

Abstract
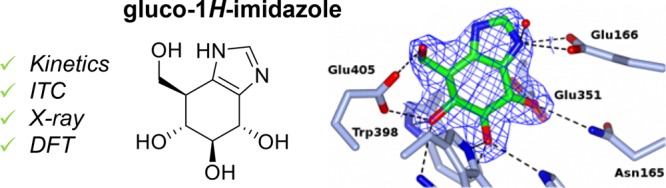
Gluco-azoles competitively inhibit glucosidases by transition-state mimicry and their ability to interact with catalytic acid residues in glucosidase active sites. We noted that no azole-type inhibitors described, to date, possess a protic nitrogen characteristic for 1H-imidazoles. Here, we present gluco-1H-imidazole, a gluco-azole bearing a 1H-imidazole fused to a glucopyranose-configured cyclitol core, and three close analogues as new glucosidase inhibitors. All compounds inhibit human retaining β-glucosidase, GBA1, with the most potent ones inhibiting this enzyme (deficient in Gaucher disease) on a par with glucoimidazole. None inhibit glucosylceramide synthase, cytosolic β-glucosidase GBA2 or α-glucosidase GAA. Structural, physical and computational studies provide first insights into the binding mode of this conceptually new class of retaining β-glucosidase inhibitors.
Glycosidases catalyze the hydrolysis of interglycosidic linkages, and glycosidase inhibitors are widely regarded as promising therapeutic entities.1 Azole-containing glycopyranoside mimics are a major class of competitive glycosidase inhibitors.2 The natural product, nagstatin (1), is a potent N-acetyl-β-d-glucosaminidase (NAG) inhibitor.3 Glucopyranose mimics bearing tetrazole (2), triazole (3, 4) and imidazole (5) moieties have been conceived as β-glucosidase inhibitors by Tatsuta and Vasella, with inhibitory potencies against sweet almond β-glucosidase ranging from low nanomolar (5) to high micromolar (4, Figure 1a).4−7 The reaction itinerary followed by many β-glucosidases proceeds via a 4H3 transition state conformation,8 matching one of the thermodynamically most favored conformations of gluco-azoles (local minima calculated for 5 are 4H3 and 3H4).9 This conformational transition state mimicry contributes considerably to the inhibitory potency of gluco-azoles. Vasella concluded, from extensive studies on gluco-azoles (including compounds 2–5), that an azole nitrogen at the position corresponding to the exocyclic oxygen in a β-glucoside substrate is essential for inhibitory potency, as this “exocyclic” nitrogen participates in lateral coordination with the catalytic acid/base of antiprotonating glucosidases.10−14 Remarkably, no azole-type inhibitors synthesized to date are protic with respect to the azole ring (such as 1H-imidazoles), and we reasoned that such positional isomers would be attractive alternatives to the reported gluco-azoles. Herein, we report on the synthesis and biochemical evaluation of gluco-1H-imidazole 6 and its analogues 7–9 as conceptually new retaining β-glucosidase inhibitors (Figure 1b).
Figure 1.

(a) Known cyclitol-azole type glycosidase inhibitors and their inhibitory potencies against porcine NAG (1)3 and sweet almond β-glucosidase (2–5).4−7 (b) 1H-Imidazoles subject of this study.
The synthesis of gluco-1H-imidazole 6 commenced with cyclohexene 10,15 which was transformed into the vicinal diamine following a modified procedure from Llebaria (Scheme 1).16 Rapid conversion of 10 was accomplished using RuCl3/NaIO4, affording a separable, near equimolar mixture of 1,2-cis-dihydroxy isomers 11a (S,S) and 11b (R,R).17 Bismesylation and subsequent azide substitution afforded diazido cyclitols 12a,b. Platinum oxide catalyzed hydrogenolysis afforded diamino cyclitols 13a,b, which were condensed with trimethyl orthoformate in hexafluoroisopropanol (HFIP) to afford imidazolines 14a,b.18 Protected 1H-imidazole 15 was obtained by oxidation of either isomer of 14 with IBX/DMSO,19 and final hydrogenolysis afforded gluco-1H-imidazole 6. Derivatization of the imidazole ring of gluco-1H-imidazoles was achieved by variation of the trimethyl orthoester employed. Thus, reaction of 13a,b with trimethyl orthovalerate followed by oxidation and deprotection furnished 2-butyl-1H-imidazole 7 via 16a,b and 17. Utilizing tetrabenzyl myo-diaminocyclitol16 as starting material yielded conduritol B-1H-imidazoles 8 and 9 (see Supporting Information (SI)).
