

Abstract
Using the simple ‘allosteron’ model, we show that it is possible, in principle, to elicit pathways by which fluctuation allostery affects self-assembly of protein complexes. We treat the cases of (i) protein fibrils and nucleation, (ii) n-mer protein complexes, and (iii) weakly attractive allosteric interactions in protein-like soft nanoscale objects that can be tuned to define exclusive self-associating families.
This article is part of a discussion meeting issue ‘Allostery and molecular machines’.
Keywords: allostery, self-assembly, ligand-binding
1. General Introduction
Effector-binding and self-assembly are vital to the function of proteins, which form part of all living organisms. Allostery, a non-local signalling and cooperativity among chemically remote sites, commonly has been related to binding-induced conformational changes: regulatory ligands trigger the transitions between ‘relaxed’ and ‘tense’ state of a protein [1,2]. However, as evidenced by a moiety of proteins that show cooperative or anti-cooperative binding without major conformational change, allostery does not imply such transitions. In this work, we investigate an alternative, but less familiar, mechanism of allosteric signalling compared with that of structural change. This mechanism exploits the modification that substrate binding, in general, applies to the amplitudes of thermal fluctuations around the mean structure of an allosteric protein. As such a restriction of random motion constitutes in turn a change in entropy, the full allosteric free energy may contain components that arise purely by this route [3].
A feature of this mechanism is that to contribute to non-local allosteric interaction, the longer-wavelength low-frequency ‘global’ modes of motion are recruited, rather than the higher-frequency and more local motions (such as side-group oscillation). Successful models of protein dynamics that capture the effect are, therefore, coarse-grained rather than atomically resolved. Specific models of particular protein systems at various degrees of coarse-graining have been constructed [4–7], which show that the orders of magnitude of real allosteric free energies can be generated by such restriction of dynamical correlations alone. An equivalent statement of the effect is a restriction of thermally accessible states on binding [8]. An explanation of allostery based on thermal fluctuations thus needs to account for the contributions of collective, non-local, vibrational modes to the binding free energies in a suitably chosen model. To compute changes of conformational entropy that arise from binding one or several ligands is a well-known, yet demanding, theoretical task in protein thermodynamics [9,10]. Similar challenges are presented to experiments, which need to identify the large-scale dynamical changes at many points on a protein on substrate-binding, and to connect them to thermodynamics. Nuclear magnetic resonance (NMR) [11], X-ray analysis of B-factors and isothermal calorimetry [12] have been deployed in comparison with modelling.
At the simplest level of theoretical approach to this phenomenon of ‘fluctuation allostery’, the use of the toy model of a protein at the coarsest possible level, with just one (harmonic) degree of freedom, has been extremely instructive. This simple unit, which can be represented without loss of generality as the scissor structure of figure 1a (although other spatial and geometric representations are equally applicable, as any normal mode within the harmonic approximation is mathematically equivalent), possesses the minimal requirements of: (i) an internal structure that supports thermal structural fluctuations, (ii) one or more potential effector-binding sites with internal rules that modify the internal fluctuations, and (iii) a route to bind to other units (figure 1b).
Figure 1.

Schematic of (a) the allosteron and (b) its binding. (Online version in colour.)
It is straightforward to calculate the free energy F(κ) of a single harmonic degree of freedom over its fluctuation spectrum, constrained by a spring constant κ. As the relevance of this treatment of the normal modes of crude protein models is restricted to low-frequency, global modes, it is appropriate to use the classical (continuous-energy) approximation to the harmonic-oscillator partition function 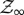 :
:
 |
1.1 |
with Boltzmann's constant kB, temperature T and a length scale λ for normalization.
Modelling the fluctuation restriction of effector-binding as a modulation of the effective spring constant for the internal mode by the dimensionless increment δ, allows a simple and direct calculation of the allosteric free energy ΔΔF of even a single degree of freedom. As a single dynamical mode of a mechanical system is typically extended across the protein, it will, in general, offer more than one potential binding sites, which are also close to anti-nodes of the global mode, where substrate-binding will, in general, affect the value of κ. If two successive effector-binding events take κ → κ + δ → κ + 2δ:
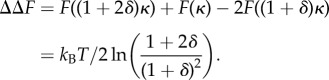 |
1.2 |
We have called this minimal fundamental building block for models of allosteric systems the ‘allosteron’1. In a natural extension of the single-mode allosteron unit, allosteric interaction between dimers can be modelled at the simplest possible level by associating two allosterons into a dimer (figure 1b), introducing a single new parameter of the harmonic coupling between them, κc. At any level of approximation within linear response, the elastic part of the Hamiltonian 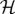 relevant to entropy-driven allostery (without contributions of momenta) is a quadratic form in the vector x of allosterons' degrees of freedom, with the (spring-constant) Hessian matrix H:
relevant to entropy-driven allostery (without contributions of momenta) is a quadratic form in the vector x of allosterons' degrees of freedom, with the (spring-constant) Hessian matrix H:
| 1.3 |
written in terms of a dimensionless form, 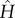 , of H in units of κ. If two allosterons with internal modes governed by spring constants κ1 and κ2 meet in association, the Hessians H1 and H2 in terms of the two single degrees of freedom (x1, x2), one per monomer, before and after their weak association are
, of H in units of κ. If two allosterons with internal modes governed by spring constants κ1 and κ2 meet in association, the Hessians H1 and H2 in terms of the two single degrees of freedom (x1, x2), one per monomer, before and after their weak association are
| 1.4 |
The entropic free energy of binding, from the introduction of the coupling interaction between two free allosterons is
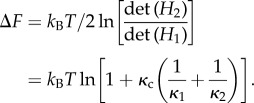 |
1.5 |
This is, of course, positive definite as it represents a reduction in internal entropy of structural fluctuations on binding. The full free energy of binding will include an enthalpic term ΔU, so ΔFbind = ΔF + ΔU, for which we will develop a simple model below, in §4.
