

Abstract
Thrombin normally cleaves fibrinogen to promote coagulation, however, binding of thrombomodulin to thrombin switches the specificity of thrombin toward protein C, triggering the anticoagulation pathway. The W215A thrombin mutant was reported to have decreased activity towards fibrinogen without significant loss of activity towards protein C. To understand how mutation of Trp215 may alter thrombin specificity, hydrogen-deuterium exchange experiments (HDXMS), accelerated molecular dynamics (AMD) simulations, and activity assays were carried out to compare the dynamics of Trp215 mutants with those of wild type (WT) thrombin. Variation in NaCl concentration had no detectable effect on the sodium-binding (220sCT) loop, but appeared to affect other surface loops. Trp215 mutants showed significant increases in amide exchange in the 170sCT loop consistent with a loss of H-bonding in this loop identified by the AMD simulations. The W215A thrombin showed increased amide exchange in the 220sCT loop and in the N-terminus of the heavy chain. The AMD simulations showed that a transient conformation of the W215A thrombin has a distorted catalytic triad. HDXMS experiments revealed that mutation of Phe227, which engages in a pi-stacking interaction with Trp215, also caused significantly increased amide exchange in the 170sCT loop. Activity assays showed only the F227V mutant had wild type catalytic activity, whereas all other mutants showed markedly lower activity. Taken together, the results explain the reduced pro-coagulant activity of the W215A mutant and demonstrate the allosteric connection between Trp215, the sodium-binding loop and the active site.
Keywords: serine protease, protein dynamics, hydrogen-deuterium exchange, allostery
Introduction
Thrombin is a dual action serine protease that binds and cleaves a variety of substrates (1). Substrate recognition is dependent on the binding of a consensus sequence at the primary specificity pockets (S1-4) within the active site, as well as binding at distinct exosites located on opposites sides of thrombin. Central to its role in the clotting cascade, thrombin distinguishes between substrates that elicit either a procoagulative or an anticoagulative response. The binding of thrombomodulin (TM) to anion binding site 1 (AEB1) of thrombin shifts the enzyme’s specificity away from procoagulative substrates, such as fibrinogen and the protease-activated receptor 1(PAR1), and towards the anticoagulative substrate, protein C (PC) (2). How TM binding to thrombin alters its substrate specificity is still not fully understood. Comparison of the X-ray crystal structures of apo-thrombin and thrombin bound to TM456, a construct of TM containing only the domains necessary for toggling thrombin’s substrate specify, shows no discernable differences between the two structures (3). Thus, TM-binding affects thrombin in ways that are invisible to X-ray crystallography.
Di Cera determined that the W215A mutant of thrombin had significantly less activity towards procoagulative substrates, yet this mutant retained most of the activity towards PC in the presence of TM (4). Interestingly, the W215I mutation had significantly less activity towards both procoagulative and anticoagulative substrates (5). Measurements of hirudin binding to three Trp215CT mutants showed that mutations at position 215CT make thrombin much less sensitive to sodium binding, offering an explanation for the decreased activity observed for procoagulative substrates (4, 6). Saturation mutagenesis of Trp215CT revealed an array of activity profiles that distinguish Trp215CT mutants in their abilities to cleave fibrinopeptide A (FpA), PAR1, and PC (5), suggesting that this residue is involved in key aspects of modulating thrombin’s activity. The dramatic effect of mutations at Trp215CT on substrate specificity identifies this residue as one of significant interest, warranting further study of how the characteristics Trp215CT mutants differ from those of WT thrombin.
Thrombin is a highly dynamic enzyme. Studies have demonstrated that the surface loops of thrombin exhibit dynamic motions (7, 8). Previous work from our lab has also connected the dynamic motions of thrombin to key catalytic regions of the protein, providing evidence for dynamic allostery (9, 10). Here we focus on Trp215CT to gain a clearer picture of how this mutation affects thrombin dynamics and activity. Due to the apparent pi-stacking interaction between Trp215CT and Phe227CT in the X-ray crystal structure of thrombin (Protein Data Base (PDB) ID code 1PPB) (11), we included mutants of Phe227CT in our study as well. We used a combination of activity assays, HDXMS to evaluate the differences in backbone dynamics, and AMD to obtain a molecular mechanism of changes in amide exchange.
HDXMS provides a measure of the backbone dynamics and solvent accessibility changes that occur on physiologically relevant time scales (12). HDXMS has already been used to compare the dynamics of apo-thrombin, PPACK-thrombin, and TM-thrombin, revealing changes in surface loop dynamics that directly impact thrombin activity (13, 14). Thus, HDXMS provides a tool for comparing the backbone dynamics of thrombin mutants with the motions native to WT thrombin.
AMD is an efficient and versatile enhanced conformational space sampling algorithm that allows for the accurate study of slow motions in bio-molecular systems up to time-scales several orders of magnitude greater than those accessible using standard classical MD methods, while still affording a fully atomistic representation of the system. The acceleration level used here has previously been shown to afford an increase in the rate of conformational space sampling by 3-4 orders of magnitude (15, 16).
We show that the W215A mutant has markedly increased dynamics in the 170sCT loop, the 220sCT loop, and the N-terminus of the heavy chain and its dynamics are less sensitive to NaCl concentration. Investigation of the pi-stacking interaction between Phe227CT and Trp215CT revealed that this interaction is critical for ordering the 170sCT loop, but Trp215CT alone is responsible for long-range allostery that impacts the 220sCT loop and the N-terminus of the heavy chain. The results of this study clarify the previously observed coupling between sodium binding and mutation of Trp215CT and reveal the dramatic changes in thrombin dynamics that result from mutations at both Trp215CT and Phe227CT.
Materials and Methods
Thrombin mutagenesis and Expression
All mutants were generated through site-directed mutagenesis using primers purchased from Integrated DNA Technologies. All thrombin constructs were expressed and refolded in E. coli, as previously described (8). Activation of thrombin was achieved by diluting properly-folded thrombin, purified through ion exchange chromatography, to 50 mL with 50 mM Tris/HCl pH 7.4, 20 mM CaCl2, 1 mg/mL PEG8000, 5% (v/v) glycerol, and 20 μg/mL E. carinatus venom (Sigma-Aldrich Car. # V8250) pretreated with AEBSF. Optimal activation times varied from 2-10hr at room temperature, determined by monitoring the conversion of pre-thrombin-2 to a-thrombin through ion exchange chromatography. Activated samples were loaded onto a MonoS cation exchange column (GE Healthcare Life Sciences), allowing for the separation of active a-thrombin from preactive (prethombin-2, meizothrombin) and autoproteolyzed forms (particularly γ-thrombin) using a gradient of 100 mM NaCl to 500 mM NaCl in 25 mM phosphate pH 6.5. Because of their inherent inactivity, thrombin mutants F227A and W215I were activated with 45 nM wild type α-thrombin present. Fully active α-thrombin is the latest eluting species from the MonoS column, however, all fully active mutants eluted earlier than WT, allowing for effective purification of F227A and W215I from the WT thrombin added for activation. Fractions containing fully activated thrombin were pooled and stored at −80 C for no longer than 1 month before use. This method of thrombin purification, which avoids dialysis, has been shown previously to result in >95% active α-thrombin. Previous NMR analysis of isotopically labeled thrombin prepared this way, demonstrated that the species present was active a-thrombin (8, 17).
