

Abstract
Exposure of normal and tumor-derived cells to TGFβ results in different outcomes, depending on the regulation of key targets. The CDK inhibitor p27Kip1 is one of these TGFβ targets and is essential for the TGFβ-induced cell cycle arrest. TGFβ treatment inhibits p27Kip1 degradation and induces its nuclear translocation, through mechanisms that are still unknown. Recent evidences suggest that SUMOylation, a post-translational modification able to modulate the stability and subcellular localization of target proteins, critically modifies members of the TGFβ signaling pathway. Here, we demonstrate that p27Kip1 is SUMOylated in response to TGFβ treatment. Using different p27Kip1 point mutants, we identified lysine 134 (K134) as the residue modified by small ubiquitin-like modifier 1 (SUMO1) in response to TGFβ treatment. TGFβ-induced K134 SUMOylation increased protein stability and nuclear localization of both endogenous and exogenously expressed p27Kip1. We observed that SUMOylation regulated p27Kip1 binding to CDK2, thereby governing its nuclear proteasomal degradation through the phosphorylation of threonine 187. Importantly, p27Kip1 SUMOylation was necessary for proper cell cycle exit following TGFβ treatment. These data indicate that SUMOylation is a novel regulatory mechanism that modulates p27Kip1 function in response to TGFβ stimulation. Given the involvement of TGFβ signaling in cancer cell proliferation and invasion, our data may shed light on an important aspect of this pathway during tumor progression.
Keywords: p27kip1, SUMOylation, TGFβ
Introduction
The cell cycle inhibitor p27Kip1 (hereafter referred to as p27) was discovered as a heat-stable inhibitor of the cyclin E-CDK2 complex in TGFβ-arrested cells (Polyak et al., 1994; Slingerland et al., 1994; Slingerland and Pagano, 2000). Subsequent studies have clarified that p27 is a tumor suppressor protein that regulates the G1 to S phase transition by binding and modulating the activity of cyclin-dependent kinases (CDKs). In G1-arrested cells, p27 expression and stability are maximal resulting in the binding and inactivation of nuclear CDKs. Following growth factor stimulation, the progressive decrease of p27 protein levels leads to full activation of cyclin E-CDK2 and cyclin A-CDK2, allowing the transition from the G1 to the S phase of the cell cycle (Slingerland and Pagano, 2000).
Transforming growth factor beta (TGFβ) signaling can exert opposing effects in cancer, such as a growth inhibitory function in early phases of tumorigenesis and the activation of a metastatic program in later stages of cancer progression (Massagué, 2008). In the first context, p27 expression is essential to induce G1 arrest in response to TGFβ treatment. Silencing of p27 in TGFβ-responsive breast or endometrial cancer cells is sufficient to overcome the G1 arrest imposed by TGFβ treatment (Donovan et al., 2002; Lecanda et al., 2009). These evidences are in complete accord with the notion that reduction of p27 levels may significantly contribute to loss of normal responsiveness to growth inhibitory stimuli during cancer progression, as demonstrated in several in vitro and in vivo models (Belletti et al., 2005). TGFβ induces cell cycle arrest by both increasing p27 protein levels and favoring its nuclear accumulation (Lecanda et al., 2007, 2009). Despite extensive evidences supporting that p27 is a key downstream molecule determining the fate of TGFβ-responsive cells, the precise molecular mechanisms whereby TGFβ regulates both p27 stability and nuclear accumulation are only poorly understood. It has been described that TGFβ blocks p27 ubiquitin-dependent nuclear degradation through the Smad2/3-mediated downregulation of two key members of the SCFSkp2 E3 ligase complex, Csk1 and Skp2. However, this mechanism seems to be necessary for the maintenance rather than the induction of p27 nuclear accumulation since the decrease of Csk1 and Skp2 follows, rather than precedes, p27 accumulation (Lecanda et al., 2009). It is clear that p27 is differently localized and phosphorylated in TGFβ-resistant compared with TGFβ-sensitive breast epithelial cells, and these modifications are associated with the failure of TGFβ to induce the inhibition of CDKs and the cell cycle arrest (Ciarallo et al., 2002), but it is much less clear whether and how TGFβ directly modifies p27 phosphorylation and/or localization.
Recent evidences suggest that SUMOylation regulates the TGFβ signaling pathway. SUMO modification significantly impacts on TGFβ type I receptor (Kang et al., 2008), and Smad proteins function (Imoto et al., 2003; Lee et al., 2003; Lin et al., 2003; Liang et al., 2004). Moreover, TGFβ directly impinge on Smad4 SUMOylation, through the stabilization of its E3 SUMO-ligase PIASxβ (Ohshima and Shimotohno, 2003).
SUMOylation is a reversible post-translational modification that consists in the covalent attachment of a SUMO molecule to one or more lysines of the target substrate. Four small ubiquitin-like modifier (SUMO) proteins are known in vertebrates. SUMOylation occurs in a stepwise manner and involves a cascade of SUMO-specific enzymes. These include an E1-activating enzyme (the Aos1/Uba2 heterodimer), the unique E2-conjugating enzyme Ubc9, and several E3 ligases, which contribute to substrate selectivity and increase the SUMOylation efficiency in vivo (Kerscher et al., 2006; Geiss-Friedlander and Melchior, 2007). The functional consequences of SUMOylation may vary and include modulation of function, subcellular localization, complex formation, and/or stability of the target proteins (Wilkinson and Henley, 2010).
Here, we test the hypothesis that SUMOylation could be involved in the regulation of p27 by TGFβ and demonstrate that TGFβ induces p27 SUMOylation on lysine 134, eventually increasing p27 stability and contributing to its nuclear accumulation and cell cycle exit.
Results
p27 is SUMOylated in vitro and in vivo and lysine 134 is the modified residue
To evaluate whether p27 could be targeted by SUMOylation, we first performed in silico analyses using SUMOplot™ (Abgent) and SUMOsp (Ren et al., 2009) prediction analysis programs (Supplementary Figure S1). These analyses revealed that several lysines (K) present in p27 protein, although non-consensus, are potential sites for SUMO attachment, and indicated K134 as the residue predicted to be SUMOylated with the highest score (Supplementary Figure S1B and C). We also observed that p27 co-immunoprecipitated with Ubc9, the sole E2 SUMO-conjugating enzyme and thus essential for SUMOylation (Supplementary Figure S1D and E), supporting the possibility that p27 was SUMOylated in vivo. By transfecting FLAG-tagged p27WT in the presence or absence of the SUMOylation machinery, we identified a slower migrating doublet of the apparent molecular weight of ∼40−45 kDa, recognized by both anti-HA and anti-FLAG antibodies, which was observed only when SUMO1 and Ubc9 were overexpressed (Figure 1A, lane 2). We next performed the same analysis with p271–170, a mutant lacking the last 28 aminoacids (Baldassarre et al., 2005; Schiappacassi et al., 2008; Berton et al., 2009) and therefore lacking two phosphorylation sites, threonines 187 (T187) and 198 (T198), that are known to be important for the regulation of p27 stability (Tsvetkov et al., 1999; Kossatz et al., 2006; Liang et al., 2007; Schiappacassi et al., 2011) and localization (Fujita et al., 2002, 2003; Motti et al., 2004). p271–170 was SUMOylated to the same extent as the WT protein (Figure 1A, lane 4), suggesting that p27 SUMOylation was not dependent on the phosphorylation of T187 or T198 and excluding K189 and K190 as SUMO modification sites. In vitro SUMOylation assay, using the recombinant histidine (His)-tagged p27WT as substrate, confirmed the appearance of higher molecular-weight bands, which were specifically recognized by an anti-p27 antibody, only when the E1 activating enzyme (SAE1/SAE2) and Ubc9 were both present (Figure 1B). In vitro SUMOylation of p27 occurred in a dose-dependent manner relative to the amount of Ubc9 added to the reaction (Figure 1B), confirming the specificity of this covalent modification.
Figure 1.