Such an approach has been used successfully as the coarsest-level model for allosteric protein dimers [4–6], in decorated form as a basis for theories of mode-coupling to local fast dynamics [5], and as a generator of fitness landscapes in the evolution of allosteric systems [12]. In spite of its extreme simplicity, this toy-model approach to protein dimers, for example, has delivered highly non-trivial insights, such as a region of its three-dimensional parameter space where a dimer may exhibit negative cooperativity [6].
Within a graded series of approximations to protein dynamics, the allosteron model sits at a similar level of coarse-graining to the rotational translational block (RTB) approximation [13]. The strong elastic inhomogeneities within proteins that render the RTB picture appropriate also suggest that the dominance, for some purposes, of a few low-frequency and long-range dynamic modes is a valid one. Allostery without structural change, driven by modulation of thermal fluctuations, which depends on long-range information transfer, is one such application. As the degree of approximation is refined, we arrive at models that resolve individual residues (such as elastic network models—ENMs [12,14–16]), and finally at all-atomistic models [12].
In this work, we apply the allosteron model to elucidate the potential contributions of internal fluctuation entropy to protein self-assembly. The concatenation of fibre-forming proteins, for example, is not usually thought of as an example of allostery. However, if we think of the two effector molecules for an allosteric protein as copies of the protein itself, then the connection is evident. There will, in general, be a modification of the internal fluctuations in a protein when it joins a self-assembling complex or fibre. In principle, this generates a non-equivalence of binding free-energies for each protein in the complex, even when these are structurally identical and when there is no mean change in structure on binding. In the following, we explore this insight, at the level of the allosteron model, in two cases: in §2, for protein fibrils of arbitrary molecular weight; in §3, for ligand binding of finite protein n-mers. Section 4 then explores another possibility identified by this approach—that of elastically tuned families of proteins that possess weak mutual and exclusive association.
2. Self-assembly of allosteron chains
Protein fibrils that consist of reversibly bound proteins may be viewed as supramolecular ‘living’ polymers that grow and break up [17–19]. At steady state, the chain lengths of polymers obey equilibrium statistics, and may therefore be controlled by the thermodynamic properties of the building blocks. These properties include an enthalpic and entropic gain or penalty of monomer association to the polymer. Because of structural, hydrophobic and electronic effects, to name a few, the free energy of monomer association to a polymer often depends on the length of the oligomer. Consequently, polymerization is often cooperative and the degree of polymerization strongly depends on the cooperativity factor [17,20–22].
The entropic and energetic contributions that lead to (cooperative) supramolecular polymerization can easily be included in the allosteron model. For instance, the association of a monomer can be entropically penalized by stiffening the monomer through an increase of the internal spring constant κ ↦ ακ with α > 0, even in the absence of coupling, i.e. Kc = 0. The entropic penalty of dimerization is then ΔSnucl = 2kB lnα, whereas the entropic penalty of elongation is only ΔSelong = kB lnα. Hence, the allosteron model captures the entropic origin for cooperativity by a statistical-mechanics description [23]. The novel feature of the allosteron model is that the internal modes of the monomers are coupled (Kc > 0), which enables entropic allosteric signalling along the backbone of the polymer. In this section, we show that coupling leads to an entropic interaction range of 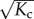 monomers, as well as an increase in the polymerization concentration by a factor of
monomers, as well as an increase in the polymerization concentration by a factor of 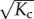 .
.
We predict these phenomena from the allosteron model by investigating the equilibrium statistics of an allosteron solution in a volume V [24,25]. The grand potential of the mixture is given by
| 2.1 |
In this expression, ρ(N) is the number density of chains of length N (note that we consider a monomer to be a chain of length N = 1), υ is the interaction volume of a monomer, ZN is the partition function of a chain of length N and μ is its chemical potential.
The partition function of the chain is obtained from the Hamiltonian where allosterons with spring constant κ bind through a binding energy ε at an entropic cost dictated by the coupling-spring constant κc. This bead-spring-like Hamiltonian is constructed by enhancing equation (1.3) by an enthalpic term ε to give 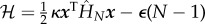 . The dimensionless Hessian is a tridiagonal matrix of which the main-diagonal elements are given by M11 = MNN = 1 + Kc and by Mnn = 1 + 2Kc for 1 < n < N. Furthermore, the first upper and lower diagonal elements equal −Kc, whereas all other matrix elements equal zero. The partition function is given by
. The dimensionless Hessian is a tridiagonal matrix of which the main-diagonal elements are given by M11 = MNN = 1 + Kc and by Mnn = 1 + 2Kc for 1 < n < N. Furthermore, the first upper and lower diagonal elements equal −Kc, whereas all other matrix elements equal zero. The partition function is given by
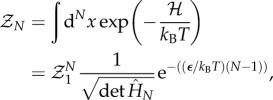 |
2.2 |
with 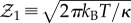 the partition function of a free monomer, see equation (1.1).
the partition function of a free monomer, see equation (1.1).
