

Abstract
Myocardial infarction (MI) is a life-threatening condition that can occur when blood flow to the heart is interrupted due to a blockage in one or more of the coronary vessels. Current treatments of MI rapidly restore blood flow to the affected myocardium using thrombolytic agents or angioplasty. Adverse effects including inflammation, tissue necrosis, and ventricular dysfunction are, however, not uncommon following reperfusion therapy. These conditions are thought to be caused by a sudden influx of reactive oxygen species (ROS) to the affected myocardium. We employed the model of left anterior descending artery ligation/reperfusion surgery in a rat model to show that ischemia/reperfusion injury is associated with the formation of toxic DNA-protein cross-links (DPCs) in cardiomyocytes. Mass spectrometry based experiments have revealed that these conjugates were formed by a free radical mechanism and involved thymidine residues of DNA and tyrosine side chains of proteins (dT-Tyr). Quantitative proteomics experiments have identified nearly 90 proteins participating in hydroxyl radical-induced DPC formation, including ROS scavengers, contractile proteins, and regulators of apoptosis. Global proteome changes were less pronounced and included increased expression of mitochondrial proteins required for aerobic respiration and biomarkers of sarcomere breakdown following ischemia/reperfusion injury. Overall, our results are consistent with a model where sudden return of oxygen to ischemic tissues induces oxidative stress, inflammation, and the formation of DNA-protein cross-links that may contribute to reperfusion injury by dysregulating gene expression and inducing cardiomyocyte death.
Keywords: ischemia-reperfusion injury, mass spectrometry based proteomics, DNA-protein cross-linking, reactive oxygen species, cardiomyocytes
Graphical abstract
Introduction
Heart disease is the leading cause of mortality for both men and women in the United States, accounting for 1 out of every 4 deaths [1,2]. Coronary heart disease disrupts the flow of oxygenated blood to the heart, potentially leading to myocardial infarction (MI) [1,2]. Current MI therapies are based on the revascularization of the injured area using thrombolytic or angioplasty techniques in an attempt to rapidly return oxygenated blood flow to the damaged myocardium. Reperfusion-induced return of oxygenated blood to the affected myocardium, however, can lead to reactivation of the electron transport chain, producing large amounts of reactive oxygen species (ROS) such as superoxide anions (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•) [3,4]. Furthermore, neutrophils accumulating in the infarcted myocardium can mediate tissue damage by releasing matrix-degrading enzymes and peroxides for downstream signaling [5–7].
ROS generated during ischemia-reperfusion injury can cause oxidative damage to cellular biomolecules including lipids, proteins, and DNA [8,9]. Several types of ROS-induced DNA damage can be formed including DNA strand breaks, nucleobase monoadducts, and covalent DNA-DNA and DNA-protein cross-links (DPCs) [10–13]. DPCs are super-bulky DNA lesions containing proteins irreversibly attached to DNA strands [12–14]. ROS-induced DPCs include covalent cross-links between ε-amino group of lysine and spirodiiminodihydantoin in DNA [15], lysine and guanidinohydantoine in DNA [16], tyrosine, and 8-oxoguanosine in DNA [15], lysine and guanine in DNA (Nε-lysine guanin-8-yl cross-links) [15–17], and tyrosine and thymidine in DNA (dT-Tyr) (Scheme 1) [18,19]. If not repaired, DPCs can induce DNA fragmentation and cell death [12–14].
Scheme 1.
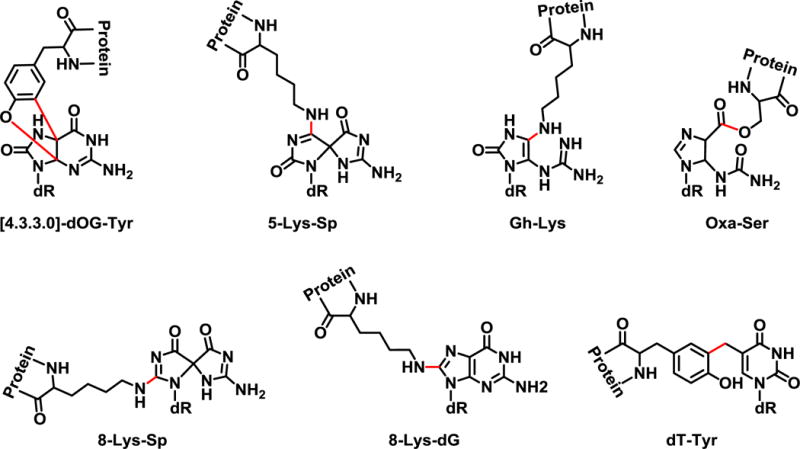
Structures of oxidant-induced DNA-protein cross-links. Chemical structures of previously identified oxidant-induced DNA-protein cross-links including; 2-amino-3-(2-amino-10-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-4,11-dioxo-3,4-dihydro-4a,9a-(epiminomethanoimino)benzofuro[2,3-d]pyrimidin-6-yl)propanoic acid ([4.3.3.0] dOG-Tyr), N6-(7-amino-1-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,9-dioxo-1,3,6,8-tetraazaspiro[4.4]nona-3,7-dien-4-yl)lysine (5-Lys-Sp), N6-(5-guanidino-1-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-2,3-dihydro-1H-imidazol-4-yl)lysine (Gh-Lys), O-(1-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-ureido-4,5-dihydro-1H-imidazole-4-carbonyl)serine (Oxa-Ser), N6-(7-amino-1-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-4,9-dioxo-1,3,6,8-tetraazaspiro[4.4]nona-2,7-dien-2-yl)lysine (8-Lys-Sp), N6-(2-amino-9-(4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-8-yl)lysine (8-Lys-dG), and 2-amino-3-(4-hydroxy-3-((1-((2R,4R,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)phenyl)propanoic acid (dT-Tyr).
In the present study, we employed left anterior descending artery (LAD) ligation/reperfusion surgery [20] to examine the formation of radical-induced DPCs in cardiomyocytes after ischemia-reperfusion injury in a rat model. A sensitive isotope dilution nanoLC-NSI+-MS/MS assay developed in our laboratory was used to quantify the formation of hydroxyl radical-mediated DPCs, while mass spectrometry based quantitative proteomics was employed to examine the proteins participating in DPC formation and to detect changes in the global cardiomyocyte proteome following ischemia-reperfusion injury.
Experimental
Chemicals and Reagents
2′-deoxythymidine, Boc-L-Tyr-OH, trifluoroacetic acid, Nuclease P1 from Penicillium citrinum, and DL-dithiothreitol were purchased from Sigma-Aldrich (St. Louis, MO). Pierce™ BCA Protein Assay kit, Pierce™ Quantitative Colorimetric Peptide Assay, Pierce Quantitative Colorimetric Peptide Assay, and TMT 6-plex reagents were purchased from Thermo Fischer Scientific (Waltham, MA). Glacial acetic acid, hydrochloric acid, and acetonitrile were obtained from Fisher Scientific (Buchs, Switzerland). Mass spectrometry grade Trypsin Gold was purchased from Promega (Madison, WI). Cell lysis solution, protein precipitation solution, and RNaseA were purchased from Qiagen (Hilden, Germany). Amicon 3K filters were purchased from Millapore (Darmstadt, Germany). Omega Nanosep 10K Omega filters were obtained from PALL Life Science (Port Washington, NY). The OxiSelect™ 8-iso-prostaglandin F2α ELISA kit was purchased from Cell Biolabs (San Diego, CA). Matrin-3 and glyceraldehyde-3-phosphate dehydrogenase primary antibody and goat anti-rabbit IgG-HRP secondary antibody were purchased from Santa Cruz Biotechnology (Dallas, TX). Superoxide dismutase [Mn] mitochondrial and pyruvate dehydrogenase E1 beta subunit primary antibody were purchased from Abcam (Cambridge, UK).
Animal safety statement
Age-matched male Sprague-Dawley rats were randomly divided into two experimental groups: left anterior descending artery (LAD) ligation surgery/reperfusion group and sham surgery control group. Animals were housed in standard rodent cages with 12-hour light-dark cycles and were given food and water ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota. All studies were conducted in accordance with the United States Department of Health and Human Services National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Animal model of myocardial infarction/reperfusion
A subset of animals within the weight range of 150–175 g underwent thoracic surgery. Anesthesia was induced with isoflurane gas (2–5% in 100% oxygen), and subsequently animals were intubated and ventilated. A thoracotomy was performed exposing the heart, and the left anterior descending (LAD) coronary artery was ligated to simulate myocardial infarction for one hour as previously described [20]. Following 1 h, the ligation was removed to restore blood flow for an additional 30 minutes to simulate reperfusion. Immediately after 30 minutes, the animals were euthanized, and the hearts excised for cardiomyocyte isolation via Langendorff reperfusion method as previously described [21]. Typical cell recovery ranged from 1.7 × 106 to 5.0 × 106 cardiomyocytes per heart, with approximately 80% cell viability. Prior to the thoracic surgery, blood was drawn from the tail vein of each animal to serve as the pre-surgery blood sample for both cardiac troponin-i (cTni) and 8-iso-prostaglandin F2α measurement assays described below. Immediately after the 30-min reperfusion, an additional blood draw occurred to serve as the post-surgery blood sample for both assays.
Troponin-I (cTni) ELISA assay
cTni levels in plasma pre- and post-LAD ligation/reperfusion was measured using the i-STAT cardiac troponin I cartridge test (Abbott, Chicago, IL) using a VetScan i-STAT 1 handheld analyzer (Abaxis, Union City, CA) following manufacturer’s protocol as described previously [22,23].
8-iso-prostaglandin F2α (8-iso-PGF2α) ELISA assay
8-iso-PGF2α levels in plasma pre- and post-LAD ligation/reperfusion was measured using the OxiSelect™ 8-iso-prostoglandin F2α ELISA kit following the manufacturer’s protocol. In short, 50 μL plasma samples (in triplicate) were treated with 2N NaOH at 45 °C for 2 hours to hydrolyze lipoprotein or phospholipids coupled to 8-iso-PGF2α. The solutions were neutralized with 10 N HCl to pH 6.0 – 8.0. The goat anti-rabbit antibody-coated 96-well plate was incubated with 100 μL anti-8-iso-PGF2α antibody (1:1000 dilution) at 25 °C for 1 hour with gentle shaking. A serial dilution of 8-iso-PGF2α standards were prepared, and 55 μL of the standards or the prepared plasma samples were combined with 55 μL of a provided 8-iso-PGF2α-HRP conjugate. The anti-8-iso-PGF2α antibody-treated plate was incubated with the prepared standards or samples at 25 °C for 1 h with gentle shaking, followed by incubation with 100 μL HRP substrate at room temperature for 30 min with gentle shaking. The HRP reaction was quenched by adding 100 μL stop solution, and the plates were immediately analyzed on a BioTek Synergy H1 microplate reader (absorbance at 450 nm).
5-hydroxymethyl-2′-deoxyuracil (5′-hm-dU)
2′-Deoxythymidine (100 mg, 0.3875 mmol) was suspended in 25 mL 0.1M phosphate buffer (pH 7.5). Sodium persulfate (21.5 mg, 0.3875 mmol) was added, and the solution was heated at 75 °C for 4 h. The reaction mixtures were separated by semi-preparative HPLC using a Zorbex XDB C18 (250 mm × 10 mm, 5 μm) column eluted with a linear gradient of water (A) and acetonitrile (B) at 3 mL/min. Solvent composition was changed from 3% B to 5% B over 30 min, then increased to 50% over 5 min, held at 50% for 2 min, decreased to 3% over 2 minutes, and finally held at 3% for 4 min for re-equilibration. Under these conditions, 5′-hm-dU eluted at 8.7 min. ESI+-MS/MS (5′-hm-dU): m/z = 259.2 [M+H]+ → 143.1[M − dR + H]+.