Scheme 1. Synthesis of 1H-Imidazoles 6 and 7.

Reagents and conditions: (a) RuCl3·3H2O (7 mol %), NaIO4, EtOAc, MeCN, H2O, 0 °C, 90 min, 11a 40%, 11b 32%; (b) MsCl, N-methyl-imidazole, Et3N, CHCl3, rt, 16 h, then NaN3, DMF, 100 °C, 16 h, 12a 74%, 12b 58%; (c) PtO2, H2, THF, rt, 16 h, 13a 80%, 13b 96%; (d) trimethyl orthoformate, HFIP, rt, 16 h, 14a 76%, 14b 87%; or trimethyl orthovalerate, HFIP, rt, 16 h, 16a 74%, 16b 78%; (e) for 15: IBX, DMSO, 45 °C, 16 h, 75% from 14a, 71% from 14b; for 17: (COCl)2, DMSO, DCM, −60 °C, 1 h, 76% from 16a; 70% from 16b; (f) Pd(OH)2/C, H2, HCl, MeOH, quant. 6; quant. 7.
We determined competitive kinetic constants and IC50 values for 1H-imidazoles 6–9 as compared with glucoimidazole 5 for inhibition of the retaining glucosidases TmGH1 from Thermotoga maritima,20TxGH116 from Thermoanaerobacterium xylanolyticum,21 sweet almond β-glucosidase (GH1), human lysosomal acid β-glucosylceramidase (GBA1, GH30), human cytosolic β-glucosylceramidase (GBA2, GH116), murine glucosylceramide synthase (GCS) and human acid α-glucosidase (GAA, GH31). Gluco-1H-imidazole 6 displayed micromolar inhibitory activity against most β-glucosidases tested, with Ki values ranging from ∼3.9 μM against GBA1 to ∼69 μM against TxGH116 (Table 1). Remarkably, given their apparent structural similarity, 1H-imidazole 6 proved to be a weaker β-glucosidase inhibitor than glucoimidazole 5, which inhibited β-glucosidases at nanomolar concentrations. Attachment of a butyl moiety, as in 7, increased the inhibitory potency of the gluco-1H-imidazole scaffold. Though this increase in potency was modest for TmGH1 and TxGH116, 7 inhibited sweet almond β-glucosidase and GBA1 with nanomolar potency. Notably, the biological role of GBA1 is breakdown of amphiphilic glucosylceramide.22 Conduritol-1H-imidazole 8 did not effectively inhibit any glucosidases tested except for GBA1, consistent with a loss of interactions at the O6 position, but also with the fact that GBA1 is effectively inhibited by conduritol B epoxide (CBE).23 As with the gluco-1H-imidazoles, addition of a butyl moiety to the conduritol-1H-imidazole scaffold increased activity against sweet almond β-glucosidase and GBA1, rendering 9 a nanomolar inhibitor of these enzymes. No compounds displayed any inhibitory activity against GBA2, GCS or α-glucosidase GAA under our assay conditions. Competitive, selective GBA1 inhibitors are considered attractive starting points for the development of pharmacological chaperones: compounds that stabilize the fold of mutant, partially malfunctioning GBA1 in Gaucher patients.24,25 Compounds 7 and 9 (as well as known glucoimidazole 5) are in our opinion attractive starting points for this purpose.
Table 1. Inhibition Constants (Ki) and IC50 Values in μM for Compounds 5–9.
| TmGH1b | TxGH116b | sweet almond GH1c | GBA1b | GBA2d | GCSd | GAAd | |
|---|---|---|---|---|---|---|---|
| 6 | 58 ± 1 | 69 ± 12 | 23 ± 1 | 3.9 ± 2 | >100 | >50 | >100 |
| 7 | 39 ± 14 | 46 ± 5 | 0.039 ± 0.006 | 0.133 ± 0.040 | >100 | >50 | >100 |
| 8 | >100d | >100d | >100d | 13.7 ± 6 | >100 | >50 | >100 |
| 9 | >100d | >100d | 0.221 ± 0.004 | 0.079 ± 0.001 | >100 | >50 | >100 |
| 5 | 0.026 ± 0.001 | 0.165 ± 0.006 | 0.067 ± 0.004e | 0.070 ± 0.005 | >100 | >50 | >100 |
Values are mean ± SD from triplicate experiments.
