

Abstract
Cisplatin (CDDP) is one of the first-line anticancer drugs indicated for use against various form of human malignancies; but, the therapeutic outcome of CDDP chemotherapy is limited due to the development of myelosuppression and genotoxicity which may lead to secondary cancer. Induction of oxidative stress in normal host cells is thought to be responsible for these adverse effects. Therefore, in search of a potential chemoprotectant, an oraganovanadium compound, viz., vanadium(III)-l-cysteine (VC-III) was evaluated against CDDP-induced clastogenicity and cytotoxicity in bone marrow cells of Swiss albino mice. CDDP was administered intraperitoneally (5mg/kg body weight [b.w.]) and VC-III was given by oral gavage (1mg/kg b.w.) in concomitant and pretreatment schedule. The results showed that VC-III administration significantly (P < 0.001) enhanced cell proliferation and inhibited apoptosis in the bone marrow niche indicating recovery of CDDP-induced myelosuppression. VC-III also significantly (P < 0.001) decreased the percentage of chromosomal aberrations, the frequency of micronuclei formation and the extent of DNA damage. The observed antigenotoxic and cytoprotective effect of VC-III was attributed to its attenuation of free radicals status and restoration of oxidised and reduced glutathione levels. These results suggest that VC-III is a potential candidate for future development as a chemoprotective agent against chemotherapy-associated primary and secondary complications.
Introduction
cis-Diaminedichloroplatinum(II), commonly known as cisplatin (CDDP), has been established as a first-line chemotherapeutic agent being used effectively against various form of neoplastic diseases either as monotherapy or in combination with other chemotherapeutics, radiation therapy and/or surgery (1,2). The antitumor efficacy of CDDP is attributed to its highly reactive hydrated platinum complex that binds to DNA and form intra- and interstrand cross-links; thereby produce subsequent interference with DNA transcription and/or DNA replication (3). However, the complete therapeutic efficacy of this drug is limited due to the development of adverse effects in the host; most severe being the bone marrow toxicity and cumulative myelosuppression (25–100% cases) (3,4). These toxic effects get resulted into sub-therapeutic dose delivery and/or discontinuation of chemotherapy, which ultimately compromise treatment outcome including disease control and survival in patients with curable malignancies (4). Beside this, the clastogenic potential of CDDP has become an alarming factor due to its deleterious effect on the genome of normal host cells (5). In patients treated long-term with CDDP, genetic damage can be observed during chemotherapy and/or many years later in terms of infertility, teratogenicity and even through development of secondary malignancies (6,7). In this regard, new strategies to defend host tissues and organs against genotoxicity of CDDP are of clinical interest and hence, development of cytoprotectants is crucial to provide protection against CDDP-induced genotoxic damage.
Vanadium, a dietary micronutrient and an ultra trace element, recently has received attention of researchers due to its wide range of pharmacological activities (8). The deficiency of this essential micronutrient leads to growth retardation, bone deformations and infertility in animals (9). Vanadium plays a major role in the regulation of hormone, glucose, lipid, bone and tooth metabolism in mammals (10). Additionally, vanadium has a profound role in DNA maintenance reactions and hence prevents genomic instability that leads to cancer (11,12). Vanadium compounds have been found to inhibit mutagenesis and carcinogenesis against a number of chemical carcinogens, viz., 1-methyl-1-nitrosourea (MNU), 7,12-dimethylbenz(a)anthracene (DMBA), 1,2-dimethylhydrazine (DMH), diethylnitrosamine (DENA), 2-acetylaminofluorene (AAF), and many more in experimental murine tumor models (13). Till date, no report is available on the effect of any vanadium compound on CDDP-induced clastogenesis and cytotoxicity in normal cells.
This led us interested to evaluate the protective role of an organovanadium compound, viz., vanadium(III)-l-cysteine (VC-III) (Figure 1) against CDDP-induced toxicity on bone marrow cell population in Swiss albino mice. The bone marrow clastogenesis was assessed by the chromosome analysis and micronucleus (MN) assay. Damage of the genomic DNA was determined by comet assay, DNA agarose gel electrophoresis and diphenylamine assay. Bone marrow cell proliferation and apoptosis were analysed by 5-bromo-2′-deoxyuridine (BrdU) labeling and terminal deoxynucleotidyl transferase (TdT) mediated dUTP nick end labeling (TUNEL) assay, respectively. In addition, oxidative stress markers such as bone marrow intracellular reactive oxygen species (ROS) production, nitric oxide (NO) generation, and levels of oxidised glutathione (GSSG) and reduced glutathione (GSH) were estimated as a probable mechanism underlying this protection.
Figure 1.

Structure of VC-III complex.
Materials and methods
Chemicals
CDDP was purchased from Cadila Health Care Limited, Goa, India. Vanadium(III) trichloride, l-cysteine, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), dihydroethidium (DHE), N-(1-napthyl) ethylenediamine dihydrochloride (NEDD), sulphanilamide, 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), ethidium bromide, Triton X-100, Giemsa modified stain solution, colchicine and diphenylamine were obtained from Sigma-Aldrich Chemicals Private Limited, Bangalore, India. Fetal bovine serum (FBS), agarose and 1kb DNA ladder were purchased from Invitrogen Bio Services India Private Limited, Bangalore, India. In situ cell death detection kit, AP and BrdU labeling and detection kit II were purchased from Roche Diagnostics India Private Limited, Kolkata, India. All the other chemicals used were of analytical grade from Merck (India) Limited, Mumbai, India.
