

Abstract
Class IIa histone deacetylases (HDACs) are transcriptional repressors whose nuclear export in the cardiac myocyte is associated with the induction of pathological gene expression and cardiac remodeling. Class IIa HDACs are regulated by multiple, functionally opposing post-translational modifications, including phosphorylation by protein kinase D (PKD) that promotes nuclear export and phosphorylation by protein kinase A (PKA) that promotes nuclear import. We have previously shown that the scaffold protein muscle A-kinase anchoring protein β (mAKAPβ) orchestrates signaling in the cardiac myocyte required for pathological cardiac remodeling, including serving as a scaffold for both PKD and PKA. We now show that mAKAPβ is a scaffold for HDAC5 in cardiac myocytes, forming signalosomes containing HDAC5, PKD, and PKA. Inhibition of mAKAPβ expression attenuated the phosphorylation of HDAC5 by PKD and PKA in response to α- and β-adrenergic receptor stimulation, respectively. Importantly, disruption of mAKAPβ-HDAC5 anchoring prevented the induction of HDAC5 nuclear export by α-adrenergic receptor signaling and PKD phosphorylation. In addition, disruption of mAKAPβ-PKA anchoring prevented the inhibition by β-adrenergic receptor stimulation of α-adrenergic-induced HDAC5 nuclear export. Together, these data establish that mAKAPβ signalosomes serve to bidirectionally regulate the nuclear-cytoplasmic localization of class IIa HDACs. Thus, the mAKAPβ scaffold serves as a node in the myocyte regulatory network controlling both the repression and activation of pathological gene expression in health and disease, respectively.
Keywords: mAKAP, HDAC5, phosphorylation, scaffold, PKD, PKA
Graphical abstract
1. Introduction
The reversible acetylation of histone proteins is a central mechanism for the regulation of gene transcription. When bound to chromatin, class IIa histone deacetylases (HDACs 4, 5, 7, and 9) repress the expression of genes associated with pathological cardiac myocyte hypertrophy, such that nuclear export of these HDACs is an important mechanism facilitating adverse remodeling in heart disease [1]. For example, class IIa HDAC nuclear export permits the expression of genes regulated by the myocyte enhancer factor 2 (MEF2) family of transcription factors, promoting cardiac myocyte hypertrophy and interstitial fibrosis and ultimately contributing to the development of heart failure. In normal physiology, class IIa HDAC-mediated repression maintains cardiac structure and function. Ablation of either the Hdac5 or Hdac9 mouse genes resulted in spontaneous cardiac hypertrophy, as well as an exaggerated response to pressure overload and to activation of the calcineurin phosphatase pro-hypertrophic signaling pathway [2, 3].
The major function of class IIa HDACs is to organize large site-specific repressor complexes, such that the export of these HDACs to the cytoplasm is key to the dissociation of co-repressors from chromatin and MEF2 activation [1]. The regulation of HDAC4/5 nuclear-cytoplasmic translocation has been extensively studied [4]. Class IIa HDACs have a N-terminal regulatory domain and a C-terminal deacetylase domain. Phosphorylation of serine residues in the N-terminal domain (HDAC4 – Ser-246/467/632; HDAC5 – Ser-259/498/661, with the latter site apparently less important) by protein kinase D (PKD) and Ca2+/calmodulin-dependent kinases creates docking sites for 14-3-3 proteins that mask the intervening HDAC4/5 nuclear localization signal and favors Crm1-dependent nuclear export [5–10]. In contrast, protein kinase A (PKA) -dependent phosphorylation of HDAC4 Ser-266 and HDAC5 Ser-279 promotes nuclear import [11–15]. Although it is unclear how this latter phosphorylation event favors nuclear localization, a HDAC5 Ser-279 phosphomimetic mutation blocked nuclear export in response to α-adrenergic or endothelin-1 receptor stimulation of adult cardiac myocytes [12, 13]. Conversely, a Ser-279 phosphoablative mutation blocked HDAC5 nuclear import in response to acute β-adrenergic or forskolin stimulation [12, 15]. Thus, PKA-dependent phosphorylation provides a mechanism for β-adrenergic receptor mediated inhibition of Gq-coupled receptor-dependent HDAC4/5 nuclear export [14]. Additional mechanisms regulating HDAC4/5 intracellular localization include oxidation of conserved cysteine resides in the deacetylase domain (HDAC4 – Cys-667/669; HDAC5 – 696/698) and HDAC4-specific, PKA-dependent proteolytic cleavage of the HDAC4 regulatory domain, both of which may be induced by β-adrenergic stimulation [4]. Together, these findings define HDAC4/5 as a node in the signal-dependent regulatory network controlling pathological cardiac remodeling. Based upon these results, it has been proposed that β-adrenergic-dependent PKA activity will oppose pathological remodeling during acute sympathetic stimulation, while loss of PKA-dependent HDAC4/5 nuclear import will contribute to remodeling in heart failure when β-adrenergic-dependent cAMP signaling is downregulated or altered in compartmentation [12, 14].
While the functions of HDAC5 phosphorylation events have been defined, it remains unclear how and where HDAC5 phosphorylation is coordinated in response to upstream stimuli. PKA signaling is highly compartmentalized in myocytes by multivalent A-Kinase Anchoring Protein (AKAP) scaffold proteins that co-localize PKA holoenzyme with relevant substrates [16]. The PKA holoenzyme tetramer is composed of two regulatory (R) and two catalytic (C) subunits in a C-R-R-C configuration. In most PKA-AKAP complexes, a X-type, four-helix bundle formed by the N-termini of the R-subunit dimer binds an amphipathic helix on the AKAP [17]. mAKAP (muscle AKAP) is a perinuclear AKAP with a canonical PKA-binding domain that is expressed in striated myocytes and neurons [18]. mAKAPβ, the alternatively-spliced mAKAP form expressed in myocytes, organizes large, multimolecular signaling complexes or “signalosomes” responsible for cardiac myocyte hypertrophy [19]. mAKAPβ signalosomes are responsive to both Gs- and Gq-coupled receptor signaling, as well as upstream IL-6 type cytokine and hypoxia signals. Through the regulated binding of multiple signaling enzymes, including mitogen-activated protein kinases and calcineurin, mAKAPβ signalosomes modulate the transcriptional activity of MEF2, NFATc, and Hif-1α transcription factors. Recently, we showed that mAKAPβ forms ternary complexes with PKD and HDAC4 [20]. In mice subjected to pressure overload mAKAPβ was required for PKD activation and PKD-dependent HDAC4 phosphorylation These results were consistent with the known association of PKD and its upstream activators phospholipase Cε and protein kinase Cε with mAKAPβ signalosomes and their role in hypertrophy [21, 22]. As mAKAPβ also binds PKA, we considered that the mAKAPβ scaffold may have a broader role in the regulation of class IIa HDACs. We now show that HDAC5 also binds mAKAPβ, forming ternary complexes with both PKA and PKD in cardiac myocytes. Accordingly, mAKAPβ expression was required for the phosphorylation of HDAC5 by both PKD and PKA in response to α-adrenergic and β-adrenergic-stimulation, respectively. Our results imply that mAKAPβ signalosomes are critical to the proper regulation of class IIa HDAC activity and localization under different physiological conditions.
