

Abstract
Cell migration is an adaptive process which depends on and responds to physical and molecular triggers. Moving cells sense and respond to tissue mechanics and induce transient or permanent tissue modifications, including extracellular matrix stiffening, compression and deformation, protein unfolding, proteolytic remodelling and jamming transitions. Here we discuss how the mechanoreciprocity of cell-tissue interactions allows cells to change position, and to define single-cell and collective movement, structural and molecular tissue organization, and cell fate decisions.
Introduction
Most cells in multicellular organisms are able to move during defined phases of tissue formation, maintenance, regeneration and immune defence, but also during diseases such as chronic inflammation and cancer1,2,3. To exert force for movement, cells interact with tissue structures such as the extracellular matrix (ECM) and other cells. The molecular organization and function of these interactions are adaptive, and vary between cell types and tissues. Although commonly studied as separate biophysical domains, ECM and cell functions are strictly interdependent and coevolve in all tissues. The resulting bi-directional crosstalk, termed dynamic reciprocity4,5 results in a gradual evolution of both the cell and the tissue through which it migrates6.
Well-defined in vitro models allow direct probing of isolated physicochemical parameters of cell migration, including the role of dimension, ECM stiffness, confinement and barrier function by the tissue, and their consequences for individual or collective cell migration7. In vivo models, such as Drosophila and zebrafish embryos and adult mice allow cross-referencing of those ECM aspects that influence cell migration in physiological and disease contexts8. These approaches have revealed that cells and engaged tissue can be regarded as multi-component viscoelastic units, subject to reciprocal mechanochemical interactions that induce, guide or limit cell migration in a context-dependent manner2,3,7. These relationships between the cell and its ECM context are inherently bi-directional, and aptly described by the term ‘mechanoreciprocity’9. We here review the force-responsive elements involved in cell-ECM interactions in the context of cell migration, summarizing the fundamental physical and molecular properties of tissues and cells that determine cell-tissue interaction and migration and we develop a framework for direct and indirect mechanoreciprocity between migrating cells and their extracellular environment. As an emerging concept, mechanoreciprocity controls the migration mode, the ECM remodelling responses and the outcomes for assembling and remodelling tissue structures.
Mechanical properties of ECM
Cells respond to tissue organization and mechanics at subcellular10, cellular11 and multicellular12 scales through interactions between the plasma membrane and the substrate This process, called mechanotransduction, involves different structural and functional parameters, here termed ‘modules’. The mechanical modules of tissues are determined by their constituent materials. Physical modules of tissues that jointly influence cell migration include ECM stiffness, confinement and topology (reviewed in2). Modules evolve and vary with cell type, tissue context and cell activation state. They depend on their spatial ECM arrangement, degree of crosslinking and other chemical modifications, as well as hydration state and stresses induced by cells or extracorporeal forces, as discussed in more detail below. Additional mechanical modules controlling cell migration include tissue porosity and nanotopology (Box 1).
Box 1. Emerging modules of tissue and cell mechanics.
Porosity
The porosity of the tissue varies from >100 μm2 between collagen fibrils in loose connective tissue and lymph nodes, to <1 μm2 between dense collagen bundles16,21. Nearly-impenetrable dense ECM impedes cell migration and requires particular abilities, such as the capacity to strongly deform the nucleus and/or to proteolytically degrade ECM and generate space111. Collagen-rich stroma and basement membrane are examples of such high-density environments158. Loose to medium-density ECM has pores that match the cell size with pore sizes around the nuclear cross-section (30-70 μm2, Fig. 1c, arrows) and represent a minimal barrier for migration at maximum speed, without requirement of tissue degradation36,79.
Nanotopology and curvature
The order of ECM macromolecules and their surface texture provide complex 3D nanopatterns. Cells discriminate aligned from disordered patterns for guidance of migration22. Engineered fibrils of 400 nm in diameter support 2-fold faster migration speed compared to 700-1200 nm fibrils159. The surface of collagen fibrils provides nanotexture by D-periodic bands160 (Fig. 1b), and globular patterns from adhering macromolecules160, yet it is unclear which level of nanoscale can be resolved by cells. The 2D structure of basement membranes is a meshwork of nanoscale pores and fibrils161 (Fig. 1b), but engineered nanoridges of comparable scales exert no apparent impact on cell migration when compared to a planar surface162. Thus, at nanoscale, moving cells likely sense protein substrate as a 3D topology, integrate curvature as either ridge-like or flat surface, and interpret basement membrane nanotopology as ‘2D’. Tissue curvature furthermore induces spatial patterning of mechanical stresses and proliferation of cell sheets, suggesting a role in enhancing proliferation and, likely, guiding migration163.
Tissue hydration
Tissue hydration is maintained by interstitial fluids, which flow between ECM macromolecules by convection. Both freely flowing and GAG-bound water fill the ECM space and regulate porosity as a dynamic equilibrium23. When vascular permeability increases during tissue trauma, inflammation or cancer, interstitial water influx increases hydrostatic pressure by multi-fold, followed by tissue swelling (edema) with increased ECM network porosity and tension by hydrostatic pushing164,165 (Fig. 1c). Edema accompanies acute and chronic tissue responses with increased migration of infiltrate leukocytes and stromal cells, and hydrostatic regulation of ECM porosity and alignment facilitates cell trafficking166. Beyond mechanical effects, edema accelerates interstitial fluid flow, which redistributes chemotactic proteins and contributes to cell guidance167.
Cell stiffness
Cell stiffness is predominantly determined by the nucleus, cortical actin, and the cytoskeleton168. Bundled actin creates higher local stiffness compared to diffuse actin structures42,43. Cell stiffness thus scales with the traction force generated by cells and lowering stiffness facilitates shape adaptation of moving cells169. Invasive cancer cells are less rigid than benign or less invasive counterpart cells169, indicating distinct cytoskeletal organization.
Material stiffness measures the amount of force required to induce a change in length. Technically, stiffness is not the same as the elastic modulus; it is common practice in tissue- and cell mechanics to use the terms interchangeably, and we here refer to both as stiffness. Stiffness depends on the composition, architecture and momentary forces acting upon the tissue. When analysed at micrometre scale, tissue stiffness varies from soft and deformable, such as brain or provisional ECM, to very stiff and non-deformable like in bundled collagen or bone (Fig. 1a). At micro- and nanoscales, ECM mechanics vary even more; single collagen fibres are multi-fold stiffer than fibrillar collagen networks13. Cells can sense substrate stiffness in the range from 0.1 to at least 25 kPa14 through integrin adhesion receptors15, and respond to stiffer substrate with preferential protrusion and alignment parallel to the substrate. Notably, this stiffness response is well established for fibroblasts and epithelial cells, but may vary for other cell types. This interaction between cellular and substrate mechanical modules is a principal component of the reciprocal relation between cell and matrix.
Figure 1. Physical ECM modules determining cell migration.
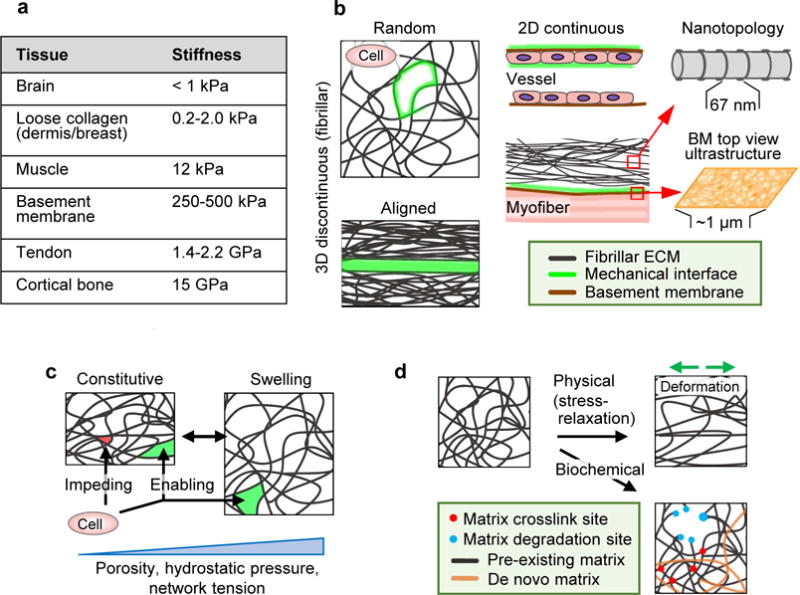
Tissue properties determining cell migration, including topology of the ECM and organization of interfaces between tissue structures. A Elastic modulus range present in macromolecular tissue structures. GAG-rich stroma surrounding cell networks, such as brain tissue, is soft (below 1.0 kPa)48; loose fibrillar type I and III collagen-based porous protein networks such as dermis and breast scale typically between 0.2 and 2 kPa111,118. Thicker, more crosslinked collagen bundles in muscle are substantially stiffer (12 kPa)48, which reaches the low GPa range in tendon153. Basement membranes consisting of type IV collagen and laminins have a stiffness in the higher kPa range, with at least double the stiffness on the epithelial side compared to the stromal side154. The stiffness of calcified tissue, including cortical bone, can go up to 15 GPa155. B Principal ECM geometries defining mechanical cell migration interfaces (green) including 1D, 2D and 3D organization and nanotopology. C Baseline porosity range, relative to cell size and deformability, and hydrostatic pressure induced reversible swelling and mechanical alterations in fibrillar ECM. D Irreversible changes induced by physical or biochemical factors. Direction of physical deformation denoted by green arrows.
The structural organisation of the ECM influences cell behaviour at different levels. ECM comprises both randomly and oriented, fibrillar or sheet-like protein polymer networks. Fibrillar networks provide both space (pores) and anchorage sites (fibrils) for moving cells, and typically pervade tissue designed to support moving cells16,17 (Fig. 1b). Tissues with highly demanding mechanical functions, such as tendons and bones, are often highly ordered, as are the collagen-rich tissues of scars and deposits resulting from chronic inflammation or fibrosis18,19. Sheet-like interfaces composed of a basement membrane adjacent to interstitial connective tissue provide linear-configured shape and efficient guidance for moving cells20,21. Cells rapidly sense and respond to these structural modules at different dimensionalities: interactions with ECM in interstitial tissues occur in three dimensions (3D), with simultaneous interaction to two-dimensional (2D) structured basement membranes (Fig. 1b), and sparse collagen fibrils sensed as one-dimensional (1D) substrate. These dimensions likely coexist in 3D tissues and jointly determine other mechanical tissue properties, including porosity and nanotopology, and their responses to external pressure and changes in hydration (Box 1)21–23.
