

Abstract
Recognition of and binding to cholesterol on the host cell membrane is an initial step in the mechanism of numerous pathogens, including viruses, bacteria, and bacterial toxins; however, a viable method of inhibiting this interaction has not yet been uncovered. Here, we describe the mechanism by which a cholesterol recognition amino acid consensus peptide interacts with cholesterol and inhibits the activity of a cholesterol-binding bacterial leukotoxin (LtxA). Using a series of biophysical techniques, we have shown that the peptide recognizes the hydroxyl group of cholesterol with nanomolar affinity and does not disrupt membrane packing, suggesting that it sits primarily near the membrane surface. As a result, LtxA is unable to bind to cholesterol or subsequently become internalized in host cells. Additionally, because cholesterol is not being removed from the cell membrane, the peptide-treated target cells remain viable over extended periods of time. We have demonstrated the use of this peptide in the inhibition of toxin activity for an antivirulence approach to the treatment of bacterial disease, and we anticipate that this approach might have broad utility in the inhibition of viral and bacterial pathogenesis.
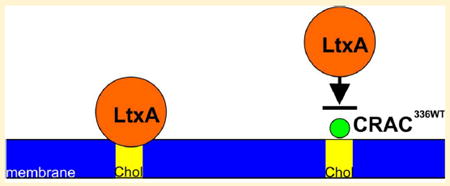
Graphical abstract
Cholesterol (Chol) is a primary component of eukaryotic cell plasma membranes1 and therefore plays an essential role in cell membrane structure and membrane protein function; numerous transmembrane proteins bind directly to Chol and/or associate with Chol-rich domains known as lipid rafts.2 Recently, an amino acid consensus motif that allows proteins to bind directly to Chol has been identified.3 This “cholesterol recognition amino acid consensus” (CRAC) motif is of the form L/V-X1-5-Y-X1-5-R/K, where X is any amino acid and the central tyrosine (Y) residue has been shown to be the most essential element of this motif for Chol binding.4
This CRAC motif has been identified and implicated in Chol binding of numerous proteins, including α-synuclein5 and β-amyloid peptide,6 which are associated with Parkinson’s disease and Alzheimer’s disease, respectively.7 Although this Chol binding has been documented and Chol levels have been linked to neurological disease, the exact mechanism of this relationship remains unknown.8 Another important class of transmembrane proteins that has been shown to bind Chol via CRAC motifs consists of G protein-coupled receptors (GPCRs), including rhodopsin, β2-adrenergic receptor, and serotonin1A receptor.9 These molecules play a significant role in signal transduction across the plasma membrane and have been implicated in numerous diseases, including cancer.10
Interestingly, the CRAC motif is also used by many pathogens to recognize Chol and has been identified in bacterial toxins, such as Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunit C (CdtC)11 and leukotoxin (LtxA),12 as well as viral proteins, including the HIV transmembrane protein gp4113,14 and the influenza virus M2 protein.15 The baculovirus GP 64 fusion protein uses a CRAC motif to bind Chol,16 and the related gB glycoproteins of vesicular stomatitis virus (VSV) G and herpes simplex virus type 1 (HSV-1) also contain the CRAC sequence, suggesting that Chol binding by a CRAC motif may play a role in fusion by these viruses, as well. For these pathogens, binding to Chol is often the first step in the pathogenic mechanism, as the toxin or virus moves from the aqueous extracellular environment to the hydrophobic membrane environment. Thus, disruption of this recognition process represents a possible method of inhibiting bacterial and viral pathogenesis.
The goal of this project was to demonstrate that a small peptide containing a CRAC motif binds to Chol and could be used to inhibit binding of a bacterial toxin to Chol. To accomplish this, we used the leukotoxin (LtxA) produced by A. actinomycetemcomitans as our model bacterial toxin. This toxin is a member of the repeats-in-toxin family of proteins17 and specifically kills immune cells of humans and Old World primates.18-20 The specificity of this activity is driven by the toxin’s association with lymphocyte function-associated antigen-1 (LFA-1), which is expressed by only human immune cells.21,22 However, in addition to its interaction with LFA-1, we and others have shown that LtxA must also bind to Chol on the plasma membrane.12,23,24 Removal of Chol or blocking of Chol binding renders the cell unsusceptible to the toxin’s activity.