We minimize the grand potential in equation (2.1) to find that we can write the solution for the distribution function as
| 2.3 |
where we have defined the free energy of aggregation 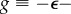
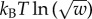 , the chemical potential
, the chemical potential 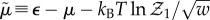 and the equilibrium constant
and the equilibrium constant 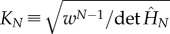 . The subsidiary definition
. The subsidiary definition  guarantees that the equilibrium constant equals unity for the monomers, N = 1, but converges to a finite value for large N (see electronic supplementary material).
guarantees that the equilibrium constant equals unity for the monomers, N = 1, but converges to a finite value for large N (see electronic supplementary material).
Indeed, from the Hamiltonian of the chain we deduce that the entropic interactions between monomers along the backbone extend over a distance of a persistence length 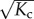 . For chains shorter than this, the stiffness may be considered large and it can be shown that
. For chains shorter than this, the stiffness may be considered large and it can be shown that 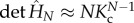 (see electronic supplementary material). For infinitely long chains, however, we find that
(see electronic supplementary material). For infinitely long chains, however, we find that 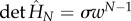 , where σ is a constant cooperativity factor: If the cooperativity factor is unity then monomer association to a chain is independent of the chain length and polymerization is ‘isodesmic’. For small values of σ, elongation is considerably easier than dimerization and polymerization is cooperative. In the present case, the value of the cooperativity factor follows from the fact that the two limits of
, where σ is a constant cooperativity factor: If the cooperativity factor is unity then monomer association to a chain is independent of the chain length and polymerization is ‘isodesmic’. For small values of σ, elongation is considerably easier than dimerization and polymerization is cooperative. In the present case, the value of the cooperativity factor follows from the fact that the two limits of  must crossover at
must crossover at 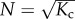 . This gives the weak dependence σ ∝ K−1/4c, which suggests that, in the absence of ligand-binding to the monomers, the allosteron model virtually predicts isodesmic polymerization.
. This gives the weak dependence σ ∝ K−1/4c, which suggests that, in the absence of ligand-binding to the monomers, the allosteron model virtually predicts isodesmic polymerization.
We confirm this by finalizing our calculation of the chain-length distribution of the polymers via a calculation of the chemical potential, μ, which is implicitly given by the mass balance 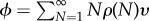 , with ϕ the overall volume fraction of the monomers [24,25]. By introducing the mass action (proportional to the experimentally controllable monomer concentration)
, with ϕ the overall volume fraction of the monomers [24,25]. By introducing the mass action (proportional to the experimentally controllable monomer concentration)  , we cast the mass balance in the form
, we cast the mass balance in the form
| 2.4 |
We numerically extract the chemical potential and insert it into equation (2.3) to obtain the length distribution of the chains.
From this distribution, we obtain the fraction of polymerized material, f, and the number-averaged degree of polymerization, 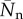 . Figure 2 shows these quantities as a function of the mass action for a coupling constant Kc ranging from 0 to 104. We have scaled the mass action using Xp = 2.34, at which half of the material is polymerized for the true isodesmic case, Kc = 0 [24]. As expected, supramolecular polymerization remains close to isodesmic: the polymerization curve maintains its symmetric S shape, and the degree of polymerization for X ≫ Xp, given by
. Figure 2 shows these quantities as a function of the mass action for a coupling constant Kc ranging from 0 to 104. We have scaled the mass action using Xp = 2.34, at which half of the material is polymerized for the true isodesmic case, Kc = 0 [24]. As expected, supramolecular polymerization remains close to isodesmic: the polymerization curve maintains its symmetric S shape, and the degree of polymerization for X ≫ Xp, given by 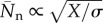 , only weakly depends on Kc through σ ≈ K−1/4c.
, only weakly depends on Kc through σ ≈ K−1/4c.
Figure 2.

Fraction of polymerized material f (left), and number-averaged degree of polymerization 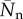 (right), as a function of the mass action X for coupling constants Kc ranging from 0 to 104. The mass action is scaled using Xp = 2.34; see main text. (Online version in colour.)
(right), as a function of the mass action X for coupling constants Kc ranging from 0 to 104. The mass action is scaled using Xp = 2.34; see main text. (Online version in colour.)