2-amino-3-(4-hydroxy-3-((1-((2R,4R,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)phenyl)propanoic acid (dT-Tyr)
5′-hm-dU (180 mg, 0.4128 mmol) was dissolved in 1 mL glacial acetic acid and 6 M HCl. Boc-L-Tyr-OH (116 mg, 0.4541 mmol) was added and the solution was refluxed (110 °C) for 1 h. Following neutralization to pH 7.0 using 1 M NaOH, the precipitate was filtered and the supernatant separated by semi-preparative HPLC on a Zorbex XDB C18 (250 mm × 10 mm, 5 μm) column eluted with a linear gradient of water (A) and acetonitrile (B). The solvent composition was held at 2% B for 5 min, increased to 30% B in 5 min, then held at 30% B for 2 min, then increased to 75% B in 1 min, held at 75% B for 1 min, decreased to 2% B in 15 min, and finally held at 2% B for 10 min for re-equilibration. Under these conditions, dT-Boc-L-Tyr-OH eluted at 19.2 min. ESI+-MS/MS (dT-Boc-L-Tyr-OH): m/z = 522.3 [M + H]+ → 406.4 [M − dR + H]+, 360.4 [M − dR − CO2H + H]+. Following HPLC purification, dT-Boc-L-Tyr-OH was dissolved in 50% TFA and incubated at room temperature for 1 hour. The solution was neutralized to pH 7.0, and the supernatant was separated by the same procedure described above. Under these conditions, dT-Tyr eluted at 10 min. ESI+-MS/MS (dT-Tyr): m/z = 422.1 [M + H]+ → 306.1 [M –dR + H]+ and 260.1[M − dR − CO2H + H]+.
DPC extraction from rat cardiomyocytes
Isolated rat cardiomyocytes (~ 1.0 × 106) were re-suspended in 1 mL of cell lysis solution (Qiagen, Hilden, Germany) and pipetted until the cells were completely lysed. The resulting lysates were treated with 40 μg RNaseA (10 μL, Qiagen) overnight at room temperature. Following incubation, each sample was treated with 750 μL of protein precipitation solution (Qiagen, Hilden, Germany) and vortexed for one minute to yield a white turbid solution. Each sample was then centrifuged at 2500 rcf for 10 minutes to pellet the protein precipitate, which was subsequently used for global proteome analyses descried below. The supernatant containing DNA and DPCs was decanted, and an equal volume of cold 100% EtOH was added to precipitate DNA/DPCs. Precipitated DNA/DPCs were washed with 70% EtOH twice and 100% EtOH, followed by re-suspending in water. DNA concentrations were determined by dG analysis as described below.
Enzymatic digestion of DPCs to tryptic peptides
DNA/DPCs (10 μg) were dissolved in 100 μL of 50 mM ammonium acetate buffer, pH 5.5 containing 5 mM ZnCl2 and heated at 90 °C for 10 min to denature the DNA. Immediately after heating, the samples were chilled in an ice bath (0 °C) to prevent DNA re-annealing. Once chilled, 2 U of nuclease P1 was added to each sample and the solutions were incubated for 4 hours at 37 °C. After 4 hours, the pH was raised to 7.0 by adding 1 μL 1 M ammonium bicarbonate, and 120 U phosphodiesterase 1 and 22 U alkaline phosphatase (Sigma, St. Louis, MO) were added to each sample. The solutions were incubated at 37 °C overnight to complete digestion of the DNA constituent of DPCs. After digestion, the solutions were transferred to Amicon 3K filters (Millapore, Darmstadt, Germany), and the free nucleosides were removed by centrifugation at 14,000 g for 10 min.
Filter-aided sample preparation (FASP) protocol for DPC protein trypsin digestion
Protein-nucleoside conjugates were washed with 100 mM HEPES three times (14,000 g for 10 minutes) and re-suspended in 100 μL 100 mM HEPES buffer. A fresh solution of 100 mM DTT/0.1% SDS in 100 mM HEPES was prepared, and 10 μL aliquot was added to each sample, followed by heating at 55 °C for 1 hour. Following heating, 5 μL of freshly prepared iodoacetamide solution (375 mM) was added, and the solution was incubated for 30 min at room temperature. Each sample was then centrifuged at 14,000 g for 10 minutes to remove excess reagents, followed by resuspension in 50 μL of 25 mM ammonium bicarbonate. Proteins were digested to tryptic peptides by incubation with 1 μg trypsin overnight at 37 °C.
FASP protocol for whole proteome trypsin digestion
The pelleted proteins from each LAD ligation/reperfusion and sham surgery sample were reconstituted in 5 mL 15 mM Tris buffer, pH 7.5/0.1% SDS and the protein concentrations were measured using the Pierce™ BCA Protein Assay kit. Equivalent amounts (100 μg) from each sample were transferred to Amicon 3K filters and washed with 100 μL 100 mM HEPES three times (14,000 g for 10 minutes). The proteins were treated with DTT and iodoacetamide as described above, followed by incubation with 1 μg trypsin overnight at 37 °C.
TMT labeling of tryptic peptides
Tryptic peptides were recovered by centrifugation at 14,000 g for 10 min and concentrated in glass MS vials with inserts to dryness. Once dry, the samples were re-suspended in 100 μL of water and peptide concentrations were determined by the Pierce Quantitative Colorimetric Peptide Assay (Thermo, Waltham, MA). For peptides from whole proteome samples, 10 μg of peptide were diluted to 35 μL of 100 mM HEPES, and the solution pH was adjusted to pH 8.0 using 100 mM NaOH. For peptides from DPC proteins, peptide amounts corresponding to 10 μg DNA were prepared as described above. To each peptide solution was added 5 μL of ACN and 10 μL of a TMT-6plex reagent (19.5 μg/μL, 40-fold excess), and the reaction mixtures were incubated at room temperature for 2 h. The reactions were quenched with 4 μL of 5% hydroxylamine for 15 minutes at room temperature. The derivatized peptides were evaporated to dryness under reduced pressure and re-suspended in 60 μL 98/2/0.1 water/ACN/formic acid. The samples were combined and desalted by stage tip procedure as follows. Stage tips were manually packed with C18 SDX packing discs and washed with 60 μL 60/40/0.1 ACN/water/formic acid (500 rcf for 5 min) and 60 μL 98/2/0.1 water/ACN/formic acid (500 rcf for 5 minutes) twice. The reconstituted samples were then loaded onto the tips (500 rcf for 5 minutes) and washed with 60 μL 98/2/0.1 water/ACN/formic (5000 rcf for 5 minutes) twice. Labeled peptides were eluted with 120 μL 60/40/0.1 ACN/water/formic acid and 60 μL 80/20/0.1 ACN/water/formic acid, then concentrated under vacuum.
NanoHPLC-NSI+-MS/MS analysis of tryptic peptides
TMT-labeled peptides were reconstituted in 20 μL 0.1% formic acid and analyzed using an LTQ Orbitrap Fusion mass spectrometer (Thermo Scientific) interfaced to a Dionex Ultimate 3000 RS autosampler and an Dionex UltiMate 3000 RSLCnano system. The samples were loaded onto a pulled-tip fused silica column with a 100 μm inner diameter packed in-house with 45 cm of 5μm Luna-C18 resin (Phenomenex, Torrence, Ca) that served both as an analytical column and as a nanospray ionization emitter. HPLC flow was held at 300 nL/min. HPLC solvents were comprised of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Solvent composition was held at 5% solvent B for 17 min, followed by an increase to 7% over 2 min, 25% over 100 min, 60% over 20 min, and finally to 95% over 1 min. The solvent composition was held at 95% for 8 min, and returned to 5% over 1 min and re-equilibration for 28 min.
The mass spectrometer was operated in a data dependent mode with dynamic exclusion enabled (repeat count: 1, exclusion duration: 15 sec). For every scan cycle, one full MS scan (m/z 320 to 2000) was collected at a resolution of 120,000 at an AGC target value of 2×105, followed by MS2 scans of as many dependent scans as possible within a cycle time of 3 seconds with HCD (normalized collision energy = 38%, isolation width = 0.7 m/z, resolution = 30,000) at an AGC target value of 1×105. Ions with a charge state of +1 were excluded from the analysis.
Protein identification
Peptide sequences were determined from their MS2 spectra using Proteome Discover 2.1 software (Thermo, Waltham, MA) and a SwissProt rat proteome database [24]. Precursor ion m/z mass tolerance was set at ± 10 ppm, fragment ion m/z tolerance at ± 0.06 Da, and up to two missed cleavages were allowed. Static modifications included carbamidomethylation (+57.02146 Da) of cysteine and TMT 6-plex (+219.163 Da) groups on lysines, tyrosines and protein N-termini. Dynamic modifications included oxidation (+15.99492 Da, M) and deamidation (+0.98402 Da, N). Identified peptides were filtered using a 1% false discovery rate and at least 1 unique peptide fragment. Isotope distributions for all six TMT reporter regions, provided by the supplier, were included in the analysis.
Relative protein quantitation
For relative quantification of DPC proteins and free proteins in cardiomyocytes from LAD surgery and sham surgery groups, reporter ion intensities (m/z 126.1277, 127.1247, 128.1344, 129.1314, 130.1411, 131.1381) were extracted using Proteome Discoverer 2.1 software. All quantitative values were rejected if not all quantitative channels were present, and we did not replace missing quantitative values with the minimum intensity. The mass tolerance for reporter ion extraction was 0.01 Da, and the signal to noise ratio of the reporter ion was at least 10.
Bioinformatic analysis of quantitative proteomics data
Whole proteome and DPC protein abundance data were processed by the Ingenuity Pathway Analysis (IPA, Qiagen, Hilden, Germany) and Cluster 3.0. To develop protein interaction networks, protein TMT abundance data were processed using a core expression analysis measuring expression fold change. Whole proteome and DPC TMT abundance data was further processed using Ingenuity Pathway Analysis (IPA) comparison analysis to identify shared protein interaction networks (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). Clustering analysis of the protein TMT abundance data was analyzed using a hierarchical clustering algorithm (uncentered correlation) [25].
DNA Quantitation by dG Analysis
To quantify DPC-containing DNA extracted from cells and to detect any potential RNA contamination, 1 μg aliquots of DNA were taken and digested to 2′-deoxynucleosides in the presence of phosphodiesterase I (120 mU), phosphodiesterase II (105 mU), DNase (35 U) and alkaline phosphatase (22 U) in 20 μL 10 mM Tris-HCl/15 mM ZnCl2 (pH 7.0) for 18 h at 37 °C. Quantitative analysis of dG in enzymatic digests was conducted by HPLC-UV on an Agilent Technologies 1100 HPLC system equipped with a diode array UV detector and an autosampler. Samples were loaded onto an Atlantis T3 C18 column (2.1 × 150 mm, 5 μm, from Waters Corporation, Milford, MA) and eluted with a gradient of 5 mM ammonium formate, pH 4.0 (A) and methanol (B). Solvent composition was changed linearly from 3 to 30% B over 15 min, increased further to 80% B over 3 min, held at 80% B for 1 min, and returned to 3% B over 2 min, where it was kept for the final 8 min of the HPLC run. UV absorbance was monitored at 260 nm. With this method, dG eluted as a sharp peak at 11.7 min (Figure S-1). dG amounts were determined by comparing HPLC peak areas to a calibration curve constructed by injecting known dG amounts.
