

Abstract
Rationale
MicroRNAs (miRs) are small, non-coding RNAs that function to post-transcriptionally regulate target genes. First transcribed as primary miR transcripts (pri-miRs), they are enzymatically processed by Drosha into premature miRs (pre-miRs) and further cleaved by Dicer into mature miRs. Initially discovered to desensitize β-adrenergic receptor (βAR) signaling, β-arrestins are now well-appreciated to modulate multiple pathways independent of G protein signaling, a concept known as biased signaling. Using the β-arrestin-biased βAR ligand carvedilol, we previously showed that β-arrestin1 (not β-arrestin2)-biased β1AR (not β2AR) cardioprotective signaling stimulates Drosha-mediated processing of six miRs by forming a multi-protein nuclear complex, which includes β-arrestin1, the Drosha microprocessor complex and a single-stranded RNA binding protein hnRNPA1.
Objective
Here, we investigate whether β-arrestin-mediated βAR signaling induced by carvedilol could regulate Dicer-mediated miR maturation in the cytoplasm and whether this novel mechanism promotes cardioprotective signaling.
Methods and Results
In mouse hearts, carvedilol indeed upregulates three mature miRs, but not their pre-miRs and pri-miRs, in a β-arrestin 1- or 2-dependent manner. Interestingly, carvedilol-mediated activation of miR-466g or miR-532-5p, and miR-674 is dependent on β2ARs and β1ARs, respectively. Mechanistically, β-arrestin 1 or 2 regulates maturation of three newly identified βAR/β-arrestin-responsive miRs (β-miRs) by associating with the Dicer maturation RNase III enzyme on three pre-miRs of β-miRs. Myocardial cell approaches uncover that despite their distinct roles in different cell types, β-miRs act as gatekeepers of cardiac cell functions by repressing deleterious targets.
Conclusions
Our findings indicate a novel role for βAR-mediated β-arrestin signaling activated by carvedilol in Dicer-mediated miR maturation, which may be linked to its protective mechanisms.
Keywords: β-arrestin-biased β-adrenergic receptor signaling, carvedilol, dicer, heart disease, microRNA biogenesis, RNA binding proteins
Graphical Abstract
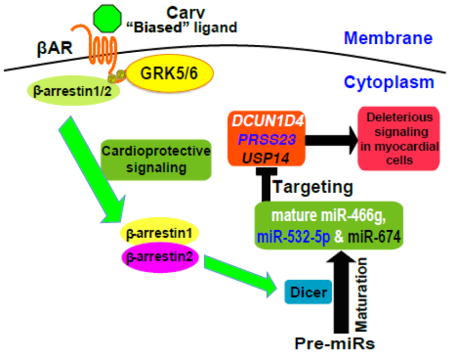
1. Introduction
MicroRNAs (miRNAs or miRs), a class of ~22 nucleotide small non-coding RNAs, govern post-transcriptional repression of target mRNAs. Many roles of miRs in cardiac physiology have been reported including the regulation of cardiomyocyte growth, contractility, and the maintenance of cardiac rhythm [1]. Furthermore, gain- and loss-of-function studies of various miRs suggested that aberrant expression of the miRs could be necessary and sometimes even sufficient for the pathogenesis of various heart diseases [2, 3], pointing towards miRs as new regulatory mechanisms and potential therapeutic targets for heart disease to complement pharmacological approaches [1].
MiR biogenesis is regulated in a complex manner, involving numerous protein-protein and protein-RNA interactions [4]. Both miR-regulator and miR-target availability often differ among cell types, tissues and especially during disease initiation and progression, responding to different upstream signaling pathways to activate distinct downstream targets. It is understood that miRs are influenced at the transcriptional level by many transcription factors. However, they are also regulated by two RNase III enzymes, Drosha and Dicer, which play dominant roles in their expression. Multiple post-transcriptional regulatory mechanisms of miR biogenesis have been identified. For example, several proteins including Cyclin D1, EGFR, and MCPIP1 modulate Dicer-mediated miR maturation [5–7]. Interestingly, a proteomic analysis assessing the global cellular interactions of the G protein-coupled receptor (GPCR) signaling mediators, β-arrestin1 and β-arrestin2, identified that β-arrestins interacted with many RNA binding proteins [8], suggesting regulatory roles of β-arrestins in miR biogenesis. We indeed showed that β-arrestin1 (not β-arrestin2) -mediated β1-adrenergic receptor (β1AR, not β2AR) signaling promotes the Drosha-mediated processing of a subset of miRs via a nuclear interaction between β-arrestin1 and the Drosha microprocessor complex [9] and that these β1AR/β-arrestin1/Drosha-responsive miRs confer cardioprotective effects [10–12].
β-arrestin1 and β-arrestin2 were initially identified as key accessory proteins involved in desensitization of GPCRs. More recent studies have shown that β-arrestins can also transduce multiple effector pathways independent of G protein signaling when GPCRs are stimulated by certain ligands, a concept known as biased signaling [13–17]. It has been proposed that GPCRs respond to different ligands much like a barcode reader, which responds to different barcodes. In this barcode hypothesis, unbiased and β-arrestin-biased ligands impart distinct patterns of receptor phosphorylation by specific GPCR kinases (GRKs), thus converting ligand-induced conformation of the receptor into selective β-arrestin functions [18–20]. For example, ligands that promote GRK2/3-mediated receptor phosphorylation lead to receptor internalization whereas ligands, such as the βAR antagonist (β-blocker) carvedilol (Carv) that promote GRK5/6-mediated receptor phosphorylation, stimulate β-arrestin signaling [18–20]. Indeed, Carv is one of three β-blockers approved for heart failure and has many documented actions including antagonism of β1AR, β2AR and α1AR as well as antioxidant effects [21, 22]. We and others previously showed that Carv also stimulates β-arrestin-mediated βAR cardioprotective signaling without activating G proteins, providing an additional mechanism for its clinical efficacy [13, 14]. In particular, using the β-arrestin-biased ligand Carv, we recently demonstrated a function of β-arrestin1 (not β-arrestin2)-mediated β1AR (not β2AR) signaling in Drosha-mediated miR processing via forming a nuclear complex with β-arrestin1, Drosha and hnRNPA1 on a subset of pri-miRs [9]. However, our understanding of whether β-arrestin-biased βAR signaling regulates other steps of miR biogenesis remains limited.
Here, we investigate whether stimulation of βARs by the β-arrestin-biased ligand Carv, can regulate Dicer-mediated miR maturation in the heart and whether this novel mechanism promotes cardioprotective signaling. Upon further verification analysis of our previous microarray data in the heart [9], we found that three additional miRs (466g, 532-5p, and 674) were upregulated by Carv stimulation and that this effect was absent in mice lacking β-arrestin1, β-arrestin2, GRK5, or GRK6. Interestingly, we observed that Carv-mediated activation of miR-466g and miR-532-5p is β2AR-dependent, but the activation of miR-674 by Carv is β1AR-dependent. While Carv did not increase the expression of both pri-miRs and pre-miRs, it enhanced the expression of mature miRs by promoting the interaction of β-arrestin 1 or 2 with the Dicer maturation RNase III enzyme. Our cardiac cell approaches further demonstrated that the three newly identified βAR/β-arrestin-responsive miRs (β-miRs) act as gatekeepers of cardiac cell functions in part by repressing the predictive or known deleterious target genes, dcun1d4, prss23 and usp14. Our data provide evidence that the β-arrestin-biased ligand Carv stimulates Dicer-mediated miR maturation, which may be an important mechanism for its cardioprotective effects.
2. Materials and Methods
2.1. Animal study approval and treatment protocol
We employed 1 to 2-day-old Sprague-Dawley rats (Envigo), 8 to 12-week-old C57BL/6 wild-type (WT; Jackson Laboratory), β-arrestin1 knockout (KO), β-arrestin2 KO, GRK5 KO, GRK6 KO, β1-adrenergic receptor (β1AR) KO, β2AR KO, or β1AR/β2AR double KO (DKO)mice, and cardiac-specific transgenic (TG) mice overexpressing WTβ1ARs or mutant β1 ARs that lack GRK phosphorylation sites (GRK β1AR TG). All animal research was reviewed and approved by the Augusta University Institutional IACUC Committees, and conformed to current NIH guidelines. The neonatal rats were euthanized by decapitation under anesthesia for cardiomyocyte and cardiac fibroblast isolation. Carvedilol (Carv; Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO) using sterile techniques and then placed in micro-osmotic pumps (Alzet model 2001; DURECT) for subcutaneous delivery into adult mice at the rate of 19mg/kg/day over a period of 7 days. In control mice, 10% (v/v) DMSO was used as vehicle. All mouse lines receiving Carv displayed no left ventricular dysfunction, and genotypic verification of KO or TG mice has been described previously [23–25]. After Carv administration, mice were euthanized by thoracotomy under 1–4% inhalant isoflurane anesthesia. Hearts were excised, flash frozen in liquid nitrogen, and subjected to RNA isolation and QRT-PCR analyses as described previously [9].