Nevertheless, the crossover concentration and crossover temperature may be affected by the entropic coupling between the monomers through the coupling parameter Kc. Indeed, in the isodesmic limit (KN = 1 for all N) the polymerization temperature is
| 2.5 |
with ΔHp ≡ ε the enthalpy of polymerization and  the entropy of polymerization. We hence find that the polymerization temperature is only weakly affected by the coupling constant. By contrast, the polymerization concentration is much more strongly affected. This quantity is given by
the entropy of polymerization. We hence find that the polymerization temperature is only weakly affected by the coupling constant. By contrast, the polymerization concentration is much more strongly affected. This quantity is given by
| 2.6 |
This shift in polymerization concentration originates from the stiffening not only of the associating monomer, but also of the monomers within a distance 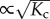 from the chain end. The interaction range, as well as the shift in polymerization concentration, may be reduced by increasing the internal spring constants of the monomers to κ ↦ ακ, which leads to Kc ↦ Kc/α. We speculate that this could be achieved by the binding of activators, such as ligands, to the monomers. The phenomena of ligand-induced dimerization and polymerization can potentially be addressed using the allosteron model. It should be noted that this generates a copolymer that consists of both activated and non-activated monomers. As we have seen, the range of entropic interaction exceeds the nearest-neighbour distance, and currently available Ising-like models cannot be applied directly [26]. We address this in forthcoming work. The importance of entropic signalling is not limited to supramolecular polymerization alone, but is also of crucial importance to the allosteric properties of proteins, as we discuss in the following.
from the chain end. The interaction range, as well as the shift in polymerization concentration, may be reduced by increasing the internal spring constants of the monomers to κ ↦ ακ, which leads to Kc ↦ Kc/α. We speculate that this could be achieved by the binding of activators, such as ligands, to the monomers. The phenomena of ligand-induced dimerization and polymerization can potentially be addressed using the allosteron model. It should be noted that this generates a copolymer that consists of both activated and non-activated monomers. As we have seen, the range of entropic interaction exceeds the nearest-neighbour distance, and currently available Ising-like models cannot be applied directly [26]. We address this in forthcoming work. The importance of entropic signalling is not limited to supramolecular polymerization alone, but is also of crucial importance to the allosteric properties of proteins, as we discuss in the following.
3. Allosteric interactions on ring complexes
In this section, we apply the model of coupled allosterons—as exploited for polymerization above—to the ligand-binding cooperativity (allostery) of polymers of fixed size. Specifically, we aim at the relative entropy changes induced by binding, conditional on prior binding events. Many proteins are known to occur as ring oligomers, such as the oxygen carrier, ferro-protein haemoglobin, or to form multi-protein complexes of ring topology. Owing to their doubly connected and intrinsically stable structure, rings are promising candidates to display entropy-driven allostery without conformational change [3]. In fact, for the hetero-tetramer haemoglobin, the cooperative O2-binding is well known [27], yet the underlying mechanism is still a matter of active research [28,29]. From a modelling point of view, rings' periodic boundary conditions suggest the possibility of generalizing results obtained for small rings to larger ones.
(a). Model
We briefly recall the main ingredients of the coupled-allosteron picture, located within the class of ENMs [14,30]: Each allosteron, called ‘unit’ in what follows for the sake of brevity, possesses an internal ‘breathing’ mode [7,31,32], modelled as a single harmonic degree of freedom, of spring constant κ (figure 1). One unit can represent a monomer, a protomer such as a helix dimer, or a protein, and interacts with its two neighbours on the ring via a harmonic mode of constant κc, of relative strength Kc ≡ κc/κ. The dimensionless Hessian matrix  that encodes the network's connectivity takes for a tetramer (four-unit) ring the form
that encodes the network's connectivity takes for a tetramer (four-unit) ring the form
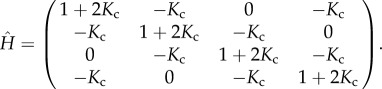 |
3.1 |
A first approach to model ligand-binding is to assume a bound unit's internal mode to change in strength, according to κ ↦ ακ. By taking this to be the only binding-induced modification of interactions, we arrive at a single-parameter model of binding. A more differentiated picture of allostery emerges from invoking a two-parameter model, in which ligand-binding is assumed to affect also the strength of the harmonic coupling to the neighbouring units, according to κc ↦ βκc. The mappings in 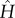 which reflect this model of binding, exemplified for unit i, take 1 ↦ α and Kc ↦ βKc in the ith diagonal entry, and multiply off-diagonal entries in both the ith row and the ith column by β.
which reflect this model of binding, exemplified for unit i, take 1 ↦ α and Kc ↦ βKc in the ith diagonal entry, and multiply off-diagonal entries in both the ith row and the ith column by β.
(b). Allosteric free energies
In order to quantify the entropy-induced cooperativity between successive binding events, we analyse differences of entropic free energies of binding, such as the allosteric free energy for two bindings,
| 3.2 |
Herein, F are total free energies computed via the determinant of 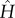 , and subscripts 0, 1 and 2 refer to the state with no ligand, one ligand, and two ligands bound, respectively.
, and subscripts 0, 1 and 2 refer to the state with no ligand, one ligand, and two ligands bound, respectively.
(c). Trimer rings
Despite its simplicity, a trimer ring may illustrate transparently the mechanism of cooperativity (of both signs) at work between states of different binding degrees. Indeed, allosteric interactions exist between the three units, pairwise neighbours in this case.