Quantitation of dT-Tyr conjugates
DPC-containing DNA (10 μg) isolated from rat cardiomyocytes as described above was dissolved in 100 μL of 10 mM Tris, pH 7.5 containing 8 Units proteinase K (10 μL, New England Biolabs, Ipswich, MA) and 50 μg of pronase (Sigma, St. Louis, MO) at 37 °C overnight to digest the protein constituents of DPCs to single amino acids. DNA was enzymatically digested to nucleosides as follows. The solutions were concentrated to dryness, and samples were re-suspended in 100 μL 10 mM Tris, pH 5.5 with 5 mM ZnCl2. Each sample was supplemented with 2.5 U of NucP1 and incubated at 37 °C for 4 hours. After 4 hours, the solution pH was raised to pH 7.0 by adding 1 μL 1 M ammonium bicarbonate, followed by adding 15 μL of 100 mM MgCl2, 160 U of PDE1, and 22 U of alkaline phosphatase (total volume 150 μL), and incubated at 37 °C overnight.
The digests were spiked with dT[15N1,13C9]-Tyr (internal standard for mass spectrometry, 200 fmol), followed by ultrafiltration through Nanosep 10K filters at 5000 g for 10 minutes. The filters were further washed with 100 μL 0.1% formic acid in water and 100 μL 0.1% formic acid. The filtrates were concentrated to dryness, re-suspended in 100 μL 0.1% acetic acid, and enriched by offline HPLC purification as follows. An Agilent Technologies HPLC system (1100 model) was used incorporating a diode array detector, an autosampler, and a fraction collector. HPLC solvents were comprised of 0.1% acetic acid in water (solvent A) and 0.1% acetic acid in acetonitrile (solvent B). The samples were loaded on a Supelcosil LC-18-DB (4.6 × 250 mm, 5 μm) column (Thermo Scientific, Waltham, MA) and eluted at a flow rate of 1 mL/min. Solvent composition was held at 2% B over 5 min, increased to 30% over 7 min, further to 75% over 1 min, held at 75% for 2 min, decreased to 2% over 1 min, and finally re-equilibrated at 2% B for 14 min. Fractions containing dT-Tyr and its isotopically labeled internal standard (12.9 – 13.9 min) were collected, dried under vacuum, and reconstituted in 0.1% formic acid (16 μL) for quantitative analysis. Due to interfering peaks, additional HPLC purification of the collected analyte and internal standard using the same method was required for some samples. To exclude the possibility of analyte carryover between runs, a blank sample (100 μL of 0.1% acetic acid in water) spiked with 200 fmol internal standard was analyzed after every third sample.
Quantitative analysis of dT-Tyr was conducted using a Dionex UltiMate 3000 RSLC nanoHPLC system (Thermo Scientific, Waltham, MA) interfaced to a TSQ Quantiva mass spectrometer (Thermo Scientific, Waltham, MA). The samples were loaded onto a pulled-tip fused silica column with a 100 μm inner diameter packed in-house with 15 cm of Synergi 4 μm Hydro RP 80Å resin (Phenomenex, Torrence, Ca) that served both as a resolving column and as a nanospray ionization emitter. HPLC solvents were comprised of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). dT-Tyr was eluted with a gradient of 1% solvent B over 6 min at a flow rate of 1.0 μL/min, followed by an increase to 30% over 10 min at a flow rate of 0.3 μL/min, then held at 30% over 2 min, then increased to 75% over 2 min, decreased to 1% over 1 min, then finally re-equilibrated at 1% for 11 min at 1 μL/min. Under these conditions, dT-Tyr and its internal standard (dT-[15N1, 13C9]Tyr) eluted at ~ 13.8 min. Electrospray ionization was achieved at a spray voltage of 2800 V and a capillary temperature of 350 °C. Collision induced dissociation was performed with Ar as a collision gas (1.0 mTorr) at a collision energy of 23 V. MS/MS parameters were optimized for maximum response during infusion of a standard solution of dT-[15N1, 13C9]Tyr.
nanoLC-NSI+-MS/MS analysis of dT-Tyr was performed in the selected reaction monitoring mode by monitoring the neutral loss of dR and the loss of dR and CO2 from protonated molecules of the analyte (m/z 422.2 [M + H]+ → 306.1 [M + H − dR]+, m/z 422.2 [M + H]+ → 260.1) and the corresponding mass transitions corresponding to 15N1,13C9-labeled internal standard (m/z 432.1 [M + H]+ → 316.1 [M + H − dR]+, m/z 432.2 [M + H]+ → 269.1). Analyte concentrations were determined using the relative response ratios calculated from nanoLC-NSI+-MS/MS peak areas in extracted ion chromatograms corresponding to dT-Tyr and its internal standard.
nanoLC-NSI+-MS/MS method validation
Calf thymus DNA (CT DNA, 50 μg, in triplicate) was spiked with increasing amounts of dT-Tyr analyte (2-500 fmol) and 200 fmol dT-[15N1,13C9]Tyr. Each sample was processed as described above. The linearity of each validation sample was determined by plotting the observed peak area ratio (analyte/internal standard) to the expected peak area ratio. The limit of detection was defined as the analyte signal at least 3 times the response as compared to the background. Accuracy and precision of the method were calculated for three samples (200 fmol, high point; 50 fmol, middle point; 10 fmol, low point). Accuracy was calculated by subtracting the average observed peak area ratio (analyte/internal standard) by the expected peak area ratio, followed by dividing the difference by the expected peak area ratio multiplied by 100. Accuracy was expressed as the % difference. Precision was calculated by dividing the standard deviation between samples by the mean peak area and multiplied by 100, expressed as % coefficient of variation (% CV).
Western Blot Analysis
Cardiomyocyte proteins (50 μg) were resolved by NuPAGE Novex 12% Bis-Tris gels (Invitrogen, Carlsbad, CA) and transferred to Invitrolon PVDF filter paper membranes (0.45 μm pore size, Life Technologies, Carlsbad, CA). The membranes were blocked for 1 h in Tris-buffered saline-Tween 20 (TBST) containing 5% (w/v) blotting grade blocker (BioRad, Hercules, CA). Following blocking, the membranes were incubated with the primary antibodies against matrin-3, superoxide dismutase [Mn], mitochondrial (SOD2), pyruvate dehydrogenase E1 component subunit beta, mitochondrial (PDH-1Eβ), troponin-T (cTni), and actin overnight at 4 °C. The membranes were rinsed with TBST and incubated with the corresponding alkaline phosphatase-conjugated antibody for 1 h at room temperature. The membranes were developed using the Clarity Western ECL substrate (BioRad) and visualized on an Odyssey 2800 Fc imager (Li-COR, Lincoln, NE) with Image Studio version 3.1 software (Li-COR). To confirm the identity of the protein constituents from cardiomyocyte DPCs, 5 μg of DNA from one LAD ligation/reperfusion and sham surgery samples were enzymatically digested to yield a nucleoside-protein conjugate. Nucleoside-protein conjugates were resolved and immunoblotted as described above.
Results
Ischemia-reperfusion injury increases plasma troponin I levels and induces ROS
To confirm that the LAD ligation procedure successfully induced myocardial infarction, plasma levels of cardiac troponin I (cTnI) were measured using a sensitive ELISA test [26,27]. cTnI is a useful biomarker of MI commonly used in the clinic to diagnose MI and acute coronary syndrome [28,29]. cTnI levels in all plasma samples collected before the thoracotomy were below the limit of detection of the assay (Table 1). The animals that underwent a sham surgery (thoracotomy only) experienced a modest increase in plasma cTnI levels to 4.4–6.6 ng/mL respectively (Table 1). In contrast, plasma levels of cTnI in animals that underwent LAD ligation/reperfusion procedure were above 50 ng/mL, confirming that they experienced an MI (Table 1).
Table 1.
Plasma cTni measurements pre- and post-surgery to confirm myocardial infarction. The cTni levels after LAD ligation/reperfusion was above the limit of detection of the assay (50 ng/mL), while the average cTni levels after the sham surgery was 5.82 ± 1.03 ng/mL.
| Experiment | Pre-Surgery cTni (ng/mL) |
Post-Surgery cTni (ng/mL) |
|---|---|---|
| LAD 1 | ND | >50 |
| LAD 2 | ND | >50 |
| LAD 3 | ND | >50 |
| Control 1 | ND | 6.57 |
| Control 2 | ND | 6.52 |
| Control 3 | ND | 4.37 |
To determine whether the ischemia/reperfusion procedure led to oxidative stress, plasma levels of 8-iso-PGF2α were measured pre- and post-procedure using the OxiSelect™ 8-iso-prostoglandin F2α ELISA assay [30–32]. 8-iso-PGF2α is an isoprostane produced by ROS-mediated peroxidation of arachidonic acid in membrane phospholipids and is commonly used as a biomarker of oxidative stress in vivo [30–32]. It is important to note that prior to the surgeries, animals where anesthetization under 100% oxygen, potentially exposing them to hyperoxic conditions and an artificial increase in ROS levels. Therefore, the baseline levels of 8-iso-PGF2α could have been elevated prior to the surgery in both control and LAD groups. We found that 8-iso-PGF2α levels slightly decreased post-thoracotomy in control group that underwent sham surgery (Controls 1 – 3) (Figure 1). In contrast, animals that underwent LAD ligation/reperfusion procedure (LAD 1 – 3) experienced a 273.8%, 357.1%, and 178.3% increase in 8-iso-PGF2α levels post-surgery compared to the pre-procedure levels, indicative of increased oxidant levels in this group (Figure 1).
Figure 1.
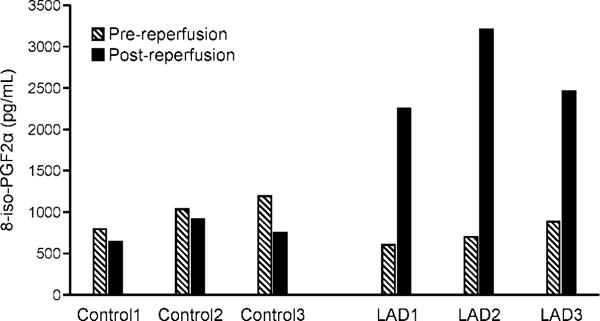
Plasma 8-iso-PGF2α measurements. Plasma 8-iso-PGF2α levels were measured pre- and post-surgery using the OxiSelect™ 8-iso-prostoglandin F2α ELISA kit. A standard curve of 200,000 ng/mL, 50,000 ng/mL, 12,500 ng/mL, 3125 ng/mL, 781ng/mL, 195 ng/mL, and 49 ng/mL was analyzed by a “log inhibitor vs response, variable slope with 4-parameters” equation to determine 8-iso-PGF2α concentration. The animals that underwent LAD ligation/reperfusion (LAD 1, 2, and 3) had a significant increase (p value < 0.05) in 8-iso-PGF2α post-surgery as compared to pre-surgery. No significant increase in 8-iso-PGF2α levels was detected in the animals that underwent the sham surgery (Controls 1 - 3).