2.2. Cell culture and transfection
HEK293 cell lines stably expressing the β1AR and β2AR have been previously described [26, 27]. The cells were transfected with cDNA of FLAG-β-arrestin1, FLAG-β-arrestin2, or HA-Dicer with Lipofectamine™ reagent (Invitrogen). Transfected cells were incubated overnight in serum-free MEM medium supplemented with 0.1% BSA, 10 mM HEPES (pH 7.4), and 1% penicillin before stimulation. Under serum starvation conditions, cells were stimulated with 1μM Carv for 4 (for RNA assays) or 20 hours (for Co-IP assays) as described previously [13].
The immortalized mouse adult atria-derived CM HL-1 cell line obtained from Dr. Claycomb was maintained as previously described [28]. The adult mouse cardiac endothelial cell (MCEC) line was purchased from Cedarlane (CLU510) and maintained according to the company’s recommendation. The cell lines were established from primary cells by transfecting SV40 T antigen and displayed normal CM or CEC characteristics and cellular markers. We used multiple batches of the cell lines subjected to 3–5 passages in culture. The primary adult mouse cardiac fibroblasts (AMCFs) were purchased from Cell Biologics, Inc (C57-6049) and maintained according to the company’s recommendation. Primary neonatal rat ventricular cardiomyocytes (NRVCs) and neonatal rat ventricular fibroblasts (NRVFs) were isolated by dissociation of 1- to 2-day-old Sprague-Dawley rats and were maintained as we published [10]. Clearly striated CMs were used within 6 hours in all experiments. The cardiac fibroblast pellet was maintained in RPMI 1640 medium with 5mM glucose, 10% FBS and antibiotics. Non-adherent cells were removed by aspiration after 4 hours and discarded. The purity of NRVCs and NRVFs was previously shown by cell type-dependent gene expression patterns [10]. In order to inhibit the expression miR-466g, miR-532-5p, and miR-674 in myocardial cells, we transfected 100nM mirVana™ miR Inhibitors (Life Technologies) specific to miR-466g (MH12523), miR-532-5p (MH11553 for mouse and MH12818 for rat), miR-674 (MH11743) and a miR inhibitor negative control (4464076) using Lipofectamine™ 2000 reagent (Invitrogen) as described previously [29]. For gain-of-function studies, we transfected the cells with a miR mimic negative control (4464058), miR-466g mirVana™ mimic (MC12523), miR-532-5p mirVana™ mimic (MC11553 for mouse and MC12818 for rat), and miR-674 mirVana™ mimic (MC11743) purchased from Life Technologies. All in vitro assays were performed 60–72 hours after transfection when maximum knockdown efficiency was reached.
2.3. In vitro simulated ischemia/reperfusion (sI/R)
Cells plated on coverslips or 6 well plates were transfected with miR inhibitors or miR mimics as aforementioned, washed, and placed in a simulated ischemia buffer that contained 118mM NaCl, 24mM NaH2CO3, 1mM NaHPO4, 2.5mM CaCl2, 1.2mM MgCl2, 20mM sodium lactate, 16mM KCl and 10mM 2-deoxyglucose (pH 6.2). Cells were then incubated in the anoxic chamber (5% CO2, 0% O2) for 1 hour followed by replacing the ischemic buffer with normal cell medium and incubating under normoxia conditions for 4 hours to complete the sI/R protocol as described [30]. Coverslips or plates were processed for qRT-PCR, immunoblotting, TUNEL staining, wound migration, BrdU staining, and tube formation assays as mentioned below.
2.4. RNA isolation and quantitative real-time RT-PCR analysis
Total RNA from cells and mouse hearts was prepared using Trizol Reagent (Invitrogen) and treated with RNase-free DNase I to remove genomic DNA as described [31, 32]. For detection of mature miRs, the TaqMan MiR Reverse Transcription Kit (a highly specific kit that generates only mature miRs, not precursors; ThermoFisher Scientific) was used to synthesize cDNA for TaqMan MiR Assays. The following probes were used to amplify and measure the amount of mature miRs by Real-Time RT-PCR: miR-467b*, 001671; miR-669p*, 465247_mat; miR-669a-3p, 463964_mat; miR-466f-3p, 241006_mat; miR-466g, 241015_mat; miR-532-5p, 001518 (mouse) or 002051 (rat); miR-671-5p, 197646_mat; miR-676, 001959; miR-674-5p, 002021; miR-208-5p, 462036_mat; miR-146a, 000468; miR-322, 001059; miR-1-1*, 462049_mat; miR-192, 000491; miR-1249, 002868; miR-290-5p, 002590; miR-698, 001632; miR-764-5p, 002031; and U6 snRNA, 001973 for endogenous controls.
cDNA for detection of primary and premature miRs was synthesized using ThermoFisher Scientific SuperScript III reverse transcriptase and random hexamer primers. Expression of primary miRs was detected using TaqMan Primary MiR assays that quantitate only primary miR transcripts (mmu-pri-miR-466g, Mm04272618_pri; mmu-pri-miR-532-5p, Mm03307612_pri; and mmu-pri-miR-674, Mm03307945_pri) and HPRT1 probe, Mm00446969_m1 for endogenous controls. Expression of premature miRs was detected using Applied Biosystems Power SYBR Green PCR Master Mix with pre-miR primers that were designed based on the miRBase database (http://microrna.sanger.ac.uk/sequences/) as successfully used [33]. The following sense and anti-sense primers were used to amplify and measure the amount of only premature miRs by Real-Time RT-PCR: mmu-pre-miR-466g, 5′-GTGTGTGCATGTGGATGTATG-3′ and 5′-GTGTGTGCATGTGTCTGTATATG-3′; mmu-pre-miR-532-5p, 5′-TTTCTCTTCCATGCCTTGAGT-3′ and 5′-TGGGTGTGGGAGGGTAAT-3′; mmu-pre-miR-674, 5′-TAGTCATCACCCTGAGCCTT-3′ and 5′-CTGTGCATACCTGAGCCTTAC-3′.
cDNA for detection of genes was synthesized using ThermoFisher Scientific SuperScript III reverse transcriptase and oligo-dT primers. Expression of genes was detected using Taqman Gene expression assays for Mm00619147_m1; mouse dcun1d4, Mm01972869_s1; mouse prss23, Rn01754671_m1: rat prss23, Rn01236255_m1; rat usp14 and Mm00446969_m1; mouse HPRT1 or Rn01527840_m1; rat HPRT1 for endogenous controls.
The following reaction components were used for each probe for mature miRs, pri-miRs, and genes: 2μL cDNA, 10μl 2X TaqMan Universal PCR Master Mix (ThermoFisher Scientific), 1μl probe, and 7μl nuclease-free water in a 20μL total volume. The following reaction components were used for each premature miR: 2μL cDNA, 10μl 2X Power SYBR Green PCR Master Mix and 4μl each primer (33pmole/μl), and 4μl nuclease-free water in a 20μL total volume. Real time PCR Reactions were amplified and analyzed in triplicate using an ABI Sequence Detection System as described previously [32]. PCR reaction conditions were as follows: Step 1: 50°C for 2 minutes, Step 2: 95°C for 10 minutes, Step 3: 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute. Expression relative to endogenous controls was calculated using 2−ΔΔCt and levels were normalized to control. We performed at least six independent experiments in triplicate using different batches of RNAs each time.
2.5. Immunoprecipitation, immunoblotting, and detection
Following stimulation with Carv, cells were washed once with 1X PBS, solubilized in 1ml of lysis buffer (5mM HEPES, 250mM NaCl, 10% glycerol, 0.5% Nonidet P-40, 2mM EDTA, and protease inhibitors) for whole cell lysates as previously described [13]. Prior to immunoprecipitation, 25μl of lysates was aliquoted into a separate tube for protein estimation. Immunoprecipitation was carried out as previously described [23, 34]. Immunoprecipitates or lysate samples were resolved by SDS-PAGE and transferred to PVDF (Bio-Rad) for immunoblotting. β-Actin, FLAG, or HA immunoblotting was carried out using monoclonal antibodies at dilutions of 1:5,000 each (Sigma-Aldrich) as previously described [13]. Dcun1d4 (rabbit, ab75595, Abcam), and usp14 (rabbit, ab137433, Abcam) antibodies were purchased and were used at dilutions of 1:1,000 each. Detection was carried out using ECL (Amersham Biosciences).
