 10.1085/jgp.22.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics. 1998;149:1633–48. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh A, Weinberger LS. Stochastic gene expression as a molecular switch for viral latency. Curr Opin Microbiol. 2009;12:460–6. doi: 10.1016/j.mib.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.St-Pierre F, Endy D. Determination of cell fate selection during phage lambda infection. Proc Natl Acad Sci U S A. 2008;105:20705–10. doi: 10.1073/pnas.0808831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golding I. Decision making in living cells: lessons from a simple system. Annu Rev Biophys. 2011;40:63–80. doi: 10.1146/annurev-biophys-042910-155227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–82. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Weinberger LS. A minimal fate-selection switch. Current opinion in cell biology. 2015;37:111–8. doi: 10.1016/j.ceb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Suel GM, Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowitz MB. Tunability and noise dependence in differentiation dynamics. Science. 2007;315:1716–9. doi: 10.1126/science.1137455. [DOI] [PubMed] [Google Scholar]
- 45.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–82. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokolik C, Liu Y, Bauer D, McPherson J, Broeker M, et al. Transcription factor competition allows embryonic stem cells to distinguish authentic signals from noise. Cell Syst. 2015;1:117–29. doi: 10.1016/j.cels.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Symonds ME. Adipose tissue biology. Springer Science & Business Media; 2011. [Google Scholar]
- 49.Ahrends R, Ota A, Kovary KM, Kudo T, Park BO, Teruel MN. Controlling low rates of cell differentiation through noise and ultrahigh feedback. Science. 2014;344:1384–9. doi: 10.1126/science.1252079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–26. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–7. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy KF, Adams RM, Wang X, Balazsi G, Collins JJ. Tuning and controlling gene expression noise in synthetic gene networks. Nucleic Acids Res. 2010;38:2712–26. doi: 10.1093/nar/gkq091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsimring LS. Noise in biology. Rep Prog Phys. 2014;77:026601. doi: 10.1088/0034-4885/77/2/026601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Austin DW, Allen MS, McCollum JM, Dar RD, Wilgus JR, et al. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–11. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- 55.Shah NA, Sarkar CA. Robust network topologies for generating switch-like cellular responses. PLoS Comput Biol. 2011;7:e1002085. doi: 10.1371/journal.pcbi.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolf DM, Arkin AP. Motifs, modules and games in bacteria. Curr Opin Microbiol. 2003;6:125–34. doi: 10.1016/s1369-5274(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 57.Brandman O, Meyer T. Feedback loops shape cellular signals in space and time. Science. 2008;322:390–5. doi: 10.1126/science.1160617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrell JE, Jr, Ha SH. Ultrasensitivity part I: Michaelian responses and zero-order ultrasensitivity. Trends Biochem Sci. 2014;39:496–503. doi: 10.1016/j.tibs.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, et al. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151:1569–80. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinberger LS, Shenk T. An HIV feedback resistor: auto-regulatory circuit deactivator and noise buffer. PLoS Biol. 2007;5:e9. doi: 10.1371/journal.pbio.0050009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012;2:a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 63.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, et al. 24 hours in the life of HIV-1 in a T cell line. PLoS pathogens. 2013;9:e1003161. doi: 10.1371/journal.ppat.1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–91. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 65.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 66.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 67.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–5. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 68.Hill AL, Rosenbloom DI, Fu F, Nowak MA, Siliciano RF. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc Natl Acad Sci U S A. 2014;111:13475–80. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sherrill-Mix S, Lewinski MK, Famiglietti M, Bosque A, Malani N, et al. HIV latency and integration site placement in five cell-based models. Retrovirology. 2013;10:90. doi: 10.1186/1742-4690-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brady T, Agosto LM, Malani N, Berry CC, O’Doherty U, Bushman F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. AIDS. 2009;23:1461–71. doi: 10.1097/QAD.0b013e32832caf28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology. 2013;139:277–84. doi: 10.1111/imm.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, et al. Dynamics of epigenetic regulation at the single-cell level. Science. 2016;351:720–4. doi: 10.1126/science.aab2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitney JB, Hill A, Sanisetty S, Penaloza-MacMaster P, Liu J, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014 doi: 10.1038/nature13594. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dahabieh MS, Ooms M, Simon V, Sadowski I. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. Journal of virology. 2013;87:4716–27. doi: 10.1128/JVI.03478-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calvanese V, Chavez L, Laurent T, Ding S, Verdin E. Dual-color HIV reporters trace a population of latently infected cells and enable their purification. Virology. 2013;446:283–92. doi: 10.1016/j.virol.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Razooky BS, Pai A, Aull K, Rouzine IM, Weinberger LS. A Hardwired HIV Latency Program. Cell. 2015;160:990–1001. doi: 10.1016/j.cell.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chavez L, Calvanese V, Verdin E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS pathogens. 2015;11:e1004955. doi: 10.1371/journal.ppat.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455:328–39. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karn J, Stoltzfus CM. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nature reviews Genetics. 2012;13:720–31. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coulon A, Chow CC, Singer RH, Larson DR. Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nature reviews Genetics. 2013;14:572–84. doi: 10.1038/nrg3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bai L, Charvin G, Siggia ED, Cross FR. Nucleosome-depleted regions in cell-cycle-regulated promoters ensure reliable gene expression in every cell cycle. Dev Cell. 2010;18:544–55. doi: 10.1016/j.devcel.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008;18:1084–91. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown CR, Boeger H. Nucleosomal promoter variation generates gene expression noise. Proc Natl Acad Sci U S A. 2014;111:17893–8. doi: 10.1073/pnas.1417527111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17454–9. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hakre S, Chavez L, Shirakawa K, Verdin E. Epigenetic regulation of HIV latency. Current opinion in HIV and AIDS. 2011;6:19–24. doi: 10.1097/COH.0b013e3283412384. [DOI] [PubMed] [Google Scholar]