The values and spacing of free energies of binding, ΔF, for the ith binding defined as ΔF ≡ Fi − F0, for no site, one site, two, and eventually all sites binding a ligand, computed within the one-parameter binding model, are plotted in figure 3a. Cumulative binding is seen to be cooperative, i.e. favoured by the differences of ΔF, for all values of α and Kc. The map of the particular allosteric free energy ΔΔF2,1 (cf. equation (3.2)) against α and Kc, in figure 3b, shows explicitly the purely cooperative allostery predicted by this model.
Figure 3.

Trimer ring, one-parameter binding model: (a) binding free energies ΔF(α) for Kc = 5. (b) Allosteric free energy ΔΔF2,1(α, Kc).
In part, this prediction may be traced back to the one-parameter model of binding, which rescales diagonal elements of the Hessian 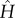 only, combined with the logarithmic dependence of the free energy on the (eigenfrequency) spectrum of
only, combined with the logarithmic dependence of the free energy on the (eigenfrequency) spectrum of 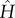 [7]. A more trimer-specific explanation relates to the mentioned degeneracy as regards nearest neighbours: Local decrease (increase) of the extent of thermal fluctuations and the associated entropy loss (gain), induced by changing one internal-mode strength, automatically carries over to all remaining units, which in a trimer are all directly coupled. Therefore, the entropy change upon altering another internal-mode strength is smaller in modulus than that for a previous binding.
[7]. A more trimer-specific explanation relates to the mentioned degeneracy as regards nearest neighbours: Local decrease (increase) of the extent of thermal fluctuations and the associated entropy loss (gain), induced by changing one internal-mode strength, automatically carries over to all remaining units, which in a trimer are all directly coupled. Therefore, the entropy change upon altering another internal-mode strength is smaller in modulus than that for a previous binding.
Within the two-parameter model of binding, we find that both cooperativity and anti-cooperativity of successive bindings (both signs of ΔΔF) can occur, depending on the set of modifiers, (α, β), and the initial ratio Kc of coupling to internal-mode strength. Maps of ΔΔF2,1(α, β) such as in figure 4, parameterized by Kc, indicate anti-cooperative binding to arise particularly for strengthened internal mode and weakened coupling. Larger values of Kc or α cause more pronounced allosteric interactions, i.e. a larger range of values of ΔΔF2,1.
Figure 4.
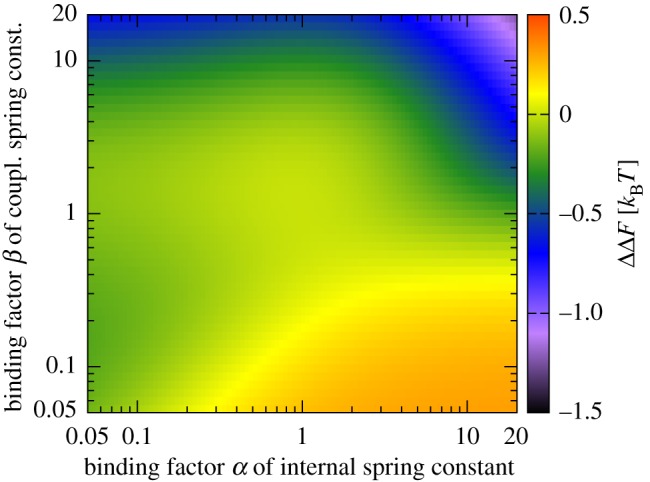
Trimer ring, two-parameter model: allosteric free energy ΔΔF2,1(α, β); initial coupling-to-internal strength ratio Kc = 2.
(d). Tetramer rings
Haemoglobin is one of the most prominent proteins of tetramer-ring topology. In contrast with binding to a trimer ring, successive binding to the four units can proceed via more than one pathway, so that both number and configuration of bound ligands [32] have to be specified in ΔF. The energy-level plot for a tetramer within the one-parameter model of binding is provided in figure 5. It reveals that two ligands at diametrical sites of the tetramer ring, either stiffening or weakening the respective internal modes, are entropically slightly less favourable than two ligands at adjacent sites. If we recall the plausibility argument for cooperativity on a trimer, via entropy-cost transfer between nearest neighbours through local change of fluctuations, the ordering of the ΔF agrees with intuition. As in the trimer case, calculations based on the one-parameter model of binding predict only positive allostery.
Figure 5.
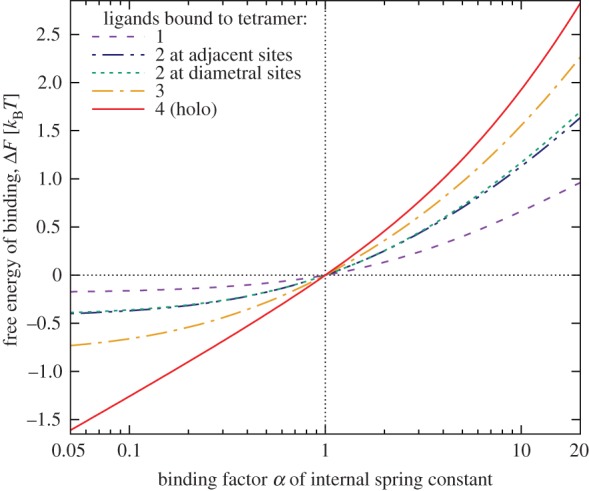
Tetramer ring, one-parameter binding model: binding free energies ΔF(α) for Kc = 5.
