

Abstract
Small cell neuroendocrine carcinoma (SCNEC) of the uterine cervix is a rare but extremely aggressive tumor. While high-risk HPV is involved at an early stage of oncogenesis in many tumors, additional driving events have been postulated to facilitate the progression of SCNECs. Identification of oncogenic drivers could guide targeted therapy of this neoplasm. Clinicopathologic features of 10 cervical SCNECs are reported. Analyses included Immunohistochemical evaluation of p16, p53, synaptophysin, and chromogranin expression; in situ hybridizations (ISH) and polymerase chain reaction (PCR) for high-risk HPV and/or HPV 18; and next generation sequencing based on a 637-gene panel. The patients ranged in age from 28 to 68 years (mean, 45.6; median, 40.5). All tumors had diffuse p16 and synaptophysin expression. All but one tumor was positive for chromogranin (extent of staining ranged from focal to diffuse). HPV 18 was detected in 6 tumors and HPV 35 in 1 tumor. At least one driver mutation was detected in 8 tumors. Four cases harbored TP53 somatic mutations, 3 of which correlated with an aberrant p53 staining pattern. Four PIK3CA mutations (p.G106A, p.N345T, p.E545K and p.E545D) were detected in 3 tumors, 2 of which also harbored TP53 mutations. Oncogenic driver mutations involving KRAS, Erbb2, c-Myc, NOTCH1, BCL6 or NCOA3 were detected in 4 tumors. Mutations in caretaker tumor suppressors PTEN, RB1, BRCA1, BRCA2, and ARID1B were also identified in 4 tumors that commonly co-harbored activating oncogenic mutations. Targeted next-generation gene sequencing identified genetic alterations involving the MAPK, PI3K/AKT/mTOR, and TP53/BRCA pathways in SCNECs. The presence of genetic alterations that are amenable to targeted therapy in SCNECs offers the potential for individualized management strategies for treatment of this aggressive tumor.
Keywords: Next-generation sequencing, Small cell neuroendocrine carcinoma, Human papillomavirus, Mutations, Uterine cervix
Introduction
Despite overall declines in incidence rates, cervical cancer is the second or third most common cancer in women with approximately 0.5 million cases worldwide. In the United States, approximately 12,990 new cases were diagnosed in 2016, with roughly 4,120 deaths.1, 2 Small cell neuroendocrine carcinoma (SCNEC) of the cervix is a rare and highly aggressive neoplasm, accounting for 1–2% (range < 5%) of all cervical tumors.3–6 Most patients present at an advanced stage at the time of hysterectomy. Compared with stage comparable squamous cell carcinoma or endocervical adenocarcinoma, SCNEC is more likely to have distant metastasis and recurrence.4, 7, 8 The main management modality of SCNEC includes surgery, chemotherapy and radiation therapy.9–11 SCNEC can recur within months after complete surgical resection with no residual disease. Despite extensive treatment, five-year survival rates for SCNEC of all stages still vary from 25% to 39%, with poorer survival in higher stage disease.12–16 Thus, there is a need for novel approaches to understand and treat this tumor.
Histologically identical to its counterparts at other sites such as the lung, SCNEC of the cervix is composed of monotonous small cells with enlarged ovoid hyperchromatic nuclei, nuclear molding, finely stippled chromatin, inconspicuous nucleoli, and scanty cytoplasm.5, 14 Abundant mitotic figures and apoptotic bodies, extensive necrosis, and crush artifacts are also characteristic features. The vast majority of SCNECs of the cervix are etiologically associated with high-risk human papillomavirus (HPV).5, 17, 18 Similar to other HPV-related malignancies,19 oncoproteins E6 and E7 are required for the initiation and upkeep of SCNECs. While high-risk HPV is involved at an early stage of oncogenesis, additional driving events, including genetic and epigenetic alterations, have been postulated to facilitate the progression of SCNECs. Loss of heterozygosity (LOH) has been reported to be a frequent genetic event in SCNECs. In one study, eight of fifteen cases displayed LOH at various 3p loci (3p14, 3p21, and 3p24) and one tumor demonstrated LOH on 17p (TP53 locus).20
Currently, cell signaling pathway inhibitors, angiogenesis inhibitors, apoptosis promoters and immunotherapies have been tested for SCNEC of the lung.21 Accordingly, the potential application of these targeted therapies to cervical SCNECs relies on identification of genetic alterations that are amenable to such therapies in these tumors. Using whole exome sequencing, a recent study demonstrated recurrent mutations in ATRX, EBRR4, and in AKT/mTOR signaling pathway genes including NF1, PTEN, RICTOR and TSC2 in SCNECs of uterine cervix.22 In another study of cervical SCNECs, sequencing of mutational hotspots within 50 cancer-related genes revealed recurrent mutations in PIK3CA, KRAS and TP53 genes. Of the 44 patients, 48% had at least 1 mutation for which some form of targeted therapy could be considered.23 However, the former study identified a mutational profile that appears to be different from that of HPV-related malignancies. The latter study only captured somatic changes in a limited panel of 50 genes which assessed only hotspot mutations. Given that the available data on SCNECs is limited and that there might be differences in the spectrum of genetic alterations encountered in different studies, we performed next generation sequencing using a larger gene panel on a set of SCNECs of the cervix collected from several institutions, with the goal of identifying genetic alterations that are amenable to targeted therapy.
Material and methods
Case selection
Cases were identified in the files of the authors’ institutions (4 from Johns Hopkins Hospital [JHH], 2 from Mayo Clinic, 2 from the University of Alabama at Birmingham, and 2 from the Ohio State University Wexner Medical Center). Histologic sections of these cases were re-reviewed by three pathologists (D.X., G. Z. and B.M.R) to confirm the diagnosis. The study was approved by the Institutional Review Board at the Johns Hopkins Hospital.
Immunohistochemistry and In Situ Hybridization (ISH)
Immunohistochemical staining was performed on formalin-fixed, paraffin-embedded tissue sections as previously described.19 Markers used included: p16 (INK4a) (mouse monoclonal, Ventana, Tucson, AZ; prediluted), synaptophysin (mouse monoclonal, Novacastra/Leica Biosystems Inc. Buffalo Grove, IL; 1:400 dilution), chromogranin (mouse monoclonal, Ventana, Tucson, AZ; prediluted), and p53 (mouse monoclonal, Ventana, Tucson, AZ; prediluted). ISHs were performed using a high-risk HPV RNA probe solution (RNAscope, Advanced Cell Diagnostics, Newark, CA; HPV types16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73, 82) and type-specific probe HPV 18 (RNAscope, Advanced Cell Diagnostics, Newark, CA). In addition, ISHs for HPV DNA (wide spectrum probe: cocktail of HPV 6, 11, 16, 18, 31, 33, 45, and type-specific probes for HPV16 and HPV18, Dako, Carpinteria, CA) had been performed on 2 tumors (cases 2 and 3) at the time of diagnosis.