2. Materials and Methods
2.1 Materials
Antibodies include mouse anti-HDAC5 (Santa Cruz sc-133225), goat anti-HDAC5 (Santa Cruz Biotechnology sc-5252), rabbit anti-phospho-HDAC5 Ser-498/661(Cell Signaling Technology 3424 that also detects phospho-HDAC4 and HDAC7), rabbit anti-phospho-PKA substrate (Cell Signaling Technology 9621), rabbit anti-mAKAP [23], mouse anti-PKA C-subunit (Santa Cruz sc-390548), rabbit anti-PKD/PKCμ (Cell Signaling Technology 90039), mouse anti-MEF2D (Santa Cruz sc-271153), mouse anti-Flag tag (Sigma A220), mouse anti-myc tag (Santa Cruz sc-40), mouse anti-HA tag agarose (Santa Cruz sc-500777), mouse anti-lamin (Santa Cruz sc-7293), mouse anti-GAPDH (Santa Cruz sc-20358), rabbit anti-mCherry (BioVision Inc 5993100), mouse anti-green fluorescent protein (GFP, Santa Cruz sc-9996), rabbit anti-GFP (Santa Cruz sc-8334), rabbit anti-ANF (US Biological, A4152-35). and mouse anti-α-actinin (Sigma A7811). ON-TARGETplus siRNA oligonucleotides were mAKAP (GAC GAA CCU UCC UUC CGA A UU) and On-TARGETplus Non-targeting siRNA #1 from Dharmacon. AKAP-IS (RRRRRRRRRRRQIEYLAKQIVDNAIQQA) and control peptides (RRRRRRRRRRRQAAYLAKQAAANAAQAA) were acquired from CHI Scientific. Kemptide (LRRASLG) was from Millipore. Adenovirus expressing HDAC5-GFP was obtained from Seven Hills Bioreagents. Expression plasmids for Flag-tagged HDAC4 (plasmid #13821), HDAC5 (plasmid #13822) and HA- and YFP-tagged PKD1 (plasmid #31527) were obtained from Addgene. The mAKAP-GFP expression plasmid and adenovirus expressing mAKAP wildtype and PKA binding site mutant lacking residues 2053–2073 were as previously described [24, 25]. pTRE expression plasmid (Clontech) was used to construct a conditional expression vector for mCherry-mAKAP 301–500 fusion protein (details available upon request).
2.2 Culture of primary neonatal rat ventricular myocytes
Myocytes were prepared as previously described [26]. Briefly, cardiac ventricles free of atria and connective tissue were isolated from 1- to 3-day-old rat pups euthanized by decapitation. Myocytes were dissociated by several cycles of trypsin treatment and serum neutralization. After dissociation, the cells were collected by centrifugation, passed through a 70-mm-mesh cell strainer to remove clumps, and pre-plated in culture dishes to remove fibroblasts. After 1 h, the medium containing the unattached myocytes was removed and the cells collected by centrifugation and plated again in 6-well dishes at a density of 500 000 myocytes/plate. Plating medium was Dulbecco’s modified Eagle’s medium (DMEM) with 17% Media 199, 1% penicillin/streptomycin (Gibco-BRL), 10% horse serum (HS) and 5% fetal bovine serum (FBS). The following day, the plates were washed and then incubated with plating media containing neither HS nor FBS (maintenance media). Myocytes were transfected with siRNA using Dharmafect 1 (Thermofisher) and with plasmids using Transfast (Promega) as recommended by the manufacturers using cells cultured in maintenance medium supplemented with 4% horse serum. Myocytes were cultured for two days following transfection and/or the addition of adenovirus to the media before analysis. For morphometry, immunocytochemistry and fluorescent imaging were performed as previously described [25]. At least 25 individual cells were measured for each condition for each biological replicate. ANF expression was determined by counting the fraction of myocytes with peri-nuclear ANF antibody staining.
2.3 Cell transfection and Immunoprecipitation assays
Vero and HEK293 cells at 50–70% confluence on 35 mm plates were transfected with Lipofectamine and 4 μg of each plasmid DNA. Cells were washed twice with PBS and then lysed with 1 ml HSE buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) supplemented with protease inhibitors (AEBSF, benzamidine, leupeptin/pepstatin). Tissue was homogenized in the same buffer. Following centrifugation at 13.2K rpm at 4° C, soluble cell lysates were incubated with 15 μL Protein-G beads and 2 μg antibody overnight at 4°C. Beads were pelleted and washed three times with HSE buffer and boiled in 25 μl 2X SDS loading buffer. Samples were separated on either 7.5% or 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were blocked in 5% milk for one hour, followed by incubation in primary antibody overnight at 4° C. Following washes, secondary antibodies (horseradish peroxidase-conjugated rabbit or mouse IgG, Santa Cruz) were incubated with the membranes (1:5000) for one hour. Signals were visualized with an enhanced chemiluminescence reagent (Pierce) and exposed to either X-ray film or an ImageQuant LAS 4000 (General Electric) imager.
2.4 Subcellular fractionation
Nuclear and cytosolic fractions were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit from Thermo-fisher. Purity of the fractions was determined by western blot for lamin A/C and GAPDH.
2.5 Glutathione S-transferase (GST) pull-down assays
Glutathione-sepharose beads charged with GST fused to PKA RIIα-subunit or GST alone were incubated with whole heart extracts (1 mg total protein in 400 μl HSE buffer) overnight at 4°C with shaking. Bound proteins were detected by immunoblotting following washing with buffer. S-tag pull-down assays were similarly performed using S-tag agarose beads and bacterially expressed proteins.
2.6 PKA activity assay
Immunoprecipitated proteins were incubated in kinase buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl2) containing 30 μM Kemptide, 100 μM [γ-32P]ATP and 1 mM cAMP for 15 min at 30°C. Reactions were terminated by spotting onto phosphocellulose strips. Filters were washed five times in 75 mM phosphoric acid and once in 95% ethanol, air dried and counted by liquid scintillation.
2.7 Statistics
Matched One- or Two-way ANOVA was performed as appropriate, followed by Tukey post-hoc testing using Graphpad Prism 7. All data are presented as mean +/− s.e.m. Repeated symbols indicate the following: * p < 0.05; ** p < 0.01; *** p < 0.001.
3. Results
3.1 mAKAPβ is a HDAC5 scaffold
We previously reported that mAKAPβ binds HDAC4 in the heart [20]. We now show that endogenous HDAC5 could also be co-immunoprecipitated with mAKAPβ antibodies using rat whole heart extracts (Fig. 1A). Conversely, like Flag-tagged HDAC4 (Sup. Fig. 1), Flag-tagged HDAC5 mediated the immunoprecipitation of GFP-tagged mAKAP by Flag-tag antibodies when the two proteins were co-expressed in heterologous cells (Fig. 1B). mAKAP binds PKD both in heterologous cells and cardiac myocytes (Sup. Fig. 2 A,B) [20, 21]. Accordingly, like for HDAC4 [20], we found that mAKAPβ was required for the association of PKD with HDAC5 as detected by co-immunoprecipitation of recombinant proteins expressed in heterologous cells (Sup. Fig. 2C) and in primary neonatal rat ventricular myocytes (Fig. 1C). Together, these data show that like HDAC4, HDAC5 forms a ternary complex with mAKAPβ and its activator PKD.
Figure 1. mAKAPβ organizes a PKD-HDAC5 Signalosome required for HDAC5 nuclear export.