Already these minimal examples of cyclic coupling demonstrate non-trivial allosteric interactions of subtle parameter dependence between units with binding sites.
4. Dynamic allostery generates tuned weak attraction
Weak association of proteins is determined by a balance of the attractive forces operative at the mutual surfaces of interaction, and entropic repulsion. The attractive forces may have for their origin van der Waals, hydrophobic or screened electrostatic effects. The entropic repulsion arises from the penalty that their association generates from the constraints it places on the amplitude of internal structural fluctuations. A general question of interest is whether this delicate balance can be ‘tuned’ in different ways. This might allow distinct families of weakly associating nanoparticles (or proteins in the biological case) which bind reversibly among themselves, but which do not associate with other, differently tuned, families. An example of how weak, non-specific interactions may affect a network of protein–protein interactions in just the way described here has recently been published by Bhattacharyya et al. [33]. In this case, native and non-native homologues of the protein DHFR, when artificially upregulated in Escherichia coli, were shown to have very different effects. The native form was able to alter abundances and functions of a network of several associated proteins, whereas the non-native form, though still functional by itself, did not possess the ‘resonance effects’ with the same network. The evidence for a network of weak allosteric interactions is in the mutual effects on function in the case of the native protein. Here, we use the simple ‘allosteron’ model, with both coupling spring constants and enthalpic surface forces derived from Lennard–Jones (L-J) potential, to explore the possibility for such entropic tuning.
Starting from the entropic free energy of binding, ΔF, given in equation (1.5), and the total free energy ΔFbind = ΔF + ΔU, we construct a simple model for both the enthalpic term, ΔU, and the spring constant κc. As a general model for weakly attractive interactions between protein surfaces we choose the L-J potential
| 4.1 |
One of the advantages of a physics-based phenomenological model such as the L-J potential is that it is able to illustrate and work with the correlation between the enthalpic binding strength  and
and  . Using the parameterized form of the interaction gives
. Using the parameterized form of the interaction gives
| 4.2 |
with 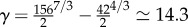 .
.
Combining these allows a single-degree-of-freedom parameterization of the set of possible L-J couplings, using ΔU as the free variable, and finding
| 4.3 |
The dimensionless free energy at the potential minimum f = ΔFbind/kBT can be written in terms of a dimensionless potential energy minimum 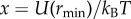 as
as
| 4.4 |
Figure 6 plots this function for various values of the renormalized coupling constant, a. Note that the asymptotic form decreases linearly with x as it must from the dominance of the enthalpic interaction, but there exist other values of x, corresponding to a ‘tuning’ of the L-J coupling to the inner elastic strength of the proteins (the κi), which lead to a family of shallow local minima. The depth and value of this minimum increases with decreasing a (figure 6). There is a lower critical value of a ≃ 1.4 below which the minimum disappears.
Figure 6.
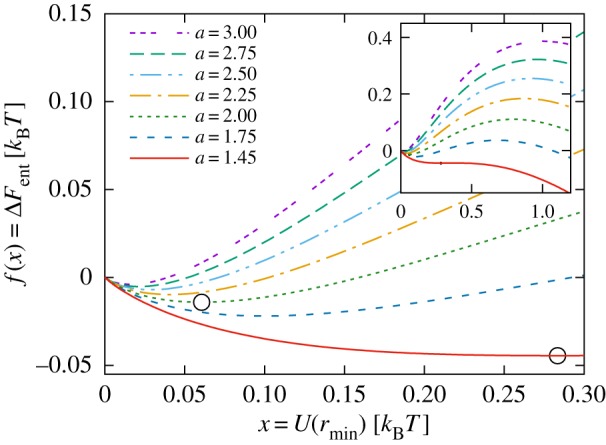
Dimensionless free-energy function f(x) from equation (4.4), for different values of a.
A word is necessary on the values of the interaction strengths. These are, of course, very small (of order kBT/50). However, this corresponds to the response of a single dynamical mode of internal motion only. In practice, there will be many internal modes that are coupled to the surface interaction, sufficient to create an associative energy of the order of kBT. Should two proteins from different families attract, the mutual free energy of attraction will have the same form as the entropy/enthalpy function within a single family, but will not be at the tuned minimum potential. This permits the emergence of interprotein potentials that generate weakly associated clusters for proteins within ‘tuned’ families exclusively.
5. Discussion
The allosteron model constitutes the maximal level of coarse-graining within models for protein dynamics that still captures the physics of thermally excited modes of deformation within proteins and their complexes. In spite of this level of simplicity, the model is nevertheless able to make non-trivial predictions about the contribution of structural fluctuations to the statistical mechanics of complexation, in terms of the ratio of coupling to internal interactions Kc. Such an extreme degree of coarse-graining, although it may clarify the role of internal entropy in protein–protein interactions, does not at first seem to promise any more specificity, such as accommodating sequence-specific or mutation effects. However, changes of protein sequence will, in general, alter the key structural elastic constants of allosteron models, so may, in principle, be parameterized. The models presented here may, therefore, be applied to real systems where effects emerge at this highly coarse-grained level. Parameterization can be achieved from finer-grained modelling at fully atomistic [4] or elastic network [12] levels.