DNA extraction
Paraffin-embedded tumor tissues, identified by H&E staining of adjacent sections (tumor elements account for more than 70% of section area), were macrodissected, and genomic DNA was extracted using a QIAamp DNA FFPE Tissue Kit with an adapted protocol (Qiagen, Valencia, CA). Briefly, slides bearing paraffin embedded tissue were baked at 68°C for 20 to 30 seconds; the tissue was deparaffinized 3 times with xylene, and residual xylene was removed by washing through serial dilutions of ethanol. The tumor tissue was separated from adjacent normal tissue and placed in a tube allowing for complete evaporation of residual ethanol. The tissue pellet was resuspended in Buffer ATL with added proteinase K. The rest of the procedure followed the manufacturer’s instruction.
Polymerase chain reaction (PCR) and Sanger sequencing
PCR-based detection and typing of HPV has been described previously. 24, 25 In brief, the samples with undetected high-risk HPV by ISHs were checked for DNA integrity by amplifying β-globin as a housekeeping gene using the following primers: PC04, 5’-CAACTTCATCCACGTTCACC-3’; GH20: 5’-GAAGAGCCAAGGACAGGTAC-3’. The size of this PCR product is 268 base pairs (bp). The DNA samples with positive test results for β-globin were subsequently studied using HPV consensus primers MY09 (5’-CGTCCMARRGGAWACTGATC-3’) and MY11 (5’-GCMCAGGGWCATAAYAATGG-3’). In the above primer sequence, M stands for A or C; R, for A or G; W, for A or T; and Y, for T or C. These primers amplified a broad spectrum of HPVs by flanking the conserved region L1 open reading frame. The PCR products (approximate 450bp) amplified by MY09/MY11 primers were diluted 10 times and used as the templates of a second PCR amplification (nested PCR) with another pair of primers, GP05 (5’-TTTGTTACTGTGGTAGATAC-3’) and GP06 (5’-GAAAAATAAACTGTAAATCA-3’), flanking an approximate 150bp fragment within the L1 open reading frame. The GP05/GP06 nested PCR products were subjected to direct DNA Sanger sequencing to analyze the HPV type. One tumor (case 1) with ISH-detected HPV 18 was used as positive control.
Targeted next generation sequencing
The targeted next generation sequencing was performed as described previously.26 Libraries were prepared using the Agilent SureSelect-XT Target Enrichment Kit. Briefly, 200–300 ng of DNA was fragmented to a size of 250–300 bp, using a Covaris M220 sonicator. The DNA fragments were end-repaired and A-tailed, then adaptors were added by ligation and the fragments were enriched by PCR (10 cycles). Each library was then hybridized to a SureSelect custom panel 2.8M bait set (Agilent) according to the manufacturer's protocol. The panel was originally designed as a clinical leukemia panel, and covered 637 genes important in the oncogenesis of both leukemia and solid tumors. The gene list is available upon request. After stringent washing the captured DNA was amplified with 12 cycles of PCR per manufacturer's protocol. The size and concentration of captured DNA custom was assessed using a Tapestation 2200 (Agilent). Captured samples were pooled and sequenced on a single HiSeq flow cell on a HiSeq 2500 (Illumina), using a 2 × 100bp PE Rapid Run v2 protocol.
Data analysis
All reads were aligned to the human genome (GRCh37.p13 /hg19), using the Burrows–Wheeler alignment (BWA) algorithm. The final Binary Alignment Map (BAM) file was used for variant calling with our custom variant caller pipeline, which called variants directly from the BAM file with multiple filters, including a filter of >4 mutant reads in both directions, a common SNP filter, and a strand bias filter. In addition, a filter based on a pool of normal FFPE tissue were applied, where variants with variant allele frequency (VAF) falling within three standard deviations from mean VAF seen in a pool of normal FFPE tissues, were filtered out. Finally, the Broad Institute Integrated Genome Viewer (IGV) was used to visually inspect all the variants called by the pipeline, variants with low quality score (median quality score <30) were excluded in this step. Two strand bias scores were calculated: the first one (SB1) considered only strand bias in variant reads: MAX (Var+, Var−)/(Var+ + Var−); the second (SB2) adjusted variant calls for inherent strand bias: (Var+/Ref+)/(Var−/Ref−). A variant call passed the strand bias filter if either SB1 ≥ 0.7 and/or 2.0 ≥ SB2 ≥ 0.5. The sequencing mean coverage exceeded 300 reads in more than 94% of all regions. Given the estimated tumor cellularity is above 90% of all the cases, we used a variant allele frequency filter of 10%. All variants were separated into 3 categories based on VAF, dbSNP and COSMIC database annotation: 1) pathogenic: loss of function mutations or hotspot mutations based on COSMIC database; 2) likely germline: 40%≤VAF≤60% with or without dbSNP annotation, but not fulfilling the criteria as pathogenic; 3) variants of uncertain significance (VUS): other variants.
Results
Clinicopathologic features of SCNEC
Clinicopathologic features are summarized in Table 1. The patients ranged in age from 28 to 68 years (mean, 45.6; median, 40.5). Tumor size ranged from 1.3 cm to 10.0 cm (mean, 5.4; median, 4.5). Tumors were composed of monotonous cells with enlarged hyperchromatic nuclei, nuclear molding, scanty cytoplasm, and numerous mitotic figures and apoptotic bodies (Figures 1A and 1B). Characteristic neuroendocrine features included finely stippled chromatin, inconspicuous nucleoli and Rosette-like structures (Figure 1C). Extensive crush artifact was frequently present. At the time of hysterectomy, lymph node metastases were identified in 5 cases. Lymph-vascular space invasion was commonly seen, even for the cases with a negative lymph node dissection. In addition to classical morphologic features, the diagnosis of SCNEC was supported by synaptophysin expression in all tumors (Table 1 and Figure 1D). In addition, 9 tumors have some degree of chromogranin expression which varied from focal to diffuse. All tumors exhibited diffuse/strong p16 expression (Table 1 and Figure 1E). By ISH, HPV 18 was detected in 5 tumors, of which 4 were detected by RNA probe (Figure 1F) and 1 by DNA probe. High-risk HPV mixed RNA probe solution (covering 18 high-risk HPV subtypes) failed to detect HPV in the 5 tumors with undetectable HPV 18. In addition, for 1 of these tumors (case 2), the wide spectrum DNA probe also failed to detect HPV. To further assess for the presence of HPV in 5 tumors with undetected high-risk HPV by ISHs, PCR, a gold standard method for detecting HPV, was employed. This analysis was technically successful in 3 of these 5 cases (amplification of β-Globin) as well as in the positive control with ISH-detected HPV 18 (case 1, Figures 2A and 2B), Two of the 3 successfully tested tumors had HPV detected by PCR, case 4 with HPV 35 (Figure 2C) and case 10 with HPV 18 (Figure 2D); case 9 had no PCR-detectable HPV (Figure 2A).