By interpreting ECM stiffness, order and porosity, migrating cells adapt their own mechanochemical repertoire of mechanically integrated cell functions, including adhesion, traction, protrusion, deformation and directional persistence. In addition to transient alterations of ECM modules through cell migration, permanent ECM remodelling takes place by enzymatic tissue remodelling (Fig. 1d). At the cellular level of integrating these cell modules, the actin cytoskeleton defines adhesion strength and cell shape to generate pulling or pushing force and induce tissue remodelling and different migration modes (Fig. 2a, b). At the molecular level, integration is mediated by mechanically responsive proteins which can unfold when pulled, and develop mechanically adhesive or signalling functions in response to tension24. Both levels of adaptive responses are discussed in more detail below.
Figure 2. Mechanical cell modules in cell migration.
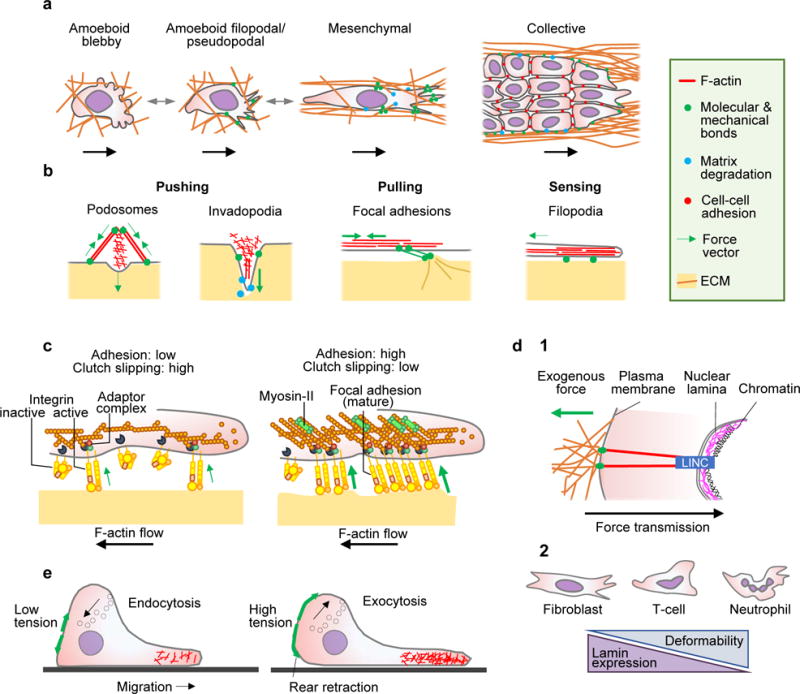
A Cell migration modes in 3D environments, including single-cell and collective migration. B Actin-rich cell surface structures mechanically interacting with tissue, including: podosomes probing the substrate vertically; invadopodia protruding and locally degrading substrate in vertical direction by releasing proteases; focal adhesions generating adhesion and pulling force; and filopodia extending along ECM structures and generating traction force in parallel. C Differential adhesion regulation. Tension generated by mechanical extracellular interactions of adhesion receptors depend upon intracellular adaptor proteins linking to the actin cytoskeleton, which flows in rearward direction (clutch). The strength and duration of adhesion receptor bonds to substrate increase with force156 and are regulated by lateral clustering of integrin adhesion receptors, which increases the number of bonds157. Diffusely distributed integrins exert weak adhesion and traction force (left). With integrin clustering, adhesions increase the number of engaged bonds to actin filaments and recruit myosin-II, providing stronger adhesion and traction force towards the substrate (right). D Nuclear mechanics. (1) Mechanical linkage between ECM, cytoskeleton, nuclear lamina and chromatin through the LINC complex (linker of nucleoskeleton and cytoskeleton) consisting of nesprins 1-4 and Sun1/2 proteins, which regulate nuclear positioning and deformation in response to cell responses to extracellular cues47. (2) Shapes and correlation between nuclear deformability and lamin expression. E Membrane tension and cell migration. Left panel, low membrane tension in partly polarized cell facilitating actin polymerization. Right panel, fully polarized cell with high membrane tension limits actin filament protrusion at the leading edge, but supports rear contraction and is counterbalanced by transport of intracellular vesicles to the plasma membrane. Green arrows, force vectors.
Cellular responses and mechanotransduction
By combining pushing and pulling, cells can adapt their shape and exhibit complex mechanocoupling responses to achieve migration (Fig. 2b). The actin cytoskeleton mediates both pushing and pulling. Polymerizing actin filaments push and protrude the membrane, under the control of Rho GTPases25,26, whereas pulling depends upon myosin motors which crosslink, bundle and contract actin filaments under the control of Rho-associated protein kinase (ROCK)27, transmitting force to the substrate28,29. Adaptor proteins play an important role in this context by connecting actin filaments towards extracellular structures, transmitting both signals and force: talin links actin filaments to focal adhesions and podosomes30, ezrin/radixin/moesin (ERM) proteins connects to the plasma membrane31; nesprins engage with the nuclear membrane32, and α-catenin and afadin bind adherens junctions33. Adaptor protein functions are dynamic, transient and contribute to pulling1,13, and further cooperate with intermediate filaments and microtubules. Moreover, adaptor proteins mediate a direct structural and mechanical interaction between cell and matrix.
Actin-based force transmission occurs at adhesion sites, mediated by integrin adhesion receptors13,34. Molecular bridges, provided predominantly by talin and vinculin34, transiently connect actin filaments with integrins and function as a ‘molecular clutch’, which translates retrograde actin flow into traction force towards the substrate35 (Fig. 2c). Strong forces result from longer-lasting actin-rich focal adhesions connected to bundled and contractile actin filaments36, and low forces are transmitted by small integrin foci or diffusely organized integrin contacts13. Adhesion maturation and regulation of traction force in mesenchymal cells depends on myosin II motors contracting actin filaments37, and involves talin28,38 and filamin-A linking integrins to actin39.
As a consequence of such plasticity of actin dynamics and adhesion regulation, actin networks and adhesion sites can respond to substrate geometry and stiffness. ECM geometry can guide actin orientation and cell alignment40: adhesion sites and actin nucleation preferentially grow along substrate edges13,36,41. At low substrate stiffness, cell adhesions are labile with cortical actin diffusely distributed13,28,42,43, but at higher substrate stiffness, adhesion strength increases, the actin cytoskeleton develops bundled and aligned filaments which contract and generate higher force42,43. For efficient migration, cells locally form and resolve adhesions, adjusting their degree of adhesion to the amount of available ligand and substrate stiffness13,24. In parallel, non-integrin adhesion systems, such as cell-surface heparan sulfate proteoglycans (HSPGs such as CD44v3, syndecans, glypicans and betaglycan) engage with fibrillar protein networks, provide additional, weaker adhesions and thereby co-regulate focal adhesion strength and cytoskeletal organization44. In addition to cell-matrix adhesion, adhesive coupling to neighbouring cells is achieved by cadherin-mediated adhesions, which connect to the actin cytoskeleton and allow stress distribution through cells, and sustain a force of approximately 100 nN perpendicular to the cell surface45. Importantly, besides regulating cell-ECM and cell-cell interactions, the actin cytoskeleton anchors the nucleus and co-regulates cell shape, volume and membrane tension.
The nucleus, the largest and stiffest organelle46, is mechanically linked to cytoskeletal filaments which determine its shape and position47, protecting nuclear content from mechanical assault. The shape and stability of the nucleus is provided by nuclear lamins, which stabilize the nuclear envelope,form a 3D network inside the nucleus46 and connect to all three cytoskeletal networks (Fig. 2d, 1). The stiffness of the nucleus ranges from 0.1 to 10 kPa48; this stiffness range is cell-type dependent and regulated by expression levels and assembly state of lamin A48. There is a direct link between nuclear mechanics and cell migration: reducing lamin A expression increases nuclear deformability (Fig. 2d, 2) and permits migration through smaller pores, whereas increased lamin A levels limit cell deformation and migration speed in 3D environments49,50 and protect the DNA from mechanical damage49,51.
There is also an interplay between the cell membrane and cytoskeletal dynamics in the migrating cell. Cytoskeletal polymerisation or hydrostatic pressure (discussed below) pushes the cell membrane outwards, leading to membrane deformation and increased tension (Fig. 2e)25. Membrane tension is defined as the surface free energy per unit area, and is measured by determining the force required to displace a membrane-bound bead with optical tweezers52. Values range between 0.2-1.6 mN/m in single cells53. Membrane tension further depends upon the net amount of membrane at the cell surface (Fig. 2e). Consequently, extension-retraction cycles in migrating cells cause and depend upon cell surface- and membrane tension regulation, and membrane tension, in turn, regulates the speed of actin polymerization by acting as a physical barrier. In protruding lamellipods, membrane of high tension is pushed by a dense actin network with filaments growing at steep angles, whereas the actin network at lower membrane tension has a lower density and contains more filaments oriented perpendicularly to the membrane54. In migrating neutrophils, increasing membrane tension impedes actin nucleation25 through phospholipase D2 (PLD2) and mammalian target of rapamycin complex 2 (mTORC2) signalling55. Conversely, low membrane tension triggers actin assembly, and enhances cell spreading and polarization56. As a consequence, any shape change during migration imposes fluctuations in membrane tension which, in turn, regulate actin networks and motility.