Thrombin activity assays
To account for the loss of A280 for all Trp mutants, enzyme concentrations were determined using BCA protein assays. Activity assay reactions occurred in 200 μL volumes containing 2.5 ng/mL human thrombin, 0.88 μM BSA, 2.93 mM CaCl2, 20 mM Tris, 200 mM NaCl, and chromogenic substrate (Diapharma, West Chester, OH). Chromogenic substrate stocks at 12.5 mg/mL in H2O were stored at −20 C, and were neutralized with Tris base before use. Chromogenic substrate dilutions were performed using a solution of 20 mM Tris and 200 mM NaCl. The thrombin-specific chromogenic substrate S-2238 was present at concentrations varying from 2.5 - 100 μM for WT, 6.25 - 200 μM for W215A, 1 - 100 μM for F227V, and 25 - 1000 μM for W215I. Activated PC (aPC)-specific chromogenic substrate S-2366 was present between 10 - 1000 μM for WT, 10 - 1000 μM for W215A, 50 - 1600 μM for F227V, and 50 - 1600 μM for W215I. All reactions were incubated at room temperature for 10 min following the addition of thrombin, before chromogenic substrate was added. Enzyme dilutions were made by diluting purified thrombin with a solution of 20 mM Tris pH 7.5, 200 mM NaCl, 5 mM CaCl2, and 15 μM BSA. Each reaction was monitored via the linear increase in absorbance at 405 nm over time, corresponding to the release of p-nitroaniline following chromogenic substrate cleavage. From the rates measured, KM and Vmax, and thereby kcat, values were determined for each substrate for each thrombin construct. All chromogenic substrate assays were repeated in triplicate.
Protein C assays were carried out as previously described (18). The TM activation of thrombin towards protein C cleavage was first performed by incubating the TM construct TM456m with purified human α-thrombin before adding protein C (Hematologic Technologies, Essex Junction, VT). TM456m was purified as previously described (14). Following a 20 min incubation with protein C, the thrombin was inactivated by addition of heparin-antithrombin-III, and the activated protein C was assayed by the addition of S-2366. Human thrombin was present at 17.4 ng/mL for each reaction, and TM456m was present at either 21.7 ng/mL, 43.5 ng/mL, 65.2 ng/mL, or 87.0 ng/mL. The rates measured in reactions from which thrombin and/or TM4546m were omitted were subtracted from the rates of the other reactions to account for any PC activation not catalyzed by the thrombin-thrombomodulin complex. The rates for each F227V reaction were divided by the rates for each WT thrombin reaction with the same amount of TM456m added, and all ratios calculated were averaged.
Fibrinogen clotting assays were performed as previously described (19). Thrombin concentrations were determined by BCA assays. The concentrations of thrombin used correspond to the amount necessary for a clot to form between 18-22 sec after the addition of fibrinogen. All clotting assays were performed in triplicate, and the average time-to-clot was used in activity determination.
Hydrogen-Deuterium Exchange Mass Spectrometry
All α-thrombin proteins were prepared for HDXMS from frozen aliquots (~7 uM). After being passed through a 0.2 micron filter, a portion was diluted to 5 μM, and 130 μL was saved for the HDXMS experiment. The remaining sample was concentrated to 10 μM using pre-rinsed 6 mL 10K MWCO Amicon concentrators, spinning at 3000 rpm in 5 min intervals at 4°C to be used for peptide identification (50 μL). In the HDXMS experiment, the sample is diluted 12-fold (see below) resulting in a final α-thrombin concentration of 420 nM.
HDXMS was performed using a Waters Synapt G2Si system with HDX technology (Waters Corporation). Deuterium exchange reactions were prepared using a Leap HDX PAL autosampler (Leap Technologies, Carrboro, NC). D2O buffer was prepared by lyophilizing 1 mL of 250 mM phosphate pH 6.5 and either 1 M NaCl for low salt experiments of 3 M NaCl for high salt experiments, before being resuspended in 10 ml 99.96% D2O immediately before use. Each deuterium exchange time point (0 min, 30 sec, 1 min, 2 min, 5 min) was measured in triplicate. For each deuteration time point, 5 μL of protein was held at 25°C for 5 min before being mixed with 55 μL of D2O buffer. The deuterium exchange was quenched for 1 min at 1°C by combining 50 μL of the deuteration reaction with 50 μL of 250 mM TCEP pH 2.5. The quenched sample was then injected in a 50 μL sample loop, followed by digestion on an in-line pepsin column (immobilized pepsin, Pierce, Inc.) at 15°C. The resulting peptides were captured on a BEH C18 Vanguard pre-column, separated by analytical chromatography (Acquity UPLC BEH C18, 1.7 uM, 1.0 × 50 mm, Waters Corporation) using a 7-85% acetonitrile in 0.1% formic acid over 7.5 min, and electrosprayed into the Waters Synapt G2Si quadrupole time-of-flight mass spectrometer. The mass spectrometer was set the collect data in the Mobility, ESI+ mode; mass acquisition range of 200-2,000 (m/z); scan time 0.4 s. Continuous lock mass correction was accomplished with infusion of leu-enkephalin (m/z = 556.277) every 30 s (mass accuracy of 1 ppm for calibration standard).
For peptide identification, the mass spectrometer was set to collect data in MSE, mobility ESI+ mode instead. Peptides masses were identified from triplicated analyses of 10 μM α-thrombin, and data were analyzed using PLGS 2.5 (Waters Corporation). Peptides masses were identified using a minimum number of 250 ion counts for low energy peptides and 50 ion counts for their fragment ions; the peptides also had to be larger than 1500 Da. The following cutoffs were used to filter peptide sequence matches: minimum products per amino acid of 0.2, minimum score of 7, maximum MH+ error of 5 ppm, a retention time RSD of 5%, and the peptides had to be present in two of the three ID runs collected. The peptides identified in PLGS were then analyzed in DynamX 3.0 (Waters Corporation). The relative deuterium uptake for each peptide was calculated by comparing the centroids of the mass envelopes of the deuterated samples with the undeuterated controls following previously published methods (20). To account for back-exchange, and systematic autosampler sample handling differences between shorter and longer deuteration times, the uptake and standard deviation values for the 30 sec and 1 min, and the 2 min and 5 min timepoints were divided by 0.67 and 0.64 respectively for every HDXMS experiment run. Data were plotted as number of deuterons incorporated vs. time (min). The Y-axis limit for each plot reflects the total number of amides within the peptide that can possible exchange. Each plot includes the peptide MH+ value, sequence, and sequential residue numbering.