2.6. Cell apoptosis by TUNEL staining
DNA fragmentation was detected in situ using TUNEL [35]. In brief, cells were incubated with proteinase K, and DNA fragments were labeled with fluorescein-conjugated dUTP using terminal deoxynucleotidyl transferase (Roche Diagnostics). The total number of nuclei was determined by manual counting of DAPI-stained nuclei in 6 random fields per coverslip (original magnification, ×200). All TUNEL-positive nuclei were counted in each coverslip. Digital photographs of fluorescence were acquired with a Zeiss microscope (ApoTome.2; Carl Zeiss) and processed with Adobe Photoshop.
2.7. Wound migration assay
Cell migration was detected as previously described [36]. In brief, 1×104 cells were plated onto each well of a 2-well Culture-Insert 35 mm μ-Dishe (81176, Ibidi, Fitchburg, WI). Once the cells were confluent, the silicone insert on each dish was removed to reveal a defined cell-free gap. The medium was replaced and images were taken at 0, 8, 12 and 24 hours. Subsequently, the distance between cell fronts was quantified in three wells of each group using Image J software. Initial open (cell free) areas (0 hour) were measured to serve as total open area, and the percentage of open area after indicated time points was calculated to determine the cell’s migratory potential.
2.8. Bromodeoxyuridine (BrdU) proliferation assay
Cell proliferation was detected in situ using BrdU as described [28]. In brief, cells were labeled with BrdU for 2 hours, and then fixed with ethanol and immunostained for BrdU incorporation using the BrdU-Labeling and Detection kit II (Roche Diagnostics) according to manufacturer’s recommendations. The total number of nuclei was determined by manual counting of DAPI-stained nuclei in 6 random fields per coverslip (original magnification, ×200). All BrdU-positive nuclei were counted in each coverslip. Digital photographs of fluorescence were acquired with a Zeiss microscope (ApoTome.2; Carl Zeiss) and processed with Adobe Photoshop.
2.9. In vitro tube formation assay
Tube formation was detected using the Matrigel matrix (354230, Corning) as described [37]. In brief, cells were harvested and 2×104 cells were seeded on the top of Matrigel-coated 96 well plates. After 2 to 4 hours, images were taken for each well to observe and to quantify in vitro tube formation. The images were then analyzed using Wimasis software (Onimagin Technologies SCA).
2.10. In silico miR target prediction analysis
We used several prediction algorithms including miRwalk, miRanda, DIANA-microT-CDS, and Targetscan [38–40]. Each of these algorithms predicts hundreds of possible targets for miR-466g, miR-532-5p, and miR-674. We focused on the targets important for deleterious signals that were predicted by at least three prediction programs.
2.11. Statistical analysis
Data are expressed as mean ± SEM from at least six independent experiments with different biological samples or mice per group. Statistical significance was determined by using two-way ANOVA for 2 variables, one-way ANOVA with Bonferroni correction for multiple comparisons or Student unpaired t-tests (GraphPad Prism version 5). A P value <0.05 was considered statistically significant.
Results
3.1. Carvedilol, a β-arrestin-biased βAR ligand induces the expression of miR-466g, miR-532-5p and miR-674 in mouse hearts
Our previous studies suggested that carvedilol (Carv), a β-blocker used clinically, stimulates β-arrestin-mediated βAR signaling [13, 14, 23], and promotes the processing of six miRs [9]. We also showed that four of these Carv-responsive miRs repress apoptotic genes and confer cardioprotective effects [10–12]. Interestingly, others have also shown that the Carv-responsive miRs are cardioprotective [30, 41, 42]. This led us to further examine whether Carv can regulate different subsets of miRs in the heart with different mechanisms. Upon further verification analysis of our microarray data [9], we identified additional three miRs (miR-466g, miR-532-5p, and miR-674) among eighteen miRs that we validated using QRT-PCR analyses, which were upregulated upon stimulation with Carv when compared to the vehicle (DMSO) control (Supplementary Table 1 and Figure 1A–C).
Figure 1. Carvedilol-mediated in vivo activation of miR-466g, miR-532-5p and miR-674 requires β-arrestin1 or β-arrestin2 and GRK5 or GRK6. Moreover, β1-adrenergic receptor is required for Carv-mediated post-transcriptional activation of miR-674, while β2-adrenergic receptor is required for Carv-mediated post-transcriptional activation of miR-466g or miR-532-5p.

A–C, WT, β-arrestin1 knockout (KO), β-arrestin2 KO, GRK5 KO, GRK6 KO, β1AR KO, β2AR KO or β1AR/β2AR double KO (DKO) mice were infused with DMSO (vehicle control) or Carv (19mg/Kg/day) for 7 days by using micro-osmotic pumps. QRT-PCR experiments were performed on RNAs isolated from mouse hearts. Three mature miRs were elevated upon Carv stimulation in WT mice. However, this induction was completely abolished in β-arrestin1 KO, β-arrestin2 KO, GRK5 KO or GRK6 KO mice, indicating an essential role of either β-arrestin and either GRK in the synthesis of mature miRs. Moreover, Carv-mediated activation of miR-466g and miR-532-5p, which is seen in WT and β1AR KO mice, was abolished in β2 AR KO and β1AR/β2AR DKO mice (A–B). Carv-mediated activation of miR-674, which is seen in WT and β2AR KO mice, was abolished in β1AR KO and β1AR/β2AR DKO mice (C). Data are shown as mean ± SEM for n=6 independent mice per group. The relative fold induction of Carv is calculated by normalizing DMSO controls for each genotype, which is indicated by vertical lines. *P<0.05 or **P<0.01 vs. DMSO. D–E, WT mice were infused with DMSO or Carv as above. QRT-PCR experiments were performed on RNAs from mouse hearts. The three pri- (D) or pre- (E) miRs were not changed significantly upon Carv stimulation in WT mice. NS: not significant. Data are shown as mean ± SEM for n=8 independent mice per group.
3.2. Increased levels of three newly identified miRs elicited by β-arrestin-biased βAR stimulation require either β-arrestin 1 or 2 and GRK 5 or 6
We next tested using knockout (KO) mouse hearts whether β-arrestin signaling and GRK5/6 phosphorylation are required for Carv-induced expression of the three newly identified miRs. We found that the Carv-mediated activation of three miRs occurred in WT mice, but was not observed in hearts from β-arrestin1 KO or β-arrestin2 KO mice and hearts lacking either GRK5 or GRK6 (Figure 1A–C). In contrast with our previous study showing the β-arrestin1 specificity on nuclear regulation in Drosha-mediated miR processing [9], these in vivo data suggest that Carv-mediated activation of three newly identified miRs is not solely dependent on β-arrestin1, but also dependent on β-arrestin2 (Figure 1A–C), and that phosphorylation by GRK 5 or 6, which promotes β-arrestin-dependent signaling [43], is also required for the activation of three newly discovered β-arrestin-responsive miRs by Carv.
3.3. Carvedilol-mediated β-arrestin-biased agonism of β1AR and β2AR induces post-transcriptional expression of miR-674 and miR-466g/miR-532-5p in vivo, respectively by promoting miR maturation
Because Carv is a β-arrestin-biased ligand for both β1AR and β2AR [13, 14], we then measured the expression of three β-arrestin-responsive miRs in hearts from β1AR KO, β2AR KO, or double receptor KO mice. Interestingly, we showed that the Carv-mediated activation of miR-466g and miR-532-5p seen in WT mice was blunted in hearts lacking β2AR (Figure 1A–B) and that the Carv-mediated activation of miR-674 seen in WT mice was blunted in hearts lacking β1AR (Figure 1C). While Carv stimulation of transgenic (TG) mice overexpressing WTβ1ARs induced an increase in expression of miR-674, hearts overexpressing a receptor that lacks GRK phosphorylation sites (GRK β1AR TG) showed no induction of miR-674 (Supplementary Figure 1). Our data suggest that miR-674 is regulated mainly by β1AR, while miR-466g and miR-532-5p are regulated mainly by β2AR.
To examine which step of miR biogenesis is regulated by β-arrestin, we measured the expression of primary transcript (pri)- and premature (pre)-miRs of miR-466g, miR-532-5p and miR-674. Carv did not increase the expression of pri-miRs and pre-miRs (Figure 1D–E), although levels of mature miRs were increased upon Carv stimulation in WT (Figure 1A–C). Taken together, these data indicate that Carv-mediated induction of the 3 mouse miRs occurs post-transcriptionally and that β-arrestin-biased signaling of βAR stimulates the maturation of a subset of miRs.
3.4. β-arrestin 1 or 2 interacts with the Dicer miR maturation enzyme
We next tested whether β-arrestin may regulate miR maturation by interacting with the Dicer miR maturation RNase III enzyme. Because we previously showed that the β-arrestin-mediated miR regulatory mechanism exists in both mouse hearts and HEK293 cells overexpressing WTβ1ARs (WTβ1AR cells) [9], we performed co-immunoprecipitation (co-IP) experiments with the total lysates of both WTβ1AR cells and WTβ2AR cells transiently overexpressing tagged-plasmids with and without Carv treatment. We observed that Carv induced an association of β-arrestin1 or β-arrestin2 with the Dicer maturation enzyme in the total lysates of WTβ1AR cells (Figure 2A–B) and WTβ2AR cells (Figure 2C–D). Our data suggest that Carv stimulation of the β1AR or the β2AR promotes the interaction between β-arrestin and Dicer to mature a subset of pre-miRs in the cytoplasm.