During cell shape change and migration, cells regulate their internal hydrostatic pressure through contraction of the actomyosin cortex, membrane tension and mechanosensitive channels. Intracellular pressures range from ~20-100 Pa, and, during cytokinesis, may rise to ~400 Pa53. High intracellular pressure pushes the plasma membrane outward in regions of low actin density, generating bleb-like protrusions or lobopodia, a hybrid of an actin-rich pseudopod and a membrane bleb57, which are crucial for migration. In confined space and channels, cancer cells polarize transmembrane ion and water channels to the leading and trailing edges, and move through anterior expansion and rear membrane shrinkage58. To form lobopodia, fibroblasts migrating in confining 3D tissue increase intracellular pressure towards the leading edge by moving the nucleus forward. This causes a rear-to-front hydrostatic pressure compartmentalization that enhances front protrusion and movement59. Both, actin protrusion and hydrostatic pressure are counteracted by membrane tension, together forming a balanced system with strong self-regulatory kinetics.
In summary, tissue and cell modules both function interdependently, forming a dynamic network of activities which occur in parallel or in series, depend upon each other and require coordination in the same cell to adhere, polarize and migrate.
Modes and mechanics of cell migration
In an ongoing process of sensing and execution, moving cells interpret multiple physical ECM parameters in parallel and translate them into an integrated response which involves multiple cell moduli and determines cell shape, polarity, stiffness, and other functions. By adjusting multiple moduli, cells regulate their cytoskeletal organization and the force balance towards ECM substrate and neighbouring cells and adapt both migration strategy and efficacy.
Cells generating low traction towards the substrate adopt rounded shapes, so called ‘amoeboid’ morphology (Fig. 2a) and displace their cell body predominantly by cortical actin flow and contractility60. Mechanotransduction through poorly assembled or non-focalized adhesions occurs by low-level friction61 or mechanical intercalation between extracellular structures by cell deformation and lateral protrusions62. When adhesion and traction forces are higher, migrating cells adopt spindle-like shapes, or ‘mesenchymal’ morphology (Fig. 2a)3,13. Here, integrin-based adhesions are more focalized and generate higher traction towards the substrate13,28,36. In 3D environments, amoeboid-moving cells tend to push, whereas mesenchymally moving cells pull on the substrate63, indicating distinct force-generating principles between migration modes. When cell-cell junctions remain intact, cells migrate collectively (Fig. 2a) and generate a combined force towards the substrate, resulting in substrate deformations beyond single-cell dimensions12,64,65. Consequently, the range of traction forces generated by moving cells is higher in collectively moving cells64,65.
The strength of traction force generated by cells is further sensitive to microenvironmental conditions, including substrate stiffness66, ligand density67, the intracellular processing of focal adhesion adaptor proteins by calpains68, and substrate organization41. By integrating these microenvironmental modules, moving cells regulate adhesion and contractility, transit between high- and low-traction force levels, and switch between migration modes69 (Fig. 2a, arrows). Switching between amoeboid and mesenchymal migration modes occurs in tumour cells in response to experimental regulation of cell protrusion and actomyosin contractility70, varying integrin availability or substrate stiffness71,72. Thus, moving cells can be viewed as multi-component viscoelastic units which constantly adjust their mechanochemical networks to navigate through heterogeneous tissue.
Contact guidance provides cell orientation by combining ligand binding and mechanocoupling with cell alignment along an anisotropic solid structure, linear or curved, which guides the moving cell body36,73. Contact guidance depends upon alignment of actin-rich protrusions and/or focal contacts along the substrate, with notable edge effects along curved or discontinuous substrate countours36. Moving fibroblasts and tumour cells form precisely aligned focal contact-like structures along the edges of fibrils of oriented collagen gels74,75 or the ridges of grooved surfaces36,76. In a cell-type dependent manner, the efficacy of contact guidance depends upon optimal spacing between parallel fibrillar or linear patterns, integrin-mediated and integrin-independent adhesions77, and myosin-mediated traction force78. The precision of guidance decreases when cell-substrate adhesion diminishes77, or when other forces counteract cell alignment such as those from cadherin-based cell-cell junctions during collective migration36. In 3D environments, contact guidance along nanotopologies is combined with cell gliding along paths of least resistance. Moving T cells precisely follow collagen fibre textures bordering complex-shaped pores79, and tumour cells align and migrate collectively along tracks of least resistance which are constitutive or pre-patterned by leader cells80,81. In all migration modes, physical and molecular ECM cues likely cooperate for contact guidance in anisotropic environments.
Stiffness directly guides cell migration in durotaxis, the movement of cells towards substrate regions of higher stiffness82. Single cells discern stiffness gradients from ~1 Pa/μm83 up to ~400 Pa/μm84 and depend on focal adhesions and actomyosin contractility to sample ECM stiffness and durotax11. In collective durotaxis, stiffness gradients are sensed through actomyosin force transmission between both ends of the cell group84. Single-cell durotaxis may depend upon differential stiffness sensing and tugging action between adhesion sites85 and stiffness-dependent differential integrin clustering86. Collective durotaxis may result from differential binding and unbinding rates of integrins and differential molecular clutch to actin flows, which jointly generate preferential mechanocoupling and movement towards stiffer higher substrate84.
Haptotaxis directs moving cells towards a gradient of increasing density of immobilized ligand, such as dendritic cell navigation towards tissue-bound chemokine CCL21 to navigate towards lymph vessels87, or cancer cell movement towards increasing ECM concentrations88. However, haptotaxis could also be a consequence of differential mechanical force coupling as both the Arp2/3 complex, which branches actin filaments in lamellipodia, and fascin, which bundles actin in filopodia, support haptotaxis along fibronectin gradients89,90 and differential actomyosin contractility defining front-rear asymmetry91.
Modelling of cell migration mechanics
This astounding parameter range for ECM and cell states, their interactions and adaptations, is challenging to fully control and co-register in cell-based experiments in vitro or in vivo, which is why often only a handful of parameters are tested. To complement wet-lab experiments, in silico modelling can recapitulate cell-based experiments and predict outcomes with broadened parameter space. Modelling allows dissection of mechanistic dependencies in time and space, which define the type, efficacy, decision making and steering of cell migration in response to tissue organization92,93. We describe principles of modelling in Box 2 and provide examples of recent efforts to model decision making in cell migration.
Box 2. In silico modeling.
Principles of modelling cell mechanics
At subcellular scales, network models, including open cell foam, beam and cable models, and tensegrity models (Table 1), combine the elastic, force-extension properties of fibrillar cytoskeletal proteins with their spatial organization170. Intracellular processes can be modelled as continua171 or discrete cell structures, including dynamic actin networks or bundles and external mechanical conditions172. Network models describe the nonlinear response of heterogeneous filamentous environments and their reciprocal interactions during cell migration. For example, the stiffness response of weakly deformed crosslinked actin networks was predicted to be a linear function of the concentration of single filaments173, confirmed by mechanical characterization of reconstituted actin protein networks in wet-lab experiments174. Network models of transient actin filament bundling, combined with cytosolic liquid models recapitulate higher-order assemblies, such as filopodia175. Whole-cell mechanical modelling requires coarser and/or multiphasic mechanical models to capture ECM orientation, bundling, and heterogeneity. The behavioural switch between collective, mesenchymal and amoeboid migration modes, for instance, is modelled as a function of cell-tissue adhesive strength176.
Importantly, distinct in silico models can be combined and integrated to reach complex mechanical, molecular and outcome predictions. For example, a coarse-grained mechanochemical model for cell, ECM and adhesions, combined with reaction diffusion equations to model mesenchymal chemotaxis, predicts complex behaviours of cancer cell invasion177; detailed network models for ECM and cytoskeleton with stochastic reaction diffusion equations reveals filopodial dynamics in leader cells172, and mechanical models for ECM with CPM reveals the nuclear deformability as rate-limiting in confined cell migration178, similar to decelerated migration in dense fibrillar collagen111.
Mechanoreciprocity in silico
To model the dynamic coevolution of migrating cells and ECM requires linking complementary modelling strategies that resolve both cell and ECM states over time. Mechanomechanical models ascribe an overall energy function to coupled FEMs for ECM and cells, physically connected by adhesion sites. The minima of this energy function correspond to (meta)stable cell-matrix equilibria and dynamics are simulated using the evolution of the energy function towards its minima. For example, combining the density and polarization of cells, their contractility and strains at cell-cell and cell-ECM contacts, and the strength and abundance of cell-ECM adhesions reveals that strain stiffening and ECM alignment precede and support the detachment of individual cells from a multicellular cluster146. Other approaches combine discrete cell-based CPM polarization and movement, traction forces and durotaxis into an adaptive ECM continuum179. Emerging hybrid network models and reaction-diffusion equations resolve the intra- and extracellular spaces down to the fibrillar level to model the production, redistribution and remodelling of ECM caused by MMPs172. Such detailed models, however, generally do not permit simulations spanning longer timescales.
Single-cell migration models predict quantitative parameters of migration from mechanical models for mechanosensory pathways, adhesion and force generation. Cell motions can be modelled as different types of Random Walks (RWs), in which a motile agent migrates with defined average velocity along its path. Inputs are rules for velocity and randomness of the trajectory and modulators such as polarization or chemical gradients (Table 1). Linking RWs with mechanical models generates diverse modes of force generation (amoeboid, pseudo/filopodial or mesenchymal) and cell shapes94,95 (see Table 1). Further, single cell durotaxis was recapitulated by a RW model which linked persistence of movement to substrate stiffness96. Cell sensitivity to substrate geometry can be modelled by representing the cytoskeleton as a series of linear, ‘Hookean’ springs responding to small deformations, which can be shortened by myosin intercalation97. The resulting traction increases cell spreading on convex substrates, but collapses laterally and thereby increases directional persistence on concave substrates97.
Table 1. Modelling strategies for mapping of cell and tissue mechanics.