Assays measured at pH 6.8 using β-p-NPG substrate.
pH 5.2 using β-2,4-DNPG substrate.
Apparent IC50.
Lit. Ki = 0.100 μM.6
One striking result displayed in Table 1 is that 1H-imidazole 6 is a comparatively much weaker retaining β-glucosidase inhibitor than glucoimidazole 5. Because the structures are closely related, we elected to investigate this result in more depth. Literature studies have shown that the inherent basicity of the azole ring in compounds 2–5 correlates to the inhibitory potency.26,27 Basicity plays a negligible role here, because the experimental pKAH values of 5 and 6 are almost identical (5: pKAH 6.2, lit. 6.1;266: pKAH 6.0; Figure S1, S2). We also explored the interaction of gluco-1H-imidazoles with TmGH1 and TxGH116 by isothermal titration calorimetry (ITC) at pH 5.8 (activity optimum11,21) and pH 6.8 (where 5 shows strongest inhibition).11 In line with the Ki data (Table 1), ITC titrations at both pH values showed nanomolar binding affinities for 5, and micromolar affinities for 6 and 7 (Table S3). The enthalpic contributions in active site binding for 1H-azoles are smaller than those observed for 5, and only partially compensated for by increased entropic contributions to the binding energy. In line with what has been reported for 5,11 the presence of a hydrophobic moiety in 7 reduced its enthalpy of binding compared to 6, which was counterbalanced by an increase in favorable entropic contributions to binding.
Next, the X-ray structure of 6 in complex with TmGH1 was determined to 1.7 Å (PDB: 5OSS), and compared to the structure of the same enzyme complexed with 5 (PDB: 2CES).11 These structures revealed the binding mode of both compounds to be similar. We observed a single molecule of 6 in the active site after ligand soaking, adopting a 4E conformation, similar to the complex of 5 and TmGH1 (Figure 2a).11 The H-bonding interactions made by 6 to active site residues were identical to those observed with 5, albeit with a slight “upward” tilt for 6 compared to 5 (∼0.4 or 0.5 Å “upward” shifts at the apical imidazole carbon, compared with ligands in chains A or B of 2CES respectively; Figure 2b, Figure S3). Crystal structures in TxGH116 at 2.1 Å resolution (Figure 2c,d) also revealed similar binding modes21 and a ∼0.5 Å “upward” tilt for 6 compared to 5 (PDB: 5OST and 5BX4, respectively). For both TmGH1 and TxGH116 complexes, B-factors in the “imidazole” portion of 6 were markedly higher when compared to the “glucose” portion of the molecule, indicating the imidazole of 6 was more disordered in the crystal structure and may be bound less strongly. No strong B-factor trend was observed for complexes with 5 (Figure S3).
Figure 2.
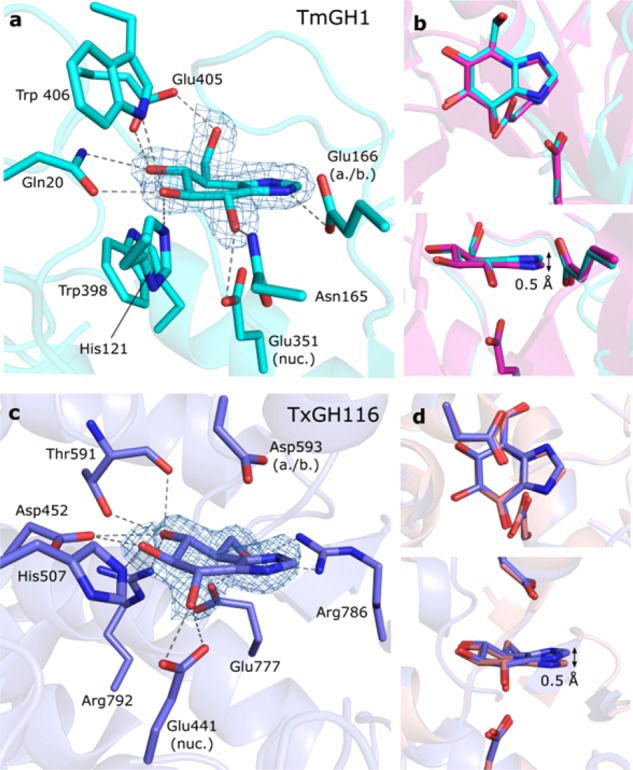
(a) Gluco-1H-imidazole 6 in complex with TmGH1, with direct H-bonding interactions shown. (b) Overlay of 5 (pink) and 6 (cyan) within the TmGH1 active site (chain B from each structure). (c) Gluco-1H-imidazole 6 in complex with TxGH116. (d) Overlay of 5 (salmon) and 6 (blue) within the TxGH116 active site. Electron densities are REFMAC maximum-likelihood/σA weighted 2Fo–Fc contoured to 0.38 (TmGH1) or 0.48 (TxGH116) e–/Å3.