Table 1.
Clinicopathologic features and mutational profile
| Case | Age | Procedure | Size (cm) |
LVI | LN | SYN | CHR | P16 | HR-HPV | P53 | Mutations | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 45 | Hysterectomy | 6.0 | Yes | No | Diffuse + | Focally + | Diffuse + | HPV 18 | Normal | Erbb2, ARID1B, BCL6 | 13 months, alive with metastastic disease |
| 2 | 60 | Hysterectomy | 9.0 | Yes | Yes | Diffuse + | Negative | Diffuse + | Not detected* | Aberrant (diffuse) | TP53, c-Myc, NCOA3 | 2 months, alive with metastastic disease |
| 3 | 29 | Hysterectomy | 1.3 | Yes | No | Diffuse + | Focally + | Diffuse + | HPV 18 | Normal | TP53, BRCA2, NOTCH1 | Not available |
| 4 | 68 | Biopsy | n/a | n/a | n/a | Diffuse + | Focally + | Diffuse + | HPV 35 | Aberrant (null) | TP53, PIK3CA, PTEN, RB1 | Not available |
| 5 | 31 | Hysterectomy | 7.5 | No | Yes | Diffuse + | Diffuse + | Diffuse + | HPV 18 | Normal | BRCA1 | 4 months, dead of disease |
| 6 | 64 | Hysterectomy | 4.5 | Yes | No | Diffuse + | Positive*** | Diffuse + | HPV 18 | Normal | PIK3CA | Not available |
| 7 | 28 | Hysterectomy | 10.0 | Yes | Yes | Diffuse + | Negative | Diffuse + | HPV 18 | Normal | KRAS | 126 months, alive with post-radiation tumor |
| 8 | 34 | Hysterectomy | 2.5 | Yes | Yes | Diffuse + | Focally + | Diffuse + | Not detected* | Normal | none detected | 11 months, dead of disease |
| 9 | 61 | Hysterectomy | 4.4 | Yes | No | Diffuse + | Positive*** | Diffuse + | Not detected** | Aberrant (diffuse) | TP53, PIK3CA | 11 months, dead of disease |
| 10 | 36 | Hysterectomy | 3.6 | Yes | Yes | Diffuse + | Focally + | Diffuse + | HPV 18 | Normal | none detected | 27 months, dead of disease |
LVI: lymph-vascular invasion; LN: lymph node metastasis; SYN: synaptophysin; CHR: chromogranin; HPV: human papillomavirus; HR: high-risk; n/a: not applicable
HPV 18 RNA probe and HR-HPV RNA mixed probe solution (covering 18 HR-HPV subtypes) failed to detect HPV (wide spectrum HPV DNA probe also failed to detect HPV in case 2); PCR amplification for HR-HPV was not successful.
HPV 18 RNA probe, HR-HPV RNA mixed probe solution (covering 18 HR-HPV subtypes), and PCR all failed to detect HPV.
By report (slide not available for review to assess staining distribution)
Figure 1.
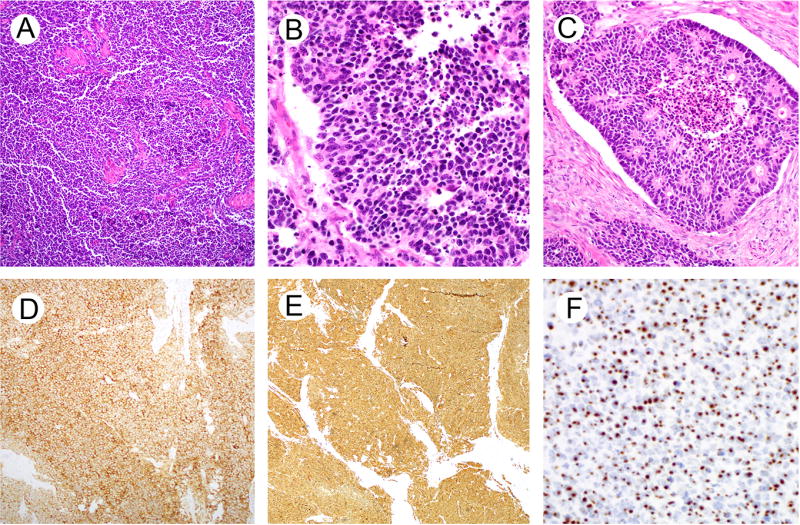
Representative histologic and immunohistochemical images of small cell neuroendocrine carcinoma (SCNEC) of the uterine cervix. The tumor displays monotonous population of cells (A, case 1) that have ovoid to angulated nuclei with molding, scanty cytoplasm, abundant mitotic and apoptotic activity (B, case 1) and rosette-like structures (C, case 10). SCNEC (case 1) shows diffuse synaptophysin (D) and p16 expression (E) and contains high-risk HPV as detected by RNA in situ hybridization (F).
Figure 2.

PCR-based detection and typing of HPV. Case 1 with ISH-detected HPV 18 was used as positive control (A, B). The samples (cases 2, 4, 8, 9, 10) with no detectable high-risk HPV by ISH were checked for DNA integrity by amplifying β-globin as a housekeeping gene (A). The DNA samples (cases 4, 9, 10) with positive test results for β-globin were subsequently studied by nested PCR using MY09/MY11 and GP05/GP06 primers. The PCR products (A) were subjected to direct DNA Sanger sequencing to analyze the HPV type. Case 4 with HPV 35 (C); case 10 with HPV 18 (D); case 9 with no PCR-detectable HPV (A).