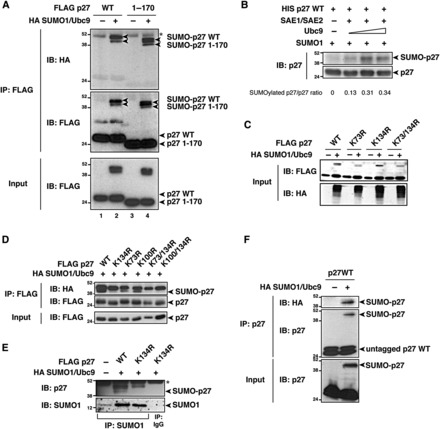
p27 is SUMOylated on K134 in vitro and in vivo. (A) Western blot analysis of FLAG-p27 and HA-SUMO1 expression in immunoprecipitates (IP) and lysates (Input) derived from 293T/17 cells transfected with FLAG-p27WT or Flag-p271–170, in the presence or absence of HA-SUMO1 and untagged Ubc9. Arrowheads indicate the unmodified and SUMO-modified p27 proteins detected using anti-HA (upper panel) or anti-FLAG (middle and lower panels) antibodies. Asterisk indicates the IgG heavy chain band. (B) Western blot analysis of p27WT recombinant protein in an in vitro SUMOylation assay. His-tagged p27 was incubated with SAE1/SAE2, SUMO, and increasing doses of Ubc9. p27 SUMOylation, expressed as a SUMOylated p27/p27 ratio, was calculated by densitometric analysis of the blots. (C) Western blot analysis of p27 and HA-SUMO1 expression in lysates (Input) derived from 293T/17 cells transfected with FLAG-p27WT or indicated FLAG-p27KR mutants in the presence or absence of HA-SUMO1 and untagged Ubc9. (D) Western blot analysis of p27 and HA-SUMO1 expression in immunoprecipitates (IP) and lysates (Input) derived from 293T/17 cells transfected with FLAG-p27WT or indicated FLAG-p27KR mutants in the presence of HA-SUMO1 and untagged Ubc9. Arrowheads indicate the unmodified and SUMO-modified p27 proteins detected using anti-HA (upper panel) or anti-FLAG (middle and lower panels) antibodies. (E) Western blot analysis of p27 and SUMO1 expression in SUMO1 (IP SUMO1) or control (IP IgG) immunoprecipitates from HeLa cells transfected with FLAG-p27WT or FLAG-p27K134R in the presence or absence of HA-SUMO1 and untagged Ubc9. Arrowheads indicate SUMO-modified p27 proteins detected using anti-p27 antibody (upper panel) or the immunoprecipitated SUMO1 (lower panel). Asterisk indicates the IgG heavy chain band. (F) Western blot analysis of p27 and HA-SUMO1 expression in lysates (Input) and immunoprecipitates (IP) derived from 293T/17 cells transfected with untagged p27WT in the presence or absence of HA-SUMO1 and untagged Ubc9. Arrowheads indicate the unmodified and SUMO-modified p27 proteins detected using anti-p27 (lower and middle panels) or anti-HA (upper panel) antibodies.
A closer evaluation of the molecular weight of SUMOylated p27 strongly suggested that p27 was mono-SUMOylated. However, the comparison of the in vitro and in vivo SUMOylation assays raised the possibility that two different lysines were modified (Figure 1A).
Our prediction analyses suggested that three lysines in p27 (K73, K100, and K134) had the highest probability to be sites of SUMOylation (Supplementary Figure S1C). We thus generated p27 mutants in which these three lysines were substituted with arginines (K to R mutants) and demonstrated that only K134 was SUMOylated, since one of the two bands recognized by anti-p27 and/or anti-HA antibodies disappeared in the p27 mutants carrying the K134R substitution (Figure 1C−E). In search for the second potential SUMOylation site (i.e. the upper band recognized by the anti-HA and/or anti-p27 antibodies), we generated p27 KR point mutants of all 13 lysines present in the p27 sequence, alone or in combination with the K134R substitution. These analyses demonstrated that only K134 was the site for SUMO modification in p27 (Supplementary Figure S2A−D), and we confirmed this using a FLAG-tagged SUMO-defective p27 mutant in which all lysine residues were substituted with arginines (FLAG-p2713KR). FLAG-p2713KR was still recognized by both anti-p27 and anti-HA (SUMO1) antibodies as a single band of ∼45 kDa (Supplementary Figure S2E), raising the possibility that one SUMOylated lysine was present in the FLAG-tag. Accordingly, the comparison between untagged and FLAG-tagged p27WT proteins demonstrated that only the latter was SUMO-modified on two different sites, while the untagged p27WT showed only one site of SUMO modification (Figure 1F and Supplementary Figure S2F). These data demonstrate that p27 is SUMOylated in vitro and in vivo and that K134 is the residue preferentially SUMO-modified.
p27 SUMOylation is increased by TGFβ stimulation
Based on the notions that the TGFβ signaling pathway is regulated by SUMOylation and on the consideration that the precise molecular mechanisms whereby TGFβ regulates p27 stability and nuclear accumulation are only poorly understood, we tested whether SUMOylation was involved in p27 regulation by TGFβ. HeLa cells transfected with FLAG-p27, HA-SUMO1, and Ubc9 were treated or not with TGFβ, and p27 SUMOylation was analyzed. TGFβ signaling induced a 3.5-fold increase in p27 SUMOylation levels (Figure 2A and Supplementary Figure S3A) and this modification was maintained up to 5 h after treatment (Figure 2A). As expected, TGFβ increased the amount of SUMOylated p27WT, while it had no effect on p27K134R (Figure 2B, lanes 3–4), which remained un-SUMOylated as the p2713KR mutant (Supplementary Figure S3B, lanes 3–4). Moreover, no SUMOylation was detected when the dominant-negative mutant of Ubc9 (Ubc9DN) (Tashiro et al., 1997) was overexpressed, both in the presence and in the absence of TGFβ (Figure 2B, lanes 5–6). To reinforce the central role of K134 in the SUMOylation of p27 following TGFβ treatment, we first substituted the two Lys of the FLAG-Tag with Arg. Yet, this modification resulted in a loss of anti-FLAG antibody recognition (Supplementary Figure S3C), preventing us from addressing whether they are non-specifically SUMOylated in the presence or absence of TGFβ. Next, we generated the HA-tagged p27WT and p27K134R proteins, and demonstrated that only p27WT was significantly SUMOylated and its SUMOylation increased by 3.8-fold after TGFβ treatment (Supplementary Figure S3D), overall confirming that K134 in p27 is the preferentially SUMOylated lysine in response to TGFβ.
Figure 2.