The underlying cause for the reduced potency of gluco-1H-imidazole 6 compared to 5 is most likely the combination of a number of factors. We propose that repositioning of the N1 atom (from the bridgehead position in 5 to the position in 6) brings two major consequences that together reduce the binding affinity of 6 compared to 5. First, considering the situation where the imidazole is in a neutral state:28 the free lone pair of the N2 atom in 5 can laterally coordinate to the acid/base residue of the bound glucosidase in typical anti-protonating glucosidases14 (although TxGH116 lacks this interaction due to the unusual placement of its acid/base residue21). This lateral positioning of N2 is maintained in 6, as observed in its complex with TmGH1 (Figure 2a). However, and in contrast to 5, 1H-imidazole 6 may undergo prototropic tautomerism (Figure 3a). Thus, though the overall pKAH values of 5 and 6 are similar, the N2 lone pair of 6 may be less available for interaction with the glucosidase acid/base, reducing the binding affinity of 6 compared to 5. Protonation of the imidazole in turn (either in solution or by proton abstraction from the acid/base residue)28 results in positive charge delocalization. Resulting charge–charge interactions with enzyme active site carboxylates are thought to contribute substantially to enzyme binding energy of azole-type inhibitors.29 We calculated the Mulliken charge on all atoms for protonated 5 and 6 by DFT. Protonation of the azole ring in 5 produces a δ+ charge on the “anomeric” carbon, which is ideally located for a charge–charge interaction with a retaining glucosidase active site nucleophile. Conversely, protonation of 6 leads to a δ+ charge largely delocalized onto the “apical” carbon atom of the imidazole, with the overall δ+ charge also being less pronounced (Figure 3b). This apical δ+ charge is located distal from the catalytic nucleophile and thus poorly positioned for charge–charge interactions, which may explain the reduced binding enthalpy observed in ITC for gluco-1H-imidazoles 6 compared to 5. The small “upward” shift and increased imidazole B-factors, observed in crystal structure complexes of 6 compared to 5 is also consistent with a weaker charge–charge interaction of the imidazole portion of 6 with the enzyme catalytic nucleophile. Interestingly, in contrast to neutral 6, glucoimidazole 5 also contains a significant δ+ character (+0.306 Mulliken charge) on the “anomeric” carbon in its neutral state (see SI).
Figure 3.
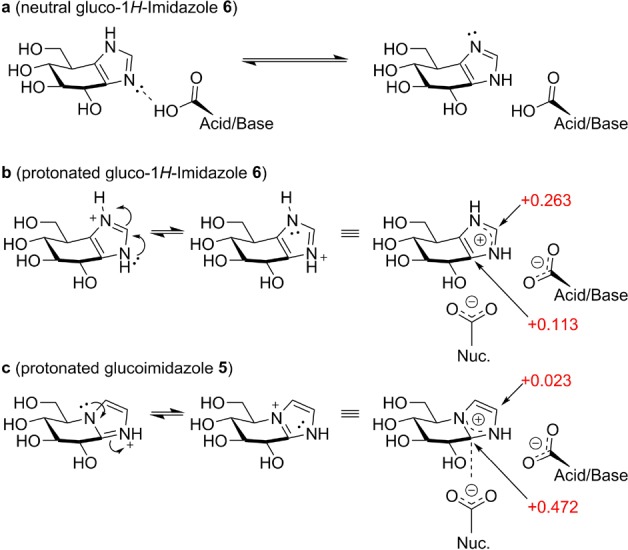
Interactions of gluco-1H-imidazole 6 and classical glucoimidazole 5 with the catalytic residues. (a) Prototropic tautomerism of 6. (b) Positive charge is delocalized onto the “apical” carbon in protonated 6. (c) In 5, positive charge is delocalized onto the anomeric equivalent carbon, ideally located for charge–charge interaction with the nucleophile residue. Mulliken charges are annotated in red.