Modelling approaches (top to bottom) are ranked from lowest to highest resolution. Combinations of modelling approaches, where different structures are resolved at different length- and time scales, are termed Multiscale Models. Models in red employ one or multiple phenomenological parameters, models in green are physics-based.
| Modelling approach* | Model scales | Key input | Key output | Modelling principle | Application |
|---|---|---|---|---|---|
| Finite Element Modelling (FEM) | Tissue, organ (mm-cm) | Mechanical moduli, constitutive relations, shape | Organ scale shape changes; mechanical rupture | Different elements that represent local mechanical properties are combined, allowing scalable modelling of heterogeneous tissue | Complex, heterogeneous, multicellular systems in tissue |
| Cellular Potts Model (CPM) | Cell, cell aggregate (μm-cm) | Cell and ECM mechanics, target volume and area, speed | Single-cell and collective dynamics, cell shape, invasiveness, vascularization | Represents cells as domains on a regular lattice, with mathematical rules prescribing their ECM contacts, interactions, proliferation and migration | Cells and cell cluster growth and migration |
| Multiphasic, active and constitutive (SGR) modelling | Cell, tissue (μm-cm) | Constitutive equations; composition, porosity and anisotropy; energy consumption | Mechanical properties, dynamics and response of entire cells | Mathematical equations dictating the flow and elastic response of cell and environment | Predicting cell deformation, relaxation and shape change under applied stress |
| Network models (numerical, analytical) | Cell, microtissue (μm-mm) | Fibril (visco-) elastic response, network architecture | Visco-elastic constitutive (dynamic) response fibrils, incl. bundling and alignment; filopodial dynamics | Fibril bending and extension properties are modelled to predict network deformation responses | Cytoskeletal mechanics, ECM mechanics |
| Coarse grained molecular dynamics (CGMD) | Fibril, bundle (nm-μm) | protein aggregate, arrangement, PMF | Mechanical response of single fibrils; effects of quaternary structural defects and crosslinking | Small particles (i.e. molecules) are aggregated and modelled as single particles, allowing modelling of larger volumes | Protein assembly and mechanical response of single protein fibrils |
| Atomistic models | Protein (≤100 nm) | Amino acid sequence, chemical composition, force fields | Potentials of mean force (PMF) for CGMD, mechanical effects of mutations and molecular damage | Numerical solution of Newton’s equations for the motions of atoms, alone and in molecules | Single protein mechanics and structure |
The collective behaviour of cell aggregates can be predicted by the cellular Potts model (CPM) and cell jamming models (CJM). CPM, supplemented with information about adhesion receptor and ligand density, predicts increasing cell and ECM strains when ECM remains space-limiting, as in tumour growth and invasion98. CJM apply the principles of jammed granular solids99 (like sand piles) to describe multicellular stiffness and collective motions100. CJM recapitulate transitions between stationary and dynamic states, similar to the ‘glass-jamming transition’, by which disordered packing of discrete units transition between stationary, solid-like and flowing, liquid-like states100,101. CJM take as input the strength of cell-cell contacts and the membrane and cortical tensions to predict cell motions. Strong connections suppress motions between cells (cells are ‘jammed’), but when migration is inducted by a stimulus cell elongation and coherent motions result. Coupling the CPM to continuum-mechanical models for 2D and 3D ECM allows to understand directional collective cell movement as a function of ECM alignment102. As a mechanical mechanism for collective cell durotaxis, a generalized clutch model predicts differential force transmission along the cell edge facing the softer substrate84. Differential strength of adhesion and the actin clutch may thus be sufficient to mediate collective durotaxis. In the future, combining physical and chemo-dynamical models with genomic and even population-based evolution models will provide a systemic understanding of the hierarchies that underpin cell fate, aggregation and migration decisions.
Mechanoreciprocity of cell-tissue interactions during cell migration
We here define mechanoreciprocity as an iterative, cyclic process in which cells modify the organization and elastic response of the environment and reciprocally adjust their behaviour9. As a consequence, cell function states and tissue topology underlie structural and molecular coevolution (Fig. 3a, 1). Mechanoreciprocity is thus an adaptive process which occurs at different time- and length scales and magnitudes, and whereby any cell-induced change of tissue composition, architecture, or tensional condition results in altered tissue mechanics, reversibly or irreversibly, locally and/or globally. At the molecular level, fibrillar ECM and cytoskeletal protein networks, including fibrillar collagen, fibrin, vimentin and neurofilaments, undergo reversible stiffening when tensile or shear forces are applied103. By pushing and pulling on ECM, moving cells induce ECM compression and densification or strain stiffening and thus exert elastic forces. Subsequently, cell detachment may be followed by viscoelastic substrate relaxation. Additionally, permanent ECM remodelling by deposition, crosslinking and degradation of ECM, may impose more long-lived responses. When two properties coevolve, three outcomes are possible (Fig. 3a, 2)104: convergence to steady state, such as quiescent cell and tissue function; periodic behaviours, such as the extension-retraction cycles in moving cells; or irregular outcomes, such as destabilized epithelial cohesion, perpetuated ECM remodelling and further deteriorating cohesion101. Thus, steady-state and oscillatory behaviours underlie predictable and often self-limiting physiological interactions, whereas chaotic coevolution may cause pathological processes.
Figure 3. Cellular and molecular mechanoreciprocity in tissue regeneration and disease.
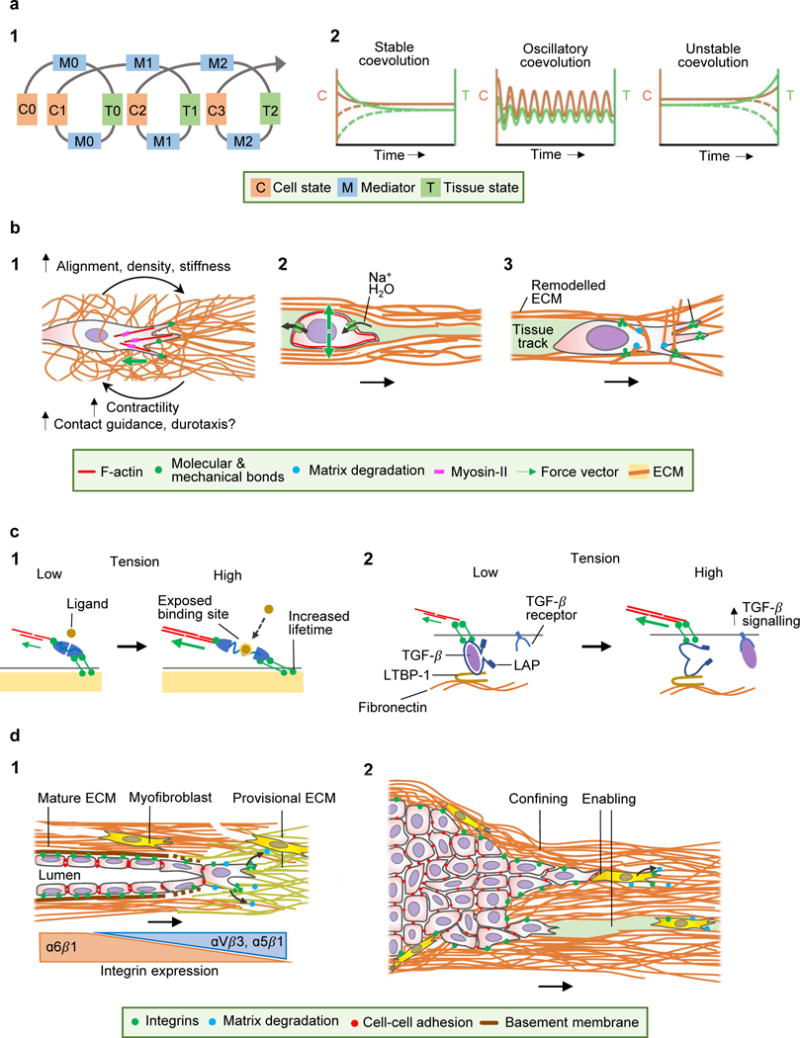
A (l) Spiral concept of mechanical cell-tissue interactions, describing coevolution of cell and tissue mechanics. Cells impose ‘mediators’ (pulling, pushing, ECM deposition, ECM degradation) and thereby alter tissue modules. Through iterative reinforcement (indicated by the spiral) or negative feedback, both cell and tissue modules undergo coevolution towards altered morphology and function. (2) Development of cell and tissue coevolution, including acquisition of a stable equilibrium (left), oscillatory coevolution with both positive and negative feedback loops (middle), or unstable coevolution, typically caused by negative feedback or disruption of co-engagement (right). The dashed lines indicate the temporal coevolution for perturbed initial conditions; in case of stable or oscillatory coevolution, the system converges to well-defined asymptotic behaviour, in the case of unstable coevolution the sensitive dependence on initial conditions typical of chaotic systems is seen. B Mechanoreciprocity in cell migration. (1) Strain stiffening of an ECM network by tension at the leading edge, creating a multi-parameter mechanochemical feed-forward loop. (2) Tissue compression by migrating cells, condensing ECM. Intracellular hydrostatic pressure is jointly maintained by stress-sensitive ion and water channels and actomyosin contractility. (3) Structural ECM remodelling. Mesenchymal migration leads to traction and fibre realignment by the leading edge, followed by pericellular proteolysis of collagen fibrils constraining the cell body, which after fibril realignment leaves behind a remodelled tissue track. C Molecular mechanoreciprocity. (1) Unfolding of mechanosensitive proteins by force. Actomyosin-contraction and tension exposes bioactive domains in adaptor proteins (e.g., talin, vinculin, p130CAS), which allows further ligands to bind and alter function in a strain-dependent manner (e.g. increased lifetime of catch-bonds). (2) Strain-dependent activation of TGF-β1. Cell adhesion and tension to ECM-tethered LAP induces a conformational change and releases TGF-β to diffuse and bind to its receptor. LTBP-1, latent TGF-β-binding protein 1. D Mechanoreciprociticy in disease. (1) Concordant immigration of fibroblasts and endothelial cells into the wound bed, which realign and degrade provisional ECM and synthesize collagen and basement membrane proteins and undergo a transition of engaged integrin systems. As outcomes, tissue alignment, density and stiffness are reciprocally linked to fibroblast function. (2) Mechanoreciprocity in cancer invasion. Dual function of ECM deposition and stiffening by myofibroblasts in sub regions, leading to encapsulation or invasion along collagen interfaces.
Epithelial, endothelial and mesenchymal cells restructure tissue while moving, and these alterations iteratively impact cell function. Beyond position change, cell migration thereby contributes to tissue building and maintenance, as discussed further below, but also to tissue regeneration and chronic disease.