Figure 2. β-arrestin1 or β-arrestin2 interacts with the Dicer miR maturation enzyme in β1-adrenergic receptor- or β2-adrenergic receptor-overexpressing cells.

A–D, WTβ1AR or WTβ2AR cells were transfected with Flag-β-arrestin1 and HA-Dicer (A or C) or Flag-α-arrestin2 and HA-Dicer (B or D). Transfected cells were serum-starved for 4 hours and stimulated with Carv for 20 hours. After Carv treatment, cell extracts were prepared and subjected to immunoprecipitation (IP) with anti-Flag, anti-HA, or non-specific IgG (control). Interaction of Dicer with β-arrestin1 or β-arrestin2 was examined by immunoblotting (IB) with anti-HA and anti-Flag.
3.5. The newly identified three βAR/β-arrestin-responsive miRs (miR-466g, -532-5p, and -674) function as protective miRs in cardiac cells
Because β-arrestin-mediated β1AR or β2AR signaling in different myocardial cells confers cardioprotective effects [23, 44–46], and the three newly identified miRs are increased by Carv-mediated βAR/β-arrestin cardioprotective pathways, we next hypothesized that these miRs may function as cytoprotective miRs. Our miR sequence analyses first revealed that miR-674 is conserved across species, while miR-532-5p is conserved between mouse and human with 1bp mismatch in non-seed (target) sequences in the rat, suggesting that the target genes & roles of these two miRs are evolutionally conserved. However, the sequences between mouse miR-466g and its human and rat counterparts were not conserved despite some homologies (Supplementary Figure 2).
To determine the importance of three newly identified β-miRs in myocardial cells under conditions of low oxygen, we used the immortalized adult mouse CM HL-1 cell line, neonatal rat ventricular CMs (NRVCs), adult mouse CFs (AMCFs), neonatal rat ventricular CFs (NRVFs), and adult mouse CECs (MCECs) as in vitro models [10, 11]. Loss-of-function approaches with all three β-miRs (for mouse cells) as well as miR-532-5p and miR-674 (for rat cells because miR-466g is mouse-specific) uncovered that knockdown of each of the three β-miRs (Supplemental Figure 3A, 3C, 4A and 4C) does not affect the apoptosis of CMs and CFs (Supplemental Figure 3B, 3D, 4B and 4D), while knockdown of miR-466g or miR-532-5p (not miR-674) increases CEC apoptosis in both normoxic and simulated ischemia/reperfusion (sI/R) conditions (Figure 3).
Figure 3. MiR-466g or miR-532-5p protects cardiac endothelial cells against apoptotic cell death.
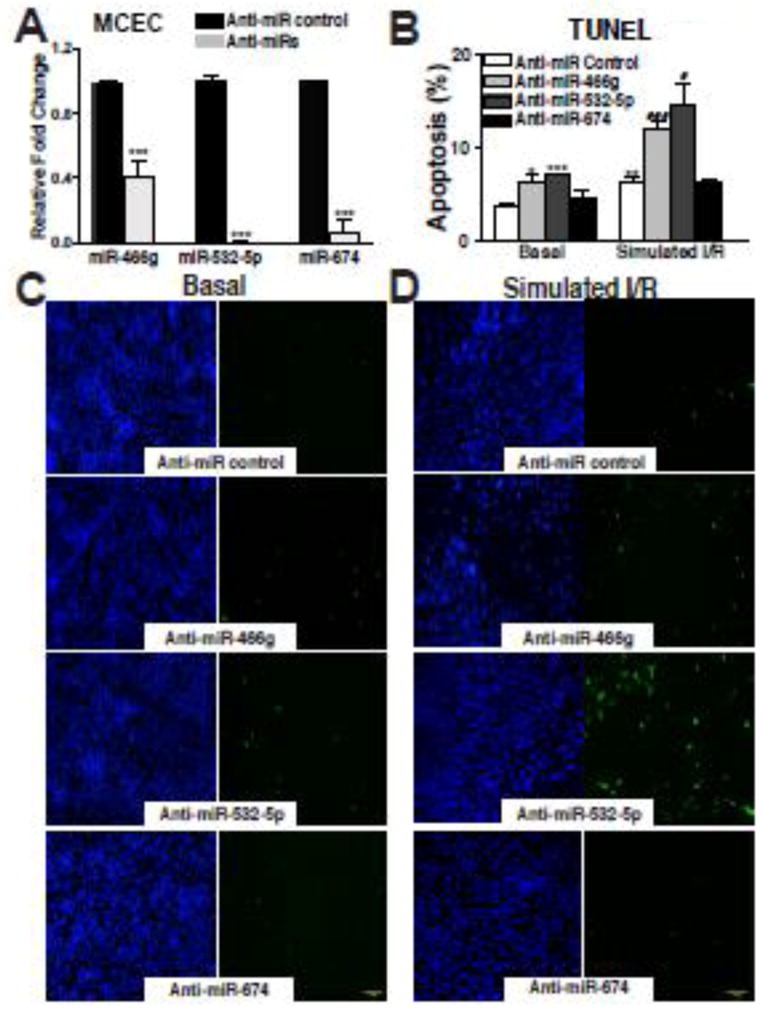
A, Mouse cardiac endothelial cells (MCECs) were transfected with anti-miR control, anti-miR-466g, anti-miR-532-5p, or anti-miR-674. Reduced expression of three miRs is shown. B–D, MCECs transfected with anti-miR control, anti-miR-466g, anti-miR-532-5p, or anti-miR-674 were subjected to in vitro simulation of I/R. TUNEL assays were then performed in both normoxic (basal) (B–C) and simulated I/R conditions (sI/R) (B and D). The percentage of TUNEL positive cells was calculated by normalizing DAPI positive cells. All data are mean ± SEM from 6 independent experiments. All images were taken in a same magnification and were shown in a same size. Scale bars=1mm. ***P<0.001 vs. anti-miR control (A). *P<0.05, **P<0.01, or ***P<0.001 vs. anti-miR control in basal. #P<0.05 or ###P<0.001 vs. anti-miR control in sI/R (B).
Because our results suggest that at least two β-miRs play a role in CECs, and our recent study showed that the expression of miR-532-5p is significantly higher in CECs than other myocardial cell types [47], we examined other functional effects of these three miRs in CECs. Knockdown of miR-466g or miR-532-5p (not miR-674) decreased CEC proliferation in the basal condition, but not in the sI/R condition where CEC functions such as proliferation are decreased compared to normoxia (Figure 4A–B and data not shown). We also observed that knockdown of miR-466g, but not miR-532-5p or miR-674, decreased CEC migration in the basal condition (Figure 4C–D). Interestingly, knockdown of miR-532-5p, but not the other two miRs, diminished tube formation (a model of angiogenesis) in CECs (Supplementary Figure 5). These results, coupled with our recent study demonstrating that knockdown of miR-532-5p in mice reduces CEC proliferation and cardiac vascularization after acute myocardial infarction (AMI) [47] suggest that miR-466g or miR-532-5p (not miR-674) functions as a gatekeeper of CEC function. Because we did not identify a potential role of miR-674 in aforementioned studies, we next used NRVFs and showed that knockdown of miR-532-5p or miR-674 decreases CF proliferation in the basal condition, but not in the sI/R condition where CF functions such as proliferation are decreased compared to normoxia (Figure 5A–B and data not shown). We also observed that knockdown of miR-674 (not miR-532-5p) decreased CF migration in the basal condition (Figure 5C–D). Together with our recent study showing that (i) the expression of miR-532-5p was significantly downregulated in CECs isolated from ischemic myocardium at 1 week post-MI, and (ii) miR-532-5p protects the heart against AMI [47], our data suggest that three newly identified β-miRs exert functional effects in myocardial cells despite their distinct actions on different cell types (mainly CECs for miR-466g and miR-532-5p, and mainly CFs for miR-674).
Figure 4. Knockdown of miR-466g decreases proliferation and migration of cardiac endothelial cells, and knockdown of miR-532-5p decreases cardiac endothelial cell proliferation.