To monitor sodium binding, the HDXMS experiments were conducted at either 100 or 300 mM NaCl. Under these conditions of the HDXMS experiment (420 nM thrombin), the sodium binding site should have been less than 80% occupied at 100mM NaCl and over 90% occupied at 300 mM NaCl (21). Our previous studies have shown that for weak binding ligands, observation of “protection” of the surface of the protein requires the binding site to be over 90% occupied (22). Therefore, we expected to see the largest difference in surface “protection” by comparing 80% bound to >90% bound sodium.
AMD simulations
Atomic coordinates for WT thrombin were obtained from the Protein Data Bank (PDB) 1.9-A X-ray crystal structure (PDB ID: 1PBB) and the active-site inhibitor was removed from the structure. For the W215A mutant, residue 215CT was manually converted from a tryptophan to an alanine residue. Each system was placed at the center of a periodically repeating box, and the simulation cell size was defined such that the distance between the edge of the simulation box and the surface of the solute was at least 12 Å. All AMD simulations were performed in explicit solvent with appropriate counter-ions to achieve cell neutrality. Bonds involving protons were constrained using the SHAKE algorithm. Electrostatic interactions were treated using the particle mesh Ewald method with a direct space sum limit of 10 Å. The ff14SB force-field was used for the solute residues, and the TIP3P water force-field was employed for solvent molecules. In the present work, we implemented a “dual boost” AMD approach (23), in which two acceleration potentials are simultaneously applied to the system: The first acceleration potential is applied to the torsion terms only, and a second, weaker acceleration is applied across the entire potential. For both WT thrombin and the W215A mutant systems, the specific torsional acceleration parameters were defined as Eb(dih) -<V0(dih)> = [4.0 kcal/mol * Number of residues], and the acceleration parameter, α(dih), was set to one-fifth of this value. The total background acceleration parameters were fixed at Eb(tot) -<V0(tot)> = α(tot) = [0.16 kcal/mol * No. atoms in simulation cell]. The specific choice of these AMD parameters was based on a previous NMR/AMD study of thrombin:PPACK (8). The results of this and other studies suggested that these acceleration parameters afforded an effective speed up in conformational space sampling of 3-4 orders of magnitude, which is in line with other studies on different systems in which AMD simulations at a similar acceleration level were directly compared to long brute-force CMD simulations (16). More details about how the acceleration is achieved in AMD are given in the Supplementary Information. The average minimum energy potentials, V0(dih) and V0(tot), were obtained from 20-ns CMD simulations performed as part of the initial equilibration procedure. It should be noted that, given the similarity of the two systems, the acceleration parameters for WT thrombin and the W215A mutant are, to all intents and purposes, identical.
For each system (WT and W215A thrombin), two AMD simulations were performed for 750,000,000 steps with a (real time) time-step of 2-fs. This is computationally equivalent to performing a 1.5μs CMD simulation. Given an effective speed up in the rate of conformational space-sampling by 3 to 4 orders of magnitude due to the application of the bias potential, we anticipate that the configurational space sampling afforded in each of the AMD simulations is associated with slow time-scale dynamics occurring on time-scales of milliseconds to tens of milliseconds, thereby identifying slow motions, including rare local unfolding/refolding events which can be experimentally probed by HDXMS in the fast limit. All simulations were performed using the AMBER14 simulation suite. For each AMD trajectory, a corrected canonical ensemble was obtained by performing the Boltzmann free energy reweighting protocol using a cumulant expansion to the second order (24).
Results
Improved HDXMS of thrombin
In order to determine the effects of sodium concentration and the W215A mutation on thrombin dynamics, HDXMS experiments were carried-out. WT and each mutant thrombin were allowed to exchange into deuterated buffer for 0-5 min followed by quenching in 100 mM tris-carboxyethylphosphine, pH 2.5, which improved sequence coverage after pepsin digestion. Fifty-one peptides were reliably identified, which covered 99.0% of the thrombin sequence. Due to the location of the mutations examined, the peptides covering the 220sCT loop varied depending on the mutation (Figure S1). To probe the dependence of the thrombin dynamics on NaCl concentration, experiments on WT and W215A thrombin were carried out at 100 mM NaCl and 300 mM NaCl.
The effect of W215A and Salt on the 220sCT Loop
We first analyzed the difference in deuterium uptake of W215A and WT thrombin in 100 mM vs. 300 mM NaCl. As previously reported by DiCera’s group, the W215A mutation renders thrombin much less sensitive to Na+ ions (4). The deuterium uptake within the W215A 220sCT loop (residues 260-275; MH+ 1673.760, 1788.801) appeared unaffected by NaCl concentration, resulting in similar uptake values at both NaCl concentrations (Figure 1). Interestingly, the 220sCT loop in WT thrombin also showed nearly identical deuterium uptake at 100 mM and 300 mM NaCl. The 220sCT loop of the W215A mutant showed notably higher deuterium uptake compared to WT thrombin, by an average of ~2 deuterons at both the high and low salt concentrations. These results indicate that the 220sCT loop is sensitive to dynamic modulation caused by changes to side chains at the base of the loop, but its dynamics appear insensitive to NaCl concentration.
Figure 1.
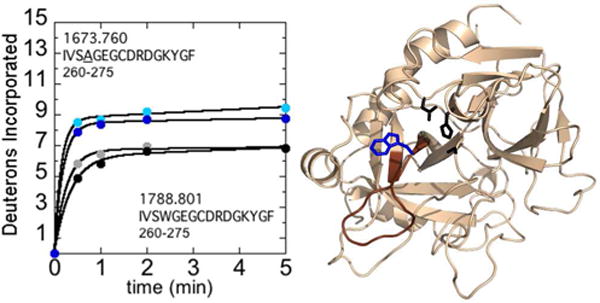
A) Deuterium incorporation into residues 212-227CT (residues 260-275; MH+ 1673.760 and 1788.801 for W215A and WT respectively) over 5 min is shown for WT thrombin at 100 mM NaCl (grey) and 300 mM NaCl (black), and for the W215A mutant at 100 mM NaCl (cyan) and 300 mM NaCl (blue). The mutant residue, is underlined in the peptide sequence shown. B) Structure of WT thrombin (PDB 1PPB) highlighting residues 212-227CT (residues 260-275; brown). The sidechains of Trp215CT (blue) and the catalytic triad (black) are shown as sticks.
Subtle effects caused by NaCl
Several other functionally interesting regions of thrombin did show a moderate dependence of deuterium uptake on NaCl concentration. The deuterium uptake in regions located near anion binding sites 1 (AEB1) and 2 (AEB2) were affected by the concentration of salt more so than by the W215A mutation. Residues 66-85CT (residues 97-117; MH+ 2586.441) showed a slight increase in deuterium uptake with decreasing salt concentration but residues 66-80CT (residues 97-112; MH+ 2014.105) showed no difference, allowing us to localize the salt effect to residues 81-85CT (residues 113-117) (Figure 2). This trend extended through residues 85-102CT (residues 117-135; MH+ 2530.295), which approaches AEB2 and includes Asp102CT of the catalytic triad. Similarly, residues 117-129CT (residues 150-162; MH+ 1513.747) showed no dependence on NaCl concentration, but residues 117-130CT (residues 150-166; MH+ 1897.984) were affected by salt indicating residues 129A-130CT (residues 163-166) of ABE2 also respond to salt concentration. Slight salt effects were observed for other surface loops including the 180sCT loop (residues 223-255; MH+ 3575.589), the 140sCT loop (residues 181-196; MH+ 1714.912), the 30sCT loop (residues 55-64; MH+ 1220.646), and the light chain (residues 1-15; MH+ 1569.737 and residues 16-31; MH+ 1934.008) (Figure S2).