Somatic mutational profile of SCNEC
Table 1 and Figure 3 summarize the pathogenic variants and variants of unknown significance (VUS) in the 10 tumors. Detailed information about these variants is described in a supplementary table. At least one pathogenic variant was detected in 8 tumors (Figure 3A). Four PIK3CA mutations, p.G106A, p.N345T, p.E545K (Figure 3B) and p.E545D, were detected in 3 tumors, of which 2 also harbored TP53 mutation. Oncogenic tyrosine kinase pathway mutations involving KRAS (p.G12V) and Erbb2 (p.R663Q) were also detected in case 7 and case 1, respectively. The tumor with Erbb2 mutation also had ARID1B (p.K2043fs) and BCL6 (p.W375C) mutations (case 1). BRCA1 (p.T367I) and BRCA2 (p.Q1187fs) mutations (Figure 3C) were also detected individually in 2 tumors (case 5 and case 3, respectively). Activating oncogenic mutations or tumor suppressor inactivating mutations in MYC, NOTCH1, NCOA3, PTEN and RB1 were identified in the tumors that also harbored other mutations. Interestingly, despite the presence of many VUS, no clinically relevant and actionable somatic mutations in the 637-gene panel were identified in 2 tumors (cases 8 and 10).These two tumors displayed classical morphologic features seen in SCNECs (Supplementary Figures 1 and 2).
Figure 3.

Somatic mutations detected by next-generation sequencing. The mutated activating oncogenic and caretaker tumor suppressor genes in SCNECs are plotted (arranged in descending order of number of mutations (A). Representative mutations of PIK3CA (missense mutation, B) and BRCA2 (deletion/frameshift mutation, C) are shown in the middle and right.
TP53 mutations in SCNEC
Four tumors harbored TP53 somatic mutations (Table 1). One tumor (case 4) had TP53 mutations (p.80fs and p.P80L) and notable mutations in 3 other genes: PIK3CA (p.G106A and p.E545D), PTEN (p.G132D and p.F241S), and RB1 (p.S751fs). This tumor demonstrated complete absence of p53 expression, consistent with the “null” pattern of aberrant/mutation-type p53 expression (Figures 4A and 4B). Insertion of nucleotides AG in the codon 80 of TP53 gene lead to frameshift mutation (Figure 4C), premature translation termination and p53 protein truncation that is not recognized by the p53 antibody. The second TP53-mutated tumor (case 2) harbored somatic mutations of TP53 (p.C238W), c-Myc (p.A199T), and NOCA3 (p.Q1239_1241del). This tumor demonstrated aberrant/mutation-type p53 over-expression, consistent with a missense mutation (Figures 5A–5C). The presence of more than 90% frequency of mutant TP53 allele was indicative of a bi-allelic pattern. A third tumor (case 9) with somatic mutations of TP53 (p.C275Y) and PIK3CA (p.N345T) also had aberrant/mutation-type p53 over-expression. Interestingly, these 3 TP53-mutated tumors were all from patients older than 60 years. HPV was detected in 1 of these TP53-mutated tumors (HPV 35 in case 4 by PCR but not ISH). Of interest, HPV 18 was detected in a tumor from a 29 year-old woman (case 3) which had TP53 p.E271Q mutation as well as somatic mutations of BRCA2 (p.Q1187fs) and NOTCH1 (p.Q2315*nonsense). Unlike the other 3 TP53-mutated tumors, the p53 expression pattern in this tumor was a normal/wild-type result.
Figure 4.
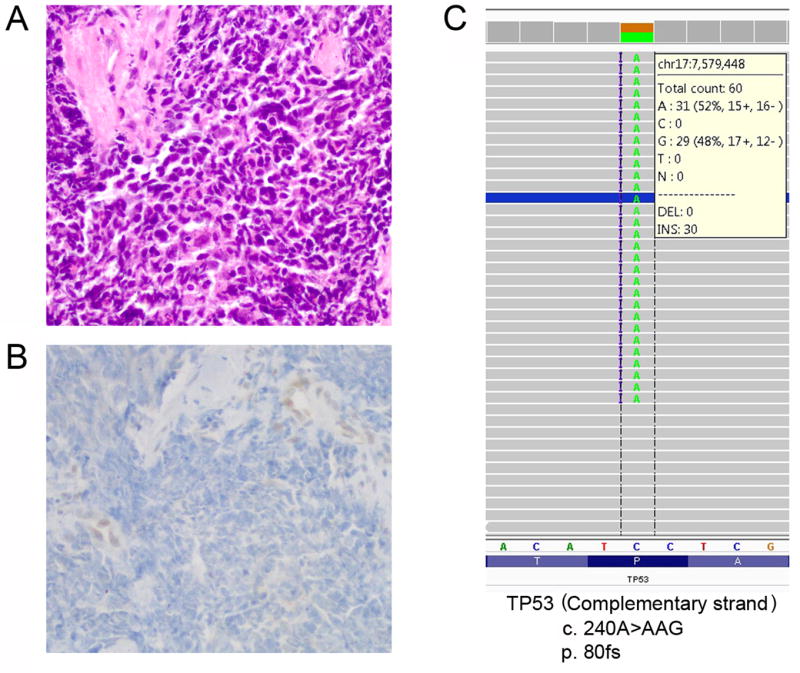
SCNEC with aberrant loss of p53 expression associated with TP53 frameshift mutation (case 4). The tumor (A, H&E) demonstrates aberrant/mutation-type complete loss of p53 expression (“null” pattern, B) because truncated p53 protein cannot be recognized by the p53 antibody. Insertion of nucleotides AG in the codon 80 of TP53 gene lead to frameshift mutation, premature translation termination and p53 protein truncation (C).
Figure 5.
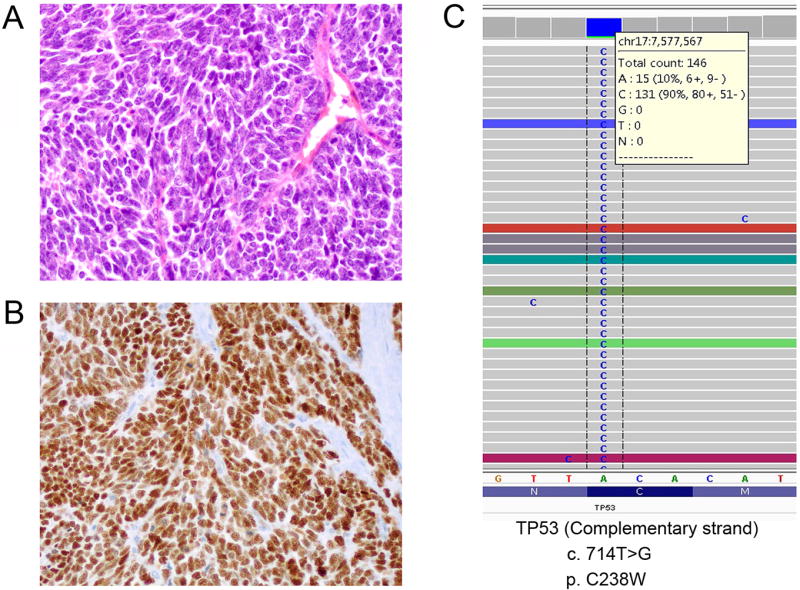
SCNEC with aberrant p53 expression associated with TP53 missense mutation (case 2, p.C238W). The tumor (A, H&E) demonstrates aberrant/mutation-type p53 over-expression (B) consistent with a missense mutation staining pattern. The presence of more than 90% frequency of mutant TP53 allele is indicative of a bi-allelic pattern (C).