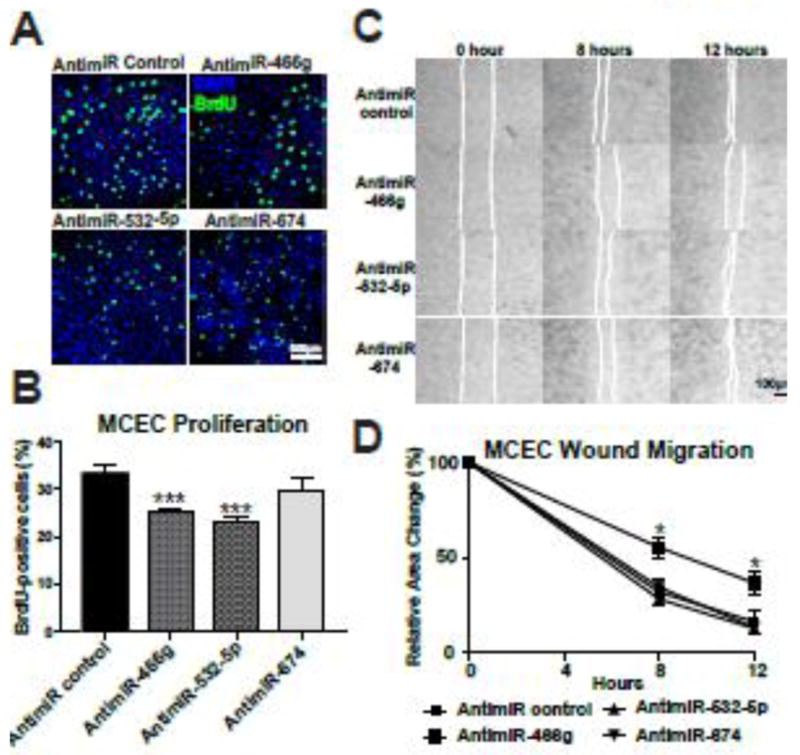
A–B, MCECs transfected with anti-miR control, anti-miR-466g, anti-miR-532-5p, or anti-miR-674 were monitored for nuclear uptake of BrdU, as an index of cell proliferation. The percentage of BrdU positive cells was calculated by normalizing DAPI positive cells. All images were taken in a same magnification and were shown in a same size. C–D, MCECs transfected with anti-miR control, anti-miR-466g, anti-miR-532-5p, or anti-miR-674 were subjected to wound migration assays. Confluent MCEC monolayers were wounded and wound closure was monitored by microscopy at the indicated times. Representative images are shown in C. Quantitative analysis of relative wound size was performed. Initial open areas (0 hour) were measured to serve as total open area, and the percentage of open area after 8 or 12 hours was calculated to determine if MCEC’s migratory potential differed between groups (B). Scale Scale bar=500μm (A) and bar=100μm (C). All data are mean ± SEM from 6 independent experiments. ***P<0.001 vs. antimiR control (B). *P<0.05 vs. other groups (D).
Figure 5. Knockdown of miR-674 decreases basal proliferation and migration of cardiac fibroblasts, and knockdown of miR-532-5p decreases basal proliferation in cardiac fibroblasts.
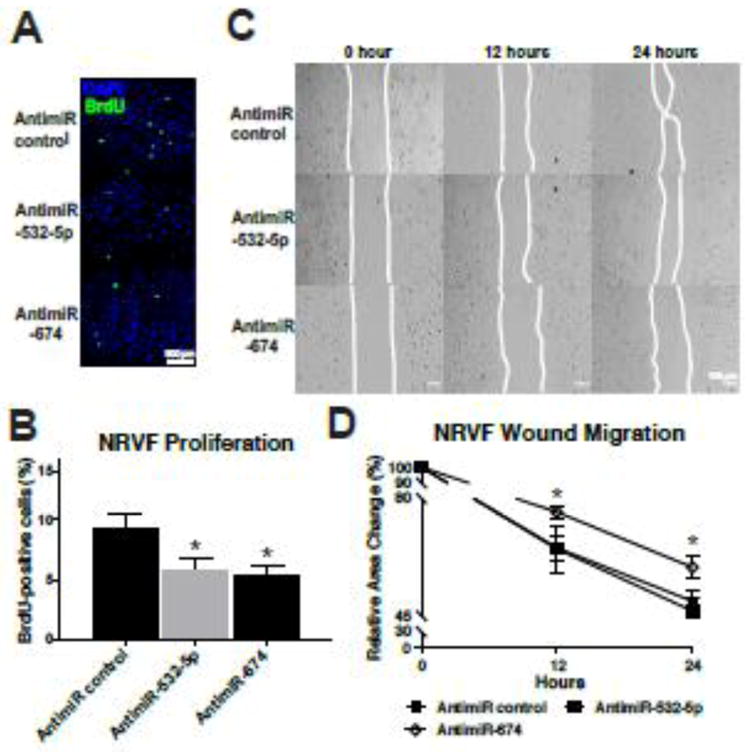
A–B, NRVFs transfected with anti-miR control, anti-miR-532-5p, or anti-miR-674 were subjected to BrdU assays as in Figure 4. The percentage of BrdU positive cells was calculated by normalizing to the number of DAPI positive cells. All images were taken in a same magnification and were shown in a same size. C–D, NRVFs transfected with anti-miR control, anti-miR-532-5p, or anti-miR-674 were subjected to wound migration assays as in Figure 4. Confluent NRVF monolayers were wounded and wound closure was monitored by microscopy at the indicated times. Representative images are shown in C. Quantitative analysis of relative wound size was performed. Initial open areas (0 hour) were measured to serve as total open area, and the percentage of open area after 12 or 24 hours was calculated to determine NRVF’s migratory potential (B). Scale bar=500μm (A) and Scale bars=100μm (C). All data are mean ± SEM from 6 independent experiments. *P<0.05 vs. antimiR control (B) or other groups (D). Note that miR-466g is not included in this rat cell experiment because miR-466g is mouse-specific.
3.6. The newly identified three βAR/β-arrestin-responsive miRs (miR-466g, -532-5p, and -674) regulate deleterious genes (dcun1d4, prss23 and usp14), respectively
In order to identify the candidate targets of three β-miRs that regulate cardiac cell functions, we first used multiple miR target prediction algorithms [38–40] and detected a substantial number of genes with putative binding sites for three β-miRs. By focusing our attention on potential target genes that were predicted by at least three bioinformatic tools, we identified dcun1d4 (defective in cullin neddylation 1 domain containing 4) for miR-466g, prss23 (protease serine 23) for miR-532-5p, and usp14 (ubiquitin specific peptidase 14: deubiquitinating enzyme) for miR-674 as genes of interest. Interestingly, two predicted target genes of three β-miRs, usp14 [48] and prss23 [47], were suggested to play deleterious roles in cardiac function. Liu N et. al. showed that usp14 is upregulated in left ventricular tissues following transverse aortic constriction in rats and in CMs exposed to AngII, and that knockdown of usp14 in CMs reduces expression of cardiac hypertrophy markers [48]. We recently showed that prss23 is a positive regulator of maladaptive endothelial-to-mesenchymal transition (EndMT) and a direct functional target of miR-532-5p in CECs and intact hearts [47].
Because miR-466g or miR-532-5p, and miR-674 have protective roles mainly in CECs and CFs, respectively, as supported by our data (Figure 3–5 and Supplementary Figure 5), we next performed loss- or gain-of-function studies in CECs and CFs to investigate whether the three targets, which were predicted by multiple algorithms, are targets of these three β-miRs. We first observed that dcun1d4 was significantly upregulated with miR-466g inhibition in normoxia (Figure 6A), but not in sI/R (1±0.12 for antimiR control vs. 1.06±0.13 for antimiR-466g. N=6). Interestingly, we also found downregulation of dcun1d4 in simulated I/R (Figure 6B). We further observed that protein levels of dcun1d4 were upregulated with miR-466g inhibition in normoxia (Supplementary Figure 6A), and that dcun1d4 was downregulated with miR-466g overexpression in normoxia, but not in sI/R (Supplementary Figure 6B–C and data not shown). These results suggest that dcun1d4 may be a CEC target of miR-466g. We next showed that prss23 was upregulated with both miR-532-5p inhibition in CECs subjected to sI/R and myocardial cells subjected to sI/R (Figure 6C–D and Supplementary Figure 7). These expression patterns of prss23 are consistent with our recent report on loss- or gain-of-function of miR-532-5p in intact hearts subjected to MI or CECs subjected to TGF-β2-mediated EndMT [47]. Our previous and current findings thus indicate that prss23 is an important direct and functional target of miR-532-5p in CECs and hearts. We also found that usp14 was upregulated with miR-674 inhibition in both normoxia and sI/R conditions (Figure 6E and data not shown). Usp14 was also upregulated in NRVFs subjected to sI/R (Figure 6F). We next observed that protein levels of usp14 were downregulated with miR-674 overexpression in normoxia, and that usp14 was downregulated with miR-674 overexpression in both normoxia (Supplementary Figure 8) and sI/R conditions (1±0.09 for miR mimic control vs. 0.19±0.04 for miR-674 mimic: P<0.01. N=6). Along with the previous report on upregulation of usp14 in stressed hearts [48], our results suggest that usp14 may be a CF target of miR-674.