Figure 2.
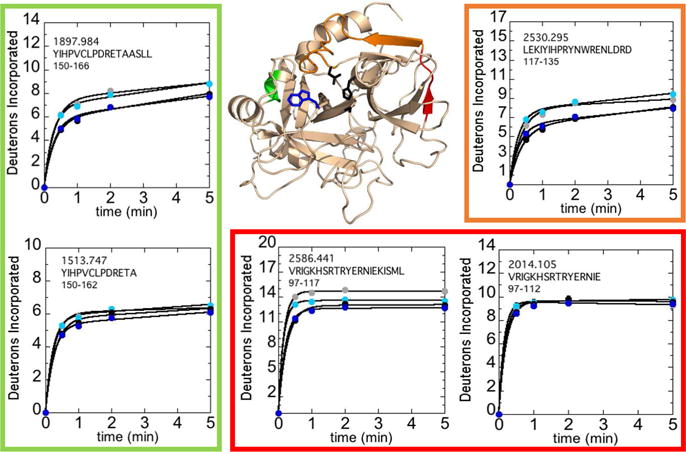
Structure of WT thrombin (PDB 1PPB) highlighting residues 81-85CT (residues 113-117; red), residues 86-102CT (residues 118-135; orange), and residues 129A-130CT (residues 163-166; green). Colored residues specify regions affected by the concentration of NaCl after subtraction of deuteron uptake of overlapping peptides 66-80CT (residues 97-112; MH+ 2014.105) from 66-85CT (residues 97-117; MH+ 2586.441) and 117-129CT (residues 150-162; MH+ 1513.747) from 117-130CT (residues 150-166; MH+ 1897.984) as well as residues 85-102CT (residues 117-135; MH+ 2530.295). The sidechains of Trp215CT (blue) and the catalytic triad (black) are shown as sticks. Deuteron incorporation into these residues over 5 min is shown for WT thrombin at 100 mM NaCl (grey) and 300 mM NaCl (black), and for the W215A mutant at 100 mM NaCl (cyan) and 300 mM NaCl (blue).
The effect of the W215A mutation and NaCl on the 170sCT loop
Unlike the 220sCT loop, both NaCl and the W215A mutation significantly affected deuterium uptake. Three peptides -residues 156-180CT (residues 197-221; MH+ 2896.550), 161-170CT (residues 202-211; MH+ 1155.619), and residues 161-181CT (residues 202-222; MH+ 2490.296) covered the 170sCT loop and portions of the C-terminal β-barrel (Figure 3). The combination of the W215A mutation and low NaCl concentration resulted in the highest exchange, and this region of the W215A mutant thrombin incorporated an average of ~4 more deuterons over 5 min as compared to WT thrombin.
Figure 3.
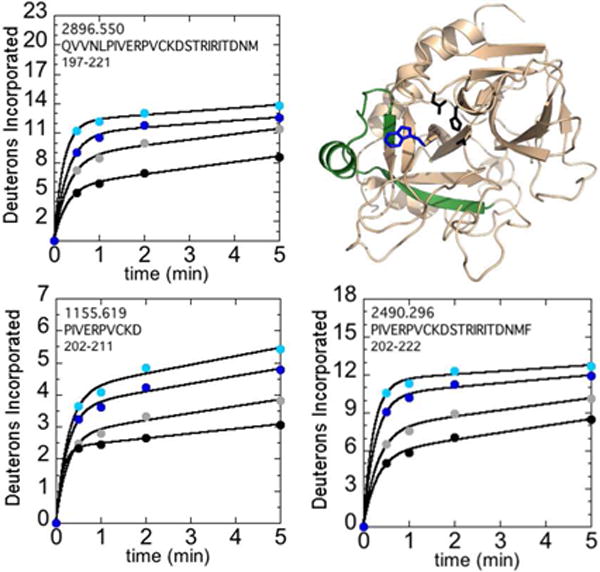
Structure of WT thrombin (PDB 1PPB) highlighting 156-181CT (residues 197-222; green). The sidechains of Trp215CT (blue) and the catalytic triad (black) are shown as sticks. Deuteron incorporation over 5 min into residues 156-180CT (residues 197-221; MH+ 2896.550), 161-170CT (residues 202-211; MH+ 1155.619), and 161-181CT (residues 202-222; MH+ 2490.296) is shown for WT thrombin at 100 mM NaCl (grey) and 300 mM NaCl (black), and for the W215A mutant at 100 mM NaCl (cyan) and 300 mM NaCl (blue).
The effect of W215A and NaCl on the N-Terminus of the heavy chain
The N-terminus of the heavy chain, residues 16-23CT (residues 37-44; MH+ 819.373) exhibited a slight increase in uptake at lower NaCl as well as in the presence of the W215A mutation (Figure 4A). Thus, the dynamics of the N-terminus of the heavy chain are also affected by NaCl and the W215A mutation.
Figure 4.
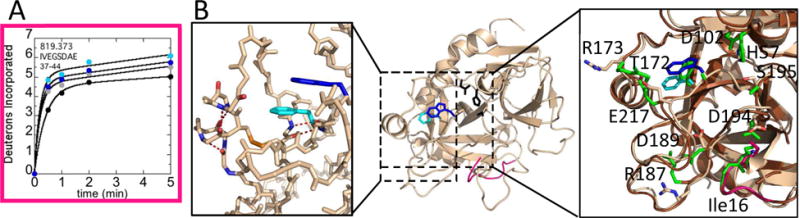
A) Deuterium incorporation over 5 min into residues 16-23CT (residues 36-44; MH+ 819.373) for WT thrombin at 100 mM NaCl (grey) and 300 mM NaCl (black), and for the W215A mutant at 100 mM NaCl (cyan) and 300 mM NaCl (blue). B) Structure of WT thrombin (wheat; PDB 1PPB) highlighting residues 16-23CT (pink). The sidechains of Trp215CT (blue), Phe227CT (cyan), and the catalytic triad (black) are shown as sticks. Accelerated MD simulations identified 5 H-bonds (red-dotted lines) within WT thrombin (left) that broke during simulations of W215A. The transient structure observed during the W215A simulation (brown) overlaying the structure of WT thrombin (right). The side chains of His57CT, Asp102CT, Thr172CT, Arg173CT, Arg 187CT, Asp 189CT, Asp194CT, Ser195CT, Trp215CT (blue), Glu217CT, and Phe227CT (cyan). The 140sCT loop is hidden for clarity. The backbone of Ile16CT (pink) is also shown as sticks. The corresponding side chains in the W215A structure, including Ala215CT, are colored green and are shown as sticks.