Animals
Adult (5–6 weeks) Swiss albino female mice (25±2g), bred in the animal colony of Chittaranjan National Cancer Institute (Kolkata, India) were used for this study. The mice were maintained at controlled temperature (22±2°C) and humidity (60±5%) under alternating light and dark conditions (12h/12h). Standard food pellets and drinking water were provided ad libitum. The experiments were carried out strictly following the guidelines of Institutional Animal Ethics Committee [Committee for the Purpose of Control and Supervision of Experiment on Animals (CPCSEA Registration No. 1774/GO/RBi/S/14/CPCSEA), India].
Synthesis of VC-III
VC-III was synthesised following the literature procedure of Papaioannou et al. (14). Vanadium(III) chloride (500mg, 3.17nM) was dissolved in dry methanol (25ml) and stirred using a magnetic stirrer at an ambient temperature under nitrogen atmosphere. Solid l-cysteine (1.16g, 9.57nM) was added to the solution in one portion. Upon stirring for another 3h, a greenish brown precipitate was formed. The precipitate was collected by filtration and washed with chilled methanol and diethyl ether and dried in vacuo over phosphorus pentoxide (15).
Experimental design
In the present study the mice were divided into five groups containing six animals (n = 6) in each group (Figure 2). Group 1 (vehicle-treated group): Each animal was given distilled water orally from Day 1 to 9 and saline water intraperitoneally from Day 1 to 5. Group 2 (only VC-III-treated group): Each animal was treated only with VC-III at a dose of 1mg/kg body weight (b.w.) orally throughout the experimental period. Group 3 (CDDP-treated group): Each animal was injected with CDDP intraperitoneally at a dose of 5mg/kg b.w. for consecutive 5 days (Day 1 to 5). Group 4 (concomitant treatment group): VC-III was administered orally at a dose of 1mg/kg b.w. in water from Day 1 to 9 and CDDP was given as in Group 3 (Day 1 to 5). Group 5 (pretreatment group): VC-III was administered orally at a dose of 1mg/kg b.w. in water 7 days prior to CDDP treatment and then continued upto Day 9 and CDDP was given as in Group 3 (Day 1 to 5). The mice were sacrificed on Day 10, 4 days after last injection of CDDP. All the experiments were performed twice.
Figure 2.

The experimental treatment schedule.
Preparation of bone marrow cell suspension
The bone marrow from the femurs of mice was flushed out with 1× Hank’s balanced buffer solution (HBSS) into a centrifuge tube. The cells were collected by centrifugation at 500 g for 10min. Cell pellets were resuspended with 1× HBSS containing 2% FBS. Then the bone marrow cellularity was determined and the samples were processed according to the methods described below.
In situ cell proliferation
Cell proliferation was measured using BrdU Labeling and Detection Kit II. The bone marrow cells were placed into BrdU-added prewarmed (37°C) cell culture medium for 60min at 37°C, 5% CO2 where the DNA of proliferating S-phase cells get labeled with BrdU. Then the cells were smeared on slides and fixed. The slides were subsequently incubated with an anti-BrdU monoclonal antibody (at 37°C for 30min), which was detected by an alkaline phosphatase-conjugated-antimouse-immunoglobulin antibody (anti-mouse-Ig-AP). The bound anti-mouse-Ig-AP was visualised using BCIP/NBT, an AP-substrate solution (16). The slides were then analysed with a light microscope (DM1000; Leica). Randomly selected cells (≥100) from 5–6 zones/slide were counted to determine the number of proliferating cells. The BrdU labeling index (BrdU LI) was determined by dividing the number of labeled cells by total number of cells counted and then multiplying by 100.
Chromosomal aberration study
Mice were injected intraperitoneally with 0.03% colchicine (1ml/100g b.w.) 90min before sacrifice. Marrow of the femur was flushed in 1% sodium citrate solution (37°C) and fixed in acetic acid/methanol (1:3). Slides were prepared by the conventional flame drying technique followed by Giemsa staining (5:24 dilution in Sorenson’s phosphate buffer) for scoring bone marrow chromosomal aberration (CA) (17). Stained slides were evaluated by observing ≥100 cells/slide with a light microscope (DM1000; Leica) at ×1000 magnification and CA was expressed in terms of percentage of aberrated metaphase plate with respect to the total observed metaphase plates.
MN assay
The bone marrow cells were collected in 1ml 0.075M KCl solution and incubated at 37°C for 10min. The tubes were centrifuged at 500 g for 10min and the pellet was carefully resuspended in, as little supernatant as possible. Two smears of bone marrow were prepared from each mouse. After air-drying, the smears were fixed in absolute methanol and stained by Giemsa (5:24 dilution in Sorenson’s phosphate buffer) (17). The slides were examined for the presence of MN at ×1000 magnification (DM1000; Leica). The number of MN were scored in ≥1000 non-overlapping, differentiated, uniformly stained cells/slide and expressed as percentage of the total cells counted.