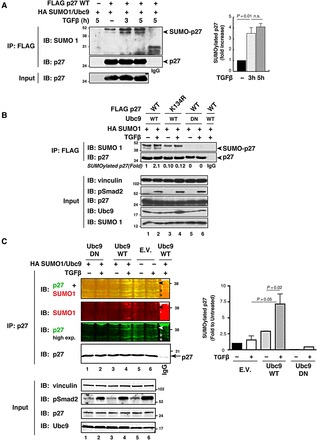
p27 SUMOylation is induced by TGFβ stimulation. (A) Western blot analysis of p27 and HA-SUMO1 expression in immunoprecipitates (IP) derived from HeLa cells transfected with FLAG-p27WT in the presence of HA-SUMO1 and untagged Ubc9, and treated or not with human recombinant TGFβ (20 ng/ml) for 3 or 5 h. Arrowheads indicated the unmodified and SUMO-modified p27 proteins detected using anti-SUMO (upper panel) or anti-p27 (lower panel) antibodies. IgG marks the lane loaded with control IP. Lysates (Input) were analyzed for the expression of p27. The levels of SUMOylated p27 in TGFβ-treated Hela cells were calculated by densitometric analysis of the bands and expressed as fold increase relative to untreated cells (right graph). (B) Western blot analysis of p27 and HA-SUMO1 expression in immunoprecipitates (IP) derived from HeLa cells transfected with FLAG-p27WT or FLAG-p27K134R in the presence of HA-SUMO1 and untagged Ubc9 wild-type (WT) or Ubc9 dominant-negative (DN), and treated or not with human recombinant TGFβ (20 ng/ml) for 3 h. Arrowheads indicate the unmodified and SUMO-modified p27 proteins detected using anti-SUMO (upper panel) or anti-p27 (lower panel) antibodies. IgG marks the lane loaded with control IP. Lysates (Input) were analyzed for the expression of p27, Ubc9, SUMO1, and Smad2 phosphorylated on Ser 465/467 (pSmad2) to evaluate the activation of the TGFβ pathway. Vinculin was used as loading control. The levels of SUMOylated p27 in TGFβ-treated HeLa cells were calculated by densitometric analysis of the bands and expressed as fold increase relative to untreated cells expressing the p27WT protein (lane 1). (C) Western blot analysis of p27 and SUMO1 expression in immunoprecipitates (IP) derived from MCF7 cells transfected or not with HA-SUMO1 and untagged Ubc9 wild-type (WT) or Ubc9 dominant-negative (DN), and treated or not with human recombinant TGFβ (20 ng/ml) for 5 h. Arrowheads indicate SUMO-modified endogenous p27 proteins detected using anti-SUMO1 (second panel) or anti-p27 (third and fourth panel) antibodies. Upper panels report the merge (yellow) of p27 (green) and SUMO1 (red) signals. The arrow indicates the unmodified IP p27. IgG marks the lane loaded with control IP. Asterisks indicate non-specific bands. Lysates (Input) were analyzed for the expression of p27, Ubc9, and Smad2 phosphorylated on Ser 465/467 (pSmad2) to confirm the activation of the TGFβ pathway. Vinculin was used as loading control. The levels of SUMOylated p27 in MCF7 cells were calculated by densitometric analysis of the bands and expressed as fold increase relative to untreated cells (right graph). Data in graphs in A and C represent mean ± SD of three independent experiments. Significance was calculated using the unpaired t-test.
To confirm our results in a more physiological condition, we tested SUMOylation of endogenous p27 protein in the TGFβ-responsive MCF7 cell line. To overcome the difficulty in identifying SUMO-modified endogenous proteins due to the transient nature of this modification, we first used a highly sensitive proteomic approach based on immunoprecipitation coupled with mass spectrometry. MCF7 cells transfected with SUMO1/Ubc9 and treated with TGFβ were immunoprecipitated with an anti-SUMO1 antibody, and the spectrum of SUMO1-associated proteins was evaluated. Among these, p27 was the one with the highest MASCOT score (Supplementary Figure S4), supporting the possibility that it was SUMOylated following TGFβ treatment. Based on this encouraging result, we next tested whether in the same conditions the SUMOylation of endogenous p27 could be revealed by immunoprecipitation and western blot analyses. Under basal condition, SUMOylated p27 was almost undetectable (Figure 2C, lane 5). However, overexpression of SUMO1/Ubc9 (Figure 2C, lane 3) or treatment with TGFβ (Figure 2C, lane 6) increased by 2-fold the levels of endogenous p27 SUMOylation, and the combination of TGFβ and SUMO1/Ubc9 overexpression was synergistically and significantly more effective (Figure 2C, lane 4 and right graph). Under the same conditions, expression of Ubc9DN completely prevented p27 SUMOylation (Figure 2C, lanes 1 and 2), as demonstrated by p27 immunoprecipitation followed by western blot analysis using either anti-p27 or anti-SUMO1 antibody (Figure 2C, the ∼37 kDa yellow band resulting from superimposition of the green anti-p27 and the red anti-SUMO1 fluorescence signals from the Odyssey Imaging System).
TGFβ-induced SUMOylation increases p27 stability by decreasing its binding to CDK2
It is known that treatment of MCF7 cells with TGFβ induces p27 protein stabilization and nuclear translocation (Massagué, 2008). We investigated whether TGFβ-induced p27 SUMOylation could be implicated in this functional regulation of p27.
First, when blocking de novo protein synthesis by treating cells expressing p27WT or p27K134R with cycloheximide (CHX) in the presence or absence of TGFβ and overexpressing the SUMO1 machinery, we observed that in the absence of TGFβ, p27WT and p27K134R had the same protein stability (Figure 3A, compare lanes 1−4 with lanes 9−12). In the presence of TGFβ, p27WT protein was more stable, while the p27K134R mutant was degraded more rapidly than in untreated cells (Figure 3A, lanes 5−8 and 13−16), suggesting that p27 SUMOylation of K134 was required for TGFβ-induced p27 protein stability. These results were confirmed by using the proteasome inhibitor MG132 along with TGFβ and CHX in cells transfected with p27WT or p27K134R (Figure 3B). Inhibition of proteasome activity in control cells induced an increase in p27WT stability that was strongly reduced by TGFβ treatment (black bars in Figure 3B graph). On the contrary, p27K134 levels particularly increased in TGFβ-treated cells (gray bars in Figure 3B graph), suggesting that p27 SUMOylation on K134 induced by TGFβ somehow prevented its ubiquitin-mediated degradation. Accordingly, the combination of TGFβ treatment and SUMO1/Ubc9 overexpression significantly increased the stability of the endogenous p27 protein (Figure 3C).
Figure 3.
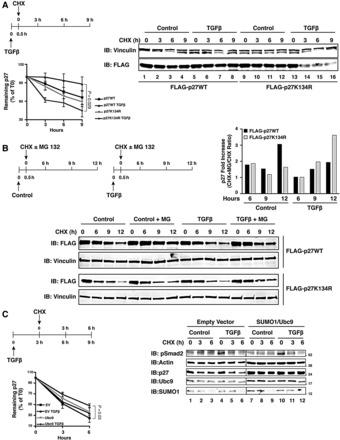
TGFβ-induced SUMOylation increases p27 protein stability. (A) Western blot analysis of FLAG-p27 expression in MCF7 cells transfected with FLAG-p27WT or FLAG-p27K134R in the presence of HA-SUMO1 and untagged Ubc9, treated or not with TGFβ (20 ng/ml) and exposed to cycloheximide (CHX, 10 µg/ml) for 9 h. Vinculin was used as loading control. The percentage of remaining normalized p27 protein (p27/vinculin ratio) relative to the level at CHX 0 h was determined by densitometric analysis of the blots (±SD) from three independent experiments (lower left graph). (B) Western blot analysis of FLAG-p27 expression in HeLa cells transfected with FLAG-p27WT or FLAG-p27K134R in the presence of HA-SUMO1 and untagged Ubc9, treated or not with TGFβ (20 ng/ml) and exposed to cycloheximide (CHX, 10 µg/ml) with or without MG-132 (10 µM) for up to 12 h. Vinculin was used as loading control. Fold increase of remaining p27 expression (relative to T0) at each time point due to MG132 treatment was determined by densitometric analysis of the blots (upper right graph). To this aim the CHX+MG-132/CHX ratio of remaining p27 expression (relative to T0) was calculated and expressed as fold induction over only CHX-treated cells. (C) Western blot analysis of endogenous p27 expression in MCF7 cells transfected or not with HA-SUMO1 and untagged Ubc9, and treated or not with TGFβ (20 ng/ml) for 3 h. Actin was used as loading control and phosphorylated Smad2 (pSer 465/467) was used as a marker of TGFβ signaling activation. The percentage of remaining normalized p27 protein (p27/actin ratio) relative to their level at CHX 0 h was determined by densitometric analysis of the blots (±SD) from three independent experiments (lower left graph).