Accelerated MD simulations of the W215A mutant thrombin
In order to complement the experimental HDXMS data, we performed extensive accelerated molecular dynamics (AMD) simulations (25, 26) on both WT thrombin and the W215A mutant (see Methods for details). AMD simulations were performed for 750,000,000 steps using a “real time” time step of 2fs, the equivalent of 1.5μs in a classical MD simulation. Because the acceleration level used here affords an increase in the rate of conformational space sampling by 3-4 orders of magnitude, the simulations presented here probe slow molecular motions occurring on timescales of milliseconds to tens of milliseconds (16).
In agreement with the experimental HDXMS data, comparative analysis of the conformational dynamics from AMD simulations of WT and W215A thrombin revealed, for the most part, very few differences between WT and the W215A mutant. The most significant difference observed was a transient destabilization of the 170sCT loop (residues 205-223) in the W215A mutant on slow timescales. Mutation of Trp215CT to Ala removes the strong ring-ring stacking interaction between Trp215CT (residue 263) and Phe227CT (residue 275), causing enhanced mobility on slow timescales of residue Phe227CT and its neighbor, Tyr228CT (residue 276). As a result, transient dislocation of H-bond interactions between residues Tyr228CT and Phe181CT (Tyr276:NH-Phe222:CO and Phe222:NH-Tyr276:CO) is observed. The loss of these H-bonds apparently results in transient unfolding of the alpha-helix residues Arg165-Ser171CT (residues 206-212) with further dislocation of the H-bonds between residues Val167CT, Glu164CT, Lys169CT, Arg165CT, and Ser171CT(residues Val208:NH-Glu205:CO, Lys210:NH-Arg206:CO, and Ser212:NH-Val208:CO). It should be noted that the local unfolding events described above are rare, and approximate free energy statistics obtained from the AMD simulations suggest that the population of this locally unfolded structural motif is less than 10%. These results are consistent with the experimental HDXMS results which showed an increased uptake of four additional deuterons in residues 156-181CT (197-222) for the W215A mutant thrombin (cf. Figure 3). The AMD simulation results suggest that the four amide protons are those of residues Arg 165CT, Val167CT, Lys169CT, and Ser171CT (residues Arg206, Val208, Lys210, and Ser212).
In addition to the transient destabilization of the 170sCT loop, the AMD simulations also identified a different transient conformation of the W215A mutant which was characterized by a dramatic shift in the side chains of Phe227CT and Ile16CT (residue 37) resulting in a misalignment of the catalytic triad (Figure 4B). A full description of the AMD simulations, including a computational analysis of the HDXMS data will be presented elsewhere (P.R.L Markwick, R. Peacock, E.A. Komives, manuscript submitted).
Determination of the role of Trp215 in stabilizing the thrombin structure
Because Trp215CT appears to be undergoing pi-stacking with Phe 227CT (11), we wondered whether the F227A mutation would have the same effects as the W215A mutation. In addition, the more conservative substitutions W215I and F227V were also prepared and all of the mutants were subjected to HDXMS under conditions where the enzyme appears to be most dynamic (100 mM NaCl). The W215A, W215I, and F227A mutants all showed similarly increased deuterium uptake in residues 156-180CT (residues 197-221; MH+ 2896.550) and residues 161-181CT (residues 202-222 MH+ 2490.296) as compared to WT thrombin (Figure 5A). Comparison of the deuterium uptake in the overlapping peptide, residues 161-170CT (202-211; MH+ 1155.619), suggested the increased deuterium uptake was at least partially occurring in the α-helix spanning residues 165-170CT (residues 206-211) in all mutants, however the W215I mutation did not destabilize to the same extent. All of the mutants also showed increased deuterium uptake in the 220sCT loop -residues 208-227CT (residues 256-275; MH+ 2152.943, MH+ 2191.954, and MH+ 2267.985 for W215A, F227A, and WT respectively), residues 212-227CT (residues 260-275; MH+ 1673.760, MH+ 1715.806, and MH+ 1788.801 for W215A, W215I, and WT respectively), and residues 212-228CT (residues 260-276; MH+ 1903.864 for F227V) -relative to wild type thrombin (Figure 5B), although the increase in uptake for F227V could not be accurately determined as there was no matching peptide in the WT dataset.
Figure 5.
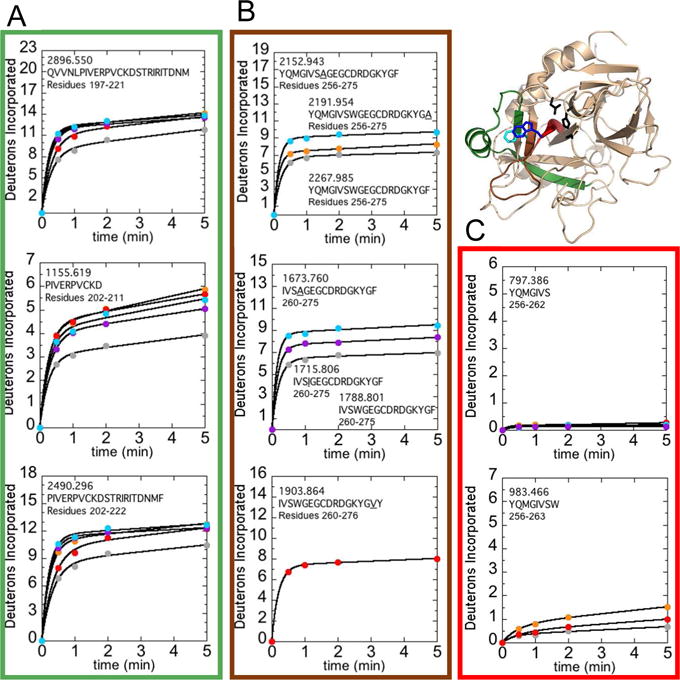
Structure of WT thrombin (PDB 1PPB) highlighting residues 156-181CT (residues 197-222; green), residues 208-215CT (residues 256-263; red), and residues 216-228CT (residues 264-276; brown). The sidechains of Trp215CT (blue), Phe227CT (cyan), and the catalytic triad (black) are shown as sticks. A) Uptake plots corresponding to residues 156-180CT (residues 197-221; MH+ 2896.550), 161-170CT (residues 202-211; MH+ 1155.619), and 161-181CT (residues 202-222; MH+ 2490.296). B) Uptake plots corresponding to residues 208-227CT (residues 256-275; MH+ 2152.943, MH+ 2191.954, and MH+ 2267.985 for W215A, F227A, and WT respectively), residues 212-227CT (residues 260-275; MH+ 1673.760, MH+ 1715.806, and MH+ 1788.801 for W215A, W215I, and WT respectively), and residues 212-228CT (residues 260-276; MH+ 1903.864 for F227V). The mutant residue, if present, is underlined in the peptide sequence shown. C) Uptake plots corresponding to residues 208-214CT (residues 256-262; MH+ 797.386) and 208-215CT (residues 256-263; MH+ 983.466). Deuterium incorporation over 5 min into the multiple peptides that cover these regions are shown for WT thrombin (grey) as well as the F227A (orange), F227V (red), W215A (cyan), and W215I (purple) mutants under experimental conditions of 100 mM NaCl.