- 88.Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends in microbiology. 2013;21:277–85. doi: 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burnett JC, Lim KI, Calafi A, Rossi JJ, Schaffer DV, Arkin AP. Combinatorial latency reactivation for HIV-1 subtypes and variants. Journal of virology. 2010;84:5958–74. doi: 10.1128/JVI.00161-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–5. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singh A, Razooky B, Cox CD, Simpson ML, Weinberger LS. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophysical journal. 2010;98:L32–4. doi: 10.1016/j.bpj.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsu MC, Dhingra U, Earley JV, Holly M, Keith D, et al. Inhibition of type 1 human immunodeficiency virus replication by a tat antagonist to which the virus remains sensitive after prolonged exposure in vitro. Proc Natl Acad Sci U S A. 1993;90:6395–9. doi: 10.1073/pnas.90.14.6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hsu MC, Schutt AD, Holly M, Slice LW, Sherman MI, et al. Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science. 1991;254:1799–802. doi: 10.1126/science.1763331. [DOI] [PubMed] [Google Scholar]
- 95.Hwang S, Tamilarasu N, Kibler K, Cao H, Ali A, et al. Discovery of a small molecule Tat-trans-activation-responsive RNA antagonist that potently inhibits human immunodeficiency virus-1 replication. J Biol Chem. 2003;278:39092–103. doi: 10.1074/jbc.M301749200. [DOI] [PubMed] [Google Scholar]
- 96.Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell host & microbe. 2012;12:97–108. doi: 10.1016/j.chom.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio. 2015;6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mediouni S, Jablonski J, Paris JJ, Clementz MA, Thenin-Houssier S, et al. Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Current HIV research. 2015;13:64–79. doi: 10.2174/1570162x13666150121111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alon U. An introduction to systems biology : design principles of biological circuits. Boca Raton, FL: Chapman & Hall/CRC; 2007. p. xvi.p. 301. [4] p. of plates pp. [Google Scholar]
- 100.Savageau MA. Biochemical systems analysis : a study of function and design in molecular biology. Reading, Mass: Addison-Wesley Pub. Co., Advanced Book Program; 1976. p. xvii.p. 379. [Google Scholar]
- 101.Black HS. Stabilized feed-back amplifiers (Reprinted from Electrical Engineering, vol 53, pg 114–120, 1934) Proceedings of the Ieee. 1999;87:379–85. [Google Scholar]
- 102.Rosenfeld N, Elowitz MB, Alon U. Negative autoregulation speeds the response times of transcription networks. J Mol Biol. 2002;323:785–93. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- 103.Cauwels A, Brouckaert P. Survival of TNF toxicity: dependence on caspases and NO. Arch Biochem Biophys. 2007;462:132–9. doi: 10.1016/j.abb.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 104.Roth J, Rummel C, Barth SW, Gerstberger R, Hubschle T. Molecular aspects of fever and hyperthermia. Neurol Clin. 2006;24:421–39. v. doi: 10.1016/j.ncl.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 105.Dwarakanath RS, Clark CL, McElroy AK, Spector DH. The use of recombinant baculoviruses for sustained expression of human cytomegalovirus immediate early proteins in fibroblasts. Virology. 2001;284:297–307. doi: 10.1006/viro.2001.0924. [DOI] [PubMed] [Google Scholar]
- 106.Mocarski ES, Shenk T, Pass RF. Cytomegaloviruses. In: Knipe DM, editor. Fields’ virology. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 2708–72. [Google Scholar]
- 107.Stinski MF, Petrik DT. Functional roles of the human cytomegalovirus essential IE86 protein. Curr Top Microbiol Immunol. 2008;325:133–52. doi: 10.1007/978-3-540-77349-8_8. [DOI] [PubMed] [Google Scholar]
- 108.Sanders RL, Clark CL, Morello CS, Spector DH. Development of cell lines that provide tightly controlled temporal translation of the human cytomegalovirus IE2 proteins for complementation and functional analyses of growth-impaired and nonviable IE2 mutant viruses. J Virol. 2008;82:7059–77. doi: 10.1128/JVI.00675-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stinski MF, Isomura H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med Microbiol Immunol. 2008;197:223–31. doi: 10.1007/s00430-007-0069-7. [DOI] [PubMed] [Google Scholar]
- 110.Macias MP, Stinski MF. An in vitro system for human cytomegalovirus immediate early 2 protein (IE2)-mediated site-dependent repression of transcription and direct binding of IE2 to the major immediate early promoter. Proc Natl Acad Sci U S A. 1993;90:707–11. doi: 10.1073/pnas.90.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gautier I, Tramier M, Durieux C, Coppey J, Pansu RB, et al. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys J. 2001;80:3000–8. doi: 10.1016/S0006-3495(01)76265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Runnels LW, Scarlata SF. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys J. 1995;69:1569–83. doi: 10.1016/S0006-3495(95)80030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.LS WEINBERGER. Methods for treating a cytomegalovirus infection. US: Google Patents; 2014. [Google Scholar]
- 114.Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci U S A. 2005;102:17448–53. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21:1102–14. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–6. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 117.Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor JM, et al. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun. 2016;7:12248. doi: 10.1038/ncomms12248. [DOI] [PMC free article] [PubMed] [Google Scholar]