In accord with the above experiments, we observed that TGFβ decreased p27WT while it increased p27K134R poly-ubiquitination (Figure 4A), although p27WT and p27K134R proteins displayed very similar stability in vitro (Supplementary Figure S5), suggesting that the affinity to members of the ubiquitination machinery was not affected by K134R substitution. In complete agreement with the previous experiments, we observed that the increased p27K134R poly-ubiquitination relied on the presence of T187 in p27, since the p27K134R/T187A and the p27T187A mutants were both barely modified by TGFβ (Supplementary Figure S6A). Moreover, in the presence of TGFβ, p27K134R was more ubiquitinated than p27WT, p27T187A, and p27K134R/T187A proteins (Figure 4B). p27 protein stability is mainly regulated by phosphorylation on T187 residue by CDK2 that allows the binding of p27 to the F-box protein Skp2, thus favoring its nuclear ubiquitin-mediated degradation (Montagnoli et al., 1999; Tsvetkov et al., 1999; Belletti et al., 2005). Intriguingly, it has been demonstrated that TGFβ treatment results in the accumulation of a p27 form not associated with cyclin/CDK2 complexes (Taipale et al., 2000). Based on this knowledge, we speculated that TGFβ-induced p27 SUMOylation interfered with p27/CDK2 binding, eventually impairing its ubiquitin-mediated degradation. To test this hypothesis, MCF7 cells were co-transfected with FLAG-tagged p27WT or p27K134R along with HA-SUMO1 and Ubc9, and were treated or not with TGFβ. Immunoprecipitation of FLAG-tagged p27 revealed a marked increase in the binding to CDK2 for non-SUMOylable p27K134R mutant compared with the WT protein (Figure 4C). Similar results were observed by using TGFβ-responsive HeLa cells (Supplementary Figure S6B, cf. lanes 2–3 with lanes 5–6). Moreover, TGFβ treatment impaired the binding between Skp2 and p27WT, having no effects on its binding to p27K134R (Figure 4D), again confirming that SUMOylation of K134 affects p27 protein stability by altering its ubiquitin-mediated degradation.
Figure 4.
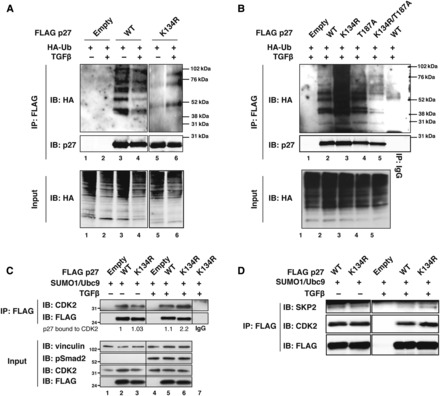
TGFβ-induced SUMOylation decreases p27 affinity for CDK2. (A) In vivo ubiquitination assay in 293T/17 cells transfected with the indicated FLAG-tagged vectors and with HA-tagged ubiquitin. Cells were then treated or not with TGFβ (20 ng/ml). Lysates were immunoprecipitated with anti-FLAG antibody and probed with anti-HA and anti-p27 antibodies. Cell lysates were probed with anti-HA antibody for the equivalent level of ubiquitin transfection in different samples (lower panel). (B) In vivo ubiquitination assay in 293T/17 cells transfected with the indicated FLAG-tagged vectors along with HA-tagged ubiquitin. Lysates were immunoprecipitated with anti-FLAG or control (IgG = Anti-V5) antibody and probed with anti-HA and anti-p27 antibodies. Cell lysates were probed with anti-HA antibody for the equivalent level of ubiquitin transfection in different samples (lower panel). (C) Co-immunoprecipitation analysis (IP: FLAG) of protein lysates derived from MCF7 cells transfected with FLAG-p27WT, FLAG-p27K134R, or FLAG empty vector in the presence of HA-SUMO1 and untagged Ubc9, and treated or not with TGFβ (20 ng/ml) for 3 h. Lysates were immunoprecipitated with anti-FLAG or control (IgG = Anti-V5) resin and probed with anti-FLAG and anti-CDK2 antibodies. Lysates (Input) were analyzed for the expression of CDK2, FLAG-p27, and Smad2 phosphorylated on Ser 465/467 (pSmad2) to confirm the activation of the TGFβ pathway. Vinculin was used as loading control. The FLAG-p27/CDK2 interaction is reported as the ratio of FLAG-p27WT bound to CDK2 based on densitometric analysis of the blots. (D) Co-immunoprecipitation analysis (IP: FLAG) of protein lysates derived from HeLa cells transfected with FLAG-p27WT, FLAG-p27K134R, or FLAG empty vector in the presence of HA-SUMO1 and untagged Ubc9, and treated or not with TGFβ (20 ng/ml) for 3 h. Lysates were immunoprecipitated with anti-FLAG or control (IgG = Anti-V5) resin and probed with anti-FLAG, anti-Skp2, and anti-CDK2 antibodies.
Next, based on the knowledge that the viral protein Gam1 specifically inhibits protein SUMOylation (Boggio et al., 2004), we verified whether inhibition of endogenous p27 SUMOylation could affect p27 accumulation after TGFβ treatment. Interestingly, Gam1 overexpression impaired the accumulation of p27 induced by TGFβ, having no effects on the levels of p27 in the absence of TGFβ (Supplementary Figure S6C and D). Overall, these results indicate that TGFβ-induced p27 SUMOylation affects p27 stability via the CDK2-mediated phosphorylation of T187 and its subsequent poly-ubiquitination.
SUMOylation is required for nuclear accumulation of p27 in response to TGFβ
As previously described (Lecanda et al., 2007, 2009), we confirmed that TGFβ induced p27 nuclear accumulation in MCF7 cells within 3 h, by western blot analysis of nuclear and cytoplasmic differential extraction (Supplementary Figure S7A) and by immunofluorescence (Supplementary Figure S7B). We next asked if SUMOylation could also be implicated in the regulation of p27 localization in response to TGFβ. To test this hypothesis, we impaired endogenous p27 SUMOylation by expressing the Ubc9DN construct. First, we checked whether Ubc9 inhibition had any effect on endogenous p27 expression in the presence or absence of TGFβ. Western blot analyses showed that impairing SUMOylation by using the Ubc9DN construct prevented TGFβ-induced p27 accumulation (Figure 5A and Supplementary Figure S8A), in accord with the stabilizing role of Ubc9 on endogenous p27 protein (Figure 3B). Next, we observed that after TGFβ treatment, p27 SUMOylation was required for its nuclear accumulation. In fact, in the presence of TGFβ, Ubc9DN decreased (Figure 5B, green arrows), while Ubc9WT increased (Figure 5B, red arrows) p27 nuclear accumulation, as demonstrated by immunofluorescence coupled with confocal analyses of MCF-7 cells transfected with the indicated Ubc9 HA-tagged expression vectors in the presence or absence of TGFβ (Figure 5B, right graph). Closer examination of transfected cells demonstrated that TGFβ (not shown) and Ubc9 expression (Supplementary Figure S8B, yellow arrows in left panels) induced the co-localization of endogenous p27 and SUMO1 in specific nuclear speckles. This co-localization was further increased by combining Ubc9 expression with TGFβ treatment (Supplementary Figure S8B, yellow arrows in middle panels), and completely prevented by expressing Ubc9DN even in the presence of TGFβ (Supplementary Figure S8B, right panels). In complete accord with these data, we demonstrated that EGFP-p27WT and p27K134R proteins were localized in the nuclear and cytoplasmic compartments at comparable extent in untreated cells (Figure 5C). However, following TGFβ exposure, EGFP-p27WT strongly accumulated in the nucleus (73% nuclear vs. 27% cytoplasmic), while the nuclear-cytoplasm distribution of EGFP-p27K134R did not significantly change relative to the levels observed in untreated cells (Figure 5C and Supplementary Figure S7). Similar results were obtained using HA-p27WT and HA-p27K134 proteins (Supplementary Figure S9), confirming that SUMOylation of K134 affected the ability of p27 to translocate and/or accumulate in the nucleus in response to TGFβ.
Figure 5.