A. Extracts prepared from adult rat hearts were used in co-immunoprecipitation assays with mAKAP antibodies and control IgG. Endogenous HDAC5 was detected by western blot. B. Extracts prepared from Vero cells co-transfected with expression plasmids for Flag-tagged HDAC5 and GFP-tagged mAKAP were used in co-immunoprecipitation assays with Flag tag antibodies. Expression of the recombinant proteins was detected using tag-specific antibodies. C. Cardiac myocytes transfected with either mAKAP-specific or control siRNA were infected with adenovirus expressing HA-tagged PKD and GFP-tagged HDAC5. Protein complexes immunoprecipitated with HDAC5 antibodies were detected with mAKAP and tag-specific antibodies. D. Cardiac myocytes transfected and infected as in C were stimulated with phenylephrine (PE, 50 μM) for 60 min. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661. E. Cytosolic and nuclear extracts were prepared from myocytes treated as in D and analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker) antibodies. *p-value vs. no drug controls; † vs. control siRNA under same treatment condition. n = 3 for each panel.
The functional relevance of mAKAPβ scaffolding for HDAC5 PKD phosphorylation and nuclear export was shown using mAKAP small interfering RNA (siRNA) oligonucleotides to deplete cardiac myocytes of endogenous mAKAPβ protein. HDAC5 phosphorylation induced by acute stimulation with the α-adrenergic agonist phenylephrine (PE) was detected using a phospho-specific antibody for HDAC5 residues 498 and 661 that has previously been shown to detect PKD-specific HDAC4/5 phosphorylation [27]. PE-induced HDAC5 Ser498/661 phosphorylation was inhibited 80% in cardiac myocytes transfected with mAKAP siRNA (Fig. 1D). PKD-catalyzed HDAC5 phosphorylation is associated with robust nuclear export that may be demonstrated by cellular fractionation (Fig. 1E) [4]. Decreased PE-induced phosphorylation in the absence of mAKAPβ expression correlated with inhibited PE-induced HDAC5 nuclear export. Together these data show that the mAKAPβ scaffold is required in cardiac myocytes both for the association of PKD with HDAC5 and for α-adrenergic receptor-induced HDAC5 phosphorylation at PKD sites and nuclear export.
3.2 Association of HDAC5 with mAKAPβ is required for α-adrenergic regulation
In order to map the HDAC5-binding domain on mAKAP, fragments of the mAKAP scaffold were co-expressed with HDAC5 in cells and assayed by co-immunoprecipitation (Fig. 2A–C). A fragment encompassing mAKAP residues 301–500 was sufficient to mediate HDAC5 binding. Consistent with the function of mAKAPβ to mediate the association of PKD with HDAC5, expression of a mCherry mAKAP 301–500 fusion protein competed the co-immunoprecipitation of both full-length mAKAP and PKD with HDAC5 (Fig. 2D). Class IIa HDACs bind directly the conserved N-terminal MADS domain of MEF2 transcription factors [28]. We have previously published that mAKAP 301–500 also binds the MEF2D MADS domain [29], raising the question of whether HDAC5 binding to mAKAP competes with or, alternatively, is enhanced by MEF2D. In order to address this question, we purified S-tagged mAKAP 301–500 and His-tagged HDAC5 and MEF2D from bacteria and performed pull-down assays (Fig. 2E). HDAC5 was brought down with S-tagged mAKAP 301–500 only in the presence of MEF2D, implying that the transcription factor is required for (or greatly enhances) the association of the class IIa HDAC with the scaffold.
Figure 2. HDAC5 is associated with a discrete mAKAP domain.

A, B. Extracts prepared from HEK293 cells co-transfected with expression plasmids for Flag-tagged HDAC5 and GFP- or mCherry tagged mAKAP fragments were used in co-immunoprecipitation assays with Flag tag antibodies. Expression of the recombinant proteins was detected using tag-specific antibodies. FP - fluorescent protein, i.e. GFP or mCherry. C. mAKAP domain structure. mAKAPβ is identical to mAKAPα 245–2314 [54]. SR - spectrin repeat domain. Binding sites are indicated for those mAKAP-binding partners whose sites have been finely mapped: PDK1, protein kinase D 1 [54]; AC5, adenylyl cyclase 5 [40]; PLCε, phospholipase C ε [22]; nesprin-1α [23]; RyR2, ryanodine receptor [55]; CaN, calcineurin [56]; RSK3, p90 ribosomal S6 kinase 3 [57]; PKA [18]; PP2A, protein phosphatase 2A [58]. D. Cardiac myocytes were infected with adenovirus expressing HA-tagged PKD, GFP-tagged HDAC5, mCherry or mCherry-tagged mAKAP 301–500. Protein complexes immunoprecipitated with HDAC5 antibodies. E. Bacterially-expressed His-tagged HDAC5 and MEF2D and S-tagged mAKAP 301–500 were used in pull-down assays with S-tag resin. S-tagged mAKAP 301–500 was detected by total protein stain of the pull-downs (Ponceau stain). n = 3 for each panel.
The identification of an anchoring disruptor peptide for HDAC5/MEF2/mAKAPβ signaling complexes allowed us to test whether HDAC5 association with the scaffold was required for its activation by α-adrenergic stimulation. Using the phospho-HDAC5 Ser-498/661 antibody, we determined that PE-induced PKD phosphorylation was blocked by expression of mCherry-mAKAP 301–500 in cardiac myocytes (Fig. 3A). Accordingly, PE-induced HDAC5 nuclear export was blocked by expression of the anchoring disruptor peptide (Fig. 3B). As HDAC5 nuclear export is associated with myocyte hypertrophy, we then assayed PE-induced myocyte hypertrophy in the presence and absence of the anchoring disruptor peptide. Morphologic hypertrophy (increased cross-section area in the presence of PE) was consistently attenuated (58% less hypertrophy) by mCherry mAKAP 301–500 (Fig. 3C). In addition, a marker for hypertrophy atrial natriuretic factor (ANF) was expressed less in the presence of the anchoring disruptor peptide. Taken together, these data support a model in which HDAC5 regulation by mAKAPβ signalosomes is important in cardiac myocytes for HDAC5 nuclear export and cellular hypertrophy in response to α-adrenergic stimulation.
Figure 3. HDAC5 association with mAKAPβ signalosomes is required for α-adrenergic-induced HDAC5 nuclear export and neonatal myocyte hypertrophy.

A. Cardiac myocytes were infected with adenovirus expressing HDAC5-GFP, PKD-HA and either mCherry-mAKAP 301–500 or mCherry control before stimulation with PE (50 μM) for 60 min. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661. B. Cytosolic and nuclear extracts were prepared from myocytes treated as in A and analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker) antibodies. For A and B, n = 3. C. Cardiac myocytes were transfected with ptTA and pTRE expression plasmids for either mCherry-mAKAP 301–500 or mCherry control before stimulation with PE (10 μM) for 2 days and staining with α-actinin antibody (green) and Hoechst nuclear stain (blue). Parallel cultures were stained for ANF expression (not shown). n = 5 separate biological replicates. *p-value vs. no drug controls; † vs. mCherry + PE. For ANF expression, 2-way matched ANOVA was significant for anchoring disruptor expression (p = 0.03 for samples with mCherry-301-500 vs. samples with mCherry control), albeit pairwise post-hoc testing comparing PE-treated control and 301–500 peptide samples did not reach statistical significance.
3.3 β-Adrenergic signaling opposes α-adrenergic-induced HDAC5 phosphorylation and nuclear export
It has been reported that via PKA, acute β-adrenergic stimulation of cardiac myocytes will inhibit the nuclear export of HDAC5 in response to Gq-coupled receptor activation [12, 14]. Accordingly, we found that co-stimulation with the β-adrenergic agonist isoproterenol (Iso) inhibited PE-induced HDAC5 Ser-498/661 phosphorylation in cardiac myocytes (Fig. 4A). Iso did not induce Ser-498/661 phosphorylation when introduced alone, and the β-adrenergic inhibition of PE-induced phosphorylation was dependent upon PKA activity, as shown using the PKA inhibitor H-89. Consistent with its effects on HDAC5 phosphorylation, Iso alone did not affect HDAC5-GFP intracellular localization, but did inhibit PE-induced HDAC5-GFP cytosolic translocation via a PKA-dependent mechanism (Fig. 4B). Together, these results confirm that acute α-adrenergic and β-adrenergic signaling oppositely regulate HDAC5 localization in cardiac myocytes.