Clinical outcome of patients with SCNEC
It has been well documented that SCNECs display highly aggressive behavior and thus, very poor prognosis. In fact, 4 patients in the current study died of disease at intervals ranging from 4 months to 27 months after hysterectomy (cases 5, 8, 9, 10). These included 1 patient with somatic mutations of TP53 and PIK3CA; 1 patient with BRCA1 mutation and 2 patients with no detectable critical mutations in this gene-panel. One patient (case 2, TP53, c-Myc, NCOA3) developed distant metastasis 2 months after hysterectomy and another patient (case 1, Erbb2, ARID1B, BCL6) had distant metastasis 13 months after hysterectomy. One patient (case 7, K-Ras mutation) developed a post-radiation pelvic bone chondroblastic osteosarcoma after a long disease-free survival (126 months). Follow-up information was not available for 3 patients.
Discussion
SCNEC of the uterine cervix is a rare but extremely aggressive tumor, with a very high mortality even among patients diagnosed with early stage disease.6, 16 Due to its rarity, there is a lack of treatment guidelines based on prospective clinical trials. Thus, given the histologic similarity to SCNEC of the lung, the therapeutic approach for cervical SCNEC, such as the combination of chemotherapeutic reagents cisplatin and etoposide, is based on that used for pulmonary tumors. Early stage cervical SCNEC is usually treated by radical hysterectomy followed by adjuvant chemotherapy or concurrent chemoradiation.27 Definitive concurrent chemoradiation, sometimes preceded by neo-adjuvant chemotherapy and followed by adjuvant chemotherapy, has been employed for locally advanced disease, and palliative chemotherapy for metastatic disease.28 However, the high recurrence rate and the very poor prognosis warrant development of novel therapeutic options to treat SCNEC of the uterine cervix, including identification of molecular alterations that are amenable to targeted therapy.
The molecular genetic investigation of SCNECs of different organs has revealed some common altered molecular pathways that can be targeted in clinical trials.21 This observation has broadened the spectrum of therapeutic options for SCNECs, regardless of the primary site of origin. While SCNECs of various sites have essentially similar histologic features, those arising in the cervix have an association with high-risk HPV and HPV 18 in particular, with HPV 18 detection rates in cervical SCNECs varying from 40% to 90%.3, 29–31 All of our tumors demonstrated diffuse p16 expression. High-risk HPV was detected in 7 of 10 tumors overall and 7 of 8 fully successfully tested tumors. HPV 18 was identified in 6 tumors (5 by ISH and 1 by PCR) and HPV 35 was detected in 1 tumor (by PCR). Since PCR amplification failed in 2 cases, the actual high-risk HPV frequency could be higher.
In this study, 3 tumors harbored TP53 somatic mutations which were correlated with an aberrant immunohistochemical staining pattern and 1 additional tumor with a TP53 mutation had a wild-type p53 expression pattern. Since the high-risk HPV viral oncoprotein E6 has the ability to neutralize the function of p53, the majority of high-risk HPV-related cervical cancers including SCNECs would be expected to have a wild-type TP53 gene. However, one study has shown that TP53 mutation occurred in 62.5% of cervical SCNECs,32 while others have not observed this.33, 34 Of note, those data derived from hotspot-based mutational analysis might underestimate the actual percentage of tumors with TP53 mutation.23 Somatic mutations of TP53 were originally thought to be required in the absence of an HPV-encoded gene product E6 that mediates loss of p53 function.35–37 Subsequent studies have demonstrated that TP53 mutation can occur regardless of HPV status.38, 39 Our study demonstrated a case with TP53 mutation and diffuse p16 expression but no detectable high-risk HPV by both ISH and PCR methods. This observation raises the possibility of two different pathways to developing SCNEC in the uterine cervix: a more common one that is high-risk HPV-driven and a less common one that is p53-driven in the absence of high-risk HPV with a similar mechanism to that of lung SCNECs. The combination of diffuse p16 expression and TP53 mutation in tumors unrelated to high-risk HPV is well-known, indicating that diffuse p16 expression by itself cannot be equated to the presence of high-risk HPV in a tumor. 40, 41 These pathways and the relationships between TP53 mutation and the presence or absence of high-risk HPV in SCNEC warrant further investigation in a larger number of tumors.
Four PIK3CA mutations G106A, N345T, E545K and E545D were detected in 3 tumors in this study. Consistent with these findings, 18% (8 of 44) of SCNECs harbored PIK3CA mutation, the most frequent alteration, in a hotspot-based study.23 As a known oncogene, PIK3CA is located at chromosome 3q26 which encodes the p110α catalytic subunit of phosphatidylinositol 3-kinase (PI3K).42 Commonly disrupted across different cancer types, PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway regulates cell proliferation, differentiation, quiescence, apoptosis, longevity and oncogenesis.43, 44 It has been demonstrated that PIK3CA plays a critical role in HPV-induced carcinogenesis, evidenced by the observation that activation of the PI3K/AKT/mTOR pathway through PIK3CA regulates various transformed phenotypes as well as growth and differentiation of HPV-immortalized cells.45 In fact, PIK3CA is thought to be one of the most frequently mutated genes in HPV-related cancers including cervical squamous cell carcinoma, endocervical adenocarcinoma and HPV-positive head and neck tumors.46–48 One patient in this cohort had an E545K mutation, one of the most common PIK3CA mutations. Other PIK3CA mutations including G106A, N345T and E545D are less common but have been reported previously. PIK3CA mutations constantly activate downstream effectors such as PDK1 and AKT that promote and sustain cellular transformation. Similarly, loss of PTEN function through somatic mutations in SCNECs causes excessive PIP3 at the plasma membrane, recruiting and activating a subset of pleckstrin homology domain–containing proteins to the cell membrane including phosphoinositide-dependent kinase-1 and AKT family members.49 A Notch1 mutation, not previously reported in SCNEC, was found in 1 tumor that did not have PIK3CA mutation. One function of mutant Notch1 is to activate c-Myc and PI3K-AKT-mTOR1 signaling through transcriptional repression of PTEN and promoting growth factor receptor signaling to PI3K-AKT.50 Taken together, the SCNECs bearing these mutations might be sensitive to mTOR or AKT inhibitor treatment, a potential targeted therapy for this highly aggressive disease.