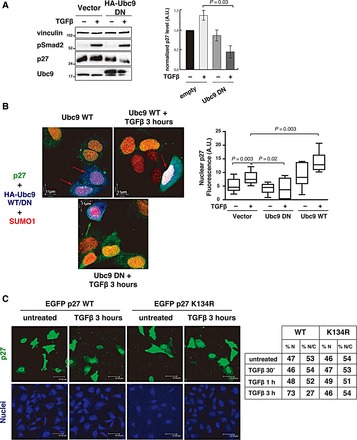
SUMOylation is necessary for TGFβ-induced p27 nuclear accumulation. (A) Western blot analysis of endogenous p27 expression level in MCF7 cells in the presence or absence of HA-tagged dominant-negative Ubc9 vector (HA-tagged Ubc9DN), and treated or not with TGFβ (20 ng/ml) for 3 h. Vinculin was used as loading control and phosphorylated Smad2 (pSer 465/467) was used as a marker of TGFβ signaling activation. Normalized p27 level (p27/vinculin ratio) relative to the level in empty vector-transfected, untreated cells was determined by densitometric analysis of the blots (±SD) (right graph). (B) Immunofluorescence analysis of p27 (green), SUMO1 (red), and Ubc9WT or Ubc9DN (blue) in MCF7 cells transiently transfected with HA-tagged Ubc9WT or Ubc9DN, and treated or not with TGFβ (20 ng/ml) for 3 h. Ubc9-expressing cells were identified using an anti-HA antibody (blue). Red arrows indicate cells expressing Ubc9WT with p27 nuclear accumulation. Green arrow indicates a cell expressing Ubc9DN without p27 nuclear accumulation. Nuclear p27 fluorescence intensity in at least 20 cells expressing or not Ubc9WT or Ubc9DN and treated or not with TGFβ was calculated using the Volocity™ Software (right graph). Significance was calculated using the Mann–Whitney unpaired t-test. (C) Immunofluorescence analysis of EGFP-p27 expression and localization (green) in HeLa cells transiently transfected with HA-SUMO1, untagged Ubc9, and EGFP-p27WT or EGFP-p27K134R mutant, and then treated with TGFβ (20 ng/ml) for 3 h. Nuclear staining is reported in blue. The percentage of cells expressing nuclear (%N) or nuclear + cytoplasmic (%N/C) EGFP-p27WT or EGFP-p27K134R before and after 30 min, 1 h, and 3 h of TGFβ treatment was determined (right table). Data were collected by using a 40× objective and scoring all transfected cells in 10 randomly selected fields per each condition.
p27 SUMOylation is required for cell cycle exit in response to TGFβ
Finally, we asked whether p27 SUMOylation in response to TGFβ had any biological effect. To answer this question, we knocked down p27 and treated MCF7 cells with TGFβ. In the absence of TGFβ, p27-silenced and control cells proliferated at a similar extent, as demonstrated by counting cells positive for the proliferation marker Ki67 that specifically labeled cycling cells (Figure 6A, lower graph). In p27 knockdown cells, TGFβ treatment partially failed to induce cell cycle exit (Figure 6A). These differences were even more evident in the presence of exogenous Ubc9 (Figure 6A and B). In control cells, Ubc9 expression reinforced the cell cycle exit induced by TGFβ (Figure 6B, left panels, red arrows), while in p27-silenced cells, it had no effects on cell cycle exit (Figure 6B, right panels, green arrows), demonstrating that p27 expression was necessary for the cell cycle arrest induced by Ubc9 and TGFβ.
Figure 6.
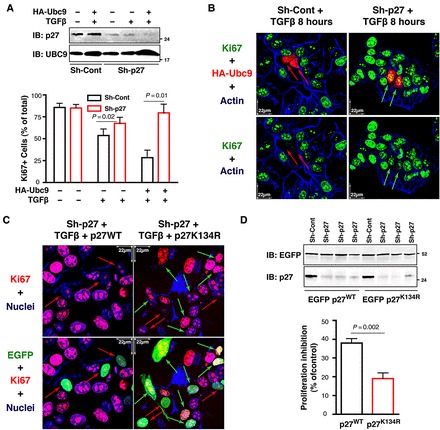
p27 SUMOylation participates in TGFβ-induced cell cycle exit. (A) Western blot analysis of p27 and Ubc9 expression in MCF7 cells stably transduced with control or p27 shRNAs, transiently transfected with HA-tagged Ubc9, and then treated with TGFβ (20 ng/ml) for 18 h (upper panel). The percentage of Ki67-positive cells in at least 50 cells expressing or not Ubc9WT and treated or not with TGFβ was determined (lower graph). Significance was calculated using the Mann–Whitney unpaired t-test. (B) Immunofluorescence analysis of Ki67 (green), Ubc9 (red), and F-Actin (blue) in cells described in A. Ubc9-expressing cells were identified using an anti-HA antibody (red). Red arrows indicate cells expressing Ubc9 and negative for Ki67, while green arrows indicate cells expressing Ubc9 and positive for Ki67. (C) Immunofluorescence analysis of Ki67 (red), EGFP-p27 (green), and nuclei (blue) in MCF7 cells stably transduced with p27 shRNAs, transiently transfected with EGFP-p27WT or EGFP-p27K134R, and then treated with TGFβ (20 ng/ml) for 18 h. Red arrows indicate cells expressing EGFP-p27 and negative for Ki67, while green arrows indicate cells expressing EGFP-p27 and positive for Ki67. (D) Western blot analysis of p27 and EGFP in the cells described in C shows the expression of EGFP-p27 proteins and endogenous p27 from three independent experiments (upper panel). The percentage of Ki67-positive cells in at least 50 cells expressing EGFP-p27WT or EGFP-p27K134R and treated with TGFβ was determined and expressed as a percent of proliferation inhibition (lower graph). Data represent mean ± SD of three independent experiments. Significance was calculated using the Mann–Whitney unpaired t-test.
These data were confirmed by expressing EGFP-p27WT or EGFP-p27K134R in p27 knockdown MCF-7 cells (Figure 6D) examining their ability to replace p27 functions in response to TGFβ (Figure 6C). Under these conditions, while the expression of EGFP-p27WT induced a clear inhibition of cell proliferation, reducing by the half the number of Ki67-positive cells after TGFβ treatment (Figure 6C, left panels, red arrows), EGFP-p27K134R failed to properly induce cell cycle exit (Figure 6C, right panels, green arrows and Figure 6D, lower graph).
Altogether, our data demonstrate that p27 is SUMOylated on K134 and this modification is important for the block of cell cycle induced by TGFβ.
Discussion
A wide range of literatures exist regarding the pathways that inactivate p27 expression and functions in cancer. Yet, much less is known regarding the mechanism by which the anti-mitogenic signals positively impinge on the p27 tumor suppressor activity. Here, we demonstrate that SUMOylation of p27, induced by the anti-mitogenic stimulus of TGFβ, is necessary for the proper regulation of its functions. This evidence is in agreement with the knowledge that SUMOylation is a post-translational modification that finely regulates protein activity, stability, and localization (Geiss-Friedlander and Melchior, 2007; Wilkinson and Henley, 2010). Whether it represents a common modification downstream from other anti-mitogenic signals able to stabilize nuclear p27, such as serum deprivation or contact inhibition, will be the matter of future investigations. In accord with our present observations, it has been reported that inhibition of SUMO-specific protease 1 (SENP1) results in increased p27 levels in colon cancer (Xu et al., 2011), corroborating the possibility that the pathway described here represents a common mechanism of p27 regulation. Our bioinformatic analyses revealed that p27 could be SUMOylated on many different lysines, even if none of the 13 lysine residues lies within the classical SUMO consensus sequence (ΨKxE/D) (Anckar and Sistonen, 2007), a possibility already described for other proteins (Gareau and Lima, 2010). Using both in vitro and in vivo SUMOylation assays, we demonstrate that p27 is modified by SUMO1 and K134 is the preferential SUMOylated residue of human p27. It is to be noted that K134 is absent in the mouse p27 protein, suggesting that different mechanisms of p27 translational control have evolved. This has been already demonstrated for the T157 phosphorylation (Liang et al., 2002), another modification involved in the regulation of p27 stability and localization after mitogenic stimulation. It is also interesting to note that a frame shift mutation involving K134 has been described in Luminal A breast cancers, where mutations in the CDKN1B gene represent driver mutations (Belletti and Baldassarre, 2012; Cancer Genome Atlas Network, 2012), and a P133T substitution, which likely disrupts the SUMOylation consensus sequence in p27, has been described in patients with familiar MEN syndrome (Lee and Pellegata, 2013). It is intriguing to note that insensitivity to the anti-proliferative effects of TGFβ represents a common feature of MEN-derived cancer cells carrying the mutation of menin (Guo and Sawicki, 2001; Kaji et al., 2001). Our findings, showing altered p27 stability and localization following TGFβ treatment when p27 is non-SUMOylable, suggest that p27 P133T mutation could affect the regulation of p27 induced by TGFβ, thereby bypassing cell cycle arrest and eventually favoring tumor progression.