Total DPC numbers are increased following ischemia-reperfusion injury
To allow for absolute quantification of ROS-induced DPCs in rat cardiomyocytes, an isotope dilution tandem mass spectrometry assay was developed for dT-Tyr conjugates (Scheme 2). This type of cross-linking involves radical-mediated reactions between the 5′-methyl of thymidine in DNA and the 3-position of tyrosine residue in proteins (Scheme 2) [18,19,33,34]. DPCs extracted from rat cardiomyocytes were enzymatically digested with nucleases and proteases to afford dT-Tyr and analyzed by isotope dilution nanoLC-NSI+-MS/MS allowing for accurate and precise quantification of hydroxyl radical-induced DNA-protein conjugates.
Scheme 2.
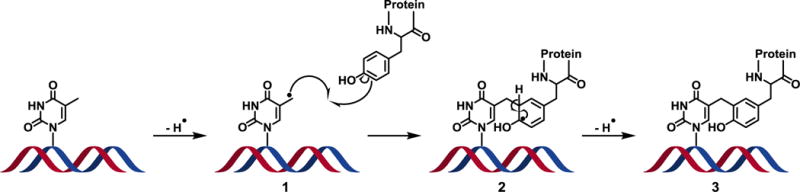
Proposed mechanism of hydroxyl radical-induced DPC formation between thymidine in DNA and tyrosine residue in proteins. Hydroxyl radicals abstract a hydrogen from the 5-methyl position of thymidine to yield a reactive thymidine radical (1), which undergoes a one-electron addition to the 3-position of a tyrosine to yield a stable methylene linkage. Subsequent hydrogen abstraction from the 3-position of tyrosine (2) rearomatizes the phenol ring to yield the stable DNA-protein cross-link (3).
Authentic standards of dT-Tyr and dT-[15N1,13C9]Tyr were prepared in our laboratory using published methodologies [35]. These standards were used to develop a sensitive and precise nanoLC-NSI+-MS/MS method for dT-Tyr using a triple quadrupole mass spectrometer. In our approach, DPC-containing DNA is spiked with known amounts of dT-[15N1,13C9]Tyr internal standard and enzymatically digested to free nucleosides. The analyte and its internal standard are enriched by offline HPLC, followed by quantitative analysis using nanoLC-NSI+-MS/MS. Selected reaction monitoring transitions included the neutral loss of dR and the simultaneous loss of dR and CO2 from protonated molecules of the analyte (m/z 422.2 [M + H]+ → 306.1 [M + H − dR]+, m/z 422.2 [M + H]+ → 260.1) and the 15N1,13C9-isotopically labelled internal standard (m/z 432.1 [M + H]+ → 316.1 [M + H − dR]+, m/z 432.2 [M + H]+ → 269.1) (Figure 2A). The new nanoLC-NSI+-MS/MS method was validated by analyzing blank DNA samples spiked with known amounts of dT-Tyr analyte and internal standard. nanoLC-NSI+-MS/MS limit of detection (LOD) was calculated as 250 amol of dT-Tyr on column, while the limit of quantification was 2 fmol on column (Figure S-2). The accuracy (% difference) of the method was calculated as 3.0%, 4.0%, and 20.0% for the high, middle, and low validation points. The precision of the method was calculated as 4.1, 4.2, and 0.0 % CV for the high, middle, and low validation points respectively.
Figure 2.
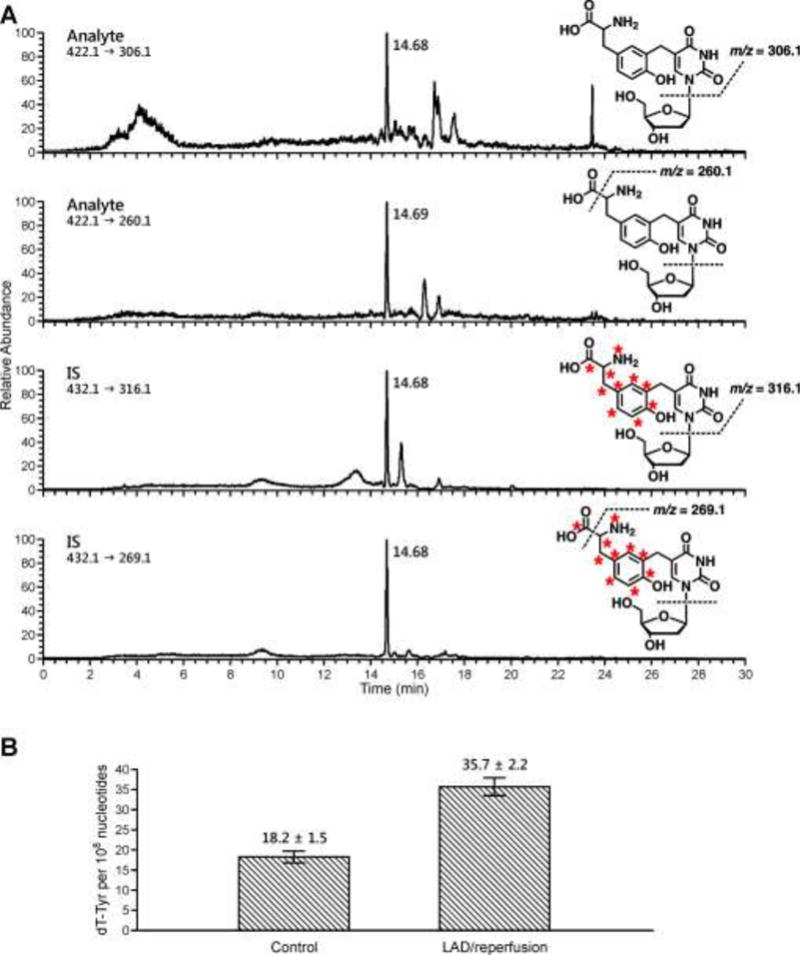
Representative nanoHPLC-NSI+-MS/MS trace of dT-Tyr observed in rat cardiomyocytes following LAD ligation/reperfusion surgery (A) and quantitative data for dT-Tyr adducts formed in rat cardiomyocytes after LAD ligation/reperfusion or sham surgery (B).
The new method was used to quantify radical-induced DPCs after LAD ligation/reperfusion compared to sham surgery (3 per group). We found that dT-Tyr amounts were significantly increased in cardiomyocytes of rats that underwent LAD ligation/reperfusion as compared to the sham group (35.7 ± 2.2 vs 18.2 ± 1.5 dT-Tyr adducts per 108 nucleotides, respectively), (Figures 2A and 2B). These findings support our hypothesis that ischemia-reperfusion injury increases the levels of hydroxyl radical-mediated DPCs in cardiomyocyte DNA.
Quantitative proteomic analysis of the protein constituents of cardiomyocyte DPCs
We next aimed to identify and quantify the proteins participating in DPC formation following LAD ligation/reperfusion surgery. Following DPC isolation from control and LAD ligation/reperfusion rat cardiomyocytes, protein constituents were analyzed by TMT quantitative proteomics technique [36]. In brief, each of the six samples (3 LAD ligation/reperfusion, 3 sham surgery) was labeled with a unique isotope tag. The samples were combined and analyzed by nanoLC-NSI+-MS/MS on an Orbitrap Fusion mass spectrometer. MS/MS sequencing of tryptic peptides was used to determine their amino acid sequences, and the proteins were identified using Proteome Discoverer 2.1 software [37]. Relative quantitation of the proteins was achieved using ion intensities of reporter ions (m/z 126.1277, 127.1247, 128.1344, 129.1314, 130.1411, 131.1381) originating from their tryptic peptides. The relative abundances of proteins in LAD ligation/reperfusion animals as compared to the sham group was determined by comparing the grouped reporter ion intensities of 126.1277, 128.1344, and 131.1381 (LAD 1-3) to the grouped reporter ion intensities of corresponding to control group (m/z 127.1247, 129.1314, and 130.1411 (controls 1-3).
The relative abundances of proteins covalently bound to DNA following LAD ligation/reperfusion were determined by comparing the grouped reporter ion intensities of 128.1344, 129.1314 and 131.1381 (LAD 1-3) to the grouped reporter ion intensities of 126.1277, 127.1247, and 130.1411 (control animals 1-3). Using these criteria, a total of 90 proteins were identified in all six samples (Table 2). The majority of the identified DPC protein constituents (80, 88.9%) increased in abundance at least 1.2-fold after LAD ligation/reperfusion as compared to sham surgery, with 21 (23.3%) of these proteins increasing in abundance at least 5-fold (Table 2). Proteins that showed the greatest change included myosins, heat shock proteins, Cu-Zn superoxide dismutases, thioredoxin, actin, and mitochondrial creatinine kinase. Of the remaining DPC proteins, 8 proteins (8.9%) did not change in abundance and one protein (1.1%, lysosome-associated membrane glycoprotein 1) decreased in abundance (Table 2).
Table 2.
Identification and quantification of proteins irreversible trapped on DNA in cardiomyocytes of rats subjected to ischemia-reperfusion or sham surgery.