KRAS mutation, c.35G>T (G12V) was identified in 1 patient who was alive for more than 10 years. Interestingly, a previous study demonstrated that KRAS mutations were present exclusively in endocervical adenocarcinomas, but not squamous cell carcinoma.47 Another study showed that KRAS mutations were seen exclusively in destructively invasive endocervical adenocarcinomas (pattern B and pattern C subgroups) and correlated with advanced stage at presentation (FIGO stage II or higher).51 KRAS mutations have been reported in SCNECs. Importantly, a patient with a KRAS mutation (c.35G>A p.G12D) was treated with the MEK inhibitor trametinib and had a complete radiologic response after 3 cycles.52 Different from the G12D mutation that activates both PI3K/AKT and MAPK pathways, the patient in the current study harbored a G12V KRAS mutation that triggers the MAPK cascade and leads to loss of the ability to bind to and signal through PI3K/AKT/mTOR.53 Theoretically, a patient whose tumor harbors a G12V KRAS mutation may respond better, compared with G12D, to MEK inhibitors.23 Another interesting somatic mutation identified in this study is ERBB2 p.R678Q mutation. This somatic mutation has been previously described in breast cancer. However, R678Q mutation showed no functional effect in an in vivo and in vitro model system.54 Therefore, a role of ERBB2 R678Q mutation in the development of SCNECs remains elusive. Similarly, further evaluation of the biological significance of somatic mutations in c-Myc, ARID1B, RB1, BCL6 and NCOA3 might provide additional molecule/pathway-based therapeutic options for SCNECs.
Unexpectedly, BRCA1 and BRCA2 somatic mutations were discovered in 2 tumors. BRCA1 p.T367I appears to be a novel mutation with unknown function. In contrast, BRCA2 c.3545_3546 TT deletion leads to frameshift mutation and translation termination. Interestingly, BRCA1 interacts with HPV oncoproteins through which E6 and E7 antagonize the ability of BRCA1 to inhibit c-Myc E-box-mediated transactivation and human telomerase reverse transcriptase promoter activity.55 The poly [ADP-ribose] polymerase (PARP) inhibitors have been successfully used for the treatment of germline BRCA-mutated ovarian cancer patients.56–58 Importantly, patients with a somatic BRCA mutation also show benefit from treatment with PARP inhibitors.59, 60 Identification of somatic BRCA mutations in patients with SCNEC allows testing of PARP inhibitors to treat this aggressive tumor.
Surprisingly, clinically relevant and actionable somatic mutations in this gene panel were not identified in 2 tumors. We suspect genetic alterations such as gene overexpression and/or inactivation, copy number variation, and DNA and/or histone-related epigenetic regulation, other than somatic mutations, may play a critical role in the oncogenic process of these tumors.
Our results are quite different from a recent study that evaluated the mutation profile for five tumor-normal paired cervical SCNECs using whole exome sequencing.22 Recurrent mutations in ATRX and ERBB4 as well as Akt/mTOR pathway genes NF1, PTEN, RICTOR, and TSC1/2 were identified in that study. Although involving the Akt/mTOR pathway, this mutation profile is different from that of the majority of high-risk HPV-related tumors which commonly contain recurrent somatic driver mutations in PIK3CA, KRAS, TP53, ERBB2 and MAPK1.19 Since the recurrent somatic TSC2, NF1 and PTEN mutations identified in that study were also present in neuroendocrine tumors of other sites, but not in high-risk HPV-related cervical tumors, the origin and genetic background of SCNEC of the cervix are thought to be different from those of cervical high-risk HPV-related tumors.22 In contrast, our findings support that the genetic landscape in the initiation and development of cervical SCNEC is similar to that of other high-risk HPV-related cervical tumors.46
In summary, utilizing a targeted next-generation gene sequencing technology, our study identified and confirmed recurrent genetic alterations involving the MAPK, PI3K/AKT/mTOR, and p53/BRCA pathways in cervical SCNECs. The presence of genetic alterations that are amenable to targeted therapy, individually or in combination, in cervical SCNECs offers the potential for individualized management strategies for treatment of this aggressive tumor.
Supplementary Material
Supplementary Figure 1. Representative histologic and immunohistochemical images of case 8. The tumor shows typical morphology of SCNEC with a monotonous population of cells and infiltrative growth pattern (A). The tumor cells have scanty cytoplasm, abundant mitotic and apoptotic activity (B) and display focal chromogranin immunoreactivity (C) and diffuse p16 expression (D).
{kind=link}
Supplementary Figure 2. Representative histologic images of case 10. The tumor displays an infiltrative growth pattern (A) and discohesive cells with molding, scanty cytoplasm, and extensive apoptosis and necrosis (B) with some rosette-like structures (C).
{kind=link}
Supplementary Table 1. Detailed information about gene mutations and variants
Acknowledgments
Funding/Support: Career Development Award by the Cervical Cancer SPORE program at Johns Hopkins (D.X.)