One of the major characteristics of SUMOylation is that, due to the reversible and transient nature of this modification, most targets are modified at a very low level; however, this is enough to achieve maximal effect, a phenomenon referred to as the ‘SUMO enigma’ (Hay, 2005). In our systems, TGFβ treatment increases the levels of SUMOylated p27 by 3−7-fold, with the level of endogenous SUMOylated p27 rising from 0.7% to ∼5% relative to the total amount of protein. This modification was sufficient to induce clear biological effects on the cell cycle in response to TGFβ.
First, TGFβ-induced p27 SUMOylation is necessary to increase its stability. We proved that p27 SUMOylation partially prevented CDK2/p27 binding, an event necessary for the phosphorylation of p27 on T187 and its subsequent recognition by the SCFSkp2 E3 ligase complex (Montagnoli et al., 1999), which leads to ubiquitination and nuclear degradation of p27. Accordingly, TGFβ decreased the binding of p27WT but not of the non-SUMOylable p27K134R mutant to Skp2. p27K134R was more ubiquitinated in vivo than the WT protein, and this effect was completely reversed by the T187A substitution. Conversely, when tested in vitro, p27WT and p27K134R displayed very similar degradation kinetics, again excluding that K134 mutation directly influenced p27 ubiquitination. Although K134 is not contained in the p27 CDK-binding domain (that spans aa 61−90), this residue lies in a highly acidic region of p27 known to influence the binding to CDKs and, in particular, to the CDK2/Cyclin A complex (Galea et al., 2008a). It is therefore possible that SUMOylation of K134 may alter the affinity of p27 for CDK2, or, alternatively, may represent a steric obstacle for CDK2 to phosphorylate the T187, anyhow reducing its phosphorylation on T187 and its proteasomal degradation and/or increasing its stability. The fact that specific inhibition of protein SUMOylation by the viral protein Gam1 affects the accumulation of endogenous p27 induced by TGFβ but not p27 expression in untreated cells strongly suggests that TGFβ governs the switch between SUMOylation and ubiquitination to regulate p27 stability.
Second, our data indicate that SUMOylation affects p27 ability to translocate and/or accumulate in the nucleus in response to TGFβ. These results support the possibility that inhibition of p27 nuclear degradation could be responsible for this effect, although it is noteworthy that K134 also lies in a region implicated in the binding of p27 to JAB1 (aa 96−151) (Tomoda et al., 1999), an interaction known to induce p27 nuclear export, suggesting that the mechanism of nuclear accumulation of p27 might also rely on a decreased nuclear export.
Our data clearly show that TGFβ induces p27 SUMOylation, but how TGFβ favors this modification has to be clarified. Two possibilities are conceivable: one is that TGFβ induces specific post-translational modification in p27 necessary for its binding to the SUMO machinery; the other is that TGFβ induces the expression or specific post-translational modification in an E3 ligase necessary to bind p27 and favoring its SUMOylation. More studies will be necessary to address this point.
Finally, we prove that p27 SUMOylation is necessary for proper cell cycle exit induced by TGFβ. Taking into account the crucial roles of p27 in controlling cell proliferation in vivo, SUMOylated p27 participating in the decision of exiting from the cell cycle represents an important step toward a better characterization of p27 tumor suppressor activities.
Our work represents the first evidence that a member of the cell cycle inhibitors is regulated by SUMOylation and highlights the possibility that SUMOylation participates in the complex regulation of the unstructured C-terminal domain of p27, which has been demonstrated to act as a flexible ‘conduit’ for signal transduction (Galea et al., 2008a, b).
Considering that both TGFβ and SUMOylation pathways are altered during tumor progression and represent promising therapeutic targets (Massagué, 2008; Bawa-Khalfe and Yeh, 2010), it is possible to anticipate that in TGFβ-dependent tumors, p27 expression and/or localization could be used as promising predictive biomarkers to identify cancer patients who may really benefit from targeting these pathways.
Materials and methods
Cell culture, transfection, and treatments
HeLa (human cervical adenocarcinoma cell line, ATCC CCL-2), MCF7 (human breast adenocarcinoma cell line, ATCC HTB-22), and 293T/17 (human embryonic kidney cell line) cells were grown in DMEM supplemented with 10% heat-inactivated FBS (Sigma). Cells were transfected with the indicated vectors using FuGENE HD Transfection Reagent (Roche). When indicated, cells were treated with 10 µg/ml cycloheximide (Sigma) for 3 h and with human recombinant TGFβ 20 ng/ml (Peprotech) for the indicated times.
For lentiviral production, 293FT cells (Invitrogen) were co-transfected, using a standard calcium phosphate precipitation, with the lentiviral shRNA constructs (pLKO) and lentiviral packaging mix (Sigma). Seventy-two hours after transfection, medium containing viral particles was used to transduce MCF7 cells. pLKO vectors encoding for control (non-target) and two different target sequences on human p27 (sh1 TRC0000039930 targeting the coding sequence and sh2 TRC0000356318 targeting the 3′UTR) were purchased from Sigma-Aldrich. Stable knockdown pools were selected by culturing transduced cells in puromycin (1 µg/ml)-containing medium. The Myc-tagged Gam1 vector has been already described (Chiocca et al., 2002).
Construction of expression vectors
The K/R p27 mutants were generated with the QuickChange XL Site-Directed Mutagenesis kit (Agilent-Stratagene), using oligonucleotides carrying the indicated mutations. All generated mutants were controlled by sequencing. The expression vectors utilized were pEGFP-C (Clontech), pFLAG-CMV 6 (Sigma), p-CMV-HA (Clontech), and pQE-30 (Qiagen).
SUMO1 cDNA was retrotranscribed from 293T/17 total RNA, using sequence-specific oligonucleotides and cloned into a pCMV-HA expression vector (Clontech). Untagged Ubc9 constructs were generated with the QuickChange XL Site-Directed Mutagenesis kit on pCMV human HA-tagged Ubc9WT and Ubc9DN (Addgene), using oligonucleotides carrying a stop codon.
Expression and purification of recombinant proteins
Production of bacterial recombinant proteins was performed essentially as previously described (Schiappacassi et al., 2011). Briefly, p27WT and p27K134R cDNAs were cloned in the pQE-30 vector (Qiagen). Escherichia coli (M15) cells were transformed with the expression plasmid to produce recombinant proteins containing Histidine (His)-tag at the N-terminus. Proteins were subsequently purified with Ni-nitrilotriaceticacid (NiNTA) resin (Qiagen).
Preparation of cell lysates, immunoblotting, and immunoprecipitation
Total cell proteins were extracted using cold RIPA lysis buffer (150 mM NaCl, 50 mM Tris–HCl (pH 8), 1% Igepal, 0.5% sodiumdeoxycholate, 0.1% SDS) plus a protease inhibitor cocktail (Complete™, Roche), 1 mM sodium orthovanadate, and 1 mM dithiothreitol. For the analysis of p27 SUMOylation levels, total cell proteins were extracted using a 1:3 ratio mix of SDS buffer I (5% SDS, 150 mM Tris–HCl (pH 6.8), 30% glycerol) and SDS buffer II (25 mM Tris–HCl (pH 8.3), 50 mM NaCl, 0.5% NP40, 0.1% SDS, 1 mM EDTA) plus a protease inhibitor cocktail (Complete™, Roche), 1 mM sodium orthovanadate, 1 mM dithiothreitol, and 20 mM N-Ethylmaleimide (Sigma). Lysates were sonicated twice for 5 sec.