| Accession | Description of Protein | % Coverage | # Peptides | # PSMs | # Unique Peptides | MW [kDa] | Fold Change (LAD/Sham) |
|---|---|---|---|---|---|---|---|
| P13437 | 3-ketoacyl-CoA thiolase, mitochondrial | 5.54 | 2 | 2 | 2 | 41.84 | 12.38 |
| P49432 | Pyruvate dehydrogenase E1 component subunit beta, mitochondrial | 13.65 | 5 | 10 | 5 | 38.96 | 9.28 |
| Q64591 | 2,4-dienoyl-CoA reductase, mitochondrial | 8.06 | 3 | 4 | 3 | 36.11 | 8.95 |
| P29117 | Peptidyl-prolyl cis-trans isomerase F, mitochondrial | 6.80 | 2 | 2 | 2 | 21.80 | 8.70 |
| P11980 | Pyruvate kinase PKM | 3.58 | 2 | 2 | 2 | 57.78 | 8.65 |
| Q9Z0V6 | Thioredoxin-dependent peroxide reductase, mitochondrial | 21.40 | 5 | 7 | 5 | 28.28 | 7.77 |
| P42123 | L-lactate dehydrogenase B chain | 23.95 | 8 | 11 | 7 | 36.59 | 7.38 |
| P35434 | ATP synthase subunit delta, mitochondrial | 13.69 | 2 | 11 | 2 | 17.58 | 6.98 |
| P13086 | Succinyl-CoA ligase [ADP/GDP-forming] subunit alpha, mitochondrial | 9.54 | 3 | 4 | 3 | 36.13 | 6.66 |
| P04642 | L-lactate dehydrogenase A chain | 9.64 | 3 | 5 | 2 | 36.43 | 6.46 |
| P14604 | Enoyl-CoA hydratase, mitochondrial | 32.41 | 9 | 22 | 9 | 31.50 | 6.42 |
| P26284 | Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial | 5.90 | 3 | 3 | 3 | 43.20 | 6.20 |
| P07895 | Superoxide dismutase [Mn], mitochondrial | 13.06 | 3 | 5 | 3 | 24.66 | 5.95 |
| Q8VHF5 | Citrate synthase, mitochondrial | 17.38 | 8 | 13 | 8 | 51.83 | 5.95 |
| P62898 | Cytochrome c, somatic | 43.81 | 5 | 8 | 5 | 11.60 | 5.94 |
| P02563 | Myosin-6 | 48.19 | 118 | 457 | 39 | 223.37 | 5.90 |
| P02564 | Myosin-7 | 42.38 | 96 | 355 | 17 | 222.95 | 5.87 |
| P15650 | Long-chain specific acyl-CoA dehydrogenase, mitochondrial | 39.07 | 17 | 60 | 17 | 47.84 | 5.60 |
| O35854 | Branched-chain-amino-acid aminotransferase, mitochondrial | 4.58 | 2 | 2 | 2 | 44.25 | 5.49 |
| P18418 | Calreticulin | 4.81 | 2 | 2 | 2 | 47.97 | 5.47 |
| O88767 | Protein deglycase DJ-1 | 8.47 | 2 | 2 | 2 | 19.96 | 5.11 |
| P51868 | Calsequestrin-2 | 10.17 | 4 | 5 | 4 | 47.81 | 5.07 |
| P04636 | Malate dehydrogenase, mitochondrial | 51.18 | 16 | 59 | 16 | 35.66 | 4.91 |
| P00507 | Aspartate aminotransferase, mitochondrial | 39.07 | 22 | 54 | 22 | 47.28 | 4.72 |
| Q01205 | Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial | 7.27 | 4 | 4 | 4 | 48.89 | 4.69 |
| P19804 | Nucleoside diphosphate kinase B | 28.95 | 4 | 5 | 4 | 17.27 | 4.55 |
| P07483 | Fatty acid-binding protein, heart | 57.89 | 8 | 34 | 8 | 14.77 | 4.53 |
| P31044 | Phosphatidylethanolamine-binding protein 1 | 25.13 | 3 | 8 | 3 | 20.79 | 4.45 |
| P04785 | Protein disulfide-isomerase | 8.64 | 4 | 4 | 4 | 56.92 | 4.40 |
| P26772 | 10 kDa heat shock protein, mitochondrial | 48.04 | 5 | 9 | 5 | 10.90 | 4.16 |
| P08461 | Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial | 14.87 | 9 | 25 | 9 | 67.12 | 4.07 |
| P16409 | Myosin light chain 3 | 73.50 | 15 | 51 | 12 | 22.14 | 3.99 |
| P70623 | Fatty acid-binding protein, adipocyte | 21.21 | 3 | 3 | 3 | 14.70 | 3.98 |
| P00564 | Creatine kinase M-type | 35.43 | 14 | 32 | 12 | 43.02 | 3.88 |
| Q9JM53 | Apoptosis-inducing factor 1, mitochondrial | 7.84 | 4 | 5 | 4 | 66.68 | 3.82 |
| Q4FZU2 | Keratin, type II cytoskeletal 6A | 4.35 | 3 | 6 | 2 | 59.21 | 3.77 |
| P48500 | Triosephosphate isomerase | 20.08 | 5 | 7 | 5 | 26.83 | 3.75 |
| P08503 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 6.41 | 3 | 4 | 3 | 46.53 | 3.71 |
| O88989 | Malate dehydrogenase, cytoplasmic | 29.34 | 11 | 19 | 11 | 36.46 | 3.70 |
| Q62812 | Myosin-9 | 1.94 | 4 | 4 | 3 | 226.20 | 3.69 |
| P04692 | Tropomyosin alpha-1 chain | 88.73 | 51 | 326 | 32 | 32.66 | 3.59 |
| P13221 | Aspartate aminotransferase, cytoplasmic | 26.15 | 9 | 12 | 9 | 46.40 | 3.58 |
| P09605 | Creatine kinase S-type, mitochondrial | 33.41 | 16 | 124 | 15 | 47.36 | 3.46 |
| P56741 | Myosin-binding protein C, cardiac-type | 17.43 | 20 | 45 | 20 | 140.67 | 3.46 |
| O35796 | Complement component 1 Q subcomponent-binding protein, mitochondrial | 11.83 | 2 | 2 | 2 | 30.98 | 3.45 |
| P58775 | Tropomyosin beta chain | 48.24 | 23 | 125 | 4 | 32.82 | 3.43 |
| P05065 | Fructose-bisphosphate aldolase A | 10.16 | 4 | 5 | 4 | 39.33 | 3.38 |
| P15999 | ATP synthase subunit alpha, mitochondrial | 26.94 | 13 | 29 | 13 | 59.72 | 3.30 |
| P06761 | 78 kDa glucose-regulated protein | 8.72 | 5 | 7 | 5 | 72.30 | 3.29 |
| P23965 | Enoyl-CoA delta isomerase 1, mitochondrial | 14.53 | 4 | 5 | 4 | 32.23 | 3.26 |
| P11507 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | 5.27 | 5 | 16 | 5 | 114.69 | 3.24 |
| P56571 | ES1 protein homolog, mitochondrial | 19.17 | 6 | 10 | 6 | 28.16 | 3.18 |
| Q68FU3 | Electron transfer flavoprotein subunit beta | 12.94 | 3 | 5 | 3 | 27.67 | 3.18 |
| P08733 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 59.64 | 10 | 40 | 10 | 18.87 | 3.15 |
| P02770 | Serum albumin | 3.45 | 2 | 7 | 2 | 68.69 | 3.10 |
| P48721 | Stress-70 protein, mitochondrial | 7.36 | 4 | 4 | 3 | 73.81 | 2.97 |
| Q62651 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | 25.99 | 9 | 11 | 9 | 36.15 | 2.91 |
| P07632 | Superoxide dismutase [Cu-Zn] | 22.73 | 3 | 5 | 3 | 15.90 | 2.77 |
| P13803 | Electron transfer flavoprotein subunit alpha, mitochondrial | 15.02 | 4 | 6 | 4 | 34.93 | 2.75 |
| Q5XIF3 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial | 31.43 | 5 | 9 | 5 | 19.73 | 2.71 |
| Q6P6R2 | Dihydrolipoyl dehydrogenase, mitochondrial | 20.04 | 9 | 15 | 9 | 54.00 | 2.69 |
| P18886 | Carnitine O-palmitoyltransferase 2, mitochondrial | 3.50 | 3 | 3 | 3 | 74.06 | 2.65 |
| D3ZHA0 | Filamin-C | 0.77 | 2 | 2 | 2 | 290.80 | 2.52 |
| P63018 | Heat shock cognate 71 kDa protein | 9.44 | 7 | 11 | 6 | 70.83 | 2.51 |
| Q9ER34 | Aconitate hydratase, mitochondrial | 19.87 | 15 | 33 | 15 | 85.38 | 2.38 |
| P16290 | Phosphoglycerate mutase 2 | 17.00 | 5 | 5 | 5 | 28.74 | 2.36 |
| P28042 | Single-stranded DNA-binding protein, mitochondrial | 11.26 | 2 | 2 | 2 | 17.44 | 2.21 |
| Q6IFW6 | Keratin, type I cytoskeletal 10 | 9.70 | 5 | 10 | 4 | 56.47 | 2.17 |
| Q6IMF3 | Keratin, type II cytoskeletal 1 | 6.24 | 4 | 6 | 3 | 64.79 | 2.08 |
| P11240 | Cytochrome c oxidase subunit 5A, mitochondrial | 21.23 | 3 | 3 | 3 | 16.12 | 2.03 |
| Q6IFV3 | Keratin, type I cytoskeletal 15 | 3.58 | 3 | 3 | 2 | 48.84 | 1.95 |
| Q9ESS6 | Basal cell adhesion molecule | 7.53 | 4 | 5 | 4 | 67.47 | 1.92 |
| P62738 | Actin, aortic smooth muscle | 47.21 | 16 | 65 | 6 | 41.98 | 1.78 |
| P01946 | Hemoglobin subunit alpha-1/2 | 15.49 | 2 | 3 | 2 | 15.32 | 1.75 |
| P04762 | Catalase | 5.88 | 3 | 5 | 3 | 59.72 | 1.65 |
| P56574 | Isocitrate dehydrogenase [NADP], mitochondrial | 5.75 | 2 | 3 | 2 | 50.94 | 1.59 |
| P10719 | ATP synthase subunit beta, mitochondrial | 17.77 | 7 | 8 | 7 | 56.32 | 1.56 |
| P32551 | Cytochrome b-c1 complex subunit 2, mitochondrial | 9.07 | 3 | 3 | 3 | 48.37 | 1.42 |
| Q920L2 | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | 2.90 | 2 | 3 | 2 | 71.57 | 1.42 |
| Q6UPE1 | Electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial | 3.73 | 2 | 2 | 2 | 68.16 | 1.41 |
| Q05962 | ADP/ATP translocase 1 | 16.11 | 5 | 6 | 3 | 32.97 | 1.38 |
| Q64119 | Myosin light polypeptide 6 | 21.85 | 4 | 8 | 1 | 16.96 | 1.14 |
| P50339 | Chymase | 9.72 | 2 | 2 | 2 | 27.55 | 1.11 |
| P51740 | Intestinal-type alkaline phosphatase 2 | 7.08 | 4 | 14 | 2 | 59.76 | 1.09 |
| P15693 | Intestinal-type alkaline phosphatase 1 | 8.33 | 4 | 21 | 2 | 58.37 | 1.07 |
| P26453 | Basigin | 7.99 | 3 | 5 | 3 | 42.41 | 1.02 |
| P02091 | Hemoglobin subunit beta-1 | 27.21 | 3 | 4 | 3 | 15.97 | 1.00 |
| P21961 | Mast cell carboxypeptidase A (Fragment) | 12.14 | 6 | 7 | 6 | 47.91 | 0.91 |
| Q01129 | Decorin | 7.34 | 3 | 4 | 3 | 39.78 | 0.91 |
| P14562 | Lysosome-associated membrane glycoprotein 1 | 4.91 | 2 | 3 | 2 | 43.94 | 0.83 |
The proteomics data for DNA-conjugated proteins was further analyzed using the Cluster 3.0 software and IPA. A subset of 25 identified DPC proteins, consisting of the most abundant proteins, underwent hierarchical clustering analysis. Significant differences in protein abundances were observed between the LAD ligation/reperfusion replicates as compared to the sham surgery group (Figure 3). Hierarchical clustering of the 25 proteins revealed tight grouping between the LAD ligation/reperfusion replicates and the sham surgery replicates, but not between the two conditions (Figure 3). IPA core analysis of the DPC proteins developed 7 protein interaction networks; organismal injury and abnormality (score 59, 26 proteins), cardiovascular system development and function (score 33, 17 proteins), free radical scavenging (score 28, 15 proteins), developmental disorder/hereditary disorder (score 26, 14 proteins), energy production or lipid metabolism (score 24, 13 proteins), and cell morphology and cellular function and maintenance (score 21, 12 proteins). Relative protein quantitation results for SOD2, PDH-1Eβ, and actin were further confirmed by western blot analysis using commercially available antibodies (Figure S-3). Overall, our results indicate that ischemia/reperfusion injury drastically increases the concentrations of proteins covalently attached to DNA (Figure 3), potentially leading to cell death and tissue damage.
Figure 3.

Heat map analysis of the TMT abundance data for 23 representative proteins identified from the DPC samples. Proteins analyzed include sarcomere proteins (8), ROS scavenging proteins (8), and the remaining most abundant proteins (7).