In conclusion, we have described a new class of competitive β-glucosidase inhibitors: the 1H-gluco-azoles. The synthetic route is flexible regarding substituents on the imidazole ring, and can likely be transferred to configurational isomers by applying this route to configurational isomers of cyclohexene 10.30 Our compounds resemble to some extent the 1H-imidazoles reported by Li and Byers,31 and Field et al.,32 whose simple, achiral molecules inhibit several glucosidases as well, though likely by a different mode of action. 1H-Imidazole 6 appeared a poorer inhibitor than compound 5, and we hypothesize this is caused by delocalization of the lone pair on the nitrogen atom due to tautomerism and/or impaired δ+ charge development at the anomeric center. Introduction of an alkyl substituent on the 1H-imidazole ring (as in 7) as well as “deletion” of the methylene carbon (as in 9) yielded potent competitive inhibitors of the human lysosomal glucosylceramidase GBA1, selective over human cytosolic GBA2 and GCS. These and related compounds we postulate have potential to be developed into pharmacological chaperone candidates for Gaucher disease and perhaps also neuropathological disorders (in particular, Parkinsonism) that are characterized by partial deficiency in GBA.33,34
Acknowledgments
We thank The Netherlands Organization for Scientific Research (NWO-CW ChemThem grant to J.M.F.G.A. and H.S.O.), the European Research Council (ERC-2011-AdG-290836 “Chembiosphing” to H.S.O., and ERC-2012-AdG-322942 “Glycopoise” to G.J.D.), Sanofi Genzyme (research grant to J.M.F.G.A. and H.S.O. and postdoctoral contract to M.A.) and Diamond Light Source (beamline I03, proposal number mx-13587) for provision of data collection facilities. G.J.D. thanks the Royal Society for the Ken Murray Research Professorship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02399.
Experimental data and procedures, enzyme kinetics, DFT calculations, crystallographic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Asano N. Glycobiology 2003, 13, 93R. 10.1093/glycob/cwg090. [DOI] [PubMed] [Google Scholar]
- Lillelund V. H.; Jensen H. H.; Liang X.; Bols M. Chem. Rev. 2002, 102, 515. 10.1021/cr000433k. [DOI] [PubMed] [Google Scholar]
- Aoyagi T.; Suda H.; Uotani K.; Kojima F.; Aoyama T.; Horiguchi K.; Hamada M.; Takeuchi T. J. Antibiot. 1992, 45, 1404. 10.7164/antibiotics.45.1404. [DOI] [PubMed] [Google Scholar]
- Tatsuta K.; Miura S.; Ohta S.; Gunji H. Tetrahedron Lett. 1995, 36, 1085. 10.1016/0040-4039(94)02460-S. [DOI] [Google Scholar]
- Ermert P.; Vasella A. Helv. Chim. Acta 1991, 74, 2043. 10.1002/hlca.19910740839. [DOI] [Google Scholar]
- Granier T.; Panday N.; Vasella A. Helv. Chim. Acta 1997, 80, 979. 10.1002/hlca.19970800329. [DOI] [Google Scholar]
- Heightman T. D.; Locatelli M.; Vasella A. Helv. Chim. Acta 1996, 79, 2190. 10.1002/hlca.19960790814. [DOI] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Curr. Opin. Struct. Biol. 2014, 28, 1. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankrathok A.; Iglesias-Fernández J.; Williams R. J.; Pengthaisong S.; Baiya S.; Hakki Z.; Robinson R. C.; Hrmova M.; Rovira C.; Williams S. J.; Ketudat Cairns J. R. ACS Catal. 2015, 5, 6041. 10.1021/acscatal.5b01547. [DOI] [Google Scholar]
- Varrot A.; Schülein M.; Pipelier M.; Vasella A.; Davies G. J. J. Am. Chem. Soc. 1999, 121, 2621. 10.1021/ja984238n. [DOI] [Google Scholar]
- Gloster T. M.; Roberts S.; Perugino G.; Rossi M.; Moracci M.; Panday N.; Terinek M.; Vasella A.; Davies G. J. Biochemistry 2006, 45, 11879. 10.1021/bi060973x. [DOI] [PubMed] [Google Scholar]
- Notenboom V.; Williams S. J.; Hoos R.; Withers S. G.; Rose D. R. Biochemistry 2000, 39, 11553. 10.1021/bi0010625. [DOI] [PubMed] [Google Scholar]