Immunoprecipitation experiments were performed using 0.5−2 mg of total cell lysate in HNTG buffer (20 mM HEPES, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100) plus the specific agarose-conjugated primary antibody (Flag-M2 affinity gel, anti-HA agarose-conjugated or anti-CDK2 antibody) and incubating overnight at 4°C. When primary antibodies were not agarose-conjugated, protein A or protein G Sepharose 4 Fast Flow (Amersham Biosciences) was added during the last 2 h of incubation.
Endogenous p27 immunoprecipitation was performed using p27 monoclonal antibody (BD, Transduction Lab). Control IPs were performed using either anti-V5 agarose-conjugated (Sigma) or anti-luciferase (Santa Cruz) antibody. Immunoprecipitated proteins were then separated in 10% SDS-polyacrylamide gels.
For immunoblotting, 40 µg of proteins were separated by 4%−20% SDS-PAGE, as previously reported (Schiappacassi et al., 2011). Primary antibodies were from Transduction Laboratories (p27, CDK2, and Ubc9), Santa Cruz (p27 C19, p27 N20, Skp2, vinculin), Sigma (FLAG, HA agarose-conjugated, SUMO1), Cell Signaling (pSer465/467 Smad2), and Covance (HA).
To calculate the fraction of SUMOylated p27, the bands corresponding to total immunoprecipitated p27 and SUMOylated p27 (only taking into account the band that could be specifically assigned to SUMO modification, i.e. the lower band of the doublet in the western blots performed with the anti-p27 antibody used to analyze different FLAG-tagged constructs), respectively, were subjected to densitometric analysis, and the percentage of SUMOylated p27 was calculated. p27 SUMOylation was then expressed as fold increase relative to untreated cells.
Mass spectrometry
In gel trypsin digestion of 1DE bands of interest was performed as previously detailed (Simula et al., 2010). Data validation was conducted by MALDI TOF peptide mass fingerprinting (PMF) on a Voyager-DE PRO Biospectrometry Workstation mass spectrometer (AB Sciex), under routine laboratory conditions (Simula et al., 2010). Database searching was done with the MASCOT search engine version 2.4 (Matrix Science Ltd), against the Swiss Prot 2012_03 database. For each gel band, the entire list of observed masses (±0.5 Da tolerance error) was compared with peptide mass values generated from the theoretical digestion of the p27 protein sequence (NP_004055.1) with trypsin by the PeptideMass Tool (http://web.expasy.org/peptide_mass/). The observed masses matched with p27 were further used for protein identification by MASCOT in order to confirm the protein identity as described (Repetto et al., 2008).
In vitro SUMOylation assay
SUMO modification of His-p27 was carried out in 30 µl reaction containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 2 mM DTT, 2 mM ATP, 270 ng SAE1/SAE2 (Boston Biochem), 300 ng Ubc9 (Boston Biochem), 4 µg SUMO1 (Boston Biochem), and 3 µg His-p27 and incubated for 2 h at 30°C. After termination in SDS-PAGE sample buffer, samples were loaded on SDS-PAGE gel and immunoblotted with the indicated antibodies.
In vitro protein degradation assay
In vitro degradation of p27 was essentially carried out as previously described (Loda et al., 1997; Schiappacassi et al., 2011). Briefly, HeLa cells were transfected with pHA-Ubiquitin vector and lysed 48 h later in ice-cold double-distilled water. The sample was frozen and thawed for three times, and then spun down to pellet debris. The supernatant was retrieved and frozen at −80°C. One microgram of His-tagged p27 was incubated at 37°C for different times in 50 µl of degradation mix containing 200 µg of lysate from HeLa cells, 50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 1 mM DTT, and 2 mM ATP. After the indicated times, reactions were stopped by adding 5× Laemmli buffer and loaded onto 4%−20% polyacrylamide gel.
Immunofluorescence
Immunofluorescence analysis was performed essentially as described before (Baldassarre et al., 2005; Schiappacassi et al., 2011). Briefly, MCF7 cells plated on coverslips and treated as indicated were fixed in PBS 4% paraformaldehyde (PFA), permeabilized in PBS 0.2% Triton X-100, and blocked in PBS 1% BSA and 10% normal goat serum. Incubation with primary antibodies, including rabbit anti-SUMO (Sigma, 8 µg/ml), goat anti-p27 C19 (Santa Cruz, 1:80), mouse anti-HA (Covance, 1:1000), and/or rabbit anti-Ki67 (Abcam, 1:200), was performed for 3 h at RT (or overnight at 4°C) in PBS 1% BSA and 1% normal goat serum. Samples were then washed in PBS and incubated with secondary antibodies for 1 h at RT. Secondary antibodies used were anti-rabbit Alexa-546, anti-mouse Alexa-546, anti-goat Alexa-488, and anti-rabbit Alexa-633 (Life Technologies). Nuclear staining was performed with TOPRO 633 (Life Technologies, 1:2000). F-Actin was stained using Alexa-633-conjugated phalloidin (Life Technologies, 1:1000). Stained cells were studied using a confocal laser-scanning microscope (TSP2 Leica) interfaced with a Leica DMIRE2 fluorescent microscope or using a Nikon Diaphot 200 epifluorescent microscope. The Volocity software (Perkin Elmer) was used to quantify fluorescence intensity (FI).
The percent of proliferation inhibition was calculated as reported (Baldassarre et al., 2005), using the formula: (% Ki67+ EGFP-negative cells) − (% Ki67+ EGFP-positive cells)/(% Ki67+ EGFP-negative cells). ‘Ki67+ EGFP-negative cells’ indicates untransfected proliferating cells, ‘Ki67+ EGPF-positive cells’ indicates proliferating p27 (WT or K134R)-expressing cells. For the Ki67 cutoff value, we calculated the mean ± SD FI/cell in each field. Then we applied a cut-off that was the mean subtracted from the SD.
Supplementary material
Supplementary Material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) IG 12854 to G.B. and AIRC IG 12075 to S.Ch., by CRO Intramural research grant to G.B., and by CRO Young Investigator Program to S.L. S.B. is a recipient of AIRC-Marie Curie Outgoing International Fellowship. S.C. is supported by an FUV (Fondazione Umberto Veronesi) fellowship.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank all members of the S.C.I.C.C. lab and the Division of Experimental Oncology 2 for scientific support and helpful discussion of the results. We thank Drs Valli De Re and Ombretta Repetto of the Proteomics Facility of the National Cancer Institute, CRO of Aviano, for the mass spectrometry analysis. We are grateful to Andrea Cia and Mara Lazzaretti for their battle in favor of cancer research, and for supporting our work.