LtxA uses a CRAC motif located between residues 334 and 340 to recognize and bind Chol.12 Previously, we showed using molecular simulations that a peptide consisting of the CRAC336 residues (CRAC336WT) interacts strongly with Chol-containing membranes, primarily at the membrane interface.25 In addition, we have shown experimentally that this same peptide inhibits the activity of LtxA against target cells.23 Here, our goal was to determine the mechanism by which the CRAC336WT peptide interacts with membranes to understand the specific mechanism by which it inhibits LtxA activity. We found that the peptide interacts near the surface of the membrane through recognition of the hydroxyl group of Chol. As a result, LtxA is unable to recognize and bind Chol, which prevents the toxin from entering the cells and, thus, killing the target cells.
MATERIALS AND METHODS
Chemicals
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-ditetradecanoyl-sn-glycero-3-phosphocholine (DMPC), and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (Chol), desmosterol (Desmo), dihydrocho-lesterol (DHC), cholesteryl chloride (CC), poly-L-lysine, and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich (St. Louis, MO). N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanol-amine (NBD-PE) and 6-dodecanoyl-2-dimethylaminonaphtha-lene (laurdan) were manufactured by Molecular Probes (Eugene, OR).
Cell Culture
THP-1 cells obtained from ATCC were maintained at 37 °C under 5% CO2 in RPMI 1640 medium containing 10% FBS and 0.05 mM 2-mercaptoethanol.
LtxA Purification
A. actinomycetemcomitans strain JP2 was grown overnight in AAGM broth26 supplemented with 12.5 μg/mL vancomycin and 75 μg/mL bacitracin. LtxA was purified from the culture supernatant using a published protocol.27 LtxA purity was confirmed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting, as shown in Figure 1, and activity was confirmed using a cytotoxicity assay.
Figure 1.
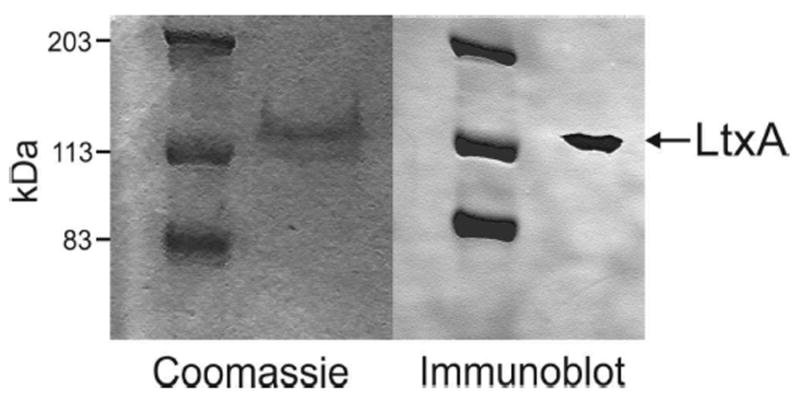
Coomassie stain and immunoblot of purified LtxA.
Peptide Synthesis
The peptides used in this work (Table 1) were prepared using 9-fluorenylmethyloxycarbonyl (Fmoc) solid-phase synthesis. Briefly, the Fmoc group of H-Rink Amide ChemMatrix resin (0.47 mmol/g) was removed with a solution of 6 wt % piperidine and 1 wt % 1-hydroxybenzotriazole monohydrate (HOBt) in dimethylformamide (DMF) for 30 min and then washed with methanol (MeOH) and dichloromethane (DCM). The Fmoc-bound amino acid (4.0 equiv) was coupled with tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) (3.9 equiv) and N,N-diisopro-pylethylamine (DIEA) (8.0 equiv) in DMF (25 mL) for 90 min followed by washing with MeOH and DCM. Subsequent Fmoc groups were removed using the same deprotection and washing steps used for the resin, and the progress of the synthesis was periodically verified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Bruker Corp., Billerica, MA). At the end of the solid-phase synthesis, the N-terminal amino acid was capped using acetic anhydride with DIEA in DMF for 30 min. The peptide was then cleaved from the resin with a solution of 2.5 vol % triisopropylsilane and 2.5 vol % water in trifluoroacetic acid (TFA) for 2 h and precipitated with cold diethyl ether.