Figure 6. MiR-466g or miR-532-5p represses dcun1d4 or prss23 in mouse cardiac endothelial cells, respectively, and miR-674 represses usp14 in neonatal rat ventricular fibroblasts.
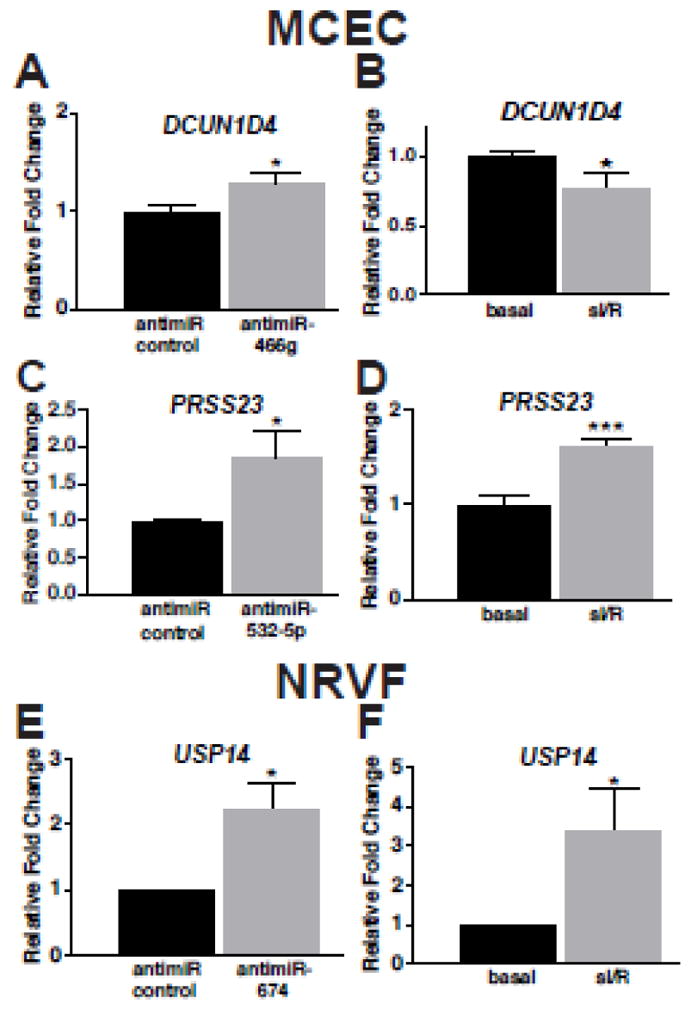
A, RNAs isolated from MCECs transfected with 100nM mirVana™ miR-466g inhibitor or 15-mer control were analyzed by QRT-PCR. Loss-of-function of miR-466g in MCECs resulted in increased dcun1d4. B, MCECs were subjected to in vitro simulation of I/R (sI/R). The expression of dcun1d4 in basal and sI/R was shown. Results are representative of 6 independent experiments with different biological samples. *P<0.05 vs. control (antimiR control or basal). C, RNAs isolated from MCECs transfected with 100nM mirVana™ miR-532-5p inhibitor or 15-mer control were analyzed by QRT-PCR. Loss-of-function of miR-532-5p in MCECs resulted in increased prss23 expression. D, MCECs were subjected to sI/R. The expression of prss23 in basal and sI/R was shown. Results are representative of 6 independent experiments with different biological samples. *P<0.05 vs. antimiR control. ***P<0.001 vs. basal. E, RNAs isolated from NRVFs transfected with miR-674 inhibitor or 15-mer control were analyzed by QRT-PCR. Loss-of-function of miR-674 in NRVFs resulted in increased usp14 expression. F, NRVFs were subjected to sI/R. The expression of usp14 in basal and sI/R was shown. Results are representative of 6 independent experiments with different biological samples. *P<0.05 vs. antimiR control or basal.
Because our data suggest that miR-466g and miR-532-5p mainly regulate CEC functions, and miR-674 mainly regulates CF functions, we lastly evaluated the expression of the three miRs and the target genes in MCECs and NRVFs treated with Carv. We found that Carv upregulated miR-466g and miR-532-5p, but downregulated their targets dcun1d4 and prss23 in MCECs subjected to normoxia, but not sI/R condition (Figure 7A–D and data not shown). We also observed that Carv upregulated miR-674, but downregulated its target usp14 in NRVFs subjected to normoxia, but not sI/R condition (Figure 7E–F and data not shown). These data indicate that the pairs of these three miRs and their targets are sensitive to Carv in MCECs and NRVFs under basal conditions. Taken together, our current data along with previous reports indicate that downregulation of deleterious dcun1d4, prss23 and usp14, which are predictive and experimentally validated targets, in part contributes to the protective actions of miR-466g, miR-532-5p and miR-674 in myocardial cells, respectively.
Figure 7. Carvedilol induces the expression of miR-466g and miR-532-5p, or miR-674, while carvedilol inhibits the expression of dcun1d4 and prss23, or usp14 in MCECs or NRVFs, respectively.

A–D, MCECs were treated with either vehicle (DMSO) or 1μM of carvedilol (Carv) for 4 hours. The expression of miR-466g, miR-532-5p, dcun1d4 and prss23 was detected using TaqMan assays. Carv elicits upregulation of miR-466g and miR-532-5p, while downregulating dcun1d4 and prss23. E–F, NRVFs were treated with either vehicle (DMSO) or 1μM of carvedilol (Carv) for 4 hours. The expression of miR-674 and usp14 was detected using TaqMan assays. Carv elicits upregulation of miR-674, while reducing the expression of usp14. N=6 in each group. *P<0.05 or **P<0.01 vs. DMSO.
4. Discussion
Here, we show an essential role of cytoplasmic β-arrestin1 or β-arrestin2 in Dicer-mediated miR maturation following stimulation by the β-arrestin-biased βAR ligand carvedilol. We demonstrate that this process results from stimulation of the β1AR or β2AR and requires β-arrestin1 or β-arrestin2 to promote Dicer-mediated maturation of three miRs (miR-466g, miR-532-5p and miR-674) in murine hearts. The molecular mechanism for this β-arrestin-mediated miR maturation involves the formation of a complex between Dicer and β-arrestin (Figure 8). Our working hypothesis for the mechanism by which β-arrestin enhances Dicer-mediated miR maturation is that phosphorylation of the β1AR or β2AR by GRK 5 or 6 mediates the association of β-arrestin 1 or 2 with the Dicer maturation enzyme on a subset of pre-miRs.
Figure 8. β-arrestin-biased agonism of β-adrenergic receptor stimulates the Dicer-mediated maturation of a subset of miRs in the cytoplasm to induce cardioprotective signaling.

The β-arrestin-biased ligand Carv, which was shown to stimulate β-arrestin-mediated cardioprotective signaling in the absence of G protein activation [13], induces the expression of three newly identified βAR/β-arrestin-responsive miRs (β-miRs) in a β1AR-, β2AR-, GRK5-, or GRK6-dependent manner. Our data also suggest that β-arrestin1 or β-arrestin2 (possibly in the hetero-oligomerization) promotes Dicer-mediated maturation of premature miR (pre-miR) into mature miRs of β-miRs by forming a novel cytoplasmic complex with the Dicer maturation enzyme. Our myocardial cell approaches also uncover that despite their distinct roles in different cell types, β-miRs act as protective miRs in part by repressing deleterious genes, dcun1d4, prss23 and usp14. P: phosphorylated; GRK: GPCR kinase.
Our sequence motif detection analyses using RBPDB (a database of RNA binding protein specificities) [49] suggest that the three βAR/β-arrestin-responsive miRs (β-miRs) identified in this study do not have known conserved sequence motifs or consensus sequences for direct binding by RNA binding proteins (RBPs) in their pre-miR regions (Supplementary Figure 2B), although additional profiling analyses in different tissues or cells and RNA structure analyses will be required to identify a potential secondary RNA structure for the recruitment of the β-arrestin/Dicer complex to pre-miR regions of all β-miRs. While we believe the direct association of β-arrestin with pre-miRs bound by Dicer to be a plausible mechanism, it is possible that activation of βAR signaling pathways downstream of β-arrestin (eg. EGFR, ERK or AKT) could regulate Dicer-mediated miR maturation via phosphorylation and thus indirectly exert regulatory effects on the activation of miR maturation [6, 50]. This idea is supported by the fact that β-arrestins do not have known RNA binding motifs. It is also possible that β-arrestin could interact with other regulators in miR biogenesis such as regulators of miR stability or components of RNA-induced silencing complex (eg. Ago2) to increase levels of the three newly identified β-miRs. Lastly, the regulation of Dicer by β-arrestin could be indirect through interaction with other RNA-binding proteins. We observed that β-arrestin 1 or 2 formed a novel complex with the general Dicer miR maturation enzyme, but how Carv promotes the maturation of the three newly identified β-miRs remains unknown. A proteomic analysis previously identified that β-arrestins potentially interact with many RBPs [8]. Therefore, additional studies such as Co-IP and RNA-CHIP assays along with loss-of-function of potential β-arrestin-interacting RBPs will be needed to further clarify the mechanism of β-arrestin in Dicer-mediated miR maturation.