Figure 4. β-adrenergic signaling opposes α-adrenergic-induced HDAC5 nuclear export.

A. Cardiac myocytes infected with HDAC5-GFP adenovirus were treated with Iso (10 μM) in the absence or presence of the PKA inhibitor H-89 (10 μM) for 15 min before stimulation with PE (50 μM) for 60 min. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661 or a HDAC5 protein antibody. B. Cytosolic and nuclear extracts were prepared from myocytes treated as in A and analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker). n = 3 for both panels. *p-value vs. no drug controls; † vs. PE; ‡ vs. PE + Iso.
3.4 HDAC5 is associated with mAKAPβ-PKA complexes in myocytes, facilitating PKA phosphorylation of HDAC5
The regulation of HDAC5 by PKA suggested that HDAC5 might be regulated by a PKA-AKAP complex. We first determined whether endogenous HDAC5 was physically associated with PKA in cardiac myocytes (Fig. 5A). HDAC5-specific immunoprecipitates contained appreciable kinase activity for the PKA substrate Kemptide that was inhibited by the PKA-specific inhibitor protein kinase I (PKI). In addition, HDAC5-GFP was detected in pull-down assays using a GST – PKA RII subunit fusion protein (GST-PKA II) and whole heart extracts (Fig. 5B). Given that mAKAP binds both PKA [18] and HDAC5, we tested whether mAKAP might recruit PKA to HDAC5. When expressed in heterologous cells, Flag-HDAC5 was associated with a small amount of PKA holoenzyme, as shown by immunoprecipitation-kinase assay (Fig. 5C). Co-expression of mAKAP-GFP increased the level of PKA activity in HDAC5 immunoprecipitates 3.7-fold, implying the presence of HDAC5-mAKAPβ-PKA protein complexes. That mAKAPβ is the primary HDAC5-associated AKAP in myocytes was shown using cardiac myocytes transfected with mAKAP siRNA (Fig. 5D). HDAC5 antibody immunoprecipitation-kinase assay detected PKA using myocytes transfected with control siRNA, but not using myocytes transfected with mAKAP siRNA that depleted the cells of mAKAPβ. In addition, HDAC5 phosphorylation was detected using a PKA substrate-specific antibody that is useful for assay of Ser-279 phosphorylation [13]. HDAC5 was PKA phosphorylated in Iso-treated myocytes only when expressing mAKAPβ (Fig. 5E). Together, these results show that mAKAPβ is the primary scaffold responsible for HDAC5-PKA association in the cardiac myocyte and that mAKAPβ is required for β-adrenergic-induced HDAC5 PKA phosphorylation in these cells.
Figure 5. HDAC5 is associated with mAKAPβ-PKA signalosomes in myocytes.

A. Extracts prepared from cardiac myocytes were used for immunoprecipitation-kinase assays. Protein complexes were immunoprecipitated with HDAC5 antibodies and control IgG. Kinase assays were performed with 1 mM cAMP, 30 μM Kemptide substrate +/− the PKA inhibitor protein kinase inhibitor (PKI). PKA catalytic subunit (PKA-C) and HDAC5 were detected in the extracts with their respective antibodies. *p-value vs. IgG; † vs. no PKI IP. B. Whole adult rat heart extracts were used in pull-down assays with bacterially-expressed GST-PKA RII-subunit fusion protein and GST control. Endogenous HDAC5 was detected with a HDAC5 antibody before and after pulldown. GST proteins in the assays were detected using Ponceau total protein stain. Arrowheads indicates the migration of GST-PKA RII and GST. C. Vero cells were co-transfected with expression plasmids for Flag-tagged HDAC5 and GFP-tagged mAKAP. Protein complexes were immunoprecipitated with HDAC5 antibodies and control IgG and kinase assays were performed as above. mAKAP-GFP, PKA catalytic subunit (PKA-C) and HDAC5 were detected in the extracts with the appropriate antibodies. *p-value vs. IgG IP; † vs. α-HDAC5 IP for HDAC5 expressing only cells; ‡ vs. α-HDAC5 IP for HDAC5 and mAKAP expressing cells. D. Cardiac myocytes were transfected with mAKAP or control siRNA oligonucleotides and infected with HDAC5-GFP adenovirus. Cellular extracts were prepared for immunoprecipitation-kinase assays as in A. *p-value vs. IgG; † vs. control siRNA. Total extract western blots for mAKAP, HDAC5, and PKA-C showed that only mAKAPβ expression was ablated by the mAKAP siRNA. E. Cardiac myocytes as in D were stimulated for 15 min with 10 μM Iso. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a PKA phospho-substrate-specific antibody that detects phosphorylated Ser-279 [13]. n = 3. * vs. control siRNA without Iso; † vs. control siRNA with Iso.
3.5 Anchored PKA is required for β-adrenergic inhibition of HDAC5 nuclear export in cardiac myocytes
The above results revealed that mAKAPβ is responsible for both PKD and PKA-dependent HDAC5 phosphorylation. In order to show the functional relevance of PKA-mAKAPβ-HDAC5 complexes without disrupting PKD scaffolding, we utilized the cell-permeant AKAP-IS type II PKA anchoring disruptor peptide to deplete mAKAPβ signalosomes of PKA [30]. AKAP-IS treatment of cardiac myocytes competed PKA binding to mAKAPβ and HDAC5 signaling complexes as shown by immunoprecipitation-kinase assay (Fig. 6A) Notably, Iso-induced HDAC5 phosphorylation detected with a PKA substrate-specific antibody was significantly attenuated in cells treated with the AKAP-IS anchoring disruptor peptide (Fig. 6B). In order to test our hypothesis that mAKAPβ facilitates the Iso and PKA-dependent inhibition of PKD HDAC5 phosphorylation, additional myocytes were treated with Iso and/or PE and the AKAP-IS peptide. As before, Iso inhibited HDAC5 Ser-498/661 phosphorylation in response to PE stimulation (Fig. 6C). Pre-treatment with AKAP-IS prevented the β-adrenergic-dependent inhibition. Likewise, AKAP-IS prevented the β-adrenergic-dependent inhibition of HDAC5 cytosolic translocation in response to α-adrenergic stimulation, as shown by subcellular fractionation (Fig. 6D).
Figure 6. Anchored PKA is required for β-adrenergic-induced HDAC5 phosphorylation and inhibition of α-adrenergic-stimulated nuclear export.

A. Cardiac myocytes were infected with HDAC5-GFP adenovirus and treated with 10 μM AKAP-IS anchoring disruptor peptide or control peptide (Ctrl Pep). PKA activity assays were performed following immunoprecipitation with mAKAP or HDAC5 antibodies or control IgG. * vs. IgG; † vs. control peptide. B. Myocytes were treated as in A before 10 min stimulation with 10 μM Iso. HDAC5-GFP immunoprecipitated with a HDAC5 antibody was detected using a PKA phospho-substrate-specific antibody that detects phosphorylated Ser-279 [13]. * vs. control peptide without Iso; † vs. control peptide with Iso. C. Myocytes infected with HDAC5-GFP adenovirus and treated with 10 μM AKAP-IS or control peptide were stimulated with 10 μM Iso for 10 min followed by 50 μM PE for 1 hr. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661 or a HDAC5 protein antibody. D. Cytosolic and nuclear extracts were prepared from myocytes treated as in C and analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker) antibodies. n = 3 for each panel. For C and D: Data are normalized to that for control peptide without Iso or PE. * vs. without Iso or PE; † vs. control peptide with PE; ‡ vs. control peptide with PE and Iso.