Comet assay
Comet assay of murine bone marrow cells were performed following a simplified protocol with slight modification (18). About 2×104 cells were suspended in 75 μl of 1.0% low melting point agarose and layered onto half-frosted slides pre-coated with a thin layer of 1.0% normal melting point agarose. A third layer of 0.5% low melting point agarose was layered on the top of the second layer. The cells were lysed for 2h at 4°C in lysing solution (pH 10.0, containing 2.5M NaCl, 100mM EDTA, 10mM Tris, 10% dimethyl sulfoxide, 1% Triton X-100). After lysis, the slides were subjected to electrophoresis in electrophoresis buffer (pH 13.1, containing 1mM EDTA, 0.3M NaOH) for 30min. After electrophoresis, the slides were neutralised with 0.4M Tris-HCl (pH 7.5). The microscopic slides were carefully dried at room temperature and stained with ethidium bromide in water (20 μg/ml; 80 μl/slide). The slides were examined at ×400 magnification under a fluorescence microscope (DM4000 B; Leica) with imaging system. Komet 5.5 software (Andor Technology) was used to take the photomicrograph of cells and to analyse various parameters of the comet. Randomly selected cells (≥100) from each slide were analysed (two slides/animals in each group). The damage is represented by DNA fragments that have migrated out of the cell nucleus during electrophoresis and formed an image of a ‘comet’ tail (19). The parameters analysed for detection of DNA damage were damaged cell (%) in each group, average tail length [migration of the DNA from the nucleus (μm)] and Olive tail moment [product of tail length and the fraction of total DNA in the tail (arbitrary units)].
DNA fragmentation by diphenylamine assay
DNA fragmentation in the bone marrow cells were carried out according to Zhivotovsky et al. (20). Briefly, 2×106 cells were lysed in lysis buffer (5mM Tris-HCl, pH 8.0, 20mM EDTA and 0.5% Triton X-100) for 30min at 4°C. The cell lysate were centrifuged at 15000 g for 15min at 4°C. Then, the supernatant containing small DNA fragments was separated from the pellet containing large pieces of DNA. The supernatant and pellet were resuspended in 10% and 5% of trichloroacetic acid, respectively, and kept overnight. Then both samples were heated at 95°C for 15min and centrifuged at 2500 g for 5min to remove proteins. Supernatant fractions were reacted with diphenylamine (DPA) for 4h at 37°C and the developing blue colour was measured at 600nm. DNA fragmentation in samples was expressed as percentage of total DNA appearing in the supernatant fraction.
DNA fragmentation by agarose gel electrophoresis
Bone marrow cells (1×106) were lysed with lysis buffer (5mM Tris-HCl, pH 8.0, 20mM EDTA and 0.5% Triton X-100) and kept on ice for 30min. The lysate was incubated with RNase A (50mg/ml) at 37°C for 2h followed by proteinase K (20mg/ml) at 37°C for 2h. The DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1) and once again with chloroform:isoamyl alcohol (24:1). The DNA was precipitated with −20°C absolute ethanol and 5M NaCl and kept at −20°C overnight. The precipitate was centrifuged at 15000 g for 15min, lyophilised and dissolved in TE buffer (10mM Tris-HCl, 1mM EDTA, pH 8.0) (20). To visualise the presence of DNA ladder, electrophoresis was performed in 1.5% agarose gel containing ethidium bromide (0.5 μg/ml); 1kb DNA ladder was run in the same gel. DNA bands were visualised and photographed in a gel documentation system (Gel Doc™ XR+ system; BioRad).
In situ cell death (apoptosis)
Apoptosis of bone marrow cells were determined by using the TUNEL method with the help of in situ cell death detection kit, according to the manufacturer’s instructions. Briefly, the cells were smeared on slides, permeabilised using 0.1% Triton X-100 and the slides were incubated with TUNEL reaction mixture containing the TdT and fluorescein-dUTP, at 37°C for 60min in a humidified chamber (21). The slides were then analysed under a fluorescence microscope (DM4000 B; Leica) and photomicrographs were taken at ×400 magnification. The apoptotic cells were identified by green fluorescence. Randomly selected cells (≥100) from 5–6 zones/slide were counted to determine the number of apoptotic cells. Apoptotic index (AI) was determined as the percentage of the labeled nuclei with respect to the total number of nuclei counted.
Intracellular free radicals generation
Intracellular ROS measurement in bone marrow cells was determined following two simplified protocol with slight modification using two probes, DHE and DCFH-DA (22). DHE is a nonfluorescent dye and freely permeable to the cell. Upon oxidation by superoxide anions (O2•−) forms the red fluorescent product ethidium (22). Cells (2×106) were loaded with 10 μM DHE and incubated in dark for 10min to allow the formation of ethidium and then analysed for fluorescence (excitation at 475nm, emission collected at 610nm) using spectrofluorimeter (Cary Eclipse; Varian).
DCFH-DA is a nonfluorescent probe that is hydrolysed by mitochondrial esterase to form DCFH. DCFH upon oxidation by H2O2 forms the fluorescent compound 2′,7′-dichlorofluorescein (DCF) (22). Cells (2×106) were loaded with 10 μM DCFH-DA and incubated in dark for 30min to allow the formation of DCF and then analysed for fluorescence (excitation at 485nm, emission collected at 529nm) using spectrofluorimeter (Cary Eclipse; Varian).
NO production in bone marrow cells (2×106) was determined by estimating the levels of stable NO metabolites, viz., nitrate (NO3−) and nitrite (NO2−) ions by reaction with Griess reagent (1% sulphanilamide, 5% phosphoric acid and 0.1% NEDD), using NaNO2 as standard (23). The absorbance was taken at 545nm using UV-visible spectrophotometer (Infinite® 200 PRO; TECAN).
GSH and GSSG level
GSH and GSSG levels in bone marrow cells (2×106) were assessed by a kinetic assay in which catalytic amounts of GSH caused a continuous reduction of DTNB to TNB at 412nm (24). Quantification was done by parallel estimation of standard curves of known GSH and GSSG concentrations.