β-arrestins not only desensitize G protein signaling but also activate a number of signaling networks by scaffolding a diverse group of signaling proteins at the GPCRs [51, 52]. The important roles of β-arrestins in regulating cytoplasmic signaling networks are now well recognized [53, 54]. For example, either β-arrestin1 or β-arrestin2 was shown to involve in β1AR-mediated cardioprotective signaling [13, 23], suggesting that β-arrestins form hetero-oligomers in the cytoplasm. Interestingly, we previously reported that only β-arrestin1 regulates Drosha-mediated miR processing in the nucleus upon β1AR stimulation [9]. This likely reflects the fact that β-arrestin1 lacks a nuclear export sequence (thus allowing for its retention in the nucleus after activation), and that β-arrestin1 monomers display increased nuclear localization [55]. Together with the possibility that β-arrestin2 could also interact with many RBPs [8], our previous finding on β-arrestin1 specificity in Drosha-mediated miR processing [9] suggests that β-arrestin2 may regulate other miR biogenesis steps (eg. miR degradation, nuclear export and dicing) rather than Drosha-mediated processing in the nucleus. The current study indeed identifies β-arrestin2 like β-arrestin1 (possibly in the hetero-oligomerization) as a regulator of Dicer-mediated miR maturation after stimulation of β1AR or β2AR, indicating that both β-arrestins play regulatory roles in miR biogenesis.
It was previously shown that β1AR uses GRK 5 or 6 and β-arrestin 1 or 2 to promote cardiomyocyte survival pathways against chronic catecholamine stimulation in the absence of G protein activation [23]. Our subsequent study suggested that Carv (known as a nonselective βAR antagonist) functions as a β-arrestin-biased ligand to promote cardioprotective signaling [13], providing an additional possible mechanism for some of its clinical efficacy. Interestingly, a meta-analysis showed that Carv did not reduce patient readmissions compared with other β-blockers despite less sudden cardiac death and all-cause mortality in patients with MI and those with heart failure [56]. Therefore, identifying additional beneficial downstream signaling pathways activated by Carv should lead to a better understanding of how biased ligands exert their cardioprotective effects. We previously showed a new mechanism that only β1AR stimulation by Carv mediates Drosha-mediated miR processing in the nucleus [9]. Interestingly, our current study also discovers a novel miR regulatory mechanism by which either β-arrestin1 or β-arrestin2 is recruited to either β1AR or β2AR after Carv stimulation for activating Dicer-mediated miR maturation in the cytoplasm.
Aberrant expression of miRs has been linked to several clinical cardiovascular diseases such as ischemia/reperfusion injury and heart failure [57–59]. Accordingly, miR mutant mice have been generated to study their functions in cardiovascular pathophysiology [60–66]. Among the five Carv/β1AR/β-arrestin1/Drosha-regulatable miRs that we identified in the heart [9], four miRs (miR-125b-5p, miR-150, miR-199a-3p and miR-214) were reported to play important roles in cardiac function [10, 12, 30, 41, 42]. Interestingly, our current results suggest that the three new Carv/βAR/β-arrestin/Dicer-responsive miRs also act as convergent hubs for cardioprotective signaling, although they do not share any common target genes. Our recent study indeed demonstrated that one of them, miR-532-5p protects the heart against AMI [47]. Although miR-466g has been shown to play a role in renal epithelial cells [67], and miR-674 has been implicated in immune responses and liver injury [68, 69], neither of these two newly identified β-arrestin-responsive miRs have been reported to play a functional role in either cardiac cells or heart tissues. Interestingly, we report novel findings that (i) knockdown of miR-466g increases CEC apoptosis, but decreases migration and proliferation of CECs, and (ii) knockdown of miR-674 decreases migration and proliferation of CFs. Based on our recent report on identification of miR-532-5p as a cardioprotective miR [47] and our current results (Figure 3–5 and Supplementary Figure 5), we postulate that β-miRs are cardioprotective miRs activated by βAR-mediated β-arrestin signaling, resulting in beneficial adaptive remodeling in the failing hearts. Further in vivo studies are warranted to investigate the function of miR-466g and miR-674 for their potential utilities in the management of cardiac disease, and the underlying mechanisms by which the two miRs confer protective effects in other relevant cell types.
In this study, we also show that the three β-miRs are upregulated by Carv-mediated cardioprotective signaling and elicit protective effects in myocardial cells despite their distinct roles in different cell types by repressing dcun1d4, prss23 or usp14. We also demonstrate that the three targets are downregulated by Carv treatment in CECs or CFs, suggesting that Carv may inhibit the targets by upregulating these three β-miRs. Interestingly, two identified targets of these three β-miRs (usp14 [48] and prss23 [47]) are known to be involved in maladaptive cardiac remodeling. We also recently demonstrated that prss23 is a direct functional target of miR-532-5p in CECs and hearts during AMI [47]. Although additional studies will be needed (i) to validate dcun1d4 or usp14 as a direct functional target of miR-466g or miR-674, respectively, and (ii) to establish mechanistic relationships between the miR/target pairs in distinct cardiac cell types and the cardioprotective effects of Carv, our bioinformatics and experimental evidences indicate that dcun1d4 is a CEC target of miR-466g, prss23 is a direct functional CEC target of miR-532-5p (supported by our recent study [47]), and usp14 is a CF target of miR-674. Altogether, our previously published and current data suggest that βAR/β-arrestin-mediated miR maturation regulatory network induced by Carv has therapeutic potential and point to the urgent need for further studies that investigate the effect of gain- or loss-of-function of this newly identified regulatory mechanism in cardiac pathophysiology.
5. Conclusion
Our data identify β-arrestin1 or β-arrestin2 as an important mediator of Dicer function to regulate miR maturation in mouse hearts and provide new insights into our understanding of how selective ligands for the βAR may modulate the metabolism of specific miRs. We postulate that the development of high affinity βAR-biased ligands, which display better efficacy for this newly discovered β-arrestin-mediated miR maturation regulatory network, may provide a new class of drugs for the treatment of cardiovascular diseases.
Supplementary Material
Highlights.
Carvedilol promotes the Dicer-mediated miR maturation.
Carvedilol-mediated miR maturation requires β-arrestin1 or 2 and β1AR or β2AR.
β-arrestin 1 or 2 forms a novel complex with the Dicer maturation enzyme.
βAR/β-arrestin-responsive miRs act as gatekeepers of myocardial cell functions.
βAR/β-arrestin-responsive miRs repress DCUN1D4, PRSS23, and USP14.
Acknowledgments
We thank Drs. Ruth Caldwell and Zheng Dong for sharing their equipment.
Sources of Funding
This work was supported by American Heart Association (AHA) Predoctoral Fellowship 16PRE30210016 to Jian-peng Teoh, National Institutes of Health (NIH) R01 HL124248 to Huabo Su, NIH R01 HL112640 and HL126949 to Neal L. Weintraub, NIH R01 HL086555 to Yaoliang Tang, NIH R01 HL134354 and AR070029 to Yaoliang Tang and Neal L. Weintraub, and American Physiological Society Shih-Chun Wang Young Investigator Award, AHA Grant-in-Aid 12GRNT12100048 and AHA Scientist Development Grant 14SDG18970040, and NIH R01 HL124251 to Il-man Kim.
Non-Standard Abbreviations and Acronyms
- βARs
β-adrenergic receptors
- β-blockers
β-adrenergic receptor antagonists
- β-miRs
β-adrenergic receptor/β-arrestin-regulated miRs
- Carv
carvedilol
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- KO
knockout
- MiRNAs or MiRs
microRNAs
- Pre-miRs
hairpin intermediate microRNAs or premature microRNAs
- Pri-miRs
long primary microRNA transcripts
- TG
transgenic
- WT
wild-type
- βAR cells
HEK 293 cells stably overexpressing βArs
Footnotes
Supplementary data are available at online.