Table 1.
Peptide Sequencesa
| peptide name | sequence | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRAC336WT | F | D | R | A | R | M | L | E | E | Y | S | K | R | F | K | K | F | G | Y |
| CRAC336SCR | F | D | R | A | R | M | Y | E | K | L | E | R | S | F | K | K | F | G | Y |
The CRAC motif is in bold type. Each peptide was acetylated at the N-terminus and amidated at the C-terminus.
Peptide Purification
The peptides were purified by reversed-phase high-performance liquid chromatography (HPLC) on a Luna prep 10 μm, 250 mm × 21.20 mm C8 column (Phenomenex, Torrance, CA) (phase A, water and 0.1% TFA; phase B, acetonitrile and 0.1% TFA) using a gradient from 95% A and 5% B to 0% A and 100% B over 60 min. The identity of the peptide was confirmed via MALDI-TOF MS (Figure S1A) and purity analyzed by reversed-phase HPLC (Figure S1B), as described. The purified peptide was then lyophilized and stored at −80 °C.
Large Unilamellar Vesicle Preparation
Liposomes were prepared using the lipid film technique28 from stock solutions of lipid and sterol in chloroform. For the ITC and CD experiments, the lipid films were hydrated with a phosphate buffer [10 mM PO4 (pH 6.5)], and for the laurdan experiments, the lipid films were hydrated with a liposome buffer [150 mM NaCl, 5 mM CaCl2, 5 mM HEPES, and 3 mM NaN3 (pH 7.4)] to produce multilamellar vesicles (MLVs). The MLVs were then extruded through a 100 nm polycarbonate Whatman membrane (GE Healthcare BioSciences, Pittsburgh, PA) with a LiposoFast extruder (AVESTIN Inc., Ottawa, ON) to create large unilamellar vesicles (LUVs).29
Giant Unilamellar Vesicle Preparation
Giant unilamellar vesicles (GUVs) were formed from a DOPC/DPPC/Chol/NBD-PE mixture (33/33/33/1 mole ratio) dissolved in a chloroform/acetonitrile solvent (90/10 volume ratio) to a final lipid concentration of 4 mg/mL. The mixture was spin-coated onto indium tin oxide (ITO)-coated glass slides (SPI, West Chester, PA) using a Laurell model WS-650-23 spin coater.30 To remove any remaining solvent, the lipid-coated slides were placed under vacuum for 30 min. A polydimethylsiloxane (PDMS) spacer was used to separate two slides and create a compartment that was filled with 18.2 MΩ/cm ultrapure water from a Milli-Q Advantage A10 system (EMD Millipore, Billerica, MA) and sealed. For 3 h at 23 °C, an electric field was applied to form GUVs.31 The GUVs were used the same day.
Isothermal Titration Calorimetry
To investigate the interaction of LtxA with Chol and the interaction of the CRAC336WT peptide with Chol and three additional sterols, isothermal titration calorimetry (ITC) was performed. ITC measurements were performed at 30 °C in a Low Volume Nano ITC instrument (TA Instruments, New Castle, DE). For measurements of LtxA affinity, 50 μL of a liposome solution (10 mM) was injected into a cell containing 100 μM LtxA. The injected liposome solution was composed of either 100% POPC or 60% POPC and 40% Chol. For measurements of CRAC336WT affinity, 50 μL of CRAC336WT (7.65 mM) was injected into a cell containing a 2 mM liposome solution, composed of 100% POPC or 60% POPC and 40% sterol, where the sterol was Chol, Desmo, DHC, or CC. A control was also run by titrating CRAC336SCR into liposome solutions of either 100% POPC or 60% POPC and 40% Chol.