- Hrmova M.; Streltsov V. A.; Smith B. J.; Vasella A.; Varghese J. N.; Fincher G. B. Biochemistry 2005, 44, 16529. 10.1021/bi0514818. [DOI] [PubMed] [Google Scholar]
- Heightman T. D.; Vasella A. T. Angew. Chem., Int. Ed. 1999, 38, 750.. [DOI] [PubMed] [Google Scholar]
- Hansen F. G.; Bundgaard E.; Madsen R. J. Org. Chem. 2005, 70, 10139. 10.1021/jo051645q. [DOI] [PubMed] [Google Scholar]
- Trapero A.; Llebaria A. ACS Med. Chem. Lett. 2011, 2, 614. 10.1021/ml200098j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola M.; Wu L.; Ferraz M. J.; Kuo C. L.; Raich L.; Breen I. Z.; Offen W. A.; Codée J. D. C.; van der Marel G. A.; Rovira C.; Aerts J. M. F. G.; Davies G. J.; Overkleeft H. S. ACS Cent. Sci. 2017, 3, 784. 10.1021/acscentsci.7b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaksar S.; Heydari A.; Tajbakhsh M.; Vahdat S. M. J. Fluorine Chem. 2010, 131, 1377. 10.1016/j.jfluchem.2010.10.002. [DOI] [Google Scholar]
- Nicolaou K. C.; Mathison C. J. N.; Montagnon T. J. Am. Chem. Soc. 2004, 126, 5192. 10.1021/ja0400382. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zeng Z.; Zeng G.; Chen M.; Zhang Y.; Zhang J.; Fang X.; Jiang M.; Lu L. Bioorg. Med. Chem. Lett. 2012, 22, 837. 10.1016/j.bmcl.2011.12.053. [DOI] [PubMed] [Google Scholar]
- Charoenwattanasatien R.; Pengthaisong S.; Breen I.; Mutoh R.; Sansenya S.; Hua Y.; Tankrathok A.; Wu L.; Songsiriritthigul C.; Tanaka H.; Williams S. J.; Davies G. J.; Kurisu G.; Cairns J. R. K. ACS Chem. Biol. 2016, 11, 1891. 10.1021/acschembio.6b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Hammed M.; Breiden B.; Schwarzmann G.; Sandhoff K. J. Lipid Res. 2017, 58, 563. 10.1194/jlr.M073510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski G. A.; Osiecki-Newman K.; Dinur T.; Fabbro D.; Legler G.; Gatt S.; Desnick R. J. J. Biol. Chem. 1986, 261, 8263. [PubMed] [Google Scholar]
- Sawkar A. R.; Cheng W.-C.; Beutler E.; Wong C.-H.; Balch W. E.; Kelly J. W. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15428. 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steet R. A.; Chung S.; Wustman B.; Powe A.; Do H.; Kornfeld S. A. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13813. 10.1073/pnas.0605928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panday N.; Vasella A. Synthesis 1999, 1999, 1459. 10.1055/s-1999-3654. [DOI] [Google Scholar]
- Panday N.; Canac Y.; Vasella A. Helv. Chim. Acta 2000, 83, 58.. [DOI] [Google Scholar]
- Wang B.; Olsen J. I.; Laursen B. W.; Navarro Poulsen J. C.; Bols M. Chem. Sci. 2017, 8, 7383. 10.1039/C7SC01540B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heightman T. D.; Vasella A.; Tsitsanou K. E.; Zographos S. E.; Skamnaki V. T.; Oikonomakos N. G. Helv. Chim. Acta 1998, 81, 853. 10.1002/hlca.19980810507. [DOI] [Google Scholar]
- Harrak Y.; Barra C. M.; Delgado A.; Castaño A. R.; Llebaria A. J. Am. Chem. Soc. 2011, 133, 12079. 10.1021/ja202610x. [DOI] [PubMed] [Google Scholar]
- Li Y.-K.; Byers L. D. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1989, 999, 227. 10.1016/0167-4838(89)90001-0. [DOI] [PubMed] [Google Scholar]
- Field R. A.; Haines A. H.; Chrystal E. J. T.; Luszniak M. C. Biochem. J. 1991, 274, 885. 10.1042/bj2740885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J. R.; Xu Y. H.; Sun Y.; Knight A. L.; McLean P. J.; Caldwell G. A.; Sidransky E.; Grabowski G. A.; Krainc D. Cell 2011, 146, 37. 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg M. E.; Burke D.; Heales S. J. R.; Cooper J. M.; Hardy J.; Wood N. W.; Schapira A. H. V. Ann. Neurol. 2012, 72, 455. 10.1002/ana.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.