Footnotes
Conflict of interest: The authors have no competing interests to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Small W, Jr, Bacon MA, Bajaj A, et al. Cervical cancer: A global health crisis. Cancer. 2017;123:2404–2412. doi: 10.1002/cncr.30667. [DOI] [PubMed] [Google Scholar]
- 3.Abeler VM, Holm R, Nesland JM, et al. Small cell carcinoma of the cervix. A clinicopathologic study of 26 patients. Cancer. 1994;73:672–677. doi: 10.1002/1097-0142(19940201)73:3<672::aid-cncr2820730328>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Satoh T, Takei Y, Treilleux I, et al. Gynecologic Cancer InterGroup (GCIG) consensus review for small cell carcinoma of the cervix. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2014;24:S102–108. doi: 10.1097/IGC.0000000000000262. [DOI] [PubMed] [Google Scholar]
- 5.Atienza-Amores M, Guerini-Rocco E, Soslow RA, et al. Small cell carcinoma of the gynecologic tract: a multifaceted spectrum of lesions. Gynecologic oncology. 2014;134:410–418. doi: 10.1016/j.ygyno.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Macdonald OK, Gaffney DK. Incidence, mortality, and prognostic factors of small cell carcinoma of the cervix. Obstetrics and gynecology. 2008;111:1394–1402. doi: 10.1097/AOG.0b013e318173570b. [DOI] [PubMed] [Google Scholar]
- 7.Chan JK, Loizzi V, Burger RA, et al. Prognostic factors in neuroendocrine small cell cervical carcinoma: a multivariate analysis. Cancer. 2003;97:568–574. doi: 10.1002/cncr.11086. [DOI] [PubMed] [Google Scholar]
- 8.Intaraphet S, Kasatpibal N, Sogaard M, et al. Histological type-specific prognostic factors of cervical small cell neuroendocrine carcinoma, adenocarcinoma, and squamous cell carcinoma. OncoTargets and therapy. 2014;7:1205–1214. doi: 10.2147/OTT.S64714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gadducci A, Carinelli S, Aletti G. Neuroendrocrine tumors of the uterine cervix: A therapeutic challenge for gynecologic oncologists. Gynecologic oncology. 2017;144:637–646. doi: 10.1016/j.ygyno.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Cohen JG, Kapp DS, Shin JY, et al. Small cell carcinoma of the cervix: treatment and survival outcomes of 188 patients. American journal of obstetrics and gynecology. 2010;203:347, e341–346. doi: 10.1016/j.ajog.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Bermudez A, Vighi S, Garcia A, et al. Neuroendocrine cervical carcinoma: a diagnostic and therapeutic challenge. Gynecologic oncology. 2001;82:32–39. doi: 10.1006/gyno.2001.6201. [DOI] [PubMed] [Google Scholar]
- 12.Conner MG, Richter H, Moran CA, et al. Small cell carcinoma of the cervix: a clinicopathologic and immunohistochemical study of 23 cases. Annals of diagnostic pathology. 2002;6:345–348. doi: 10.1053/adpa.2002.36661. [DOI] [PubMed] [Google Scholar]
- 13.Ganesan R, Hirschowitz L, Dawson P, et al. Neuroendocrine Carcinoma of the Cervix: Review of a Series of Cases and Correlation With Outcome. International journal of surgical pathology. 2016;24:490–496. doi: 10.1177/1066896916643385. [DOI] [PubMed] [Google Scholar]
- 14.Howitt BE, Kelly P, McCluggage WG. Pathology of Neuroendocrine Tumours of the Female Genital Tract. Current oncology reports. 2017;19:59. doi: 10.1007/s11912-017-0617-2. [DOI] [PubMed] [Google Scholar]
- 15.Straughn JM, Jr, Richter HE, Conner MG, et al. Predictors of outcome in small cell carcinoma of the cervix--a case series. Gynecologic oncology. 2001;83:216–220. doi: 10.1006/gyno.2001.6385. [DOI] [PubMed] [Google Scholar]
- 16.Kurman RJ, Carcangiu ML, Herrington CS, et al. WHO Classification of Tumors of Female Reproductive Organs. Lyon: IARC; 2014. [Google Scholar]
- 17.Horn LC, Lindner K, Szepankiewicz G, et al. p16, p14, p53, and cyclin D1 expression and HPV analysis in small cell carcinomas of the uterine cervix. International journal of gynecological pathology : official journal of the International Society of Gynecological Pathologists. 2006;25:182–186. doi: 10.1097/01.pgp.0000185406.85685.df. [DOI] [PubMed] [Google Scholar]
- 18.Wang HL, Lu DW. Detection of human papillomavirus DNA and expression of p16, Rb, and p53 proteins in small cell carcinomas of the uterine cervix. The American journal of surgical pathology. 2004;28:901–908. doi: 10.1097/00000478-200407000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Xing D, Schoolmeester JK, Ren Z, et al. Lower Female Genital Tract Tumors With Adenoid Cystic Differentiation: P16 Expression and High-risk HPV Detection. The American journal of surgical pathology. 2016;40:529–536. doi: 10.1097/PAS.0000000000000565. [DOI] [PubMed] [Google Scholar]
- 20.Mannion C, Park WS, Man YG, et al. Endocrine tumors of the cervix: morphologic assessment, expression of human papillomavirus, and evaluation for loss of heterozygosity on 1p,3p, 11q, and 17p. Cancer. 1998;83:1391–1400. doi: 10.1002/(sici)1097-0142(19981001)83:7<1391::aid-cncr17>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Abidin AZ, Garassino MC, Califano R, et al. Targeted therapies in small cell lung cancer: a review. Therapeutic advances in medical oncology. 2010;2:25–37. doi: 10.1177/1758834009356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho SY, Choi M, Ban HJ, et al. Cervical small cell neuroendocrine tumor mutation profiles via whole exome sequencing. Oncotarget. 2017;8:8095–8104. doi: 10.18632/oncotarget.14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frumovitz M, Burzawa JK, Byers LA, et al. Sequencing of mutational hotspots in cancer-related genes in small cell neuroendocrine cervical cancer. Gynecologic oncology. 2016;141:588–591. doi: 10.1016/j.ygyno.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evander M, Edlund K, Boden E, et al. Comparison of a one-step and a two-step polymerase chain reaction with degenerate general primers in a population-based study of human papillomavirus infection in young Swedish women. Journal of clinical microbiology. 1992;30:987–992. doi: 10.1128/jcm.30.4.987-992.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang SW, Lee YS, Chen TA, et al. Human papillomavirus in oral leukoplakia is no prognostic indicator of malignant transformation. Cancer epidemiology. 2009;33:118–122. doi: 10.1016/j.canep.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Gondek LP, Zheng G, Ghiaur G, et al. Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia. 2016;30:1916–1920. doi: 10.1038/leu.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang KL, Chang TC, Jung SM, et al. Primary treatment and prognostic factors of small cell neuroendocrine carcinoma of the uterine cervix: a Taiwanese Gynecologic Oncology Group study. European journal of cancer. 2012;48:1484–1494. doi: 10.1016/j.ejca.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 28.Zivanovic O, Leitao MM, Jr, Park KJ, et al. Small cell neuroendocrine carcinoma of the cervix: Analysis of outcome, recurrence pattern and the impact of platinum-based combination chemotherapy. Gynecologic oncology. 2009;112:590–593. doi: 10.1016/j.ygyno.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Herrington CS, Graham D, Southern SA, et al. Loss of retinoblastoma protein expression is frequent in small cell neuroendocrine carcinoma of the cervix and is unrelated to HPV type. Human pathology. 1999;30:906–910. doi: 10.1016/s0046-8177(99)90243-5. [DOI] [PubMed] [Google Scholar]