The thermodynamics of each reaction were determined by fitting curves of the raw heats to models within NanoAnalyze version 3.5.0. As shown in Figure S2A, interactions between full-length LtxA or either peptide with 100% POPC membranes were fit using the independent model, in which each protein/peptide can bind to n POPC molecules. Interactions between full-length LtxA or either peptide with membranes composed of POPC and sterol were fit using the multiple-sites model, with a ratio of sites of 1.5/1, corresponding to a lipid composition of 60% POPC and 40% sterol, where each protein/peptide can bind to n POPC molecules and m sterol molecules. The equations used for the fits performed by the independent model and the multiple-sites model utilize experimentally known quantities (total sample concentrations and individual heats measured) and have been detailed previously.32
Peptide Structural Changes
To determine the structure of CRAC336WT after binding to Chol and other sterols, circular dichroism (CD) spectra were recorded using a Jasco J-815 CD spectrometer (Jasco Inc., Easton, MD). Spectral scans were performed from 240 to 190 nm, with a scanning speed of 20 nm/min and a bandwidth of 1.0 nm, in 10 mM phosphate buffer using a peptide concentration of 0.25 mg/mL. A 0.01 cm path-length quartz cuvette was used for the measurements. To ensure that the spectra represent the structure of only bound peptide, unbound peptide was removed using centrifugal filters (Amicon 30000 MWCO, EMD Millipore) after 30 min.33,34 CD spectra were processed in OriginPro2015. The secondary structure was determined with DICHROWEB using CONTIN/LL and either the SP175 reference set (for solutions containing only peptide) or the SMP180 reference set (for solutions containing peptides and liposomes).35-40
Membrane Packing Assay
To investigate bilayer packing, a generalized polarization (GP) assay was performed. Laurdan was incorporated into liposomes containing 100% POPC, 80% POPC and 20% sterol, or 60% DMPC and 40% Chol. The liposomes were then incubated with either liposome buffer, CRAC336WT, or CRAC336SCR at a lipid/peptide ratio of 50/1 at 23 °C for 15 min before the sample was excited at 340 nm using a Quantamaster 400 spectrofluorometer (PTI Horiba, Edison, NJ). The GP was calculated using the following equation:
| (1) |
where I440 and I490 are the fluorescence emission intensities at 440 and 490 nm, respectively.
Fluorescent LtxA Labeling
LtxA was labeled with Alexa Fluor 555 NHS Ester (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions, with one modification. Specifically, after LtxA was labeled, it was purified using a 40000 MWCO Zeba Spin Desalting column (Pierce Biotechnology, Rockford, IL).
Confocal Microscopy
To stabilize THP-1 cells onto ibiTreat μ-Dishes (Ibidi, Martinsried, Germany), the cells were differentiated into tissuelike macrophages using cell culture medium supplemented with PMA (100 ng/mL) over 72 h. These cells were then incubated with 2 drops/mL NucBlue Live ReadyProbes reagent (Molecular Probes) for 20 min to label the cell nuclei. Adherence of GUVs to ibiTreat μ-Dishes was facilitated by treating the dishes with poly-L-lysine.
Each μ-Dish, containing either THP-1 cells or GUVs was treated with CRAC336WT, CRAC336SCR, or PBS for 30 min to allow binding of CRAC336WT to occur before LtxA was added. Next, each μ-Dish was incubated with 30 ng of AF555-LtxA for 30 min. The molar peptide/toxin ratio in each dish was 100/1. Imaging was conducted using a Nikon C2si+ confocal microscope equipped with a LU-N4S laser unit and a 60× oil objective (NA = 1.4). The images were processed using Elements version 4.3, Nikon’s imaging software suite, and Fiji.41
CRAC336WT Long-Term Cell Toxicity Assay
THP-1 cells (ATCC) were maintained at 37 °C under 5% CO2 in RPMI 1640 medium containing 10% FBS, 0.05 mM 2-mercaptoethanol, and 40 μg/mL (15.9 μM) CRAC336WT peptide, alongside THP-1 cells grown in peptide-free medium. Over a period of 65 days, cell viability was measured every 2–3 days using a Trypan blue assay.