Disclosures
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. The Journal of clinical investigation. 2013;123(1):11–8. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu H, Fan GC. Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc Res. 2012;94(2):284–92. doi: 10.1093/cvr/cvr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nature reviews Genetics. 2010;11(9):597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 4.Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38(3):323–32. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Yu Z, Wang L, Wang C, Ju X, Wang M, Chen K, Loro E, Li Z, Zhang Y, Wu K, Casimiro MC, Gormley M, Ertel A, Fortina P, Chen Y, Tozeren A, Liu Z, Pestell RG. Cyclin D1 induction of Dicer governs microRNA processing and expression in breast cancer. Nat Commun. 2013;4:2812. doi: 10.1038/ncomms3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, Hsu JL, Wu Y, Lam YC, James BP, Liu X, Liu CG, Patel DJ, Hung MC. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497(7449):383–7. doi: 10.1038/nature12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki HI, Arase M, Matsuyama H, Choi YL, Ueno T, Mano H, Sugimoto K, Miyazono K. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol Cell. 2011;44(3):424–36. doi: 10.1016/j.molcel.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104(29):12011–6. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, Traynham CJ, Pironti G, Mao L, Su H, Johnson JA, Koch WJ, Rockman HA. beta-arrestin1-biased beta1-adrenergic receptor signaling regulates microRNA processing. Circulation research. 2014;114(5):833–44. doi: 10.1161/CIRCRESAHA.114.302766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Ramesh G, Kim IM. MicroRNA-150 Protects the Mouse Heart from Ischemic Injury by Regulating Cell Death. Cardiovasc Res. 2015;106(3):387–97. doi: 10.1093/cvr/cvv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park KM, Teoh JP, Wang Y, Broskova Z, Bayoumi AS, Tang Y, Su H, Weintraub NL, Kim IM. Carvedilol-responsive microRNAs, miR-199a-3p and -214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2016;311(2):H371–83. doi: 10.1152/ajpheart.00807.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL, Kim IM. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2017;114:72–82. doi: 10.1016/j.yjmcc.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, Rockman HA. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(38):14555–60. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104(42):16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100(19):10782–7. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, Lark MW. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335(3):572–9. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 17.Patel CB, Noor N, Rockman HA. Functional selectivity in adrenergic and angiotensin signaling systems. Mol Pharmacol. 2010;78(6):983–92. doi: 10.1124/mol.110.067066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4(185):ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–97. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heitzler D, Durand G, Gallay N, Rizk A, Ahn S, Kim J, Violin JD, Dupuy L, Gauthier C, Piketty V, Crepieux P, Poupon A, Clement F, Fages F, Lefkowitz RJ, Reiter E. Competing G protein-coupled receptor kinases balance G protein and beta-arrestin signaling. Mol Syst Biol. 2012;8:590. doi: 10.1038/msb.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruffolo RR, Jr, Feuerstein GZ. Pharmacology of carvedilol: rationale for use in hypertension, coronary artery disease, and congestive heart failure. Cardiovasc Drugs Ther. 1997;11(Suppl 1):247–56. doi: 10.1023/a:1007735729121. [DOI] [PubMed] [Google Scholar]
- 22.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49(16):1722–32. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 23.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117(9):2445–58. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, Rockman HA. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;297(4):H1377–86. doi: 10.1152/ajpheart.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010;189(3):573–87. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278(37):35403–11. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 27.Naga Prasad SV, Laporte SA, Chamberlain D, Caron MG, Barak L, Rockman HA. Phosphoinositide 3-kinase regulates beta2-adrenergic receptor endocytosis by AP-2 recruitment to the receptor/beta-arrestin complex. J Cell Biol. 2002;158(3):563–75. doi: 10.1083/jcb.200202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramakrishna S, Kim IM, Petrovic V, Malin D, Wang IC, Kalin TV, Meliton L, Zhao YY, Ackerson T, Qin Y, Malik AB, Costa RH, Kalinichenko VV. Myocardium defects and ventricular hypoplasia in mice homozygous null for the Forkhead Box M1 transcription factor. Dev Dyn. 2007;236(4):1000–13. doi: 10.1002/dvdy.21113. [DOI] [PubMed] [Google Scholar]
- 29.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66(4):2153–61. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 30.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. The Journal of clinical investigation. 2012;122(4):1222–32. doi: 10.1172/JCI59327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem. 2005;280(23):22278–86. doi: 10.1074/jbc.M500936200. [DOI] [PubMed] [Google Scholar]
- 32.Kim IM, Wolf MJ, Rockman HA. Gene deletion screen for cardiomyopathy in adult Drosophila identifies a new notch ligand. Circulation research. 2010;106(7):1233–43. doi: 10.1161/CIRCRESAHA.109.213785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–4. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem. 2009;284(30):20375–86. doi: 10.1074/jbc.M109.005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3(125):ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teoh JP, Park KM, Wang Y, Hu Q, Kim S, Wu G, Huang S, Maihle N, Kim IM. Endothelin-1/endothelin A receptor-mediated biased signaling is a new player in modulating human ovarian cancer cell tumorigenesis. Cell Signal. 2014;26(12):2885–95. doi: 10.1016/j.cellsig.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124(6):720–30. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 38.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. Rna. 2008;14(6):1012–7. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 40.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 41.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–81. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Ha T, Zou J, Ren D, Liu L, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc Res. 2014;102(3):385–95. doi: 10.1093/cvr/cvu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liggett SB. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci Signal. 2011;4(185):pe36. doi: 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- 44.Grisanti LA, Gumpert AM, Traynham CJ, Gorsky JE, Repas AA, Gao E, Carter RL, Yu D, Calvert JW, Garcia AP, Ibanez B, Rabinowitz JE, Koch WJ, Tilley DG. Leukocyte-Expressed beta2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation. 2016;134(2):153–67. doi: 10.1161/CIRCULATIONAHA.116.022304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grisanti LA, Traynham CJ, Repas AA, Gao E, Koch WJ, Tilley DG. beta2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc Natl Acad Sci U S A. 2016;113(52):15126–15131. doi: 10.1073/pnas.1611023114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carr R, 3rd, Schilling J, Song J, Carter RL, Du Y, Yoo SM, Traynham CJ, Koch WJ, Cheung JY, Tilley DG, Benovic JL. beta-arrestin-biased signaling through the beta2-adrenergic receptor promotes cardiomyocyte contraction. Proc Natl Acad Sci U S A. 2016;113(28):E4107–16. doi: 10.1073/pnas.1606267113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayoumi AS, Teoh JP, Aonuma T, Yuan Z, Ruan X, Tang Y, Su H, Weintraub NL, Kim IM. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc Res. 2017;113(13):1603–14. doi: 10.1093/cvr/cvx132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu N, Chai R, Liu B, Zhang Z, Zhang S, Zhang J, Liao Y, Cai J, Xia X, Li A, Liu J, Huang H, Liu S. Ubiquitin-specific protease 14 regulates cardiac hypertrophy progression by increasing GSK-3beta phosphorylation. Biochem Biophys Res Commun. 2016;478(3):1236–41. doi: 10.1016/j.bbrc.2016.08.100. [DOI] [PubMed] [Google Scholar]
- 49.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39(Database issue):D301–8. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monk CE, Hutvagner G, Arthur JS. Regulation of miRNA transcription in macrophages in response to Candida albicans. PLoS ONE. 2010;5(10):e13669. doi: 10.1371/journal.pone.0013669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62(2):305–30. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9(5):373–86. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 54.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308(5721):512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 55.Ma L, Pei G. Beta-arrestin signaling and regulation of transcription. J Cell Sci. 2007;120(Pt 2):213–8. doi: 10.1242/jcs.03338. [DOI] [PubMed] [Google Scholar]
- 56.Dinicolantonio JJ, Lavie CJ, Fares H, Menezes AR, O’Keefe JH. Meta-Analysis of Carvedilol Versus Beta 1 Selective Beta-Blockers (Atenolol, Bisoprolol, Metoprolol, and Nebivolol) Am J Cardiol. 2013;111(5):765–9. doi: 10.1016/j.amjcard.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 57.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circulation research. 2007;100(3):416–24. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 58.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170(6):1831–40. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22(23):3242–54. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ, Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell. 2009;17(5):662–73. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129(2):303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 63.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15(2):261–71. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135(24):3989–93. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 65.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23(18):2166–78. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress -dependent cardiac growth and gene expression by a microRNA. Science. 2007;316(5824):575–9. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 67.Jacobs ME, Kathpalia PP, Chen Y, Thomas SV, Noonan EJ, Pao AC. SGK1 regulation by miR-466g in cortical collecting duct cells. Am J Physiol Renal Physiol. 2016;310(11):F1251–7. doi: 10.1152/ajprenal.00024.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin J, Chen YT, Xia J, Yang Q. MiR674 inhibits the neuraminidase-stimulated immune response on dendritic cells via down-regulated Mbnl3. Oncotarget. 2016;7(31):48978–48994. doi: 10.18632/oncotarget.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su K, Wang Q, Qi L, Hua D, Tao J, Mangan CJ, Lou Y, Li L. MicroRNA-674-5p/5-LO axis involved in autoimmune reaction of Concanavalin A-induced acute mouse liver injury. Toxicol Lett. 2016;258:101–7. doi: 10.1016/j.toxlet.2016.06.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.