Evidence for exposure of the Trp215 backbone amide
Although increased deuterium uptake in peptides spanning the 220sCT loop was observed for the Trp215CT and Phe227CT mutants, examination of the deuterium uptake into two overlapping peptides spanning residues 208-215CT (residues 256-263) in WT, F227A and F227V allowed assignment of a significant portion of the effect to the backbone amide at position 215CT (residue 263). By subtracting the deuterium uptake into the peptide spanning residues 208-214CT (residues 256-262; MH+ 797.386) from that of the peptide spanning residues 208-215CT (residues 256-263; MH+ 983.466), it was possible to determine that a full additional amide was exchanged into position 215CT in the F227A mutant and a stepwise increase in deuterium uptake with decreasing hydrophobic character in the sidechain of residue 227CT (Figure 5C). Thus, the hydrophobicity of the residue at position 227 affects the exchange of the amide group at position 215.
Understanding the HDXMS results in light of thrombin catalytic activity
Because HDXMS showed that mutation of W215A, W215I, F227A, and F227V affected the dynamics of the loops surrounding the thrombin active site, the catalytic activity of these mutants was characterized using chromogenic substrates that target only the active site. The catalytic ability of W215A, W215I, and F227A towards the coagulative substrates fibrinopeptide A (FPA) and protease-activated receptor 1 (PAR1), and the anticoagulative substrate protein C (PC) have already been characterized (5). When expressing and purifying the various mutants as fusion constructs containing 18 residues from prothrombin, we noticed that W215A and F227V were able to catalyze the self-cleavage of the propeptide which occurs after cleavage of the activation peptide by ecarin. W215I and F227A required the addition of WT thrombin to affect the self-cleavage. Consistent with its ability to self-cleave, the F227V mutant retained significant activity towards all substrates tested compared to WT thrombin (Table 1). Although W215A could self-cleave, its activity was decreased to less than 10% of WT thrombin in chromogenic substrate assays as well as towards fibrinogen. The mutants W215I and F227A, lost their ability to self-activate, and were not studied further due to their lack of observed activity.
Table 1.
Activity of the various thrombin mutants.
| Construct | S elf-Activating? | s/sWT S-2238 | s/sWT S-2366 | Fibrinogen Activation units relative to WT thrombin | Protein C activation rate relative to WT thrombin |
|---|---|---|---|---|---|
| WT | Yes | 1.00+/−0.16 | 1.00+/−0.03 | 1 | 1 |
| W215A | Yes | 0.053+/− 0.007 | 0.038+/− 0.003 | 0.034+/−0.002 | n.d. |
| W215I | No | 0.017+/− 0.001 | 0.013+/− 0.003 | n.d. | n.d. |
| F227A | No | n.d. | n.d. | n.d. | n.d. |
| F227V | Yes | 1.00+/−0.15 | 0.66+/−0.06 | 0.86+/−0.06 | 0.66+/−0.07 |
s/sWT is the ratio of kcat/KM for the mutant divided by the kcat/KM for the wild type. Fibrinogen activation units correspond to the amount of thrombin required to clot fibrinogen in 20 sec. This amount for WT thrombin is 100 ng or 3 pmol. Protein C activation was determined as described in the Methods section.
We compared two chromogenic substrates, the S-2238, which has a bulky Phe at P3 and is thought to mimic fibrinogen, vs. S-2366, which has a smaller pyroGlu at P3 and is thought to mimic protein C and PAR1. Though WT thrombin was able to cleave both chromogenic substrates, each mutant preferentially cleaved the procoagulant mimicking substrate S-2238, which is best exemplified by F227V (Table 1).
Discussion
The effect of NaCl on the dynamics of thrombin
Even though the sodium binding site was identified between the 220sCT and 180sCT loops (21, 27), our HDXMS results did not show a difference in dynamics in these loops at NaCl concentrations between 100 mM NaCl and 300 mM NaCl for WT thrombin. These concentrations were chosen to maximize the difference in surface “protection” by a ligand with a millimolar binding constant (22). Other regions did show differences in exchange at 100 vs 300 mM NaCl indicating that differences could be observed under the experimental conditions. The region of thrombin that showed the most significant sensitivity to NaCl concentration was the 170sCT loop (residues 197-222; 156-181CT), which did not show an appreciable difference in conformation between the sodium-free and sodium bound forms according to crystallographic evidence (28). Mutation of Trp215CT did cause a significant increase in the dynamics of the 220sCT loop indicating that its motions can, in fact, be altered. Di Cera has previously reported that mutation of Trp 215CT appears to decrease the Na-binding affinity of thrombin as indicated by hirudin binding (4). Our results suggest that Trp215CT is coupled to the 220sCT loop but that Na affects the conformational states of the 170s loop and the N-terminus of the heavy chain which would also be expected to alter activity and hirudin binding.
Other effects of NaCl concentration observed were almost entirely localized to the surface loops of thrombin, in both WT thrombin and the W215A mutant. Interaction of monovalent ions interact with regions of thrombin distinct from the 220sCT and 180sCT loops was proposed previously based on MD simulations (29) and Xiao et al. showed that the surface loops of thrombin displayed a smaller ensemble of conformations upon interaction with sodium ions (30). In fact, we show decreased amide exchange in the surface loops at higher NaCl concentrations consistent with these simulation results. Our results also showed a marked sodium-induced stabilization of the 90sCT loop (residues 117-135), which contains Asp102CT, and this may actually be the major underlying cause of observed effects of sodium ions on thrombin activity.
The effect of mutations at Trp215 and Phe227 on local structure
Just as a lower concentration of NaCl leads to increased deuterium uptake in the 170sCT loop and the β-strands that flank it, so does the interruption of the pi-stacking interaction between the side chain at positions 215CT and 227CT. When the side chain at either position was replaced with Val or Ile, the 170sCT loop was slightly more stabilized than for the alanine mutants. Analysis of the deuterium uptake into residues 156-181CT (residues 197-222) in conjunction with the hydrogen bonds broken during the AMD simulations of W215A localized the amides that are likely undergoing increased amide exchange in the Trp215CT and Phe227CT mutants to those within the 170sCT loop itself.
Uniqueness of the Trp215CT-Phe227CT interaction
Trp215CT is nearly perfectly conserved across the chymotrypsin family of serine proteases, although Phe227CT is not. Val or Ile is found at position 227CT for all members in the coagulation protease family except for thrombin. It was recently shown that the 170sCT loop in murine urokinase-type plasminogen activator (muPA), which contains a Val at position 227CT, exists in equilibrium between two distinct, highly dynamic conformations that require the Cys168CT-Cys182CT bond to “adopt different rotational conformers to allow” for interconversion (31). This result is consistent with our observation that the F227V mutant has higher exchange in the 170sCT loop. Conventional molecular dynamics simulations analyzed using a Markov state model showed that trypsin, which also has a Val at position 227CT, samples conformational states where the side chain of Trp215CT is rotated towards the active site (32). In fact, there are many apparently stabilizing interactions that Trp215CT makes with residues in the 170sCT loop including interaction with Ile174CT. Additional interactions within the 170sCT loop that don’t directly involve Trp215CT include the interaction between Thr172CT and Glu217CT. Given that the T172A, I174A, E217A, and F227A all exhibit the same ~10-fold decrease in activity towards FpA and PAR1, while retaining protein C activation (5), we may speculate that these side chains all work together to optimize thrombin’s procoagulative function.