The results obtained using the PKA anchoring disruptor peptide were corroborated using a RNAi-rescue strategy and expression of a full-length mAKAP PKA-binding site mutant (ΔPKA) lacking the PKA-binding amphipathic helix at mAKAP residues 2053–2073 (Fig. 2C) [18]. In order to avoid potential dominant negative effects, expression of wildtype and ΔPKA mutant mAKAP protein was performed such that the recombinant myc-tagged proteins were expressed following co-transfection with mAKAP siRNA oligonucleotides at levels similar to endogenous mAKAPβ in control cardiac myocytes (Fig. 7A). Immunoprecipitation-kinase assay for PKA activity confirmed that RNAi-rescue with wildtype myc-mAKAP, but not ΔPKA mutant resulted in HDAC5 association with PKA (Fig. 7B). Likewise, RNAi-rescue with wildtype myc-mAKAP, but not ΔPKA mutant permitted Iso induction of HDAC5 phosphorylation by PKA in myocytes (Fig. 7C). Notably, RNAi-rescue with wildtype myc-mAKAP, but not ΔPKA mAKAP mutant, conferred Iso-dependent inhibition of PE-induced HDAC5 Ser-498/661 phosphorylation and nuclear export (Fig. 7D,E). Together with the results obtained with the AKAP-IS peptide, these data show that mAKAPβ-anchored PKA is required for β-adrenergic-induced HDAC5 phosphorylation by PKA in myocytes, as well as β-adrenergic-dependent inhibition of α-adrenergic HDAC5 nuclear export.
Figure 7. mAKAPβ-bound PKA is required for β-adrenergic-induced HDAC5 phosphorylation and inhibition of α-adrenergic-stimulated nuclear export.

Cardiac myocytes transfected with either mAKAP-specific or control siRNA were infected with adenovirus expressing myc-tagged wildtype mAKAP or a full-length PKA binding site mutant (ΔPKA) lacking residues 2053–2073 [24]. A. Western blot showing endogenous and myc-tagged mAKAP protein expression confirming RNAi-rescue. B. PKA activity assays were performed following immunoprecipitation with HDAC5 antibodies or control IgG. * vs. IgG; † vs. myc-mAKAP. C. Myocytes were treated for 10 min with 10 μM Iso. HDAC5-GFP immunoprecipitated with a HDAC5 antibody was detected using a PKA phospho-substrate-specific antibody that detects phosphorylated Ser-279 [13]. * vs. no drug; † vs. myc-mAKAP. D. Myocytes were stimulated with 10 μM Iso for 10 min followed by 50 μM PE for 1 hr. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661. E. Cytosolic and nuclear extracts were prepared from myocytes treated as in D and analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker) antibodies. For D and E: Data are normalized to that for control peptide without Iso or PE. * vs. no drug; † vs. PE; ‡ vs. myc-mAKAP with PE and Iso. n = 3 for each panel.
3.6 Acute and chronic β-adrenergic stimulation differentially affects HDAC5 in the myocyte
It has been noted that chronic pre-treatment with forskolin to induce prolonged cAMP elevation does not inhibit Gq-mediated HDAC5 nuclear export [12]. We have found that, similarly, β-adrenergic stimulation with Iso only inhibited HDAC5 PKD-site phosphorylation when given for 1 hour, but not 24 hours priors to PE α-adrenergic stimulation (Fig. 8A). In addition, 24 hours of Iso pre-treatment was unable to inhibit PE-induced HDAC5 nuclear export (Fig. 8B). Remarkably, 24 hours, but not 1 hour of Iso treatment promoted both HDAC5 phosphorylation at the PKD sites and HDAC5 nuclear export in the absence of α-adrenergic stimulation (Fig. 8C,D). These data are consistent with a model in which chronic β-adrenergic stimulation remodels the cardiac myocyte signaling network to promote, rather than inhibit HDAC5 nuclear export, as discussed further below.
Figure 8. In contrast to acute stimulation, chronic β-adrenergic stimulation promotes HDAC5 nuclear export.

Cardiac myocytes were infected with adenovirus expressing GFP-tagged HDAC5 and stimulated with 10 μM Iso and/or 50 μM PE for 1 or 24 hrs. A and C. HDAC5-GFP was immunoprecipitated with a HDAC5 antibody and detected using a phospho-specific antibody for Ser-498/661 or a HDAC5 protein antibody. B and D. Cytosolic and nuclear extracts were analyzed by western blotting with HDAC5, lamin (nuclear marker), or GAPDH (cytosolic marker) antibodies. n = 3 for each panel. A and B: * vs. no drug; † vs. PE at 1 hr; ‡ vs. PE and Iso at 1 hr. C and D: * vs. no drug; † vs. Iso at 1 hr.
4. Discussion
Scaffold proteins confer efficiency and specificity in intracellular signal transduction by mediating the association of specific substrates with their upstream regulatory enzymes [31]. Class IIa HDACs are regulated by multiple post-translational modifications that control their ability to repress gene expression related to cardiac remodeling [4]. The central finding of this study is that mAKAPβ is a scaffold in the cardiac myocyte that confers HDAC5 association with and regulation in an oppositional manner by PKD and PKA. Using siRNA, anchoring disruptor peptides, and RNAi-rescue with a binding site mutant, we show that mAKAPβ scaffolding is functionally relevant in the cardiac myocyte to PKD-dependent HDAC5 nuclear export and the inhibition of this process by acute β-adrenergic signaling [12, 13]. Located mainly, if not exclusively, at the nuclear envelope in both stimulated and unstimulated adult and neonatal ventricular myocytes by binding to the outer nuclear membrane protein nesprin-1α [20, 23], mAKAPβ signalosomes are poised for the regulation of nuclear gene expression by upstream signaling. Coordination of both PKD and PKA class IIa HDAC phosphorylation is consistent with a role for mAKAPβ signalosomes in the fine-tuning of gene expression responsible for matching ventricular mass to systemic demands for cardiac output [20].
Pathological myocyte hypertrophy is promoted by Gq-coupled receptor signaling that can activate PKD through protein kinase C and diacylglycerol-dependent mechanisms [21, 32]. The Smrcka lab has recently described a novel mechanism for local activation of PKD at mAKAPβ: Gβγ activation of mAKAPβ-bound phospholipase Cε that catalyzes diacylglycerol production from phosphatidylinositol 4-phosphate on adjacent Golgi apparatus [21, 33]. We now reveal that PKD phosphorylation and nuclear export of HDAC5 is mAKAPβ-dependent in PE-stimulated neonatal rat ventricular myocytes (a useful model for pathological myocyte hypertrophy despite the cardioprotective role of α-adrenergic signaling in vivo [34]). HDACs are regulated in a dynamic fashion as repressor complexes reversibly associate with chromatin, the fraction of HDACs present within the nucleus depending upon the current signaling state of the cell [12, 15]. In this regard, HDAC molecules constantly approach the nuclear envelope as they cross back and forth into the nucleus. We suggest that class IIa HDACs shuttling in and out of the nucleus dock transiently at mAKAPβ where they are subject to hypertrophic agonist-induced PKD phosphorylation, affecting the overall equilibrium of HDAC nuclear export and import. mAKAPβ signalosomes are not, however, solely responsible for class IIa HDAC regulation, as HDAC4/5 nuclear export is also induced by AKAP-lbc-PKD signalosomes, nuclear Ca2+/calmodulin-dependent protein kinases, and oxidation of HDAC5 cysteine residues [7, 35, 36]. Taken together, ours and others’ findings suggest that scaffold proteins and signaling enzymes within multiple cellular compartments regulate HDAC nuclear/cytoplasmic localization, such that overall HDAC5 function is determined by the net activity of multiple signaling pathways acting in concert within the myocyte. Nevertheless, mAKAPβ signalosomes apparently serve as a critical node in this regulatory network. Accordingly, inhibition of mAKAPβ signalosome formation using a MEF2D/HDAC5 anchoring disruptor peptide inhibited PE-induced hypertrophy.