Quantitative proteomic analysis of global cardiomyocyte proteome
To determine whether the observed increases in DNA-protein cross-linking are linked to global changes in protein abundance in response to LAD ligation/reperfusion injury, the entire cardiomyocyte proteome from control and MI/reperfusion groups were analyzed using the same TMT quantitative proteomics technique described above [36]. A total of 359 proteins were identified in all six samples (Table S-1). Of these, the majority (N = 288, 80.2%) did not change in abundance after LAD ligation/reperfusion as compared to the sham surgery (Table S-1). Of the remaining 71 proteins, 48 proteins were found to increase in abundance at least 1.2-fold after LAD ligation/reperfusion and 23 proteins decreased in abundance under the same conditions (Table S-1). The majority of the proteins that increased in abundance following LAD ligation/reperfusion (N =20, 41.7%) were mitochondrial proteins including malate dehydrogenase, enoyl-CoA hydratase, cytochrome C oxidase, and NADH dehydrogenase (Table S-1, Figure 4). This is not unexpected, as the sudden switch from hypoxic conditions to normoxic conditions when the blood flow to the affected area is restored increases the expression of proteins required for aerobic respiration [8]. The remaining proteins were categorized as membrane/extracellular proteins (14, 29.2%), sarcomere proteins (5, 10.4%), cytoplasmic proteins (4, 8.3%), nuclear proteins (4, 8.3%), and endoplasmic reticulum proteins (1, 2.1%) (Figure S-4A). Among the proteins that decreased in abundance following LAD ligation/reperfusion, 39.1% (9) were nuclear, followed by cytoplasmic (6, 26.1%), mitochondrial (3, 13.0%), cell membrane (3, 13.0%), and endoplasmic reticulum (2, 8.7%) (Figure S-4B). Relative protein quantitation results for GAPDH and PDH-1Eβ were further confirmed by western blot analysis using commercially available antibodies (Figure S-3).
Figure 4.
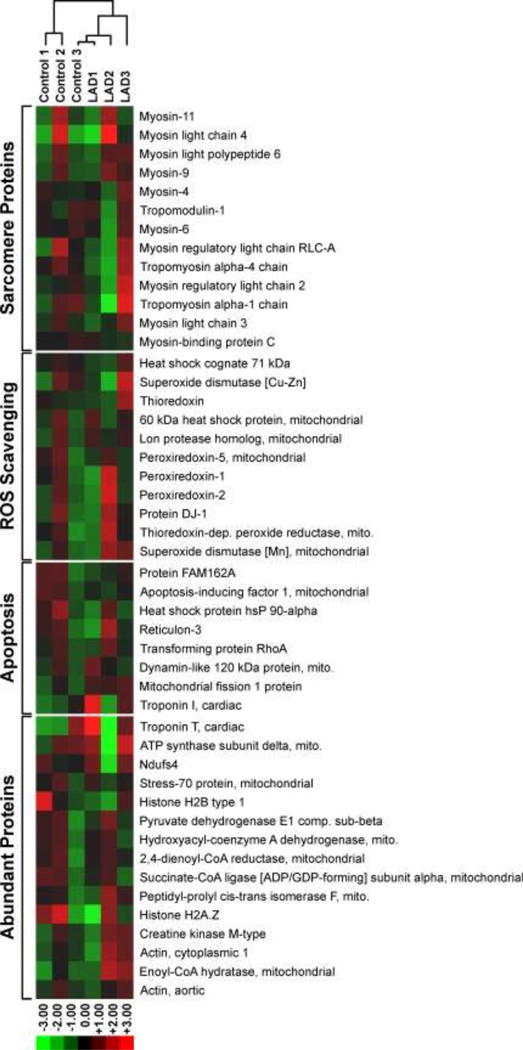
Heat map analysis of the TMT abundance data for 47 representative proteins identified in the whole proteome. Proteins analyzed include sarcomere proteins (13), ROS scavenging proteins (11), apoptosis regulating proteins (7), and the remaining most abundant proteins (16).
The proteomics data were further processed using the Cluster 3.0 software and IPA. A subset of 50 proteins, consisting of 12 sarcomeric proteins, 10 ROS-scavenging proteins, 10 apoptotic proteins, and 15 abundant proteins, underwent hierarchical clustering analysis. Differences in protein abundances were observed across all the LAD ligation/reperfusion and sham replicates, and hierarchical clustering revealed no obvious grouping among the replicates (Figure 3). Overall, the cluster analysis results illustrated the slight increase in the abundance of ROS-scavenging proteins protein abundance after LAD ligation/reperfusion as compared to sham surgery, and no observable difference in the remaining proteins investigated. IPA core analysis of the identified proteome proteins developed 17 protein interaction networks, with metabolic disease/disorder (score 52, 29 proteins), free radical scavenging (score 49, 28 proteins), and energy production/nucleic acid metabolism (score 37, 23 proteins) the highest scored pathways.
A comparison of the quantitative proteomics results for free cardiomyocyte proteins and proteins covalently trapped on DNA revealed a total of 78 proteins (86.7%) that were identified in both experiments. Of the 78 proteins shared between the two experiments, 14 proteins increased in abundance after LAD ligation/reperfusion compared to sham surgery in both whole protein and DPC samples, while the rest were only increased for the cross-linked proteins, but not the global proteome (Figure 5). Overall, these results indicate little correlation between global protein amounts and DPC formation, suggesting that oxidative cross-linking between proteins and DNA is induced by the influx of hydroxyl radicals during reperfusion of cardiac tissues following ischemia.
Figure 5.
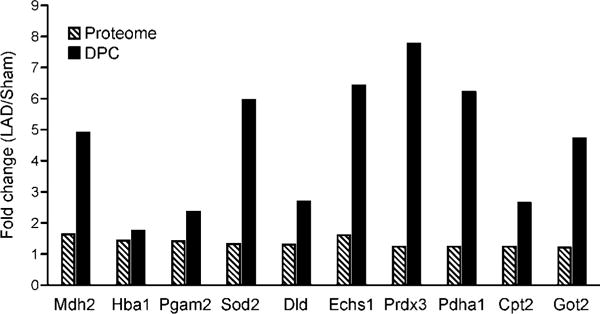
Changes in protein abundances in DPCs (solid black) vs whole proteome (striped bars). The protein abundances (LAD ligation/sham) in the whole proteome and DPCs of 10 representative proteins were compared. Protein names correspond to gene symbols: Mdh2, malate dehydrogenase, mitochondrial; Hba1, hemoglobin subunit-alpha ½; Pgam2, phosphoglycerate mutase 2; Sod2, superoxide dismutase [Mn], mitochondrial; Dld, dihydrolipoyl dehydrogenase, mitochondrial; Echs1, enoyl-CoA hydratase, mitochondrial; Prdx3, thioredoxin-dependent peroxide reductase, mitochondrial; Cpt2, carnitine O-palmitoyltransferase 2, mitochondrial; Got2, aspartate aminotransferase, mitochondrial.
Discussion
DNA-protein crosslinks are super-bulky DNA adducts capable of interfering with crucial biological functions such DNA replication and transcription [13]. Recent studies in our laboratory have shown that DPC lesions completely block human DNA polymerases [38–41]. Similarly, Nakano et al reported that DPCs over 14.1 kDa in size located on the translocating strand completely block DNA unwinding by human helicases (mini chromosome maintenance Mcm467 subcomplex), while DPCs between 5 to 14.1 kDa severely inhibit helicase progression [42]. Recently, our laboratory found that bacterial RNA polymerases were blocked by protein lesions conjugated to the N7 position of guanine (Ji and Tretyakova, unpublished data).
Covalent DPCs readily form in cells via a radical mechanism in the presence of reactive oxygen species. Izzotti et al previously reported age-related DPC accumulation in the brain, liver, and heart of 12 or 24-month-old mice [43]. In mouse myocardium, DPCs accounted for 2.2 ± 0.3% of total DNA in 12-month old mice and 3.4 ± 0.5% of total DNA in 24-month old mice [43]. Furthermore, there was a strong correlation between the levels of measured 8-oxoguanine and DPCs (r = 0.800, P<0.0001), indicating the DPC formation was related to ROS exposure [43]. However, protein identities have remained unknown, and no absolute quantitation of DPCs was conducted.
Every year, 735,000 people in the United States suffer myocardial infarction [1,2]. Although reperfusion surgery can successfully restore the blood flow to affected myocardium, a sudden influx of oxygenated blood leads to a spike in ROS and reperfusion injury, which is associated with acute cardiomyocyte necrosis, and potentially chronic heart disease [8,44,45]. An estimated 40–50% of the final MI damage has been attributed to reperfusion injury [8]. However, the exact mechanism of myocardium damage following ischemia-reperfusion event has not been elucidated. We hypothesized that the influx of hydroxyl radicals and other reactive oxygen species caused by ischemia/reperfusion leads to the formation of toxic DNA-protein cross-links, contributing to reperfusion injury.
In the present study, we employed a well-established animal model of ischemia/reperfusion (LAD ligation/reperfusion) to investigate DPC formation in cardiomyocytes following ischemia-reperfusion injury [46]. Animals in treatment group that underwent LAD ligation/reperfusion exhibited greatly elevated plasma levels of cTnI (>50 ng/mL), confirming severe MI (Table 1). Furthermore, plasma levels of the oxidative stress biomarker 8-iso-PGF2α were increased 3-5-fold in animals that underwent the LAD surgery and reperfusion as compared to sham surgery (Figure 1), although pre-surgery 8-iso-PGF2α may have been elevated because anesthetization created a hyperoxic environment. Taken together, our results shown in Table 1 and Figure 1 confirm that animals in the LAD group experienced MI/reperfusion injury that led to an increase in oxidants production in the heart.
Two separate approaches were employed to investigate DPC formation in rat cardiomyocytes following ischemia-reperfusion injury. Absolute numbers of free radical induced DPCs were established using the sensitive and accurate isotope dilution nanoLC-NSI+-MS/MS method for thymine-tyrosine conjugates recently developed in our laboratory (Figure 2). Protein identities were established using quantitative proteomics, which was coupled with analysis of the global proteome in order to detect any system-wide changes in protein levels. It was necessary to employ quantitative proteomics in this case because background levels of DPCs are found in healthy heart tissues as a result of aerobic respiration.
In order to determine the absolute numbers of hydroxyl radical-induced DPC in cardiomyocytes following ischemia/reperfusion injury, DPC-containing DNA was digested with nucleases and proteinases to single monomer level, and the amounts of thymidine-tyrosine conjugates (dT-Tyr, Scheme 2) were determined by isotope dilution nanoLC-NSI+-MS/MS using 13C labeled internal standard synthesized in our laboratory (Figure 2A). dT-Tyr conjugate numbers were significantly increased in cardiomyocytes of rats that underwent ischemia-reperfusion injury as compared to sham controls (Figure 2B). Altman et al previously measured 3-[(1,3-dihydro-2,4-dioxopyrimidin-5-yl jmethyl]-L-tyrosine (Thy-Tyr) adducts in SP2/0 murine cells treated with Fe(II) using a gas chromatography mass spectrometry assay [47]. Treatment with 1 mM Fe(II) yielded 32 Thy-Tyr adducts per 105 nucleotides [47], nearly a 1000-fold increase over our highest observed dT-Tyr levels. They were also able to detect up to 15 Thy-Tyr adducts per 106 nucleotides in control cells [47], which are once again significantly higher than what was observed in our sham surgery samples. Taken together, this indicates that both cell culture conditions and treatment of cells with Fe(II) may expose cells to levels of hydroxyl radicals that are not physiologically relevant [48].
Proteins covalently attached to DNA in rat cardiomyocytes were identified and quantified using quantitative proteomics. These analyses have revealed a highly significant increase in radical-induced DPC formation following LAD ligation/reperfusion surgery as compared to sham surgery (Table 2, Figure 3). Quantitative proteomic analysis of DNA-conjugated proteins has identified a total of 374 proteins: of these, 37 proteins were upregulated at least 1.2-fold after LAD ligation/reperfusion as compared to sham surgery and 16 proteins were downregulated at least 1.2-fold under the same conditions (Table S-1).