RESULTS
LtxA and CRAC336WT Have a Strong Affinity for Chol
A total of six experiments were performed to investigate the thermodynamics of binding of LtxA to POPC and Chol, as well as binding of CRAC336WT and CRAC336SCR to POPC and Chol. These experiments were designed to be conducted at a lipid composition at which no phase separation is expected, so that the thermodynamic properties of binding to Chol could be extracted without the additional complication of phase separation.42-44
To obtain the thermodynamic properties of binding of LtxA to membranes, liposomes composed of either 100% POPC or 60% POPC and 40% Chol were titrated into a solution of LtxA, and the data were fit using the independent model or the multiple-binding sites model, respectively (Figure S2A). The heats of injection and the lines of best fit are shown in Figure S2B, and the thermodynamic constants obtained are listed in Table 2. The equilibrium dissociation constant (KD) of association of LtxA with POPC was determined to be 8.75 × 10−4 M, and its negative entropic value suggests that desolvation effects from the hydrophobic interactions between LtxA and POPC dominate the reaction.45-47 The KD for the interaction between LtxA and Chol was determined to be 2.31 × 10−10 M, which was 6 orders of magnitude more favorable than the affinity of LtxA for POPC. As shown in Table 2, the Gibbs free energy (ΔG) of the interaction between LtxA and Chol is much more favorable than that between LtxA and POPC. The entropic (ΔS) contribution to ΔG is similar for both reactions, but the enthalpic (ΔH) contribution from the binding of LtxA to Chol is much more favorable than that for binding of LtxA to POPC, indicating that more and/or stronger noncovalent bonds are formed between LtxA and Chol relative to LtxA and POPC.46,48,49
Table 2.
Thermodynamic Parameters of Interactions of LtxA, CRAC336WT, and CRAC336SCR with Chola
| KD (M) | ΔH (kJ/mol) | ΔS (kJ mol−1 K−1) | −TΔS (kJ/mol) | ΔG (kJ/mol) | |
|---|---|---|---|---|---|
| LtxA and POPC | 8.75 × 10−4 | 15.24 | 0.109 | −32.99 | −17.8 |
| LtxA and Chol | 2.31 × 10−10 | −29.76 | 0.086 | −26.15 | −55.9 |
| CRAC336WT and POPC | 3.81 × 10−3 | 32.58 | 0.155 | −46.8 | −14.2 |
| CRAC336WT and Chol | 5.05 × 10−8 | 2.81 | 0.152 | −46.2 | −43.4 |
| CRAC336SCR and POPC | 5.36 × 10−5 | −7.65 | 0.057 | −17.27 | −24.9 |
| CRAC336SCR and Chol | 4.07 × 10−4 | −5.45 | 0.047 | −14.24 | −19.7 |
The affinity and thermodynamic properties were obtained using the independent or two-site model, as described in Materials and Methods.
To determine the thermodynamic properties of binding of CRAC336WT and CRAC336SCR to Chol, one of the peptides was titrated into a solution of liposomes composed of either 100% POPC or 60% POPC and 40% Chol. The data were fit using the independent model or the multiple-binding sites model, respectively (Figure S2A). The heats of injection and the lines of best fit are shown in panels C and D of Figure S2, and the thermodynamic constants obtained are listed in Table 2.
The results listed in Table 2 indicate that the CRAC336WT peptide interacts weakly with membranes composed of 100% POPC, with a dissociation constant of 3.81 × 10−4 M. The affinity of this peptide for membranes containing 40% Chol was 5.05 × 10−8 M, 4 orders of magnitude higher than the affinity of the peptide for POPC. The free energy (ΔG) is more favorable for binding of CRAC336WT to Chol than to POPC, and like that for LtxA, this difference is due to differences in the enthalpic rather than entropic contributions to the free energy.
Because the CRAC336SCR peptide lacks an intact CRAC sequence, we hypothesized that this peptide would have minimal affinity for Chol and could therefore be used in this work as a negative control. Using ITC, we found that the affinity of the CRAC336SCR peptide for POPC membranes was reduced relative to that of CRAC336WT; in addition, the presence of Chol in the membrane did not enhance the affinity of CRAC336SCR for the membrane (Table 2), demonstrating that this peptide has no affinity for Chol.