The residue at position 215CT may flip into the active site
Multiple other studies have shown that the Trp215CT side chain of thrombin can occlude the active site upon mutation of residues near the active site such as D102N (33), Δ146-149e (34), Y225P (35), and N143P (36). In MD simulations, WT thrombin adopts a similar occluded conformational state in a sodium-free environment (30). When analyzing the effects of W215A, W215I, and F227V on the activity of thrombin towards substrates, we observed that the activity of W215I towards both chromogenic substrates was strikingly diminished compared to the W215A mutant. The F227A mutant also could not self-activate and this could be due to a reconfiguration of the Trp215CT side chain into the active site preventing optimal activity. In contrast, the F227V mutant is able to self-activate, cleave fibrinogen, and activate PC with near WT activity. A consistent model that explains these data is that a Val at position 227CT, as is found in all other coagulation proteases, is sufficient to stabilize the Trp215CT side chain from flipping into the active site whereas an Ala at position 227CT is not. Additionally, the Ile215CT interaction with Phe227CT is not as optimal as the interaction of Trp215CT with Val227CT resulting in the repositioning of Ile215CT into the active site blocking activity. These data show the importance of introducing multiple conservative substitutions for understanding the role of specific enzyme residues as well as the importance of correlating data from HDXMS with enzyme activity.
The HDXMS data presented here show that the backbone amide of Trp215CT in WT thrombin does exchange with deuterated solvent, but at a rate far slower than nearly all other regions of the enzyme. The presence of Phe227CT in thrombin may be an evolutionarily significant event allowing the S1 site to more often be open for substrate binding. Consequently, the catalytic triad of thrombin would be stabilized to a greater degree than is observed for other chymotrypsin-like proteases.
Long-range effects of mutation of Trp215
Mutations at Trp215CT appeared to have a greater effect on deuterium exchange into the 220sCT loop than mutations at 227CT suggesting that the W215A mutation destabilized residues in the 220sCT loop to a greater extent than the F227A mutation. Although mutations at Phe227CT led to the increased exchange into the backbone amide of Trp215CT, only mutations at Trp215CT appeared to notably affect amide exchange into the whole 220sCT loop.
Our HDXMS results also showed that the W215A mutant had significantly higher exchange into the N-terminus of the heavy chain, and this effect was greater than for any of the other mutants tested (Figure 4A and S3). The AMD simulations of the W215A mutant showed a transient structure in which the side chain of Phe227CT (residue 275) was rotated into the position that the Trp215CT side chain formerly occupied. Huntington and Esmon also observed a shift in the position of Phe227CT in the crystal structure of the S195A mutant (37). In our AMD simulations of the W215A mutant, the side chain of Arg173CT (residue 214) also rotated to form a new interaction with Glu217CT (residue 265). Interestingly, the structure of the W215A/E217A double mutant thrombin (38, 39) showed the 214-219CT segment collapsed into the S1 pocket similar to the structures of D102N (33), Δ146-149e (34), Y225P (35), and N143P (36). Our AMD results suggest that removal of the Trp215CT side chain transforms the backbone of residues 214-219CT into a much less structured segment, stabilized only when the side chain of Arg173CT makes contact with Glu217CT. Kinetic analysis of R173A by Di Cera et al. showed no significant deviation of activity towards either procoagulative or anticoagulative substrates as compared to WT thrombin (5), but it is possible that the Arg173-Glu217CT interaction stabilizes the 214-219CT segment only when Trp215CT is also mutated. This would explain why the double W215A/E217A mutant shows a collapsed S1 pocket in X-ray crystal structures (38). Our results thus provide the first experimental evidence of the allosteric connections between position 215CT and the active site first proposed by Di Cera (33, 38).
The AMD results show that the structural effects of the altered 214-219CT segment are accompanied by a shift in the backbone hydrogen bonding to the N-terminal amino group of Ile16CT (residue 37) and a reorientation of the Ile16CT side chain. In addition, the side chains of Asp194CT (residue 240) and Ser195CT (residue 241) are altered resulting in improper alignment of the serine hydroxyl group with His57CT (residue 79) (Figure 4B). Finally, Asp189CT (residue 235) at the base of the S1 pocket is also rotated almost 180° away from its position in the wild type enzyme and forms a new H-bond with Arg187CT (residue233) which is now located in the sodium binding pocket.
The work presented here identifies the distinct changes in the structure of thrombin that result from both changes in the concentration of NaCl and the disruption of the Trp215CT-Phe227CT pi-stacking interaction. Our results explain how the 170sCT loop, which makes up part of ABE2, may act as another potential regulatory region of thrombin. Additionally, the similar effects of NaCl concentration and the disruption of the Trp215CT-Phe227CT pi interaction on the 170sCT loop of thrombin opens the door for questions regarding whether sodium ions help modulate this pi interaction. Most importantly, the combination of HDXMS, which provides evidence of structural changes that are difficult to elucidate by crystallographic methods alone, with AMD simulations, provides a clearer understanding of allostery in thrombin. Here we show that mutation of Trp215CT rebalances the ensemble of states to populate structures in which the 170sCT loop is unfolded and structures in which the catalytic triad and S1 pocket are perturbed. Future experiments that investigate the dynamics of thrombin will be imperative for developing a full picture regarding complex enzyme regulation.
Supplementary Material
Acknowledgments
This work was supported by NIH grant HL070999. RP acknowledges support from the Molecular Biophysics Training Grant T32 GM008326.
Footnotes
To accommodate readers who use one of several different numbering schemes for thrombin, we report residues in the chymotrypsin numbering which is used in PDB files of all serine proteases in which loops are denoted 60A, 60B, etc. The chymotrypsin numbering is denoted with a subscript CT, and is followed by the sequential numbering, which is more useful for describing HDXMS data, given in parentheses.