A large fraction of the oxidatively cross-linked proteins (43.3%) were mitochondrial in nature, including cytochrome C oxidase, pyruvate dehydrogenase, and enoyl-CoA hydratase (Table 2). Since our DPC extraction methodology does not discriminate chromosomal DNA from mitochondrial DNA, it is possible that these proteins are covalently trapped on mitochondrial DNA. Mitochondria increase the production of superoxide anions, hydrogen peroxide, and hydroxyl radicals in response to ischemia/reperfusion injury [3,4,49,50]. For example, all four complexes of the electron transport chain can produce reactive superoxide anions, and ischemic damage to complex I and III is believed to enhance the production of superoxides released during reperfusion injury [4,51,52]. Furthermore, NAD-dependent mitochondrial pyruvate dehydrogenase (PDH), a protein identified in the DPC samples, can produce both O2− and H2O2 as enzymatic byproducts [53,54]. The mitochondrial NADH/NAD+ ratio is elevated post-ischemia, leading to an increased enzymatic activity NAD-dependent dehydrogenases such as PDH and an increase in O2− and H2O2 byproducts [53]. Therefore, it is not surprising that mitochondrial proteins are exposed to increased oxidant levels and are thus susceptible to becoming covalently trapped on mitochondrial DNA.
Additionally, many of the identified DPC protein constituents are known to be present in both the mitochondria or cytoplasm and the nucleus. For example, pyruvate dehydrogenase E1 components, which increased in DPC abundance 9.2-fold after LAD ligation/reperfusion compared to sham surgery, are classified as mitochondrial proteins. However, mitochondrial stress stimulates the translocation of pyruvate dehydrogenases to the nucleus to generate nuclear acetyl CoA, which is required for histone acetylation and the activation of transcription [55].
Ironically, ROS scavenging enzymes, thioredoxin-dependent peroxide reductase, superoxide dismutase [Mn], superoxide dismutase [Cu-Zn], and catalase have been found to participate in cross-linking to DNA following reperfusion injury (Table 2). Although these ROS scavenging enzymes are induced in response to oxidative stress, superoxide dismutases and catalase are known to release hydroxyl radicals as a byproduct of peroxide metabolism [4,56,57]. Hydroxyl radicals are extremely reactive species capable of abstracting hydrogen (H•) from nearby DNA bases [58–60]. The resulting radical intermediates can react with nearby dismutase or catalase enzyme, yielding a DPC with the ROS scavenging protein.
The observed increase in oxidative DNA-protein cross-linking (Figure 3) cannot be attributed to changes in global protein levels following reperfusion injury. Only modest increases in protein biomarkers of cardiomyocyte injury after LAD ligation/reperfusion compared to sham surgery, while the majority of the proteome was unchanged (Table S-1, Figure 4). Increased levels of troponin-T have been established as a specific biomarker of myocardial damage and the breakdown of the contractile apparatus [61–63]. Furthermore, previous studies have observed the degradation and release of other sarcomere proteins including troponin-I, myosin light chain 1, and tropomyosins after LAD ligation/reperfusion in rats [64]. Our observation of increased global levels of the sarcomere proteins troponin-T, troponin-I, myosin-3, myosin light chain 4, and actin following LAD ligation/reperfusion is also consistent with the breakdown of the contractile apparatus after reperfusion injury, and may indicate damaged sarcomere proteins are translocated to the cytoplasm of cardiac cells as well as into the extracellular plasma.
Our results for global proteome changes are consistent with previous studies that utilized quantitative proteomics techniques to measure changes in protein expression after LAD ligation or LAD ligation/reperfusion injury compared to sham surgery [63,65]. For example, De Celle et al utilized two-dimensional gel electrophoresis quantification and mass spectrometry-based proteomics to identify 8 proteins (troponin T, heat shock protein 20 and 27, heterogeneous nuclear ribonucleoprotein K, pyruvate dehydrogenase E1-β, annexin A3, adenylate kinase 1, and serum amyloid p-component precursor) that were upregulated after LAD ligation/reperfusion and two proteins (myosin heavy chain α and catechol-o-methyltransferase) that were downregulated after LAD ligation/reperfusion [63]. Our quantitative proteomics analysis also observed upregulation of troponin T, pyruvate dehydrogenase E1-β, and annexin-3 after LAD ligation/reperfusion. Furthermore, we observed upregulation of two other heat shock proteins (10 kDa heat shock protein and 60 kDa heat shock protein mitochondrial) and adenylate kinase 2 (Figure 4, Table S-1).
In another study, Liu et al utilized label-free spectral counting analysis to identify 15 mitochondrial proteins that became upregulated after LAD ligation as compared to a sham surgery [65]. Our current study identified all of these mitochondrial proteins, with six proteins (amine oxidase, mitochondrial 2-oxogluterate/malate carrier protein, ATP synthase subunit gamma, aldehyde dehydrogenase, dihydrolipoyllysine residue acetyltransferase component of pyruvate dehydrogenase complex, and electron transfer flavoprotein subunit alpha) upregulated after LAD ligation/reperfusion [65]. These changes in mitochondrial protein expression are indicative of the ischemia-induced mitochondrial dysfunction and dysregulation of the electron transport chain.
To our knowledge, our study is the first to provide conclusive evidence for the increased oxidative DPC formation following ischemia/reperfusion injury. Our results suggest that oxidative DNA-protein cross-linking formation is a direct consequence of a myocardial infarction/reperfusion injury. Furthermore, the quantitative nanoLC-NSI+-MS/MS assay for DNA-protein cross-links reported here has the potential to improve the development of preventative therapies aimed to reduce reperfusion injury. Previous experimental and clinical investigations of antioxidant therapies have reported mixed or inconsistent results [66,67]. This partially may be caused by the lack of specificity and sensitivity of assays used to measure oxidant production [68–70]. Hydroxyl radical-induced dT-Tyr conjugates could serve as a novel biomarker of oxidant-induced tissue damage, providing a more accurate measure of the efficacy of future therapies designed to reduce oxidant levels brought by reperfusion.
Supplementary Material
Highlights.
Ischemia/reperfusion mediated DNA-protein cross-linking was investigated.
We employed left anterior descending artery ligation/reperfusion surgery rat model.
Quantitative proteomics identified 90 proteins involved in ROS-induced cross-linking.
Cross-linking occurred by a free radical mechanism between thymidine and tyrosine.
Acknowledgments
We thank Aimee Rinas (Thermo Fischer Scientific) for her contribution to the development of the FASP procedure, TMT labeling procedure, and assisting with nanoLC-NSI-MS/MS analysis, Rosa Viner (Thermo Fischer Scientific) for her help in the development of the nanoHPLC-NSI+-MS/MS procedure for analyzing TMT-labeled peptides, Ryan Bomgarden (Thermo Fischer Scientific) for his contributions to the DPC sample preparation procedure for TMT labeleing, and Robert Carlson (University of Minnesota) for creating the graphics for this article.
Funding:
This work was supported by the U.S. National Institute of Environmental Health Sciences [grant number R01 ES-023350].
Abbreviations
- 8-iso-PGF2α
8-iso-prostaglandin F2α
- Ar
argon
- cTni
troponin-I
- DPC
DNA-protein cross-link
- dT-Tyr
2-amino-3-(4-hydroxy-3-((1-((2R,4R,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)phenyl)propanoic acid
- EtOH
ethanol
- GO
gene ontology
- H2O2
hydrogen peroxide
- HCl
hydrochloric acid
- h
hour
- LAD
left anterior descending artery
- min
minute
- O2−
superoxide
- OH•
hydroxide anion
- PDH-1Eβ
pyruvate dehydrogenase E1 beta subunit
- ROS
reactive oxygen species
- SOD2
superoxide dismutase [Mn]
- TMT
thermo mass tags
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, et al. Executive Summary: Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80(5):780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 4.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Pathophysiological role of neutrophils in acute myocardial infarction. Thromb Haemost. 2013;110(3):501–514. doi: 10.1160/TH13-03-0211. [DOI] [PubMed] [Google Scholar]
- 6.Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280(6):H2649–H2657. doi: 10.1152/ajpheart.2001.280.6.H2649. [DOI] [PubMed] [Google Scholar]
- 7.Lefer DJ, Shandelya SM, Serrano CV, Jr, Becker LC, Kuppusamy P, Zweier JL. Cardioprotective actions of a monoclonal antibody against CD-18 in myocardial ischemia-reperfusion injury. Circulation. 1993;88(4 Pt 1):1779–1787. doi: 10.1161/01.cir.88.4.1779. [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avkiran M, Marber MS. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39(5):747–753. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- 10.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Cross-linking of 2-deoxyribonolactone and its beta-elimination product by base excision repair enzymes. Biochemistry. 2003;42(8):2449–2455. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 11.Guan L, Greenberg MM. Irreversible inhibition of DNA polymerase beta by an oxidized abasic lesion. J Am Chem Soc. 2010;132(14):5004–5005. doi: 10.1021/ja101372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat Res. 2005;589(2):111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Tretyakova NY, Groehler A, Ji S. DNA-Protein cross-links: formation, structural identities, and biological outcomes. Acc Chem Res. 2015;48(6):1631–1644. doi: 10.1021/acs.accounts.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, Salem AM. Repair and biochemical effects of DNA-protein crosslinks. Mutat Res. 2011;711(1-2):113–122. doi: 10.1016/j.mrfmmm.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Xu X, Muller JG, Ye Y, Burrows CJ. DNA-protein cross-links between guanine and lysine depend on the mechanism of oxidation for formation of C5 vs C8 guanosine adducts. J Am Chem Soc. 2008;130(2):703–709. doi: 10.1021/ja077102a. [DOI] [PubMed] [Google Scholar]
- 16.Johansen ME, Muller JG, Xu X, Burrows CJ. Oxidatively induced DNA-protein cross-linking between single-stranded binding protein and oligodeoxynucleotides containing 8-oxo-7,8-dihydro-2′-deoxyguanosine. Biochemistry. 2005;44(15):5660–5671. doi: 10.1021/bi047580n. [DOI] [PubMed] [Google Scholar]
- 17.Perrier S, Hau J, Gasparutto D, Cadet J, Favier A, Ravanat JL. Characterization of lysine-guanine cross-links upon one-electron oxidation of a guanine-containing oligonucleotide in the presence of a trilysine peptide. J Am Chem Soc. 2006;128(17):5703–5710. doi: 10.1021/ja057656i. [DOI] [PubMed] [Google Scholar]
- 18.Dizdaroglu M, Gajewski E, Reddy P, Margolis SA. Structure of a hydroxyl radical induced DNA-protein cross-link involving thymine and tyrosine in nucleohistone. Biochemistry. 1989;28(8):3625–3628. doi: 10.1021/bi00434a071. [DOI] [PubMed] [Google Scholar]