Statistical analysis
All data were expressed as mean ± SD, n = 6 mice per group. The mean values were statistically analysed by one-way analysis of variance using GraphPad Prism (version 5.0; GraphPad Software, Inc., CA, USA) followed by appropriate post hoc test (Dunnett’s multiple comparison test). Significant difference was indicated when the P value was <0.05.
Results
Effect on bone marrow cellularity
In this study, the mice treated with CDDP showed significantly (P < 0.001) reduced number of viable cells in femoral bone marrow (Figure 3A). But treatment with the test compound significantly (P < 0.001) increased the viable bone marrow cells count by 65.78 and 97.13%, in case of concomitant treatment and pretreatment group, respectively.
Figure 3.

VC-III induced protection against CDDP-induced myelosuppression. Histograms show (A) bone marrow cellularity and (B) BrdU LI (%) in different groups after CDDP administration. Data were represented as mean ± SD, n = 6. **Significantly (P < 0.001) different from Group 1 and ##significantly (P < 0.001) different from Group 3. Representative photomicrographs of cell proliferation assay showing (C) non-proliferating cells and (D) proliferating cells (indicated by BCIP/NBT staining), ×400 magnification, scale bar = 25 μm.
Modulation of bone marrow cell proliferation
BrdU LI in the bone marrow cells of vehicle-treated group was found to be 51.82% (Figure 3B‒D), which was decreased distinctly due to the cytotoxic effect of CDDP in Group 3 to 24.18% (P < 0.001). The oraganovanadium compound significantly (P < 0.001) reversed the inhibitory effect of CDDP on cellular proliferation and raised BrdU LI to 35.81% in concomitant treatment schedule and to 40.48% in pretreatment schedule.
Prevention of CDDP-induced clastogenicity
Animals treated with CDDP showed significantly (P < 0.001) high proportion of CA of 36.28% (Table 1, Figure 4A) compared to relatively low frequency of chromosomal aberrations of 6.47% in vehicle-treated group. The frequency of CA was 22.34 and 16.42%, respectively, in case of concomitant and pretreatment group, which were significantly (P < 0.001) much lower compared to the CDDP-treated group.
Table 1.
Attenuation of chromosomal aberration (%), micronuclei formation (%) and DNA damage (comet assay) in bone marrow cells by VC-III complex in CDDP-treated mice
| Groups | Chromosomal aberration (%) | Micronuclei (%) | DNA damage (comet assay) | ||
|---|---|---|---|---|---|
| Damaged cell (%) | Comet tail length (μm) | Olive tail moment | |||
| Group 1 | 6.47±0.91 | 0.22±0.02 | 8.15±1.04 | 2.62±0.38 | 0.35±0.06 |
| Group 2 | 6.29±0.74 | 0.20±0.01 | 7.59±0.89 | 2.25±0.25 | 0.26±0.03 |
| Group 3 | 36.28±2.71** | 1.31±0.06** | 54.46±4.22** | 32.20±2.83** | 9.52±0.68** |
| Group 4 | 22.34±2.40## | 0.68±0.03## | 21.27±2.31## | 12.73±1.48## | 2.18±0.26## |
| Group 5 | 16.42±1.33## | 0.53±0.04## | 18.51±2.09## | 10.38±1.11## | 1.79±0.19## |
Data were represented as mean ± SD, n = 6. **Significantly (P < 0.001) different from Group 1; ##Significantly (P < 0.001) different from Group 3.
Figure 4.

VC-III mediated prevention of CDDP-induced clastogenesis. (A) Metaphase complements of bone marrow cells showing structural aberrations, ×1000 magnification, scale bar = 10 μm. Arrows indicate break (B), gap (G), sister-chromatid union (SCU) and terminal deletion (TD). (B) Representative photomicrograph of Giemsa-stained bone marrow slide showing MN (indicated by black arrow), ×1000 magnification, scale bar = 10 μm.
Similarly, mice treated with CDDP showed significantly (P < 0.001) high MN frequency of 1.31±0.06 compared to Group 1 mice (0.22±0.02) (Table 1, Figure 4B). Treatment with the organovanadium compound in concomitant and pretreatment schedule was able to minimize MN frequency to a significant (P < 0.001) level of ~0.68±0.03 and 0.53±0.04, respectively, compared to CDDP-treated mice.
Protection from CDDP-induced DNA damage
Comet assay was carried out to examine DNA damage in bone marrow cells (Figure 5A‒E) and for this purpose damaged cell (%), average tail length (μm) and Olive tail moment were analysed (Table 1). Large round head and no tail was observed in the bone marrow cells of Group 1 mice. But CDDP treatment resulted in long comet tail formation due to DNA damage in significantly (P < 0.001) large number of cell population. DNA with diffused head and scattered tail was also observed. Concomitant treatment and pretreatment with the vanadium compound significantly (P < 0.001) reduced the number of damaged cells by 60.96 and 66.09%, respectively, compared to Group 3 mice. VC-III administration also mitigated comet tail length and Olive tail moment in comparison to CDDP-treated mice.
Figure 5.