We have previously published that HDAC4 forms ternary complexes with mAKAPβ and active PKD [20]. In Kritzer et al., we showed that active PKD was increased in the hearts of mice subjected to both short term and long term pressure overload and in which cardiac hypertrophy was evident [20]. Conditional AKAP6 (mAKAP) cardiac myocyte-specific gene deletion, that attenuated pathological cardiac remodeling and conferred a survival benefit in the face of long term pressure overload, prevented PKD activation and HDAC4 phosphorylation on PKD sites as assayed with phospho-Ser-246 and phospho-Ser-467/632 antibodies [20]. In contrast to HDAC4, the increase in HDAC5 phosphorylation on PKD sites 2 and 16 weeks after transverse aortic constriction survival surgery did not reach statistical significance in that study, precluding a determination of mAKAPβ dependence. (The only exception was the statistically significant, but small 1.4-fold increase in HDAC5 Ser-259 phosphorylation 16 weeks after transverse aortic constriction that was not mAKAPβ-dependent.) Differential regulation of HDAC4 and HDAC5 phosphorylation and nuclear export in cardiac myocytes has been previously reported [36, 37], as well as in other cell types such as the skeletal myocyte [38]. Future studies will be required to identify the pathophysiologic states and/or timing relevant to mAKAPβ-PKD activation of HDAC5 in vivo. In a report by Taglieri, et al., showing the regulation of PKD by the AKAP-lbc scaffold protein, HDAC5 was found to more highly PKD phosphorylated during pressure overload [39], potentially due to a difference in mouse strain or extent of disease from Kritzer, et al.
PKD and PKA phosphorylate HDAC5 with opposite functional consequences. Our data support a model in which by bringing together HDACs and PKA mAKAPβ also facilitates the negative regulation of class IIa HDAC nuclear export by acute β-adrenergic stimulation (Figs. 5–7). mAKAPβ organizes signalosomes that include all of the machinery necessary for the local regulation of cAMP signaling, including type 5 (but not type 6) adenylyl cyclase, PKA, and type 4D3 phosphodiesterase [26, 40]. We found that depletion of mAKAPβ, as well as replacement of wildtype mAKAPβ with a mAKAPβ PKA-binding site mutant, inhibited both the association of PKA with HDAC5 and PKA-dependent HDAC5 phosphorylation in the cardiac myocyte. These results suggest that mAKAPβ is the primary AKAP in the myocyte responsible for HDAC5 phosphorylation by PKA and were surprising given the known regulation of HDAC5 by AKAP-lbc [7, 39, 41]. However, while AKAP-lbc has been shown to bind PKD and regulate HDAC5 phosphorylation by that kinase [7, 39, 41], AKAP-lbc and HDAC5 have not yet been co-immunoprecipitated, raising the possibility that they do not stably bind (precluding AKAP-lbc-mediated PKA-HDAC5 co-immunoprecipitation) and/or that HDAC5 is only phosphorylated by PKD after release from the scaffold away from any associated PKA. Accordingly, AKAP-lbc-bound PKA has not yet been shown to phosphorylate HDAC5. It is perhaps informative that mAKAP siRNA inhibited phosphorylation of HDAC5 by PKA more completely than by PKD (Cf. Figs. 1D and 5E).
The sole PKA site on HDAC5 is Ser-279 (designated Ser-280 in Ha, et al [13]) within the conserved sequence KVAERRSSPLLRRK, such that mutation of that site prevents detection of the protein using the “PKA substrate specific” antibody used herein to detect HDAC5 phosphorylation after isoform-specific immunoprecipitation [13, 20]. As this sequence is identical in HDAC4 and as HDAC4 binds mAKAPβ [20], mAKAPβ presumably facilitates the PKA-dependent phosphorylation of that deacetylase as well. Interestingly, the PKA site is present also in HDAC9, but not HDAC7, suggesting a broader role for PKA and potentially mAKAPβ in the regulation of class IIa HDACs. Experiments using both HDAC5 Ser-279 phosphomimetic and phosphoablative mutant proteins in both adult and neonatal cardiac myocytes have shown the relevance of this residue to HDAC5 nuclear-cytoplasmic translocation [12, 13, 15]. In addition, the phosphorylation of Ser-279 correlates with the robust inhibition by Iso of PE-induced HDAC5 S498/661 phosphorylation in myocytes and HDAC5 nuclear retention (Fig. 4) [12, 36]. Weeks, et al, recently reported that HDAC5 phosphorylation detected using a new antibody specific for phosphorylated Ser-279 was inhibited, not increased by stimulation with Iso alone, despite Iso promoting PKA-dependent nuclear retention [15]. In their experiments, decreased Ser-279 phosphorylation was accompanied by decreased phosphorylation of the PKD sites, which they attribute to the induction of phosphatase activity in newly identified PP2A-B55α-HDAC5 complexes [15]. In our experiments, we did not detect a change in the phosphorylation of HDAC5 PKD sites or increased HDAC5 nuclear localization in the presence of Iso alone, potentially reflecting a greater basal HDAC5 PKD-site phosphorylation in adult myocytes [12, 14, 15]. Taken together, one may reconcile ours and others’ results by concluding that Ser-279 phosphorylation is only relevant to blocking HDAC5 nuclear export when there is a simultaneous, strong PKD-activating stimulus. One should note that it is not entirely clear whether the primary biochemical mechanism by which Ser-279 phosphorylation promotes nuclear import is the inhibition of PKD site phosphorylation. In contrast to our results, Ha et al. found that Iso-induced Ser-279 phosphorylation inhibited HDAC5 binding to 14-3-3, nuclear export, and MEF2-dependent gene expression without inhibiting HDAC5 PKD-site phosphorylation [13].