- 19.Gajewski E, Fuciarelli AF, Dizdaroglu M. Structure of hydroxyl radical-induced DNA-protein crosslinks in calf thymus nucleohistone in vitro. Int J Radiat Biol. 1988;54(3):445–459. doi: 10.1080/09553008814551821. [DOI] [PubMed] [Google Scholar]
- 20.Smith SA, Mammen PP, Mitchell JH, Garry MG. Role of the exercise pressor reflex in rats with dilated cardiomyopathy. Circulation. 2003;108(9):1126–1132. doi: 10.1161/01.CIR.0000084538.40542.56. [DOI] [PubMed] [Google Scholar]
- 21.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51(3):288–298. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apple FS, Murakami MM, Christenson RH, Campbell JL, Miller CJ, Hock KG, Scott MG. Analytical performance of the i-STAT cardiac troponin I assay. Clin Chim Acta. 2004;345(1-2):123–7. doi: 10.1016/j.cccn.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 23.Apple FS, Ler R, Chung AY, Berger MJ, Murakami MM. Point-of-care i-STAT cardiac troponin I for assessment of patients with symptoms suggestive of acute coronary syndrome. Clin Chem. 2006;52(2):322–5. doi: 10.1373/clinchem.2005.060293. [DOI] [PubMed] [Google Scholar]
- 24.O’Donovan C, Martin MJ, Gattiker A, Gasteiger E, Bairoch A, Apweiler R. High-quality protein knowledge resource: SWISS-PROT and TrEMBL. Brief Bioinform. 2002;3(3):275–284. doi: 10.1093/bib/3.3.275. [DOI] [PubMed] [Google Scholar]
- 25.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 26.Zagorski J, Gellar MA, Obraztsova M, Kline JA, Watts JA. Inhibition of CINC-1 decreases right ventricular damage caused by experimental pulmonary embolism in rats. J Immunol. 2007;179(11):7820–6. doi: 10.4049/jimmunol.179.11.7820. [DOI] [PubMed] [Google Scholar]
- 27.Dries JL, Kent SD, Virag JA. Intramyocardial administration of chimeric ephrinA1-Fc promotes tissue salvage following myocardial infarction in mice. J Physiol. 2011;589(Pt 7):1725–40. doi: 10.1113/jphysiol.2010.202366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daubert MA, Jeremias A. The utility of troponin measurement to detect myocardial infarction: review of the current findings. Vasc Health Risk Manag. 2010;6:691–699. doi: 10.2147/vhrm.s5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al Saleh A, Alazzoni A, Al Shalash S, Ye C, Mbuagbaw L, Thabane L, Jolly SS. Performance of the high-sensitivity troponin assay in diagnosing acute myocardial infarction: systematic review and meta-analysis. CMAJ Open. 2014;2(3):E199–E207. doi: 10.9778/cmajo.20130074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuhara K, Nakashima T, Abe M, Masuda T, Hamada H, Iwamoto H, Fujitaka K, Kohno N, Hattori N. Suplatast tosilate protects the lung against hyperoxic lung injury by scavenging hydroxyl radicals. Free Radic Biol Med. 2017;106:1–9. doi: 10.1016/j.freeradbiomed.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Friedenreich CM, Pialoux V, Wang Q, Shaw E, Brenner DR, Waltz X, Conroy SM, Johnson R, Woolcott CG, Poulin MJ, et al. Effects of exercise on markers of oxidative stress: an Ancillary analysis of the Alberta Physical Activity and Breast Cancer Prevention Trial. BMJ Open Sport Exerc Med. 2016;2(1):e000171. doi: 10.1136/bmjsem-2016-000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arany I, Hall S, Reed DK, Reed CT, Dixit M. Nicotine Enhances High-Fat Diet-Induced Oxidative Stress in the Kidney. Nicotine. Tob Res. 2016;18(7):1628–1634. doi: 10.1093/ntr/ntw029. [DOI] [PubMed] [Google Scholar]
- 33.Dizdaroglu M, Gajewski E. Structure and mechanism of hydroxyl radical-induced formation of a DNA-protein cross-link involving thymine and lysine in nucleohistone. Cancer Res. 1989;49(13):3463–3467. [PubMed] [Google Scholar]
- 34.Margolis SA, Coxon B, Gajewski E, Dizdaroglu M. Structure of a hydroxyl radical induced cross-link of thymine and tyrosine. Biochemistry. 1988;27(17):6353–6359. doi: 10.1021/bi00417a024. [DOI] [PubMed] [Google Scholar]
- 35.Charlton TS, Ingelse BA, Black DS, Craig DC, Mason KE, Duncan MW. A covalent thymine-tyrosine adduct involved in DNA-protein crosslinks: synthesis, characterization, and quantification. Free Radic Biol Med. 1999;27(3-4):254–61. doi: 10.1016/s0891-5849(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 36.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 37.Lemeer S, Hahne H, Pachl F, Kuster B. Software tools for MS-based quantitative proteomics: a brief overview. Methods Mol Biol. 2012;893:489–99. doi: 10.1007/978-1-61779-885-6_29. [DOI] [PubMed] [Google Scholar]
- 38.Wickramaratne S, Mukherjee S, Villalta PW, Scharer OD, Tretyakova NY. Synthesis of sequence-specific DNA-protein conjugates via a reductive amination strategy. Bioconjug Chem. 2013;24(9):1496–1506. doi: 10.1021/bc400018u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeo JE, Wickramaratne S, Khatwani S, Wang YC, Vervacke J, Distefano MD, Tretyakova NY. Synthesis of site-specific DNA-protein conjugates and their effects on DNA replication. ACS Chem Biol. 2014;9(8):1860–1868. doi: 10.1021/cb5001795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickramaratne S, Boldry EJ, Buehler C, Wang YC, Distefano MD, Tretyakova NY. Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine. J Biol Chem. 2015;290(2):775–787. doi: 10.1074/jbc.M114.613638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickramaratne S, Ji S, Mukherjee S, Su Y, Pence MG, Lior-Hoffmann L, Fu I, Broyde S, Guengerich FP, Distefano M, et al. Bypass of DNA-Protein Cross-links Conjugated to the 7-Deazaguanine Position of DNA by Translesion Synthesis Polymerases. J Biol Chem. 2016;291(45):23589–23603. doi: 10.1074/jbc.M116.745257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakano T, Miyamoto-Matsubara M, Shoulkamy MI, Salem AM, Pack SP, Ishimi Y, Ide H. Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J Biol Chem. 2013;288(7):4649–4658. doi: 10.1074/jbc.M112.419358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izzotti A, Cartiglia C, Taningher M, De Flora S, Balansky R. Age-related increases of 8-hydroxy-2′-deoxyguanosine and DNA-protein crosslinks in mouse organs. Mutat Res. 1999;446(2):215–223. doi: 10.1016/s1383-5718(99)00189-8. [DOI] [PubMed] [Google Scholar]
- 44.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987;84(5):1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hashmi S, Al Salam S. Acute myocardial infarction and myocardial ischemia-reperfusion injury: a comparison. Int. J Clin Exp Pathol. 2015;8(8):8786–8796. [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, Alloush J, Beck E, Weisleder N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J Vis Exp. 2014;86 doi: 10.3791/51329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altman SA, Zastawny TH, Randers-Eichhorn L, Cacciuttolo MA, Akman SA, Dizdaroglu M, Rao G. Formation of DNA-protein cross-links in cultured mammalian cells upon treatment with iron ions. Free Radic Biol Med. 1995;19(6):897–902. doi: 10.1016/0891-5849(95)00095-f. [DOI] [PubMed] [Google Scholar]
- 48.Halliwell B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett. 2003;540(1-3):3–6. doi: 10.1016/s0014-5793(03)00235-7. [DOI] [PubMed] [Google Scholar]
- 49.Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114(3):524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lenaz G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol. 2012;942:93–136. doi: 10.1007/978-94-007-2869-1_5. [DOI] [PubMed] [Google Scholar]
- 52.Chen HJ, Chiu WL, Lin WP, Yang SS. Investigation of DNA-protein cross-link formation between lysozyme and oxanine by mass spectrometry. Chembiochem. 2008;9(7):1074–1081. doi: 10.1002/cbic.200700686. [DOI] [PubMed] [Google Scholar]
- 53.Adam-Vizi V, Tretter L. The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int. 2013;62(5):757–763. doi: 10.1016/j.neuint.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 54.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289(12):8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158(1):84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- 56.Yim MB, Chock PB, Stadtman ER. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proc Natl Acad Sci U S A. 1990;87(13):5006–5010. doi: 10.1073/pnas.87.13.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goyal MM, Basak A. Hydroxyl radical generation theory: a possible explanation of unexplained actions of mammalian catalase. Int J Biochem Mol Biol. 2012;3(3):282–289. [PMC free article] [PubMed] [Google Scholar]
- 58.Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl radicals and DNA base damage. Mutat Res. 1999;424(1-2):9–21. doi: 10.1016/s0027-5107(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 59.Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013;5(2) doi: 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991;281(1-2):9–19. doi: 10.1016/0014-5793(91)80347-6. [DOI] [PubMed] [Google Scholar]
- 61.Remppis A, Ehlermann P, Giannitsis E, Greten T, Most P, Muller-Bardorff M, Katus HA. Cardiac troponin T levels at 96 hours reflect myocardial infarct size: a pathoanatomical study. Cardiology. 2000;93(4):249–53. doi: 10.1159/000007034. [DOI] [PubMed] [Google Scholar]
- 62.Metzler B, Hammerer-Lercher A, Jehle J, Dietrich H, Pachinger O, Xu Q, Mair J. Plasma cardiac troponin T closely correlates with infarct size in a mouse model of acute myocardial infarction. Clin Chim Acta. 2002;325(1-2):87–90. doi: 10.1016/s0009-8981(02)00296-6. [DOI] [PubMed] [Google Scholar]
- 63.De Celle T, Vanrobaeys F, Lijnen P, Blankesteijn WM, Heeneman S, Van Beeumen J, Devreese B, Smits JF, Janssen BJ. Alterations in mouse cardiac proteome after in vivo myocardial infarction: permanent ischaemia versus ischaemia-reperfusion. Exp Physiol. 2005;90(4):593–606. doi: 10.1113/expphysiol.2005.030296. [DOI] [PubMed] [Google Scholar]
- 64.Van Eyk JE, Powers F, Law W, Larue C, Hodges RS, Solaro RJ. Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation. Circ Res. 1998;82(2):261–71. doi: 10.1161/01.res.82.2.261. [DOI] [PubMed] [Google Scholar]
- 65.Liu T, Chen L, Kim E, Tran D, Phinney BS, Knowlton AA. Mitochondrial proteome remodeling in ischemic heart failure. Life Sci. 2014;101(1-2):27–36. doi: 10.1016/j.lfs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Effect of 48-h intravenous trimetazidine on short- and long-term outcomes of patients with acute myocardial infarction, with and without thrombolytic therapy; A double-blind, placebo-controlled, randomized trial. The EMIP-FR Group. European Myocardial Infarction Project–Free Radicals. Eur Heart J. 2000;21(18):1537–1546. doi: 10.1053/euhj.1999.2439. [DOI] [PubMed] [Google Scholar]
- 67.Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discov Med. 2011;11(57):106–114. [PubMed] [Google Scholar]
- 68.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86(5):1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 69.Logan A, Cocheme HM, Li Pun PB, Apostolova N, Smith RA, Larsen L, Larsen DS, James AM, Fearnley IM, Rogatti S, et al. Using exomarkers to assess mitochondrial reactive species in vivo. Biochim Biophys Acta. 2014;1840(2):923–930. doi: 10.1016/j.bbagen.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 70.Kaludercic N, Deshwal S, Di Lisa F. Reactive oxygen species and redox compartmentalization. Front Physiol. 2014;5:285. doi: 10.3389/fphys.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.