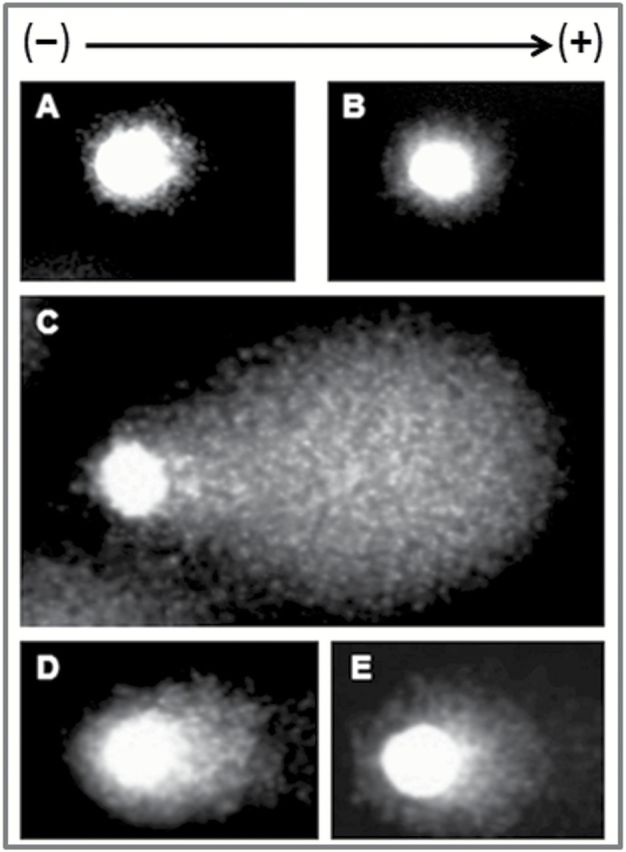
VC-III attenuated DNA damage induced by CDDP in murine bone marrow cells. Photomicrographs of DNA damage (comet assay) found in different groups, ×400 magnification. (A) Intact DNA with no tail in Group 1, (B) no DNA damage observed in Group 2, (C) highly damaged DNA with scattered tail migration in Group 3, (D) less migration of DNA with short comet tail in Group 4 and (E) minimal migration of DNA with small tail in Group 5.
Attenuation of CDDP-induced DNA fragmentation
Genomic DNA fragmentation in bone marrow cells was found to be 7.26% in Group 1 (Figure 6B). CDDP administration caused a significantly (P < 0.001) greater rate (47.18%) of DNA fragmentation in Group 3 mice. In case of concomitant treatment and pretreatment group, the percentages of DNA fragmentation were significantly (P < 0.001) reduced to 25.16 and 22.39%, respectively. Similar to the spectrophotometric DNA fragmentation assay, the electrophoretogram generated by agarose gel electrophoresis showed CDDP treatment resulted in a remarkable oligonucleosome length degradation of DNA, characterised by mixed laddering and smearing of DNA fragments (lane 4) (Figure 6A). Treatment with VC-III well mitigated CDDP-induced DNA fragmentation, as evidenced by marked reduction in DNA ladder formation.
Figure 6.

VC-III reduced CDDP-induced DNA fragmentation and apoptosis in bone marrow niche. (A) Agarose gel electrophoresis of mouse bone marrow genomic DNA. Lane 1: 1kb DNA ladder; lane 2: Group 1; lane 3: Group 2; lane 4: Group 3; lane 5: Group 4; lane 6: Group 5. (C) Representative photomicrograph of TUNEL assay performed in bone marrow cells, × 200 magnification. Apoptotic cells (TUNEL label cells) are indicated by white arrows whereas non-apoptotic cells are indicated by white broken arrows. To the right, histograms show quantification of (B) DNA fragmentation (%) and (D) apoptotic index (%) in mice bone marrow cells after CDDP administration. Data were represented as mean ± SD, n = 6. **Significantly (P < 0.001) different from Group 1 and ##significantly (P < 0.001) different from Group 3.
Reduction of CDDP-induced apoptosis
AI in the bone marrow cells of vehicle-treated group was found to be 4.17% (Figure 6C and D), which was increased markedly due to the cytotoxic effect of CDDP in Group 3 to 34.93% (P < 0.001). The test compound significantly (P < 0.001) inhibited CDDP-induced apoptotic cell death to 15.85% in concomitant treatment schedule and to 12.03% in pretreatment schedule.
Amelioration of free radicals status
Administration of CDDP resulted in a significant (P < 0.001) increase in ROS and NO levels in bone marrow cells of Group 3 mice, in comparison to the vehicle-treated group (Figure 7A‒C), when estimated by DHE, DCFH-DA and Griess reagent, respectively. Concomitant treatment with VC-III resulted in significant (P < 0.001) reduction in O2•−, H2O2 and NO levels by 21.44, 26.48 and 19.24% in comparison to the CDDP-treated group, respectively. In the pretreatment group, the reduction in O2•−, H2O2 and NO levels was found to be 27.83% (P < 0.001), 34.50% (P < 0.001) and 26.91% (P < 0.001), respectively, compared to CDDP-treated mice.
Figure 7.

VC-III modulated CDDP-induced alteration of cellular redox status in bone marrow cells of mice. VC-III reduced (A) O2•− level, (B) H2O2 level, (C) NO level, (D) GSSG level and enhanced (E) GSH level in bone marrow cells. Data were represented as mean ± SD, n = 6. **Significantly (P < 0.001) different from Group 1, #significantly (P < 0.01) different from Group 3 and ##significantly (P < 0.001) different from Group 3.
Restoration of GSH and GSSG levels
CDDP administration resulted in significant (P < 0.001) depletion of GSH level along with significant (P < 0.001) rise in GSSG level in Group 3 mice bone marrow cells compared to Group 1 (Figure 7D and E). Concomitant treatment and pretreatment with VC-III increased GSH level by 33.22% (P < 0.01) and 54.95% (P < 0.001), respectively, in comparison to Group 3. Additionally, concomitant treatment and 7 days pretreatment with the vanadium compound significantly (P < 0.001) reduced the elevated level of GSSG by 24.32 and 36.04%, respectively, compared to Group 3.