- 30.Stoler MH, Mills SE, Gersell DJ, et al. Small-cell neuroendocrine carcinoma of the cervix. A human papillomavirus type 18-associated cancer. Am J Surg Pathol. 1991;15:28–32. doi: 10.1097/00000478-199101000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Masumoto N, Fujii T, Ishikawa M, et al. P16 overexpression and human papillomavirus infection in small cell carcinoma of the uterine cervix. Hum Pathol. 2003;34:778–783. doi: 10.1016/s0046-8177(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 32.Wistuba II, Thomas B, Behrens C, et al. Molecular abnormalities associated with endocrine tumors of the uterine cervix. Gynecologic oncology. 1999;72:3–9. doi: 10.1006/gyno.1998.5248. [DOI] [PubMed] [Google Scholar]
- 33.Ishida GM, Kato N, Hayasaka T, et al. Small cell neuroendocrine carcinomas of the uterine cervix: a histological, immunohistochemical, and molecular genetic study. International journal of gynecological pathology : official journal of the International Society of Gynecological Pathologists. 2004;23:366–372. doi: 10.1097/01.pgp.0000139637.01977.61. [DOI] [PubMed] [Google Scholar]
- 34.Pao CC, Kao SM, Chen JH, et al. State of mutational alterations of p53 and retinoblastoma susceptibility genes in papillomavirus-negative small cell cervical carcinomas. Journal of surgical oncology. 1994;57:87–93. doi: 10.1002/jso.2930570204. [DOI] [PubMed] [Google Scholar]
- 35.Kaelbling M, Burk RD, Atkin NB, et al. Loss of heterozygosity on chromosome 17p and mutant p53 in HPV-negative cervical carcinomas. Lancet. 1992;340:140–142. doi: 10.1016/0140-6736(92)93214-8. [DOI] [PubMed] [Google Scholar]
- 36.Kim YT, Thomas NF, Kessis TD, et al. p53 mutations and clonality in vulvar carcinomas and squamous hyperplasias: evidence suggesting that squamous hyperplasias do not serve as direct precursors of human papillomavirus-negative vulvar carcinomas. Human pathology. 1996;27:389–395. doi: 10.1016/s0046-8177(96)90113-6. [DOI] [PubMed] [Google Scholar]
- 37.Paquette RL, Lee YY, Wilczynski SP, et al. Mutations of p53 and human papillomavirus infection in cervical carcinoma. Cancer. 1993;72:1272–1280. doi: 10.1002/1097-0142(19930815)72:4<1272::aid-cncr2820720420>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 38.Scholes AG, Liloglou T, Snijders PJ, et al. p53 mutations in relation to human papillomavirus type 16 infection in squamous cell carcinomas of the head and neck. International journal of cancer. 1997;71:796–799. doi: 10.1002/(sici)1097-0215(19970529)71:5<796::aid-ijc17>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 39.Tommasino M, Accardi R, Caldeira S, et al. The role of TP53 in Cervical carcinogenesis. Human mutation. 2003;21:307–312. doi: 10.1002/humu.10178. [DOI] [PubMed] [Google Scholar]
- 40.Yemelyanova A, Ji H, Shih Ie M, et al. Utility of p16 expression for distinction of uterine serous carcinomas from endometrial endometrioid and endocervical adenocarcinomas: immunohistochemical analysis of 201 cases. The American journal of surgical pathology. 2009;33:1504–1514. doi: 10.1097/PAS.0b013e3181ac35f5. [DOI] [PubMed] [Google Scholar]
- 41.O'Neill CJ, McBride HA, Connolly LE, et al. Uterine leiomyosarcomas are characterized by high p16, p53 and MIB1 expression in comparison with usual leiomyomas, leiomyoma variants and smooth muscle tumours of uncertain malignant potential. Histopathology. 2007;50:851–858. doi: 10.1111/j.1365-2559.2007.02699.x. [DOI] [PubMed] [Google Scholar]
- 42.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 43.Fruman DA, Chiu H, Hopkins BD, et al. The PI3K Pathway in Human Disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 45.Henken FE, Banerjee NS, Snijders PJ, et al. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Molecular cancer. 2011;10:71. doi: 10.1186/1476-4598-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright AA, Howitt BE, Myers AP, et al. Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer. 2013;119:3776–3783. doi: 10.1002/cncr.28288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Koneva LA, Virani S, et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:4735–4745. doi: 10.1158/1078-0432.CCR-16-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cellular signalling. 2014;26:149–161. doi: 10.1016/j.cellsig.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 51.Hodgson A, Amemiya Y, Seth A, et al. Genomic abnormalities in invasive endocervical adenocarcinoma correlate with pattern of invasion: biologic and clinical implications. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2017 doi: 10.1038/modpathol.2017.80. [DOI] [PubMed] [Google Scholar]
- 52.Lyons YA, Frumovitz M, Soliman PT. Response to MEK inhibitor in small cell neuroendocrine carcinoma of the cervix with a KRAS mutation. Gynecologic oncology reports. 2014;10:28–29. doi: 10.1016/j.gore.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. Journal of the National Cancer Institute. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer discovery. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Fan S, Meng Q, et al. BRCA1 interaction with human papillomavirus oncoproteins. The Journal of biological chemistry. 2005;280:33165–33177. doi: 10.1074/jbc.M505124200. [DOI] [PubMed] [Google Scholar]
- 56.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 57.Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet. Oncology. 2017;18:1274–1284. doi: 10.1016/S1470-2045(17)30469-2. [DOI] [PubMed] [Google Scholar]
- 58.Matulonis UA, Penson RT, Domchek SM, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Annals of oncology : official journal of the European Society for Medical Oncology. 2016;27:1013–1019. doi: 10.1093/annonc/mdw133. [DOI] [PubMed] [Google Scholar]
- 59.Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. The Lancet. Oncology. 2016;17:1579–1589. doi: 10.1016/S1470-2045(16)30376-X. [DOI] [PubMed] [Google Scholar]
- 60.Dougherty BA, Lai Z, Hodgson DR, et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget. 2017;8:43653–43661. doi: 10.18632/oncotarget.17613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Representative histologic and immunohistochemical images of case 8. The tumor shows typical morphology of SCNEC with a monotonous population of cells and infiltrative growth pattern (A). The tumor cells have scanty cytoplasm, abundant mitotic and apoptotic activity (B) and display focal chromogranin immunoreactivity (C) and diffuse p16 expression (D).
Supplementary Figure 2. Representative histologic images of case 10. The tumor displays an infiltrative growth pattern (A) and discohesive cells with molding, scanty cytoplasm, and extensive apoptosis and necrosis (B) with some rosette-like structures (C).
Supplementary Table 1. Detailed information about gene mutations and variants