Cells can generate traction force upon ECM networks, reversibly deform ECM architecture and locally increase stiffness and ligand density42,65 to transiently impact cell function. Moving epithelial and mesenchymal cells locally stiffen collagen networks by up to 1 kPa42,65, and similar stiffening can induce invasive behaviour in breast cancer cells105. Beyond stiffening, moving cells induce ECM network alignment and densification in the direction of force64,65, and this augments the cellular force response and cell stiffness42 (Fig. 3b, 1). Cell-induced strain stiffening depends upon β1 integrin and actomyosin-mediated mechanocoupling, as well as activation of focal adhesion kinase (FAK), p130Cas and nuclear myocardin-related transcription factor-A (MRTF-A)64. In concert, strain stiffening with locally increased ligand density may reinforce duro-/haptotaxis and contact guidance75,82,106,107, as a cell-autonomous mechanical mechanism for directional self-steering64.
Furthermore, moving cells push against tissue structures. Actin-based cell protrusions, including the leading edge and podosomes, generate protrusion forces and compress deformable substrate very locally30. Larger, cell-scale tissue deformation occurs when the moving cell body and particularly the nucleus move in 3D confined space and push against mechanically confining boundaries20,21 (Fig. 3b, 2). T-cells crawling through 3D tissue deform their cell body, but also displace ECM fibrils in a mechanically integrated process79. Tumour cells moving along engineered microtracks in 3D collagen push against and condense the collagen interface, without degrading collagen by matrix metalloproteinases (MMPs), and thereby widen the track in which they move20. Within expanded tracks moving cells can rearrange as a collective strand20, representing a mechanically reciprocal step towards self-organization.
The ECM can also be remodelled in a non-reversible fashion by biochemial modification and external mechanical stresses. Examples include proteolytic ECM degradation by MMPs108 and ECM deposition and crosslinking109 to first alter and then stabilize the altered structure (Fig. 1d). In addition, contractile cells can permanently deform, densify and align ECM networks between cells by stress-induced tension110, alter ECM porosity and nanotopology, ligand type and density, and these changes reciprocally define whether and how the cell migrates111,112. Contact-dependent regions of cell confinement by ECM structures, including fibrillar barriers, are preferentially cleaved by MT1-MMP, and loose ends of fibrils become realigned along adjacent structures112,113 (Fig. 3b, 3). As a consequence, a path largely cleared of ECM allows cells to move through originally much denser, impenetrable tissue (pores <5-10 μm2)111. The cleared ECM path represents a confining interface for contact guidance of follower cells and transition to collective movement112,114. For cell passage through basement membranes, localized ECM remodelling by podosomes, invadopodia and stromal cells likely cooperate with mechanical pushing and pulling to form a structural gap through which the cell migrates115,116. Breaching the basement membrane is critical in vascular sprouting117, and supports cancer metastasis to distant organs118. Proteolytic movement thus introduces ECM remodelling, with functional consequences for the cell itself and follower cells.
Further, forces occurring between cell surface and ECM may conformationally unfold strain-sensitive ECM, adhesion and cytoskeletal proteins24. When deformed, strain-sensitive proteins expose previously cryptic epitopes and alter function, such as the number of exposed adaptor sites, enzymatic activity, or signalling state. Strain-sensitive cytoskeletal adapter proteins include talin38 and p130Cas119, and increasing load can prolong the substrate bond lifetime of integrins120, actomyosin121, and cadherin-catenin complexes122 (Fig. 3c, 1). When stretched by cells engaging α5β1 and αvβ3 integrins, anchored fibronectin fibrils unmask previously cryptic adhesion sites for further integrin binding and fibronectin assembly123. Through a similar mechanical process, integrin αvβ6 exerts tension upon the prodomain of latent transforming growth factor beta 1 (TGF-β1), which liberates TGF-β1 from latency-associated peptide (LAP), an anchor protein covalently linked to ECM124. TGF-β is released by as little as 40 nN pulling force125, and its release is facilitated on pre-strained ECM, such as during tissue remodelling by myofibroblasts126 (Fig. 3c, 2). Tissue stiffening enhances TGF-β activation and TGF-β activates cytoskeletal contractility, and both events cooperate to additional release of TGF-β. Thus, mechanical protein unfolding is fundamental in converting forces into biochemical signalling and, again, actomyosin based force transmission, in a reciprocal cycle24.
In stationary epithelia with isotropic force distribution, connected cells adopt polygonal shape and cease migration with high cell density (‘jamming transition’). Physical or molecular stimuli may, however, initiate collective migration of confluent epithelia with cell-cell junctions retained3,6, and transition from stationary to collectively migrating states is supported when cell-cell adhesions are strong101. Dysfunctional regulation of cortical tension, observed in freshly isolated asthmatic epithelium, facilitates unjamming with extensive cell flows and disrupted epithelial stability101. Besides cell-cell interaction stability, confined space may force loosely connected cells to establish cell-cell junctions, undergo a partial jamming transition and move collectively along joint paths80. The jamming transition concept provides a multi-parameter framework defining transitions of cytoskeletal interactions across cell boundaries and pressure conditions during collective movements in confined tissue space.
Mechanoreciprocity in disease
Tissue regeneration initiated by, for instance trauma or inflammation aims to reinstall the integrity and function of epithelia, connective tissue and blood vessels. After wounding, provisional fibrin- or fibronectin-rich ECM is colonized by fibroblasts and endothelial cells from adjacent intact tissue which jointly recreate vascularized connective tissue127 (Fig. 3d, 1). Interstitial fibroblasts secrete proteases which dissolve the provisional ECM while depositing fibrillar collagen networks128. Initially loose collagen networks become aligned by contact-dependent collagenolysis mediated by MT1-MMP129. Concurrent with collagen deposition, fibroblasts co-engage collagen-binding α1β1, α2β1 and α11β1 integrins, together with fibrin- and fibronectin-binding αVβ3, αVβ5, α5β1 and αVβ1130, and these multi-ligand engagements mediate focal adhesion strengthening, stress fibre formation, and contractility to reinstall collagen bundling and tissue tension13,39,131. This leads to a step-wise transition from randomly textured fibrin to comparably ordered collagen ECM, while fibroblasts transit from migratory and secretory to resident and contractile state128 (Fig. 3a, 2: stable coevolution). Concurrently, endothelial cells collectively sprout into the wound bed from intact neighbouring vessels. Endothelial tip cells are initiated by proteolytic invadopodia which focally degrade the vascular basement membrane of intact vessels to enable cell penetration through the gap of least resistance117. Tip cells engage with fibrin predominantly via αVβ3, α5β1 integrins132, realign ECM by tension133, and engage MT1-MMP for fibrin and collagen degradation114,134. Follower cells connect through VE-cadherin and tight junctions127, move actively133 and deposit basement membrane along the cell-ECM interface117,135 (Fig. 3d, 1). Thus, endothelial sprouts create their own path of least resistance while depositing substrate for collective guidance.
After acute trauma, this canonical program to tissue regeneration is self-limiting, reaching an equilibrium of stable microanatomy of ECM and vascularization. However, when the defect persists in chronic wounds, perpetuation of fibrosis and/or inflammation can result in divergent outcomes. When the myofibroblast response dominates, collagen deposition and stiffening may perpetuate, as in vascular fibrosis in atherosclerosis136, the foreign body response18 or the reactive tumour stroma (see below). Network modelling of pulmonary fibrosis suggests that collagen deposition by preactivated fibroblasts and stiffening of lung tissue coevolve, with fibrosis as outcome137. When (sub)acute inflammation dominates, perpetuated proteolytic ECM remodelling and collagen degradation may ultimately destroy tissue (ulceration)138. Fibrotic encapsulation versus lytic tissue degeneration thus represent differently composed mechanoreciprocal progression and distinct outcomes of ECM remodelling.
Mechanoreciprocity is also relevant for cancer invasion and metastasis. In cancers, the growing lesion and the reactive tumour stroma coevolve as a self-propagating neo-tissue, including inflammation, fibrosis, neoangiogenesis and cancer cell migration6. As organizers of ECM remodelling, myofibroblasts become activated by tumour and stromal cell-derived cytokines139. Myofibroblasts deposit and crosslink collagen and other ECM proteins by lysyl oxidase (LOX), and thereby stiffen ECM118. High ECM stiffness potentiates mechanical and molecular reprogramming of cancer cells by enhancing growth factor signalling118; invadopodia activity140, tension, deformation and remodelling of basement membrane to support cell transmigration in vitro116 and metastasis in vivo141 and integrin signalling118, which supports survival and stemness. ECM stiffening further enhances TGF-β activation by myofibroblasts, in parallel to increased release of chemokines and matrisome proteins126,128. In stiff environments, excess TGF-β aggravates myofibroblast differentiation15, enhances fibrosis and diversifies cancer cell invasion plasticity by favouring single-cell dissemination139. Myofibroblasts and tumour cells jointly rearrange tissue topology, by aligning and bundling collagen, creating new corridors of single-cell width19 to guide cancer cells along paths of least resistance142,143 and favour partial cell jamming and transition to collective invasion80,112 (Fig. 3d, 2). Besides collagen, fibronectin is deposited by both cancer cells and myofibroblasts which provides a bi-modal scaffold for contact guidance of epithelial cancer cells through integrin signaling107 and for latent TGF-β1 activation by moving tumour cells using α6β4 integrin77. In other sub regions, tumour cells may be prevented from invasion by a collagen capsule. The capsule acts as a barrier and increases the intra-tumour pressure, limits blood vessel sprouting and blood supply and impedes delivery of systemic therapy144,145. In silico, ECM alignment and stiffening and contractile stresses during cell migration reinforce each other reciprocally, with intermediate ECM stiffness as most conducive to invasion146. Similarly, combining cellular Potts with fibrous ECM modelling predicts a biphasic cell response to collagen density, ECM stiffness and pore size, with optimum cancer migration and persistence at intermediate level of each module102. Thus, cancer cell invasion depends on both reciprocal interactions and fibrosis which, depending on topology, reprogram tumour sub regions to either promote migration or prevent it through fibrotic encapsulation (Fig. 3d, 2).