Discussion
The bone marrow is the primary site of the body where hematopoietic stem cells and more mature blood cells lineage progenitors reside and differentiate in an adult cell population (25). Due to the proliferating nature, the bone marrow cells are very much sensitive to clastogenic chemicals and susceptible to DNA damage (26). This damage, particularly in such undifferentiated cell population of bone marrow, is dangerous because it can lead to mutations and genetic rearrangements. If such cells survive and proliferate, the risk of secondary cancer such as leukaemia increases (6,27–29). The current study demonstrated that the test compound VC-III was neither clastogenic nor cytotoxic at the tested dose. Moreover, it provided adequate protection to murine bone marrow cells against CDDP-induced myelosuppression as evident from cellularity of femoral bone marrow. Inhibition of cell proliferation is one of the major causes of CDDP-induced myelotoxicity and related complications. In this study, cell proliferation was markedly suppressed by CDDP as reflected by BrdU labeling assay. In this assay, BrdU gets incorporated into DNA in place of thymidine and BrdU-labeled cells serve as a measure of DNA synthesis or cell proliferation (30). Treatment with the oraganovanadium compound effectively reversed CDDP-induced inhibition of bone marrow cell proliferation. This indicates that the compound may provide protection to the early progenitor cells and pluripotent stem cells of the bone marrow niche.
Developments of CA and MN have been commonly used as sensitive indicators in the clastogenic assays of a drug (3). During proliferation if a clastogenic agent is administered it may act during the cell division and cause chromosomal damage, such as break (31). The mitotic plate of CDDP-treated mice showed high incidence of aberrations; among which most common were stretching, chromatid break and gap. CDDP administration in mice also showed high MN frequency in bone marrow cells. This suggests increased rate of chromosome loss or fragmentation during earlier nuclear division upon CDDP treatment. Treatment with the test compound significantly minimised the incidence of CA and MN in the bone marrow niche. These observations clearly suggest the preventive role of VC-III against the clastogenic potential of CDDP.
The extent of DNA damage was estimated by comet assay, DNA agarose gel electrophoresis and DPA assay which allows the detection of diverse kinds of DNA damage (32,33). Results from the comet assay showed increase in the extent of DNA damage in CDDP-treated murine bone marrow cells as evident from significant elevation in damage cell (%), comet tail length and Olive tail moment. Administration of CDDP also resulted into DNA ladder formation indicating rise in DNA fragmentation and it was quantitatively confirmed in terms of colourimetric DPA assay. Treatment with VC-III substantially mitigated CDDP-induced DNA damage and its associated parameters in comet assay. VC-III administration also reduced fragmented DNA (%). The electrophoretogram of genomic DNA agarose gel electrophoresis showed similar trend of protection exerted by the test compound. These results undoubtedly indicates that the oraganovanadium compound possess potent genoprotective efficacy.
Finally, apoptotic cell death was evaluated in this study by means of TUNEL assay. This assay has been designed to detect cells that undergo internucleosomal DNA cleavage which is a hallmark of apoptosis (34). CDDP-treated mice showed high proportion of TUNEL-positive cells. This may be due to the direct cytotoxicity or extensive induction of DNA damage that leads to cell death (5). Treatment with the vanadium compound diminished this apoptotic cell death and conferred cytoprotection to the host.
In this present work the mechanism of VC-III-mediated protection of murine bone marrow cells from CDDP-induced toxicity need to be studied. It is well known that CDDP treatment has been found to increase generation of free radicals, such as O2•−, H2O2, •OH, and NO in the host (4). Accumulation of these free radicals may cause damage to cellular genome and other critical biomolecules which ultimately leads to genotoxicity and secondary malignancies (35,36). In the present study, in order to evaluate whether the observed genoprotective and cytoprotective effect was due to the modulation of cellular antioxidant system, oxidative stress markers such as generation of ROS, NO and GSSG, GSH levels were determined.
In the present work, CDDP administration significantly increased O2•−, H2O2 and NO level in the bone marrow cells of Group 3 mice as measured by DHE, DCFH-DA and Griess reagent, respectively. On the other hand, organovanadium complex VC-III prevented CDDP-induced generation of free radicals and oxidative stress, which could be attributed to free radical scavenging activity of the test compound. CDDP treatment also caused significant depletion of GSH level and increase in GSSG level in Group 3 mice. Reduction of GSH level was ascribed to the utilization of GSH to combat CDDP-generated free radicals which also clarified the elevation of its oxidised form. Treatment with VC-III restored the GSH and GSSG level and normalised the condition. These results suggest that the protection by VC-III may be mediated through the modulation of cellular antioxidant system. These observations were in line with previous study in which VC-III was reported to have potent antioxidant efficacy (15).
In conclusion, this study demonstrates for the first time that an organovanadium compound possesses a protective role in the abatement of CDDP-induced genotoxicity and myelosuppression in mice. One probable justification of VC-III-mediated protection is that simultaneous treatment with VC-III would allow inception of free radicals generated by CDDP before they reach DNA and induce damage. In addition, vanadium-mediated induction of the DNA repair mechanism could possibly take place (37,38). Therefore, VC-III can be a promising chemoprotective agent to overcome CDDP-induced myelosuppression and simultaneous treatment failure; and may be useful to avert secondary malignancy and to reduce the risk for abnormal reproductive outcomes in cancer patients and medical personnel exposed to CDDP.