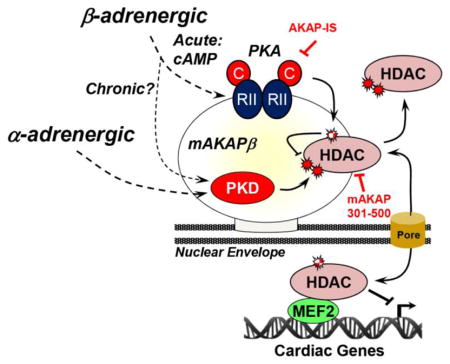
By binding PKA, PKD, and HDACs, mAKAPβ at the nuclear envelope can coordinate the bidirectional regulation of class IIa HDAC cytoplasmic-nuclear localization, providing a model for the differential regulation of HDACs in health and disease (Graphical Abstract). Sympathetic β1-adrenergic signaling via norepinephrine is responsible for acutely increasing cardiac contractility in exercise and in flight-or-fight responses. Under these conditions, adverse remodeling is not activated and MEF2-dependent transcription should be repressed by class IIa HDACs [12]. mAKAPβ-dependent PKA phosphorylation of HDACs presumably contributes to this repression by opposing HDAC export that might be induced by increased contractile Ca2+ transients. In contrast, in cardiovascular disease, Gq-coupled receptor signaling is elevated, and cardiac remodeling (myocyte hypertrophy, increased cell death and interstitial fibrosis, and altered metabolism and gene expression) ensues [12, 42]. Notably, in heart failure total myocardial cAMP levels and PKA-dependent phospholamban phosphorylation are decreased [43, 44], consistent with the idea that β-adrenergic regulation of cardiac contractility is progressively down-regulated in heart disease. Despite this down-regulation, however, we and others have published that chronic Iso stimulation will induce myocyte hypertrophy in vitro [24], and the clinical efficacy of β-blockers attests to the role of β-adrenergic signaling in pathological remodeling [45]. Many of the pro-remodeling effects of chronic β-adrenergic stimulation are attributable to PKA-independent, Ca2+/calmodulin-dependent protein kinase signaling that is also upstream of class IIa HDACs [46, 47]. Nevertheless, PKA overexpression will also down-regulate SERCA2a expression in vitro and induce dilated cardiomyopathy in vivo [14, 48]. In addition, we reported that Iso-induced myocyte hypertrophy in vitro was dependent both upon mAKAPβ expression and PKA-anchoring by the scaffold (as shown by RNAi-rescue), correlating with activation of calcineurin-NFAT signaling [24]. We show now that 24 hours of β-adrenergic stimulation was sufficient in vitro to alter the response of the myocytes with regards to HDAC5 regulation, changing β-adrenergic signaling from promoting nuclear import to promoting nuclear export (Fig. 8). How exactly mAKAPβ-associated PKA signaling is “remodeled” to permit HDAC5 nuclear export and to promote hypertrophy during states of chronic β-adrenergic stimulation require further investigation. Overall, we favor a model in which chronic catecholaminergic simulation in disease results in molecular remodeling of the myocyte signaling network, such that bulk cAMP signaling related to contractility is progressively down-regulated, while specific cAMP pools regulating remodeling become activated. This model is consistent with data relating to the activation in remodeling of other cAMP effectors, such as Epac, and the intracellular redistribution of phosphodiesterases and β-adrenergic receptors in heart failure [42, 49, 50]. Based upon both our previously published and our newly presented results, we propose that at perinuclear mAKAPβ signalosomes, PKA is relevant to both opposing remodeling during “normal,” transient catecholamine surges and to promoting remodeling during pathological, chronic catecholamine elevation.
Much of the previous literature regarding mAKAPβ has focused on how this conserved protein is deleterious in disease [19]. Aside from evidence that mAKAPβ conditional gene deletion will reduce the “physiologic” hypertrophy induced in swimming mice [20], our new finding that mAKAPβ-bound PKA will oppose HDAC5 activation during acute β-adrenergic stimulation provides the first mechanism supporting mAKAPβ’s importance in the normal heart, justifying its conservation throughout vertebrate evolution. The bidirectional regulation of class IIa HDACs by mAKAP signalosomes is likely to be relevant to not only cardiac myocytes, but also skeletal myocytes and neurons. Like HDACs 4, 5, and 9, mAKAP is expressed primarily in the heart, skeletal muscle and brain [4, 18]. In skeletal myocytes, β-adrenergic stimulation induces HDAC4 PKA phosphorylation and HDAC4 and HDAC5 nuclear influx, potentially comprising a mechanism to avoid MEF2-dependent remodeling during strenuous exercise [11]. In addition, mAKAPβ is required for MEF2 activation in skeletal myocytes and for myotube formation and muscle regeneration following injury [29, 51]. Thus, it is likely that in skeletal muscle, mAKAPβ also confers bidirectional class IIa HDAC regulation. In neurons, HDAC5 nuclear export is required for axon regeneration following injury [52]. We recently published that mAKAPα, the alternatively-spliced form of mAKAP in neurons, is similarly required for both the survival and neurite outgrowth of retinal ganglion cells in vitro and for retinal ganglion cell survival in vivo following optic nerve crush [53]. Together, these findings suggest that mAKAP signalosomes are central to the response to stress in excitable cells, providing critical infrastructure for the fine-tuned regulation of gene expression in health and disease.
Supplementary Material
Highlights.
mAKAPβ is a scaffold for HDAC5, PKD, and PKA signalosomes in cardiac myocytes.
mAKAPβ facilitates the signal-dependent phosphorylation of HDAC5 by both PKD and PKA.
mAKAPβ signalosomes regulate HDAC5 nuclear export in response to hypertrophic stimuli.
mAKAPβ-PKA anchoring permits β-adrenergic inhibition of HDAC5 nuclear export.
Acknowledgments
Funding
This work was supported, in whole or in part, by the State of Connecticut Department of Public Health Grant 2014-0133 (K.D.K.) and National Institutes of Health Grants HL126825 (K.D.K. and M.S.K.) and HL126950 (M.S.K).
Abbreviations
- HDAC
histone deacetylase
- Iso
isoproterenol
- mAKAP
muscle A-kinase anchoring protein
- MEF2
myocyte enhancer factor-2
- PE
phenylephrine
- PKA
cAMP-dependent protein kinase A
- PKD
protein kinase D
Footnotes
Disclosures
Drs. Dodge-Kafka, Li, and Kapiloff are co-inventors of patented intellectual property concerning the targeting of mAKAP for the treatment of heart failure. Dr. Kapiloff owns equity in Anchored RSK3 Inhibitors, LLC, and Cardiac RSK3 Inhibitors, LLC, companies interested in developing mAKAP targeted therapies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xie M, Hill JA. HDAC-dependent ventricular remodeling. Trends Cardiovasc Med. 2013;23(6):229–35. doi: 10.1016/j.tcm.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–88. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24(19):8467–76. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weeks KL, Avkiran M. Roles and post-translational regulation of cardiac class IIa histone deacetylase isoforms. J Physiol. 2015;593(8):1785–97. doi: 10.1113/jphysiol.2014.282442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408(6808):106–11. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24(19):8374–85. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol Cell. 2008;32(2):169–79. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci U S A. 2000;97(26):14400–5. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci U S A. 2000;97(14):7835–40. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, Bazett-Jones DP, Yang XJ. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol Cell Biol. 2000;20(18):6904–12. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Schneider MF. Opposing HDAC4 nuclear fluxes due to phosphorylation by beta-adrenergic activated protein kinase A or by activity or Epac activated CaMKII in skeletal muscle fibres. J Physiol. 2013;591(14):3605–23. doi: 10.1113/jphysiol.2013.256263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang CW, Lee L, Yu D, Dao K, Bossuyt J, Bers DM. Acute beta-adrenergic activation triggers nuclear import of histone deacetylase 5 and delays G(q)-induced transcriptional activation. J Biol Chem. 2013;288(1):192–204. doi: 10.1074/jbc.M112.382358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2010;107(35):15467–72. doi: 10.1073/pnas.1000462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sucharov CC, Dockstader K, Nunley K, McKinsey TA, Bristow M. beta-Adrenergic receptor stimulation and activation of protein kinase A protect against alpha1-adrenergic-mediated phosphorylation of protein kinase D and histone deacetylase 5. J Card Fail. 2011;17(7):592–600. doi: 10.1016/j.cardfail.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weeks KL, Ranieri A, Karas A, Bernardo BC, Ashcroft AS, Molenaar C, McMullen JR, Avkiran M. beta-Adrenergic Stimulation Induces Histone Deacetylase 5 (HDAC5) Nuclear Accumulation in Cardiomyocytes by B55alpha-PP2A-Mediated Dephosphorylation. J Am Heart Assoc. 2017;6(4) doi: 10.1161/JAHA.116.004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol. 2012;52(2):351–8. doi: 10.1016/j.yjmcc.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20(7):1651–62. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapiloff MS, Schillace RV, Westphal AM, Scott JD. mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci. 1999;112(Pt 16):2725–36. doi: 10.1242/jcs.112.16.2725. [DOI] [PubMed] [Google Scholar]