Conclusions and outlook
The reciprocity of cell and tissue mechanics generates a situation whereby virtually every step of cell and tissue biology depends upon mechanochemical events. For example, a purely mechanical signal can initiate a developmental program. In the developing Xenopus embryo, emigration of neural crest cells is triggered by a mechanical tension signal, which cooperates with preceding EMT signals and chemokine signals present in the stroma, yet neither stimulus alone suffices to induce delamination147. To distil cause-consequence relationships from multi-parametric wet-lab analyses, advanced statistics is required to discriminate the role of cell migration from integrated growth, survival and therapy response programs148 and such parametric data will allow in silico modelling to successfully combine large-scale tissue analysis with micro- and nano-topological models.
Besides cell migration and tissue remodelling, other intracellular processes respond to mechanical stimuli, including gene transcription, cell differentiation and metabolism149. Thus, force-sensitive adhesion signalling may cooperate with mechanosensitive ion channels, chromatin, transcription factors and the protein trafficking machinery. Current strategies of the mechanobiology field, as delineated in this review, focus on understanding complex mechanistic relationships, but ultimately aim to deliver novel insight and rationale for interference strategies. Examples include dampening adhesion signalling or actomyosin contractility, for instance by FAK or Rho kinase inhibitors148,150,151, to interrupt the ECM stiffness-induced cell programming. However, beyond mechanical functions, molecular effectors also contribute to signalling networks, and the mechanobiology in complex disease models may be complicated by parallel or counteracting networks. As an example, collagen-crosslinking enzymes including LOX increase ECM stiffness which enhances cancer invasion and metastasis109,118, however LOX can also dampen oncogenic signalling and limit neoplastic progression152. Combating chronic tissue stiffening and remodelling effectively may require bi- or multimodal intervention, such as co-targeting of neoangiogenesis and macrophage influx to prevent detrimental fibrotic scarring near biomedical implants18. In summary, mechanical mechanisms feed molecular processes, and vice versa, to co-direct cell and tissue homeostasis and pathology. Thus, the framework of mechanoreciprocity mandates us to integrate biomedical disciplines to enhance diagnostic and therapeutic workflows and improve disease control.
Acknowledgments
We thank Mirjam Zegers for proofreading the manuscript. The PF laboratory is supported by the European Research Council (617430-DEEPINSIGHT), NWO-Vici (918.11.626), Horizon 2020 consortium MULTIMOT (634107-2), the Cancer Genomics Center, NIH-U54 CA210184-01, the MD Anderson Cancer Center Moon Shot program, the Radboud Nanomedicine Alliance, and from CS by funds from NWO-FOM (E1012M, E1009M, E1013M) and the TU/e Institute for Complex Molecular Systems.
References
- 1.Gardel ML, et al. Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration. Annu Rev Cell Dev Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol. 2014;15:813–824. doi: 10.1038/nrm3897. [DOI] [PubMed] [Google Scholar]
- 3.Te Boekhorst V, Preziosi L, Friedl P. Plasticity of Cell Migration In Vivo and In Silico. Annu Rev Cell Dev Biol. 2016;3228361:1–28. doi: 10.1146/annurev-cellbio-111315-125201. [DOI] [PubMed] [Google Scholar]
- 4.Bornstein P, McPherson J, Sage H. In: Pathobiology of the endothelial cell. Nossel H, Vogel H, editors. New York: Academic Press; 1982. pp. 215–228. [Google Scholar]
- 5.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 6.Friedl P, Alexander S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Paul CD, Hung WC, Wirtz D, Konstantopoulos K. Engineered Models of Confined Cell Migration. Annu Rev Biomed Eng. 2016;18:159–180. doi: 10.1146/annurev-bioeng-071114-040654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Almeida PG, Pinheiro GG, Nunes AM, Gonçalves AB, Thorsteinsdóttir S. Fibronectin assembly during early embryo development: A versatile communication system between cells and tissues. Dev Dyn. 2016;245:520–535. doi: 10.1002/dvdy.24391. [DOI] [PubMed] [Google Scholar]
- 9.Paszek MJ, Weaver VM. The tension mounts: Mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 10.Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 2012;151:1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tambe DT, et al. Collective cell guidance by cooperative intercellular forces. Nat Mater. 2011;10:469–475. doi: 10.1038/nmat3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat Commun. 2015;6:8720. doi: 10.1038/ncomms9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 15.Park JS, et al. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials. 2011;32:3921–3930. doi: 10.1016/j.biomaterials.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf K, et al. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol. 2009;20:931–941. doi: 10.1016/j.semcdb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cicchi R, et al. From molecular structure to tissue architecture: Collagen organization probed by SHG microscopy. J Biophotonics. 2013;6:129–142. doi: 10.1002/jbio.201200092. [DOI] [PubMed] [Google Scholar]
- 18.Dondossola E, et al. Examination of the foreign body response to biomaterials by nonlinear intravital microscopy. Nat Biomed Eng. 2016;1:7. doi: 10.1038/s41551-016-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Provenzano PP, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilina O, Bakker GJ, Vasaturo A, Hoffman RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. Phys Biol. 2011;8:029501–029501. doi: 10.1088/1478-3975/8/1/015010. [DOI] [PubMed] [Google Scholar]
- 21.Weigelin B, Bakker GJ, Friedl P. Intravital third harmonic generation microscopy of collective melanoma cell invasion. IntraVital. 2012;1:32–43. doi: 10.4161/intv.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grossman M, et al. Tumor cell invasion can be blocked by modulators of collagen fibril alignment that control assembly of the extracellular matrix. Cancer Res. 2016;76:4249–4258. doi: 10.1158/0008-5472.CAN-15-2813. [DOI] [PubMed] [Google Scholar]
- 23.Maytin EV. Hyaluronan: More than just a wrinkle filler. Glycobiology. 2016;26:553–559. doi: 10.1093/glycob/cww033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu X, Margadant F, Yao M, Sheetz M. Molecular Stretching Modulates Mechanosensing Pathways. Protein Sci. 2017;26:1337–1351. doi: 10.1002/pro.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houk AR, et al. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell. 2012;148:175–188. doi: 10.1016/j.cell.2011.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Beningo KA, Hamao K, Dembo M, li Wang Y, Hosoya H. Traction forces of fibroblasts are regulated by the Rho-dependent kinase but not by the myosin light chain kinase. Arch Biochem Biophys. 2006;456:224–231. doi: 10.1016/j.abb.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elosegui-Artola A, et al. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat Cell Biol. 2016;18:540–548. doi: 10.1038/ncb3336. [DOI] [PubMed] [Google Scholar]
- 29.Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev. 2014;94:235–63. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- 30.Kronenberg NM, et al. Long-term imaging of cellular forces with high precision by elastic resonator interference stress microscopy. Nat Cell Biol. 2017;19:864–872. doi: 10.1038/ncb3561. [DOI] [PubMed] [Google Scholar]
- 31.McClatchey AI. ERM proteins at a glance. J Cell Sci. 2014;127:3199–3204. doi: 10.1242/jcs.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mellad JA, Warren DT, Shanahan CM. Nesprins LINC the nucleus and cytoskeleton. Curr Opin Cell Biol. 2011;23:47–54. doi: 10.1016/j.ceb.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57:815–855. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Case LB, Waterman CM. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol. 2015;17:955–963. doi: 10.1038/ncb3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1:761–772. doi: 10.1016/0896-6273(88)90124-9. [DOI] [PubMed] [Google Scholar]
- 36.Ray A, et al. Anisotropic forces from spatially constrained focal adhesions mediate contact guidance directed cell migration. Nat Commun. 2017;8:14923. doi: 10.1038/ncomms14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riveline D, et al. Focal contacts as mechanosensors: Externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–1185. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rio A, et al. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gehler S, et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starke J, Wehrle-Haller B, Friedl P. Plasticity of the actin cytoskeleton in response to extracellular matrix nanostructure and dimensionality. Biochem Soc Trans. 2014;42:1356–1366. doi: 10.1042/BST20140139. [DOI] [PubMed] [Google Scholar]
- 41.Sun X, et al. Asymmetric nanotopography biases cytoskeletal dynamics and promotes unidirectional cell guidance. 2015;1 doi: 10.1073/pnas.1502970112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall MS, et al. Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs. Proc Natl Acad Sci U S A. 2016;113:14043–14048. doi: 10.1073/pnas.1613058113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast Adaptation and Stiffness Matching to Soft Elastic Substrates. Biophys J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moon JJ, et al. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J Cell Physiol. 2005;203:166–176. doi: 10.1002/jcp.20220. [DOI] [PubMed] [Google Scholar]
- 45.Maruthamuthu V, Sabass B, Schwarz US, Gardel ML. Cell-ECM traction force modulates endogenous tension at cell–cell contacts. Proc Natl Acad Sci. 2011;108:4708–4713. doi: 10.1073/pnas.1011123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGregor AL, Hsia CR, Lammerding J. Squish and squeeze-the nucleus as a physical barrier during migration in confined environments. Curr Opin Cell Biol. 2016;40:32–40. doi: 10.1016/j.ceb.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khatau SB, et al. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci. 2009;106:19017–19022. doi: 10.1073/pnas.0908686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swift J, et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harada T, et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–682. doi: 10.1083/jcb.201308029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowat AC, et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem. 2013;288:8610–8618. doi: 10.1074/jbc.M112.441535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denais CM, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Derenyi I, Julicher F, Prost J. Formation and Interaction of Membrane Tubes. Phys Rev Lett. 2002;88:238101. doi: 10.1103/PhysRevLett.88.238101. [DOI] [PubMed] [Google Scholar]