When applied as a building block of supramolecular polymers, the allosteron model is capable of not only capturing cooperativity in the usual way, but also predicting entropic allosteric signalling along the backbone of the chain. When a monomer associates to the chain, a number of 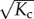 of monomers near the chain end are entropically penalized, which causes an increase in the polymerization concentration by a factor
of monomers near the chain end are entropically penalized, which causes an increase in the polymerization concentration by a factor 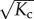 . We find that a reduction in the polymerization concentration may be triggered through ‘activation’ of the monomers, e.g. by stimuli such as ligand-binding that modify the coupling constant to Kc ↦ Kc/α.
. We find that a reduction in the polymerization concentration may be triggered through ‘activation’ of the monomers, e.g. by stimuli such as ligand-binding that modify the coupling constant to Kc ↦ Kc/α.
We have also investigated ligand-binding to allosteron complexes that represent ring-type proteins. In this case, binding is assumed to modify the strengths of both internal and coupling interactions and predicts a palette of cooperative binding behaviour even for small rings. In particular, maps of the allosteric free energy for two binding events in the parameter space of the two spring modifiers show regions of negative allostery and an intriguing non-monotonic dependence. Further studies along these lines might extend to inter-ring coupling, possibly adding to the understanding of allostery without conformational change reported for chaperonins [34].
When an allosteron coupling model is derived from a specific model for intermolecular potentials, we find the possibility to ‘tune’ the surface attraction and the internal elasticity of soft nanoparticles, in general, and proteins in particular. Simultaneous tuning of the dimensionless interparticle coupling Kc and the enthalpy of binding creates ‘families’ of particles that possess weak mutual associativity (and thereby potential allosteric activity of other, enzymatic, kinds).
The application of the allosteron model to the three cases of small, specifically bound clusters, large specifically bound fibrils and non-specific binding has for all revealed subtle phenomena that are open to experimental investigation.
Supplementary Material
Acknowledgments
T.C.B.M. thanks E. Shakhnovich (Harvard) for the valuable discussions about his work on DHFR protein networks, which motivated the calculation of §4.
Endnote
The word ‘allosteron’ was coined by the author T.C.B.M. for presentation at the Royal Society Discussion Meeting ‘Allostery and molecular machines’, DATE 2017.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
The authors acknowledge the Engineering and Physical Sciences Research Council (grant no. EP/N031431/1) for funding of this project.
References
- 1.Monod J, Wyman J, Changeux JP. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118. ( 10.1016/S0022-2836(65)80285-6) [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP, Edelstein SJ. 2005. Allosteric mechanisms of signal transduction. Science 308, 1424–1428. ( 10.1126/science.1108595) [DOI] [PubMed] [Google Scholar]
- 3.Cooper A, Dryden DTF. 1984. Allostery without conformational change: a plausible model. Eur. Biophys. J. 11, 103–109. ( 10.1007/BF00276625) [DOI] [PubMed] [Google Scholar]
- 4.Hawkins RJ, McLeish TCB. 2004. Coarse-grained model of entropic allostery. Phys. Rev. Lett. 93, 098104 ( 10.1103/PhysRevLett.93.098104) [DOI] [PubMed] [Google Scholar]
- 5.Hawkins RJ, McLeish TCB. 2006. Coupling of global and local vibrational modes in dynamic allostery of proteins. Biophys. J. 91, 2055–2062. ( 10.1529/biophysj.106.082180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toncrova H, McLeish TCB. 2010. Substrate-modulated thermal fluctuations affect long-range allosteric signaling in protein homodimers: exemplified in CAP. Biophys. J. 98, 2317–2326. ( 10.1016/j.bpj.2010.01.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLeish TCB, Rodgers TL, Wilson MR. 2013. Allostery without conformation change: modelling protein dynamics at multiple scales. Phys. Biol. 10, 056004 ( 10.1088/1478-3975/10/5/056004) [DOI] [PubMed] [Google Scholar]
- 8.Hilser VJ, Wrabl JO, Motlagh HN. 2012. Structural and energetic basis of allostery. Annu. Rev. Biophys. 41, 585–609. ( 10.1146/annurev-biophys-050511-102319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tidor B, Karplus M. 1994. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J. Mol. Biol. 238, 405–414. ( 10.1006/jmbi.1994.1300) [DOI] [PubMed] [Google Scholar]
- 10.Gilson MK, Given JA, Bush BL, McCammon JA. 1997. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys. J. 72, 1047–1069. ( 10.1016/S0006-3495(97)78756-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzeng SR, Kalodimos CG. 2012. Protein activity regulation by conformational entropy. Nature 488, 236–240. ( 10.1038/nature11271) [DOI] [PubMed] [Google Scholar]