- 19.Passariello CL, Li J, Dodge-Kafka K, Kapiloff MS. mAKAP-a master scaffold for cardiac remodeling. J Cardiovasc Pharmacol. 2015;65(3):218–25. doi: 10.1097/FJC.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kritzer MD, Li J, Passariello CL, Gayanilo M, Thakur H, Dayan J, Dodge-Kafka K, Kapiloff MS. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circulation Heart failure. 2014;7(4):663–72. doi: 10.1161/CIRCHEARTFAILURE.114.001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV. Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153(1):216–27. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286(26):23012–21. doi: 10.1074/jbc.M111.231993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pare GC, Easlick JL, Mislow JM, McNally EM, Kapiloff MS. Nesprin-1alpha contributes to the targeting of mAKAP to the cardiac myocyte nuclear envelope. Exp Cell Res. 2005;303(2):388–99. doi: 10.1016/j.yexcr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118(Pt 23):5637–46. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Vargas MA, Kapiloff MS, Dodge-Kafka KL. Regulation of MEF2 transcriptional activity by calcineurin/mAKAP complexes. Exp Cell Res. 2013;319(4):447–54. doi: 10.1016/j.yexcr.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20(8):1921–30. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinnett-Smith J, Ni Y, Wang J, Ming M, Young SH, Rozengurt E. Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling. Am J Physiol Cell Physiol. 2014;306(10):C961–71. doi: 10.1152/ajpcell.00048.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6(2):233–44. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 29.Vargas MA, Tirnauer JS, Glidden N, Kapiloff MS, Dodge-Kafka KL. Myocyte enhancer factor 2 (MEF2) tethering to muscle selective A-kinase anchoring protein (mAKAP) is necessary for myogenic differentiation. Cell Signal. 2012;24(8):1496–503. doi: 10.1016/j.cellsig.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, Scott JD. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A. 2003;100(8):4445–50. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langeberg LK, Scott JD. Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol. 2015;16(4):232–44. doi: 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avkiran M, Rowland AJ, Cuello F, Haworth RS. Protein kinase d in the cardiovascular system: emerging roles in health and disease. Circ Res. 2008;102(2):157–63. doi: 10.1161/CIRCRESAHA.107.168211. [DOI] [PubMed] [Google Scholar]
- 33.Malik S, deRubio RG, Trembley M, Irannejad R, Wedegaertner PB, Smrcka AV. G protein betagamma subunits regulate cardiomyocyte hypertrophy through a perinuclear Golgi phosphatidylinositol 4-phosphate hydrolysis pathway. Mol Biol Cell. 2015;26(6):1188–98. doi: 10.1091/mbc.E14-10-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen BC, O’Connell TD, Simpson PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011;51(4):518–28. doi: 10.1016/j.yjmcc.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116(3):675–82. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haworth RS, Stathopoulou K, Candasamy AJ, Avkiran M. Neurohormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms. Circ Res. 2012;110(12):1585–95. doi: 10.1161/CIRCRESAHA.111.263665. [DOI] [PubMed] [Google Scholar]
- 37.Monovich L, Vega RB, Meredith E, Miranda K, Rao C, Capparelli M, Lemon DD, Phan D, Koch KA, Chapo JA, Hood DB, McKinsey TA. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010;584(3):631–7. doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Randall WR, Schneider MF. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J Cell Biol. 2005;168(6):887–97. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taglieri DM, Johnson KR, Burmeister BT, Monasky MM, Spindler MJ, DeSantiago J, Banach K, Conklin BR, Carnegie GK. The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J Mol Cell Cardiol. 2014;66:27–40. doi: 10.1016/j.yjmcc.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009;284(35):23540–6. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol Cell. 2004;15(6):889–99. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 42.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327(5973):1653–7. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 43.Bristow MR, Minobe W, Rasmussen R, Larrabee P, Skerl L, Klein JW, Anderson FL, Murray J, Mestroni L, Karwande SV, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89(3):803–15. doi: 10.1172/JCI115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling--targets for heart failure therapy. Nature reviews Cardiology. 2012;9(12):717–33. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bristow MR. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circ Res. 2011;109(10):1176–94. doi: 10.1161/CIRCRESAHA.111.245092. [DOI] [PubMed] [Google Scholar]
- 46.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123(1):37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Grone HJ, Katus HA, Olson EN, Backs J. Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation. 2014;130(15):1262–73. doi: 10.1161/CIRCULATIONAHA.114.006185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89(11):997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 49.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc’h F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res. 2005;97(12):1296–304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 50.Perera RK, Nikolaev VO. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol (Oxf) 2013;207(4):650–62. doi: 10.1111/apha.12077. [DOI] [PubMed] [Google Scholar]
- 51.Lee SW, Won JY, Yang J, Lee J, Kim SY, Lee EJ, Kim HS. AKAP6 inhibition impairs myoblast differentiation and muscle regeneration: Positive loop between AKAP6 and myogenin. Sci Rep. 2015;5:16523. doi: 10.1038/srep16523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155(4):894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Cameron EG, Li J, Stiles TL, Kritzer MD, Lodhavia R, Hertz J, Nguyen T, Kapiloff MS, Goldberg JL. Muscle A-Kinase Anchoring Protein-alpha is an Injury-Specific Signaling Scaffold Required for Neurotrophic- and Cyclic Adenosine Monophosphate-Mediated Survival. EBioMedicine. 2015;2(12):1880–7. doi: 10.1016/j.ebiom.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michel JJ, Townley IK, Dodge-Kafka KL, Zhang F, Kapiloff MS, Scott JD. Spatial restriction of PDK1 activation cascades by anchoring to mAKAPalpha. Mol Cell. 2005;20(5):661–72. doi: 10.1016/j.molcel.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153(4):699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Negro A, Lopez J, Bauman AL, Henson E, Dodge-Kafka K, Kapiloff MS. The mAKAPbeta scaffold regulates cardiac myocyte hypertrophy via recruitment of activated calcineurin. J Mol Cell Cardiol. 2010;48(2):387–94. doi: 10.1016/j.yjmcc.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Kritzer MD, Michel JJ, Le A, Thakur H, Gayanilo M, Passariello CL, Negro A, Danial JB, Oskouei B, Sanders M, Hare JM, Hanauer A, Dodge-Kafka K, Kapiloff MS. Anchored p90 ribosomal S6 kinase 3 is required for cardiac myocyte hypertrophy. Circ Res. 2013;112(1):128–39. doi: 10.1161/CIRCRESAHA.112.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dodge-Kafka KL, Bauman A, Mayer N, Henson E, Heredia L, Ahn J, McAvoy T, Nairn AC, Kapiloff MS. cAMP-stimulated protein phosphatase 2A activity associated with muscle A kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase PDE4D3. J Biol Chem. 2010;285(15):11078–86. doi: 10.1074/jbc.M109.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.