- 53.Fischer-Friedrich E, Hyman AA, Jülicher F, Müller DJ, Helenius J. Quantification of surface tension and internal pressure generated by single mitotic cells. Sci Rep. 2015;4:6213. doi: 10.1038/srep06213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller J, et al. Load Adaptation of Lamellipodial Actin Networks. Cell. 2017;171:188–200.e16. doi: 10.1016/j.cell.2017.07.051. [DOI] [PubMed] [Google Scholar]
- 55.Diz-Muñoz A, et al. Membrane Tension Acts Through PLD2 and mTORC2 to Limit Actin Network Assembly During Neutrophil Migration. PLoS Biol. 2016;14:e1002474. doi: 10.1371/journal.pbio.1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raucher D, Sheetz MP. Cell spreading and lamellipodial extension rate is regulated by membrane tension. J Cell Biol. 2000;148:127–136. doi: 10.1083/jcb.148.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. 2012;197:439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stroka KM, et al. Water permeation drives tumor cell migration in confined microenvironments. Cell. 2014;157:611–623. doi: 10.1016/j.cell.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petrie RJ, Koo H, Yamada KM. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science. 2014;345:1062–1065. doi: 10.1126/science.1256965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lämmermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol. 2009;21:636–644. doi: 10.1016/j.ceb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 61.Álvarez-González B, et al. Three-dimensional balance of cortical tension and axial contractility enables fast amoeboid migration. Biophys J. 2015;108:821–832. doi: 10.1016/j.bpj.2014.11.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lämmermann T, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 63.Yip AK, Chiam KH, Matsudaira P. Traction stress analysis and modeling reveal that amoeboid migration in confined spaces is accompanied by expansive forces and requires the structural integrity of the membrane–cortex interactions. Integr Biol. 2015;7:1196–1211. doi: 10.1039/c4ib00245h. [DOI] [PubMed] [Google Scholar]
- 64.Gjorevski N, Piotrowski AS, Varner VD, Nelson CM. Dynamic tensile forces drive collective cell migration through three-dimensional extracellular matrices. Sci Rep. 2015;5:11458. doi: 10.1038/srep11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Helvert S, Friedl P. Strain Stiffening of Fibrillar Collagen during Individual and Collective Cell Migration Identified by AFM Nanoindentation. ACS Appl Mater Interfaces. 2016;8:21946–21955. doi: 10.1021/acsami.6b01755. [DOI] [PubMed] [Google Scholar]
- 66.Jannat RA, Dembo M, Hammer DA. Traction forces of neutrophils migrating on compliant substrates. Biophys J. 2011;101:575–584. doi: 10.1016/j.bpj.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reinhart-King CA, Dembo M, Hammer DA. The dynamics and mechanics of endothelial cell spreading. Biophys J. 2005;89:676–689. doi: 10.1529/biophysj.104.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Undyala VV, et al. The calpain small subunit regulates cell-substrate mechanical interactions during fibroblast migration. J Cell Sci. 2008;121:3581–3588. doi: 10.1242/jcs.036152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Renkawitz J, et al. Adaptive force transmission in amoeboid cell migration. Nat Cell Biol. 2009;11:1438–1443. doi: 10.1038/ncb1992. [DOI] [PubMed] [Google Scholar]
- 70.Sanz-Moreno V, et al. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 71.Ehrbar M, et al. Elucidating the role of matrix stiffness in 3D cell migration and remodeling. Biophys J. 2011;100:284–293. doi: 10.1016/j.bpj.2010.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hegerfeldt Y, et al. Collective Cell Movement in Primary Melanoma Explants: Plasticity of Cell-Cell Interaction, beta1-Integrin Function, and Migration Strategies. Cancer Res. 2002;62:2125–2130. [PubMed] [Google Scholar]
- 73.Matthes T, Gruler H. Analysis of cell locomotion-Contact guidance of human polymorphonuclear leukocytes. Eur Biophys J. 1988;15:343–357. doi: 10.1007/BF00254722. [DOI] [PubMed] [Google Scholar]
- 74.Kubow KE, Conrad SK, Horwitz AR. Matrix microarchitecture and myosin II determine adhesion in 3D matrices. Curr Biol. 2013;23:1607–1619. doi: 10.1016/j.cub.2013.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dickinson RB, Guido S, Tranquillo RT. Biased cell migration of fibroblasts exhibiting contact guidance in oriented collagen gels. Ann Biomed Eng. 1994;22:342–356. doi: 10.1007/BF02368241. [DOI] [PubMed] [Google Scholar]
- 76.Meyle J, Gültig K, Nisch W. Variation in contact guidance by human cells on a microstructured surface. J Biomed Mater Res. 1995;29:81–88. doi: 10.1002/jbm.820290112. [DOI] [PubMed] [Google Scholar]
- 77.Gopal S, et al. Fibronectin-guided migration of carcinoma collectives. Nat Commun. 2017;8:14105. doi: 10.1038/ncomms14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Driscoll MK, Sun X, Guven C, Fourkas JT, Losert W. Cellular contact guidance through dynamic sensing of nanotopography. ACS Nano. 2014;8:3546–3555. doi: 10.1021/nn406637c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolf K, Müller R, Borgmann S, Bröcker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 2003;102:3262–3269. doi: 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
- 80.Haeger A, Krause M, Wolf K, Friedl P. Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta. 2014;1840:2386–2395. doi: 10.1016/j.bbagen.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 81.Nam KH, et al. Multiscale Cues Drive Collective Cell Migration. Sci Rep. 2016;6:29749. doi: 10.1038/srep29749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vincent LG, Choi YS, Alonso-Latorre B, Del Álamo JC, Engler AJ. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol J. 2013;8:472–484. doi: 10.1002/biot.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sunyer R, et al. Collective cell durotaxis emerges from long-range intercellular force transmission. Science. 2016;353:1157–1161. doi: 10.1126/science.aaf7119. [DOI] [PubMed] [Google Scholar]
- 85.Elosegui-Artola A, et al. Rigidity sensing and adaptation through regulation of integrin types. Nat Mater. 2014;13:631–7. doi: 10.1038/nmat3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paszek MJ, Boettiger D, Weaver VM, Hammer DA. Integrin clustering is driven by mechanical resistance from the glycocalyx and the substrate. PLoS Comput Biol. 2009;5 doi: 10.1371/journal.pcbi.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber M, et al. Interstitial Dendritic Cell Guidance by Haptotactic Chemokine Gradients. Science. 2013;339:328–332. doi: 10.1126/science.1228456. [DOI] [PubMed] [Google Scholar]
- 88.McCarthy JB, Furcht LT. Laminin and fibronectin promote the haptotactic migration of B16 mouse melanoma cells in vitro. J Cell Biol. 1984;98:1474–1480. doi: 10.1083/jcb.98.4.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson HE, et al. F-actin bundles direct the initiation and orientation of lamellipodia through adhesion-based signaling. J Cell Biol. 2015;208:443–455. doi: 10.1083/jcb.201406102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu C, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Autenrieth TJ, et al. Actomyosin contractility and RhoGTPases affect cell-polarity and directional migration during haptotaxis. Integr Biol. 2016;8:1067–1078. doi: 10.1039/c6ib00152a. [DOI] [PubMed] [Google Scholar]
- 92.Painter KJ. Modelling cell migration strategies in the extracellular matrix. J Math Biol. 2009;58:511–543. doi: 10.1007/s00285-008-0217-8. [DOI] [PubMed] [Google Scholar]
- 93.Schlüter DK, Ramis-Conde I, Chaplain MAJ. Computational modeling of single-cell migration: The leading role of extracellular matrix fibers. Biophys J. 2012;103:1141–1151. doi: 10.1016/j.bpj.2012.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwarz US, Safran SA. Physics of adherent cells. Rev Mod Phys. 2013;85:1327. [Google Scholar]
- 95.Mogilner A, Keren K. The Shape of Motile Cells. Curr Biol. 2009;19:R762–R771. doi: 10.1016/j.cub.2009.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Novikova EA, Raab M, Discher DE, Storm C. Persistence-driven durotaxis: Generic, directed motility in rigidity gradients. 2015;78103:1–5. doi: 10.1103/PhysRevLett.118.078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.He X, Jiang Y. Substrate curvature regulates cell migration. Phys Biol. 2017;14:35006. doi: 10.1088/1478-3975/aa6f8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szabó A, Merks RMH. Cellular Potts Modeling of Tumor Growth, Tumor Invasion, and Tumor Evolution. Front Oncol. 2013;3:1–12. doi: 10.3389/fonc.2013.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Hecke M. Jamming of soft particles: geometry, mechanics, scaling and isostaticity. J Phys Condens Matter. 2010;22:33101. doi: 10.1088/0953-8984/22/3/033101. [DOI] [PubMed] [Google Scholar]
- 100.Bi D, Lopez JH, Schwarz JM, Manning ML. A density-independent rigidity transition in biological tissues. Nat Phys. 2015;11:1074–1079. [Google Scholar]
- 101.Park JA, et al. Unjamming and cell shape in the asthmatic airway epithelium. Nat Mater. 2015;14:1040–1048. doi: 10.1038/nmat4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scianna M, Preziosi L, Wolf K. A Cellular Potts Model simulating cell migration on and in matrix environments. Math Biosci Eng. 2013;10:235–261. doi: 10.3934/mbe.2013.10.235. [DOI] [PubMed] [Google Scholar]
- 103.Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA. Nonlinear elasticity in biological gels. Nature. 2005;435:191–194. doi: 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- 104.Schneider KR, Britton NF. Reaction-diffusion Equations and Their Application to Biology. Academic Press; London: 1986. p. 277. Biometrical J. 31, 720 (1989) [Google Scholar]
- 105.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 106.Carey SP, et al. Local extracellular matrix alignment directs cellular protrusion dynamics and migration through Rac1 and FAK. Integr Biol (Camb) 2016;8:821–835. doi: 10.1039/c6ib00030d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oudin MJ, et al. Tumor cell–driven extracellular matrix remodeling drives haptotaxis during metastatic progression. Cancer Discov. 2016;6:516–531. doi: 10.1158/2159-8290.CD-15-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–33. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim J, et al. Stress-induced plasticity of dynamic collagen networks. Nat Commun. 2017;8:842. doi: 10.1038/s41467-017-01011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wolf K, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201:1069–84. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wolf K, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 113.Kirmse R, Otto H, Ludwig T. Interdependency of cell adhesion, force generation and extracellular proteolysis in matrix remodeling. J Cell Sci. 2011;124:1857–1866. doi: 10.1242/jcs.079343. [DOI] [PubMed] [Google Scholar]