Funding
Indian Council of Medical Research (ICMR) , New Delhi, India (3/2/2/58/2011/NCD-III, 45/36/2008/PHA-BMS); University Grant Commission (UGC), New Delhi, India (18-12/2011(ii)EU-V).
Acknowledgements
The authors would like to acknowledge Prof. (Dr) Jaydip Biswas, Director, Chittaranjan National Cancer Institute, for his support in this study. The authors are also thankful to Dr Manas Ranjan Ray, Head, Department of Experimental Hematology, Chittaranjan National Cancer Institute, for his guidance on the micronucleus assay.
Conflict of interest statement: None declared.
References
- 1. Marcu L. G. (2013) Improving therapeutic ratio in head and neck cancer with adjuvant and cisplatin-based treatments. Biomed Res. Int., 2013, 817279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. NCI. (2014) Cancer Drug Information: Cisplatin. National Cancer Institute at the National Institutes of Health http://www.cancer.gov/cancertopics/druginfo/cisplatin (accessed December 04, 2014).
- 3. Attia S. M. (2010) The impact of quercetin on cisplatin-induced clastogenesis and apoptosis in murine marrow cells. Mutagenesis, 25, 281–288. [DOI] [PubMed] [Google Scholar]
- 4. Das B., Antoon R., Tsuchida R., et al. (2008) Squalene selectively protects mouse bone marrow progenitors against cisplatin and carboplatin-induced cytotoxicity in vivo without protecting tumor growth. Neoplasia, 10, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rjiba-Touati K., Ayed-Boussema I., Skhiri H., Belarbia A., Zellema D., Achour A., Bacha H. (2012) Induction of DNA fragmentation, chromosome aberrations and micronuclei by cisplatin in rat bone-marrow cells: protective effect of recombinant human erythropoietin. Mutat. Res., 747, 202–206. [DOI] [PubMed] [Google Scholar]
- 6. Attia S. M. (2012) Influence of resveratrol on oxidative damage in genomic DNA and apoptosis induced by cisplatin. Mutat. Res., 741, 22–31. [DOI] [PubMed] [Google Scholar]
- 7. IARC Working Group on the Evaluation of Carcinogenic Risks to Human. (2012) Pharmaceuticals, Volume 100 A, A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks. Hum., 100, 1‒–401. [PMC free article] [PubMed] [Google Scholar]
- 8. Rehder D. (2012) The potentiality of vanadium in medicinal applications. Future Med. Chem., 4, 1823–1837. [DOI] [PubMed] [Google Scholar]
- 9. Mukherjee B., Patra B., Mahapatra S., Banerjee P., Tiwari A., Chatterjee M. (2004) Vanadium–an element of atypical biological significance. Toxicol. Lett., 150, 135–143. [DOI] [PubMed] [Google Scholar]
- 10. Nath R. (2000) Micronutrients and Trace Elements. A.P.H. Publishing Corporation, New Delhi, pp. 337‒–346. [Google Scholar]
- 11. French R. J., Jones P. J. (1993) Role of vanadium in nutrition: metabolism, essentiality and dietary considerations. Life Sci., 52, 339–346. [DOI] [PubMed] [Google Scholar]
- 12. Nunes G. G., Bonatto A. C., de Albuquerque C. G., et al. (2012) Synthesis, characterization and chemoprotective activity of polyoxovanadates against DNA alkylation. J. Inorg. Biochem., 108, 36–46. [DOI] [PubMed] [Google Scholar]
- 13. Bishayee A., Waghray A., Patel M. A., Chatterjee M. (2010) Vanadium in the detection, prevention and treatment of cancer: the in vivo evidence. Cancer Lett., 294, 1–12. [DOI] [PubMed] [Google Scholar]
- 14. Papaioannou A., Manos M., Karkabounas S., Liasko R., Evangelou A. M., Correia I., Kalfakakou V., Pessoa J. C., Kabanos T. (2004) Solid state and solution studies of a vanadium(III)-L-cysteine compound and demonstration of its antimetastatic, antioxidant and inhibition of neutral endopeptidase activities. J. Inorg. Biochem., 98, 959–968. [DOI] [PubMed] [Google Scholar]
- 15. Basu A., Bhattacharjee A., Roy S. S., Ghosh P., Chakraborty P., Das I., Bhattacharya S. (2014) Vanadium as a chemoprotectant: effect of vanadium(III)-L-cysteine complex against cyclophosphamide-induced hepatotoxicity and genotoxicity in Swiss albino mice. J. Biol. Inorg. Chem., 19, 981–996. [DOI] [PubMed] [Google Scholar]
- 16. Ellwart J., Dörmer P. (1985) Effect of 5-fluoro-2’-deoxyuridine (FdUrd) on 5-bromo-2’-deoxyuridine (BrdUrd) incorporation into DNA measured with a monoclonal BrdUrd antibody and by the BrdUrd/Hoechst quenching effect. Cytometry, 6, 513–520. [DOI] [PubMed] [Google Scholar]
- 17. Klein C. B., Broday L., Costa M. (2001) Mutagenesis assays in mammalian cells. Curr. Protoc. Toxicol., Chapter 3, Unit3.3. [DOI] [PubMed] [Google Scholar]
- 18. Dhawan A., Mathur N., Seth P. K. (2001) The effect of smoking and eating habits on DNA damage in Indian population as measured in the Comet assay. Mutat. Res., 474, 121–128. [DOI] [PubMed] [Google Scholar]