References
- 1.Bode W. The structure of thrombin: a janus-headed proteinase. Semin Thromb Hemost. 2006;32:16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- 2.Esmon CT. Regulation of blood coagulation. Biochim Biophys Acta. 2000;1477:349–360. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 3.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 4.Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- 5.Marino F, Pelc LA, Vogt A, Gandhi PS, Di Cera E. Engineering thrombin for selective specificity toward protein C and PAR1. J Biol Chem. 2010;285:19145–19152. doi: 10.1074/jbc.M110.119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayala Y, Di Cera E. Molecular recognition by thrombin. Role of the slow– >fast transition, site-specific ion binding energetics and thermodynamic mapping of structural components. J Mol Biol. 1994;235:733–746. doi: 10.1006/jmbi.1994.1024. [DOI] [PubMed] [Google Scholar]
- 7.Lechtenberg BC, Johnson DJ, Freund SM, Huntington JA. NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc Natl Acad Sci U S A. 2010;107:14087–14092. doi: 10.1073/pnas.1005255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuglestad B, Gasper PM, Tonelli M, McCammon JA, Markwick PR, Komives EA. The dynamic structure of thrombin in solution. Biophys J. 2012;103:79–88. doi: 10.1016/j.bpj.2012.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasper PM, Fuglestad B, Komives EA, Markwick PR, McCammon JA. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc Nat Acad Sci. 2012;109:21216–21222. doi: 10.1073/pnas.1218414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handley LD, Fuglestad B, Stearns K, Tonelli M, Fenwick RB, Markwick PR, Komives EA. NMR reveals a dynamic allosteric pathway in thrombin. Sci Rep. 2017;7:39575. doi: 10.1038/srep39575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 A crystal structure of human alpha-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. The EMBO Journal. 1989;8:3467. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Truhlar SM, Croy CH, Torpey JW, Koeppe JR, Komives EA. Solvent accessibility of protein surfaces by amide H/2H exchange MALDI-TOF mass spectrometry. J Am Soc Mass Spectrom. 2006;17:1490–1497. doi: 10.1016/j.jasms.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Koeppe JR, Seitova A, Mather T, Komives EA. Thrombomodulin tightens the thrombin active site loops to promote protein C activation. Biochemistry. 2005;44:14784–14791. doi: 10.1021/bi0510577. [DOI] [PubMed] [Google Scholar]
- 14.Handley LD, Treuheit NA, Venkatesh VJ, Komives EA. Thrombomodulin binding selects the catalytically active form of thrombin. Biochemistry. 2015;54:6650–6658. doi: 10.1021/acs.biochem.5b00825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cervantes CF, Markwick PR, Sue SC, McCammon JA, Dyson HJ, Komives EA. Functional dynamics of the folded ankyrin repeats of I kappa B alpha revealed by nuclear magnetic resonance. Biochemistry. 2009;48:8023–8031. doi: 10.1021/bi900712r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamenik AS, Kahler U, Fuchs JE, Liedl KR. Localization of millisecond dynamics: Dihedral entropy from accelerated MD. J Chem Theory Comput. 2016;12:3449–3455. doi: 10.1021/acs.jctc.6b00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baerga-Ortiz A, Rezaie AR, Komives EA. Electrostatic dependence of the thrombin-thrombomodulin interaction. J Mol Biol. 2000;296:651–658. doi: 10.1006/jmbi.1999.3447. [DOI] [PubMed] [Google Scholar]
- 18.White CE, Hunter MJ, Meininger DP, White LR, Komives EA. Large-scale expression, purification and characterization of small fragments of thrombomodulin: the roles of the sixth domain and of methionine 388. Protein Engineering, Design and Selection. 1995;8:1177–1187. doi: 10.1093/protein/8.11.1177. [DOI] [PubMed] [Google Scholar]
- 19.Fenton JW. Thrombin. Annals New York Acad Sci. 1986;485:5–15. doi: 10.1111/j.1749-6632.1986.tb34563.x. [DOI] [PubMed] [Google Scholar]
- 20.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 22.Mandell JG, Baerga-Ortiz A, Akashi S, Takio K, Komives EA. Solvent accessibility of the thrombin-thrombomodulin interface. J Mol Biol. 2001;306:575–589. doi: 10.1006/jmbi.2000.4416. [DOI] [PubMed] [Google Scholar]
- 23.Hamelberg D, de Oliveira CA, McCammon JA. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J Chem Phys. 2007;127:155102. doi: 10.1063/1.2789432. [DOI] [PubMed] [Google Scholar]
- 24.Miao Y, Sinko W, Pierce L, Bucher D, Walker RC, McCammon JA. Improved Reweighting of Accelerated Molecular Dynamics Simulations for Free Energy Calculation. J Chem Theory Comput. 2014;10:2677–2689. doi: 10.1021/ct500090q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 26.Markwick PR, McCammon JA. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys Chem Chem Phys. 2011;13:20053–20065. doi: 10.1039/c1cp22100k. [DOI] [PubMed] [Google Scholar]
- 27.Zhang E, Tulinsky A. The molecular environment of the Na+ binding site of thrombin. Biophys Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]
- 28.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 29.Kurisaki I, Takayanagi M, Nagaoka M. Toward Understanding Allosteric Activation of Thrombin: A Conjecture for Important Roles of Unbound Na+ Molecules around Thrombin. J Phys Chem B. 2015;119:3635–3642. doi: 10.1021/jp510657n. [DOI] [PubMed] [Google Scholar]
- 30.Xiao J, Melvin RL, Salsbury FR. Mechanistic insights into thrombin’s switch between “slow” and “fast” forms. Phys Chem Chem Phys. 2017;19:24522–24533. doi: 10.1039/c7cp03671j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kromann-Hansen T, Lange EL, Sørensen HP, Hassanzadeh-Ghassabeh G, Huang M, Jensen JK, Muyldermans S, Declerck PJ, Komives EA, Andreasen PA. Discovery of a novel conformational equilibrium in urokinase-type plasminogen activator. Sci Rep. 2017;7:3385. doi: 10.1038/s41598-017-03457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plattner N, Noé F. Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models. Nat commun. 2015;6 doi: 10.1038/ncomms8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Nat Acad Sci. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bah A, Carrell CJ, Chen Z, Gandhi PS, Di Cera E. Stabilization of the E* form turns thrombin into an anticoagulant. J Biol Chem. 2009;284:20034–20040. doi: 10.1074/jbc.M109.012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niu W, Chen Z, Gandhi PS, Vogt AD, Pozzi N, Pelc LA, Zapata F, Di Cera E. Crystallographic and kinetic evidence of allostery in a trypsin-like protease. Biochemistry. 2011;50:6301–6307. doi: 10.1021/bi200878c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niu W, Chen Z, Bush-Pelc LA, Bah A, Gandhi PS, Di Cera E. Mutant N143P reveals how Na+ activates thrombin. J Biol Chem. 2009;284:36175–36185. doi: 10.1074/jbc.M109.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huntington JA, Esmon CT. The molecular basis of thrombin allostery revealed by a 1.8 A structure of the “slow” form. Structure. 2003;11:469–479. doi: 10.1016/s0969-2126(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 38.Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, Di Cera E. The anticoagulant thrombin mutant W215A/E217A has a collapsed primary specificity pocket. J Biol Chem. 2004;279:39824–39828. doi: 10.1074/jbc.M407272200. [DOI] [PubMed] [Google Scholar]
- 39.Gandhi PS, Page MJ, Chen Z, Bush-Pelc L, Di Cera E. Mechanism of the anticoagulant activity of thrombin mutant W215A/E217A. J Biol Chem. 2009;284:24098–24105. doi: 10.1074/jbc.M109.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.