- 114.Stratman AN, et al. Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood. 2009;114:237–247. doi: 10.1182/blood-2008-12-196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: A diversity of strategies. J Cell Biol. 2014;204:291–302. doi: 10.1083/jcb.201311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Glentis A, et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun. 2017;8:924. doi: 10.1038/s41467-017-00985-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Seano G, et al. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat Cell Biol. 2014;16:931–41. doi: 10.1038/ncb3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Levental KR, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sawada Y, et al. Force Sensing by Mechanical Extension of the Src Family Kinase Substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guo B, Guilford WH. Mechanics of actomyosin bonds in different nucleotide states are tuned to muscle contraction. Proc Natl Acad Sci. 2006;103:9844–9849. doi: 10.1073/pnas.0601255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buckley CD, et al. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 2014;346:1254211. doi: 10.1126/science.1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klotzsch E, et al. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc Natl Acad Sci. 2009;106:18267–18272. doi: 10.1073/pnas.0907518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dong X, et al. Force interacts with macromolecular structure in activation of TGF-β. Nature. 2017;542:55–59. doi: 10.1038/nature21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buscemi L, et al. The single-molecule mechanics of the latent TGF-β1 complex. Curr Biol. 2011;21:2046–2054. doi: 10.1016/j.cub.2011.11.037. [DOI] [PubMed] [Google Scholar]
- 126.Klingberg F, et al. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J Cell Biol. 2014;207:283–297. doi: 10.1083/jcb.201402006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hinz B. The role of myofibroblasts in wound healing. Curr Res Transl Med. 2016;64:171–177. doi: 10.1016/j.retram.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 129.Hotary KB, et al. Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and -independent processes. J Exp Med. 2002;195:295–308. doi: 10.1084/jem.20010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koivisto L, Heino J, Häkkinen L, Larjava H. Integrins in Wound Healing. Adv wound care. 2014;3:762–783. doi: 10.1089/wound.2013.0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mohammadi H, et al. Filamin A Mediates Wound Closure by Promoting Elastic Deformation and Maintenance of Tension in the Collagen Matrix. J Invest Dermatol. 2015;135:2852–2861. doi: 10.1038/jid.2015.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Laurens N, et al. Single and combined effects of alphavbeta3- and alpha5beta1-integrins on capillary tube formation in a human fibrinous matrix. Angiogenesis. 2009;12:275–285. doi: 10.1007/s10456-009-9150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Du Y, et al. Three-Dimensional Characterization of Mechanical Interactions between Endothelial Cells and Extracellular Matrix during Angiogenic Sprouting. Sci Rep. 2016;6:21362. doi: 10.1038/srep21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yana I, et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J Cell Sci. 2007;120:1607–1614. doi: 10.1242/jcs.000679. [DOI] [PubMed] [Google Scholar]
- 135.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lan TH, Huang XQ, Tan HM. Vascular fibrosis in atherosclerosis. Cardiovasc Pathol. 2013;22:401–407. doi: 10.1016/j.carpath.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 137.Oliveira CLN, Bates JHT, Suki B. A network model of correlated growth of tissue stiffening in pulmonary fibrosis. New J Phys. 2014;16 doi: 10.1088/1367-2630/16/6/065022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sutcliffe JES, et al. Changes in the extracellular matrix surrounding human chronic wounds revealed by 2-photon imaging. Int Wound J. 2017 doi: 10.1111/iwj.12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Odenthal J, Takes R, Friedl P. Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis. 2016;37:1–12. doi: 10.1093/carcin/bgw098. [DOI] [PubMed] [Google Scholar]
- 140.Alexander NR, et al. Extracellular Matrix Rigidity Promotes Invadopodia Activity. Curr Biol. 2008;18:1295–1299. doi: 10.1016/j.cub.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chang TT, Thakar D, Weaver VM. Force-dependent breaching of the basement membrane. Matrix Biol. 2017;57–58:178–189. doi: 10.1016/j.matbio.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goetz JG, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gaggioli C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 144.Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nia HT, et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat Publ Gr. 2016;1:1–11. doi: 10.1038/s41551-016-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahmadzadeh H, et al. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proc Natl Acad Sci. 2017;114:E1617–E1626. doi: 10.1073/pnas.1617037114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barriga E, Franze K, Charras G, Mayor R. How mechanics orchestrate morphogenesis: Mesodermal stiffening triggers neural crest migration in vivo. Mech Dev. 2017;145:S46. [Google Scholar]
- 148.Vennin C, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. 2017;9:eaai8504. doi: 10.1126/scitranslmed.aai8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Miroshnikova YA, Nava MM, Wickstrom SA. Emerging roles of mechanical forces in chromatin regulation. J Cell Sci. 2017;130:2243–2250. doi: 10.1242/jcs.202192. [DOI] [PubMed] [Google Scholar]
- 150.Sadok A, et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015;75:2272–2284. doi: 10.1158/0008-5472.CAN-14-2156. [DOI] [PubMed] [Google Scholar]
- 151.Hirata E, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nilsson M, Adamo H, Bergh A, Halin Bergström S. Inhibition of Lysyl Oxidase and Lysyl Oxidase-Like Enzymes Has Tumour-Promoting and Tumour-Suppressing Roles in Experimental Prostate Cancer. Sci Rep. 2016;6:19608. doi: 10.1038/srep19608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hansen P, Kongsgaard M, Kovanen V, Suetta C, Aagaard P. Mechanical properties and collagen cross-linking of the patellar tendon in old and young men. J Appl Physiol. 2009;107:880–886. doi: 10.1152/japplphysiol.00291.2009. [DOI] [PubMed] [Google Scholar]
- 154.Halfter W, et al. New concepts in basement membrane biology. FEBS J. 2015;282:4466–4479. doi: 10.1111/febs.13495. [DOI] [PubMed] [Google Scholar]
- 155.Zioupos P, Currey J. Changes in the Stiffness, Strength, and Toughness of Human Cortical Bone With Age. Bone. 1998;22:57–66. doi: 10.1016/s8756-3282(97)00228-7. [DOI] [PubMed] [Google Scholar]
- 156.Marshall BT, et al. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–3. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 157.Cluzel C, et al. The mechanisms and dynamics of αvβ3 integrin clustering in living cells. J Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus-independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Meehan S, Nain AS. Role of suspended fiber structural stiffness and curvature on single-cell migration, nucleus shape, and focal-adhesion-cluster length. Biophys J. 2014;107:2604–2611. doi: 10.1016/j.bpj.2014.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Holmes DF, et al. Corneal collagen fibril structure in three dimensions: Structural insights into fibril assembly, mechanical properties, and tissue organization. Proc Natl Acad Sci U S A. 2001;98:7307–7312. doi: 10.1073/pnas.111150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liliensiek SJ, Nealey P, Murphy CJ. Characterization of endothelial basement membrane nanotopography in rhesus macaque as a guide for vessel tissue engineering. Tissue Eng Part A. 2009;15:2643–2651. doi: 10.1089/ten.tea.2008.0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pot SA, et al. Nanoscale topography-induced modulation of fundamental cell behaviors of rabbit corneal keratocytes, fibroblasts, and myofibroblasts. Investig Ophthalmol Vis Sci. 2010;51:1373–1381. doi: 10.1167/iovs.09-4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nelson CM, et al. Emergent patterns of growth controlled by multicellular form and mechanics. Proc Natl Acad Sci U S A. 2005;102:11594–11599. doi: 10.1073/pnas.0502575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hatami-Marbini H, Etebu E, Rahimi A. Swelling pressure and hydration behavior of porcine corneal stroma. Curr Eye Res. 2013;38:1124–32. doi: 10.3109/02713683.2013.809769. [DOI] [PubMed] [Google Scholar]
- 165.Majno G, Palade GE, Schoefl GI. STUDIES ON INFLAMMATION II. The Site of Action of Histamine and Serotonin along the Vascular Tree: A Topographic Study. J Biophys Biochem Cytol. 1961;11(3):607–626. doi: 10.1083/jcb.11.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- 167.Shields JD, et al. Autologous Chemotaxis as a Mechanism of Tumor Cell Homing to Lymphatics via Interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell. 2007;11:526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 168.Ingber DE, Wang N, Stamenović D. Tensegrity, cellular biophysics, and the mechanics of living systems. Reports Prog Phys. 2014;77:46603. doi: 10.1088/0034-4885/77/4/046603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Swaminathan V, et al. Mechanical Stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011;71:5075–5080. doi: 10.1158/0008-5472.CAN-11-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Broedersz CP, MacKintosh FC. Modeling semiflexible polymer networks. Rev Mod Phys. 2014;86:995. [Google Scholar]
- 171.Bischofs IB, Schwarz US. Cell organization in soft media due to active mechanosensing. Proc Natl Acad Sci. 2003;100:9274–9279. doi: 10.1073/pnas.1233544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim MC, Whisler J, Silberberg YR, Kamm RD, Asada HH. Cell Invasion Dynamics into a Three Dimensional Extracellular Matrix Fibre Network. PLoS Comput Biol. 2015;11:1–29. doi: 10.1371/journal.pcbi.1004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.MacKintosh FC, Käs J, Janmey PA. Elasticity of semiflexible biopolymer networks. Phys Rev Lett. 1995;75:4425–4428. doi: 10.1103/PhysRevLett.75.4425. [DOI] [PubMed] [Google Scholar]
- 174.Gardel ML. Elastic Behavior of Cross-Linked and Bundled Actin Networks. Science. 2004;304:1301–1305. doi: 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- 175.Mofrad MRK, Kamm RD. In: Engineering. Mofrad MRK, Kamm RD, editors. Cambridge University Press; 2006. pp. 71–128.pp. 152–169. [DOI] [Google Scholar]
- 176.Preziosi L, Vitale GA. Multiphase Model of Tumor and Tissue Growth Including Cell Adhesion and Plastic Reorganization. Math Model Methods Appl Sci. 2011;21:1901–1932. [Google Scholar]
- 177.Ribeiro FO, Gómez-Benito MJ, Folgado J, Fernandes PR, García-Aznar JM. Computational model of mesenchymal migration in 3D under chemotaxis. Comput Methods Biomech Biomed Engin. 2017;20:59–74. doi: 10.1080/10255842.2016.1198784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chauvière A, Preziosi L, Verdier C. Cell mechanics: from single scale-based models to multiscale modeling. CRC Press; 2010. [Google Scholar]
- 179.van Oers RFM, Rens EG, LaValley DJ, Reinhart-King CA, Merks RMH. Mechanical Cell-Matrix Feedback Explains Pairwise and Collective Endothelial Cell Behavior In Vitro. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]