- 19. Abid-Essefi S., Zaied C., Bouaziz C., Salem I. B., Kaderi R., Bacha H. (2012) Protective effect of aqueous extract of Allium sativum against zearalenone toxicity mediated by oxidative stress. Exp. Toxicol. Pathol., 64, 689–695. [DOI] [PubMed] [Google Scholar]
- 20. Zhivotovsky B., Samali A., Orrenius S. (2001) Determination of apoptosis and necrosis. Curr. Protoc. Toxicol., Chapter 2, Unit 2.2. [DOI] [PubMed] [Google Scholar]
- 21. Gavrieli Y., Sherman Y., Ben-Sasson S. A. (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol., 119, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson J. P. (2001) Oxidative metabolism. Curr. Protoc. Cytom., Chapter 9, Unit 9.7. [DOI] [PubMed] [Google Scholar]
- 23. Coşkun S., Gönül B., Ozer C., Erdoğan D., Elmas C. (2007) The effects of dexfenfluramine administration on brain serotonin immunoreactivity and lipid peroxidation in mice. Cell Biol. Toxicol., 23, 75–82. [DOI] [PubMed] [Google Scholar]
- 24. Rahman I., Kode A., Biswas S. K. (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc., 1, 3159–3165. [DOI] [PubMed] [Google Scholar]
- 25. Acton Q. A. (2013) Bone Marrow Cells—Advances in Research and Application: 2013 Edition. ScholaryEditions, Atlanta, Georgia, pp. 31‒–114. [Google Scholar]
- 26. Antunes L. M. G., Darin J. D. C., Bianchi M. A. de. P. (1999) Anticlastogenic effect of vitamin C on cisplatin in vivo. Genet. Mol. Biol., 22, 415‒–417. [Google Scholar]
- 27. Travis L. B., Curtis R. E., Boice J. D., Jr, Platz C. E., Hankey B. F., Fraumeni J. F., Jr (1996) Second malignant neoplasms among long-term survivors of ovarian cancer. Cancer Res., 56, 1564–1570. [PubMed] [Google Scholar]
- 28. Pedersen-Bjergaard J., Daugaard G., Hansen S. W., Philip P., Larsen S. O., Rørth M. (1991) Increased risk of myelodysplasia and leukaemia after etoposide, cisplatin, and bleomycin for germ-cell tumours. Lancet, 338, 359–363. [DOI] [PubMed] [Google Scholar]
- 29. Travis L. B., Curtis R. E., Storm H., et al. (1997) Risk of second malignant neoplasms among long-term survivors of testicular cancer. J. Natl Cancer Inst., 89, 1429–1439. [DOI] [PubMed] [Google Scholar]
- 30. Das R. K., Hossain S. K., Bhattacharya S. (2005) Diphenylmethyl selenocyanate inhibits DMBA-croton oil induced two-stage mouse skin carcinogenesis by inducing apoptosis and inhibiting cutaneous cell proliferation. Cancer Lett., 230, 90–101. [DOI] [PubMed] [Google Scholar]
- 31. Arafa S. A., AboEl-Ela M. M., Kassem H. S., Kassem H. S. (2008) Study of the genotoxic effect of cyclophosphamide on albino mice bone marrow polychromatic erythrocytes and the protective effect of captopril. Bull. Alex. Fac. Med., 44, 821‒–828. [Google Scholar]
- 32. Diab K. A. E., Elmakawy A. I., Abd-Elmoneim O. M., Sharaf H. A. (2012) Assessment of genotoxicity and histopathological changes induced by polyethylene glycol (PEG6000) in male mice. J. Cytol. Histol., 3, 153. [Google Scholar]
- 33. Attia S. M., Bakheet S. A. (2013) Effect of dihydrokainate on the capacity of repair of DNA damage and apoptosis induced by doxorubicin. Mutagenesis, 28, 257‒–261. [DOI] [PubMed] [Google Scholar]
- 34. Geske F. J., Nelson A. C., Lieberman R., Strange R., Sun T., Gerschenson L. E. (2000) DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ., 7, 393–401. [DOI] [PubMed] [Google Scholar]
- 35. Bakheet S. A., Attia S. M., Al-Rasheed N. M., et al. (2011) Salubrious effects of dexrazoxane against teniposide-induced DNA damage and programmed cell death in murine marrow cells. Mutagenesis, 26, 533–543. [DOI] [PubMed] [Google Scholar]
- 36. Bhattacharjee A., Basu A., Ghosh P., Biswas J., Bhattacharya S. (2014) Protective effect of selenium nanoparticle against cyclophosphamide induced hepatotoxicity and genotoxicity in Swiss albino mice. J. Biomater. Appl., 29, 303–317. [DOI] [PubMed] [Google Scholar]
- 37. Chakraborty T., Chatterjee A., Saralaya M. G., Chatterjee M. (2006) Chemopreventive effect of vanadium in a rodent model of chemical hepatocarcinogenesis: reflections in oxidative DNA damage, energy-dispersive X-ray fluorescence profile and metallothionein expression. J. Biol. Inorg. Chem., 11, 855–866. [DOI] [PubMed] [Google Scholar]
- 38. Sankar Ray R., Roy S., Ghosh S., Kumar M., Chatterjee M. (2004) Suppression of cell proliferation, DNA protein cross-links, and induction of apoptosis by vanadium in chemical rat mammary carcinogenesis. Biochim. Biophys. Acta, 1675, 165–173. [DOI] [PubMed] [Google Scholar]