References
- Anckar J., Sistonen L. (2007). SUMO: getting it on. Biochem. Soc. Trans. 35, 1409–1413. [DOI] [PubMed] [Google Scholar]
- Baldassarre G., Belletti B., Nicoloso M.S., et al. (2005). p27Kip1-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 7, 51–63. [DOI] [PubMed] [Google Scholar]
- Bawa-Khalfe T., Yeh E.T. (2010). SUMO losing balance: SUMO proteases disrupt SUMO homeostasis to facilitate cancer development and progression. Genes Cancer 1, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belletti B., Baldassarre G. (2012). New light on p27kip1 in breast cancer. Cell Cycle 11, 3701–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belletti B., Nicoloso M.S., Schiappacassi M., et al. (2005). p27kip1 functional regulation in human cancer: a potential target for therapeutic designs. Curr. Med. Chem. 12, 1589–1605. [DOI] [PubMed] [Google Scholar]
- Berton S., Belletti B., Wolf K., et al. (2009). The tumor suppressor functions of p27kip1 include control of the mesenchymal/amoeboid transition. Mol. Cell. Biol. 29, 5031–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggio R., Colombo R., Hay R.T., et al. (2004). A mechanism for inhibiting the SUMO pathway. Mol. Cell 16, 549–561. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocca S., Kurtev V., Colombo R., et al. (2002). Histone deacetylase 1 inactivation by an adenovirus early gene product. Curr. Biol. 12, 594–598. [DOI] [PubMed] [Google Scholar]
- Ciarallo S., Subramaniam V., Hung W., et al. (2002). Altered p27Kip1 phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor β-mediated G1 arrest. Mol. Cell. Biol. 22, 2993–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J.C., Rothenstein J.M., Slingerland J.M. (2002). Non-malignant and tumor-derived cells differ in their requirement for p27Kip1 in transforming growth factor-β-mediated G1 arrest. J. Biol. Chem. 277, 41686–41692. [DOI] [PubMed] [Google Scholar]
- Fujita N., Sato S., Katayama K., et al. (2002). Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 277, 28706–28713. [DOI] [PubMed] [Google Scholar]
- Fujita N., Sato S., Tsuruo T. (2003). Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 278, 49254–49260. [DOI] [PubMed] [Google Scholar]
- Galea C.A., Nourse A., Wang Y., et al. (2008a). Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27Kip1. J. Mol. Biol. 376, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea C.A., Wang Y., Sivakolundu S.G., et al. (2008b). Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity and signaling conduits. Biochemistry 47, 7598–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau J.R., Lima C.D. (2010). The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss-Friedlander R., Melchior F. (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956. [DOI] [PubMed] [Google Scholar]
- Guo S.S., Sawicki M.P. (2001). Molecular and genetic mechanisms of tumorigenesis in multiple endocrine neoplasia type-1. Mol. Endocrinol. 15, 1653–1664. [DOI] [PubMed] [Google Scholar]
- Hay R.T. (2005). SUMO: a history of modification. Mol. Cell 18, 1–12. [DOI] [PubMed] [Google Scholar]
- Imoto S., Sugiyama K., Muromoto R., et al. (2003). Regulation of transforming growth factor-β signaling by protein inhibitor of activated STAT, PIASy through Smad3. J. Biol. Chem. 278, 34253–8. [DOI] [PubMed] [Google Scholar]
- Kaji H., Canaff L., Lebrun J.J., et al. (2001). Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type β signaling. Proc. Natl Acad. Sci. USA 98, 3837–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.S., Saunier E.F., Akhurst R.J., et al. (2008). The type I TGF-β receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 10, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher O., Felberbaum R., Hochstrasser M. (2006). Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180. [DOI] [PubMed] [Google Scholar]
- Kossatz U., Vervoorts J., Nickeleit I., et al. (2006). C-terminal phosphorylation controls the stability and function of p27Kip1. EMBO J. 25, 5159–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecanda J., Parekh T.V., Gama P., et al. (2007). Transforming growth factor-β, estrogen, and progesterone converge on the regulation of p27Kip1 in the normal and malignant endometrium. Cancer Res. 67, 1007–1018. [DOI] [PubMed] [Google Scholar]
- Lecanda J., Ganapathy V., D'Aquino-Ardalan C., et al. (2009). TGFβ prevents proteasomal degradation of the cyclin-dependent kinase inhibitor p27kip1 for cell cycle arrest. Cell Cycle 8, 742–756. [DOI] [PubMed] [Google Scholar]
- Lee M., Pellegata N.S. (2013). Multiple endocrine neoplasia type 4. Front. Horm. Res. 41, 63–78. [DOI] [PubMed] [Google Scholar]
- Lee P.S., Chang C., Liu D., et al. (2003). Sumoylation of Smad4, the common Smad mediator of transforming growth factor-β family signaling. J. Biol. Chem. 278, 27853–27863. [DOI] [PubMed] [Google Scholar]
- Liang J., Zubovitz J., Petrocelli T., et al. (2002). PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 8, 1153–1160. [DOI] [PubMed] [Google Scholar]
- Liang M., Melchior F., Feng X.H., et al. (2004). Regulation of Smad4 sumoylation and transforming growth factor-β signaling by protein inhibitor of activated STAT1. J. Biol. Chem. 279, 22857–22865. [DOI] [PubMed] [Google Scholar]
- Liang J., Shao S.H., Xu Z.X., et al. (2007). The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9, 218–224. [DOI] [PubMed] [Google Scholar]
- Lin X., Liang M., Liang Y.Y., et al. (2003). Activation of transforming growth factor-β signaling by SUMO-1 modification of tumor suppressor Smad4/DPC4. J. Biol. Chem. 278, 18714–9. [DOI] [PubMed] [Google Scholar]
- Loda M., Cukor B., Tam S.W., et al. (1997). Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med. 3, 231–234. [DOI] [PubMed] [Google Scholar]
- Massagué J. (2008). TGFβ in cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A., Fiore F., Eytan E., et al. (1999). Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 13, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motti M.L., De Marco C., Califano D., et al. (2004). Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle 3, 1074–1080. [PubMed] [Google Scholar]
- Ohshima T., Shimotohno K. (2003). Transforming growth factor-β-mediated signaling via the p38 MAP kinase pathway activates Smad-dependent transcription through SUMO-1 modification of Smad4. J. Biol. Chem. 278, 50833–50842. [DOI] [PubMed] [Google Scholar]
- Polyak K., Kato J.Y., Solomon M.J., et al. (1994). p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Ren J., Gao X., Jin C., et al. (2009). Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics 9, 3409–3412. [DOI] [PubMed] [Google Scholar]
- Repetto O., Rogniaux H., Firnhaber C., et al. (2008). Exploring the nuclear proteome of Medicago truncatula at the switch towards seed filling. Plant J. 56, 398–410. [DOI] [PubMed] [Google Scholar]
- Schiappacassi M., Lovat F., Canzonieri V., et al. (2008). p27Kip1 expression inhibits glioblastoma growth, invasion and tumor-induced neoangiogenesis. Mol. Cancer Ther. 7, 1164–1175. [DOI] [PubMed] [Google Scholar]
- Schiappacassi M., Lovisa S., Lovat F., et al. (2011). Role of T198 modification in the regulation of p27Kip1 protein stability and function. PLoS One 6, e17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simula M.P., Cannizzaro R., Canzonieri V., et al. (2010). PPAR signaling pathway and cancer-related proteins are involved in celiac disease-associated tissue damage. Mol. Med. 16, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingerland J.M., Pagano M. (2000). Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17. [DOI] [PubMed] [Google Scholar]
- Slingerland J.M., Hengst L., Pan C.H., et al. (1994). A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor β-arrested epithelial cells. Mol. Cell. Biol. 14, 3683–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Tiihonen E., Heiskanen A., Laiho M. (2000). Accumulation of a form of p27Kip1 not associated with Cdk-cyclin complexes in transforming growth factor-β-arrested Mv1Lu cells. Exp. Cell Res. 259, 107–116. [DOI] [PubMed] [Google Scholar]
- Tashiro K., Pando M.P., Kanegae Y., et al. (1997). Direct involvement of the ubiquitin-conjugating enzyme Ubc9/hus5 in the degradation of IkBα. Proc. Natl Acad. Sci. USA 94, 7862–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda K., Kubota Y., Kato J. (1999). Degradation of the cyclin-dependent kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398, 160–165. [DOI] [PubMed] [Google Scholar]
- Tsvetkov L.M., Yeh K.H., Lee S.J., et al. (1999). p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr. Biol. 9, 661–664. [DOI] [PubMed] [Google Scholar]
- Wilkinson K.A., Henley J.M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Li J., Zuo Y., et al. (2011). SUMO-specific protease 1 regulates the in vitro and in vivo growth of colon cancer cells with the upregulated expression of CDK inhibitors. Cancer Lett. 309, 78–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.