- 12.Rodgers TL, Townsend PD, Burnell D, Jones ML, Richards SA, McLeish TCB, Pohl E, Wilson MR, Cann MJ. 2013. Modulation of global low-frequency motions underlies allosteric regulation: demonstration in CRP/FNR family transcription factors. PLoS Biol. 11, e1001651 ( 10.1371/journal.pbio.1001651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tama F, Sanejouand YH. 2001. Conformational change of proteins arising from normal mode calculations. Protein Eng. Des. Sel. 14, 1–6. ( 10.1093/protein/14.1.1) [DOI] [PubMed] [Google Scholar]
- 14.Tirion MM. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77, 1905–1908. ( 10.1103/PhysRevLett.77.1905) [DOI] [PubMed] [Google Scholar]
- 15.Bahar I, Atilgan AR, Demirel MC, Erman B. 1998. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 80, 2733–2736. ( 10.1103/PhysRevLett.80.2733) [DOI] [Google Scholar]
- 16.Rodgers TL, Burnell D, Townsend PD, Pohl E, Cann MJ, Wilson MR, McLeish TCB. 2013. ΔΔPT: a comprehensive toolbox for the analysis of protein motion. BMC Bioinformatics 14, 183 ( 10.1186/1471-2105-14-183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Greef TFA, Smulders MMJ, Wolffs M, Schenning APHJ, Sijbesma RP, Meijer EW. 2009. Supramolecular polymerization. Chem. Rev. 109, 5687–5754. ( 10.1021/cr900181u) [DOI] [PubMed] [Google Scholar]
- 18.Rest C, Kandanelli R, Fernández G. 2015. Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions. Chem. Soc. Rev. 44, 2543–2572. ( 10.1039/C4CS00497C) [DOI] [PubMed] [Google Scholar]
- 19.Besenius P. 2017. Controlling supramolecular polymerization through multicomponent self-assembly. J. Polym. Sci. A Polym. Chem. 55, 34–78. ( 10.1002/pola.28385) [DOI] [Google Scholar]
- 20.Goldstein RF, Stryer L. 1986. Cooperative polymerization reactions: analytical approximations, numerical examples, and experimental strategy. Biophys. J. 50, 583–599. ( 10.1016/S0006-3495(86)83498-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaefer C, Voets IK, Palmans ARA, Meijer EW, van der Schoot P, Besenius P. 2012. Controlling the cooperativity in the supramolecular polymerization of ionic discotic amphiphiles via electrostatic screening. ACS Macro Lett. 1, 830–833. ( 10.1021/mz300218e) [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni C, Meijer EW, Palmans ARA. 2017. Cooperativity scale: a structure–mechanism correlation in the self-assembly of benzene-1,3,5-tricarboxamides. Acc. Chem. Res. 50, 1928–1936. ( 10.1021/acs.accounts.7b00176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saroleá-Mathot L. 1953. Thermodynamic and spectroscopic properties of associated solutions. Trans. Faraday Soc. 49, 8–20. ( 10.1039/TF9534900008) [DOI] [Google Scholar]
- 24.van der Schoot PPAM. 2005. Theory of supramolecular polymerization. In Supramolecular polymers, 2nd edn (ed. Ciferri A.), pp. 77–106. Boca Raton, FL: CRC Press. [Google Scholar]
- 25.van der Schoot P. 2009. Nucleation and cooperativity in supramolecular polymers. In Engineering aspects of self-organising materials (ed. Koopmans RJ.) (Advances in Chemical Engineering no. 35), pp. 45–77. London, UK: Academic Press. [Google Scholar]
- 26.Jabbari-Farouji S, van der Schoot P. 2012. Theory of supramolecular co-polymerization in a two-component system. J. Chem. Phys. 137, 064906 ( 10.1063/1.4742192) [DOI] [PubMed] [Google Scholar]
- 27.Bohr C, Hasselbach K, Krogh A. 1904. [About a biologically vital impact that the blood's osmotic pressure of carbonic acid exerts on the blood's binding of oxygen]. Skand. Arch. Physiol. 15, 401–412 (in German). [Google Scholar]
- 28.Ciaccio C, Coletta A, De Sanctis G, Marini S, Coletta M. 2008. Cooperativity and allostery in haemoglobin function. IUBMB Life 60, 112–123. ( 10.1002/iub.6) [DOI] [PubMed] [Google Scholar]
- 29.Eaton WA, Henry ER, Hofrichter J, Mozarelli A. 1999. Is cooperative oxygen binding by hemoglobin really understood? Nat. Struct. Biol. 6, 351–358. ( 10.1038/7586) [DOI] [PubMed] [Google Scholar]
- 30.Bahar I, Lezon TR, Bakan A, Shrivastava IH. 2010. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem. Rev. 110, 1463–1497. ( 10.1021/cr900095e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkins RJ. 2005. Coarse-grained models of dynamic allostery in proteins. PhD thesis: University of Leeds. [Google Scholar]
- 32.Toncrova H. 2010. Coarse-grained models of biomolecule dynamics and allostery. PhD thesis: University of Leeds. [Google Scholar]
- 33.Bhattacharyya S, Bershtein S, Yan J, Argun T, Gilson AI, Trauger SA, Shakhnovich EI. 2016. Transient protein-protein interactions perturb E. coli metabolome and cause gene dosage toxicity. eLife 5, e20309 ( 10.7554/eLife.20309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng W, Brooks BR, Thirumalai D. 2007. Allosteric transitions in the chaperonin GroEL are captured by a dominant normal mode that is most robust to sequence variations. Biophys. J. 93, 2289–2299. ( 10.1529/biophysj.107.105270) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article has no additional data.