Comparison of the free energy values and affinity constants of LtxA and CRAC336WT for membranes (Table 2) indicates that both LtxA and CRAC336WT have a significantly greater affinity for Chol than for POPC. Furthermore, the similarity between the thermodynamics of the interaction of CRAC336WT and LtxA with Chol suggests that the affinity of LtxA for Chol is driven primarily by the toxin’s CRAC motif.
The Secondary Structure of CRAC336WT Is Altered upon Binding to Chol
The favorable enthalpy change observed in the ITC experiment upon binding of CRAC336WT to Chol indicates that this reaction results in more and/or stronger noncovalent bonds between the peptide and Chol than between the peptide and POPC. To investigate whether conformational changes are involved in this difference in bonding, we conducted a CD experiment.
The mean residue ellipticity (MRE), which measures the molar circular dichroism of each residue within the peptide, was calculated for each CD spectrum (Figure S3). The MRE was measured for CRAC336WT in solution and after interaction of the peptide with membranes composed of either 100% POPC or 60% POPC and 40% Chol (Figure 2). In solution, CRAC336WT is composed of approximately 25% α-helices and 25% β-sheets, with the remaining structure containing random coils or disordered regions. This result is consistent with our previous molecular dynamics simulation of CRAC336WT, which produced a similar solution structure.25
Figure 2.
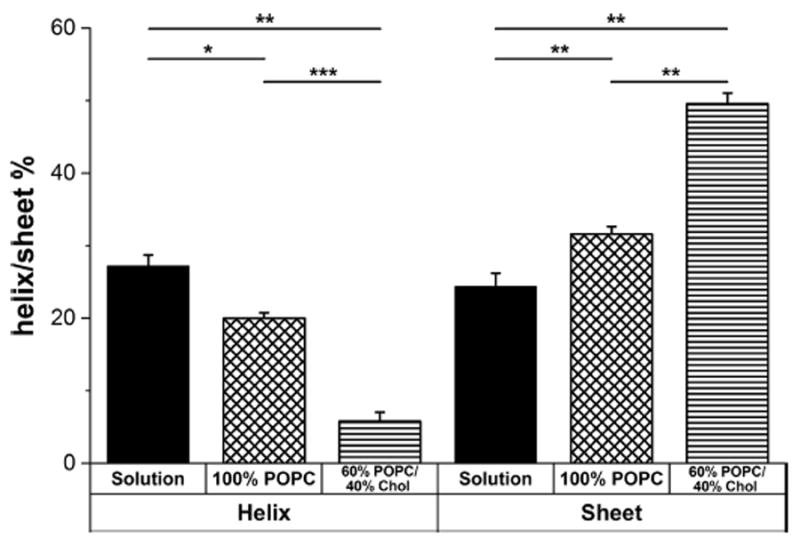
Predicted secondary structure of CRAC336WT in solution or in membranes composed of either 100% POPC or 60% POPC and 40% Chol, determined with DICHROWEB using CONTIN/LL and the SP175 or SMP180 reference set. The bar graph is split into an α-helical structure section (left) and a β-sheet structure section (right). Each bar graph represents data averaged over three independent experiments. The level of significance was determined using an unpaired two-sample t test. ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05; NS, P > 0.05.
As the peptide moved from solution to a POPC membrane, the helicity decreased slightly and the fraction of β-sheet structure increased slightly. The structural changes as the peptide moved from solution to a Chol-containing membrane were much more pronounced, with a large decrease in helicity and a large increase in the level of β-sheet structure, indicating that at least some of the differences in the enthalpic contributions to free energy observed by ITC are due to conformational changes in the peptide upon binding to Chol.
The CRAC336WT Membrane Affinity Depends on Sterol Structure
To characterize recognition by CRAC336WT of Chol in the membrane, we performed ITC experiments using liposomes composed of POPC and one of four sterols. As shown in Figure 3A, each sterol varied only slightly in structure from Chol, allowing us to determine if CRAC336WT recognition of the sterol occurs at the head, body, or tail of the molecule. Relative to Chol, desmosterol (Desmo) has an altered tail, dihydrocholesterol (DHC) has an altered A ring, and cholesteryl chloride (CC) has an altered headgroup. The results, shown in Figure S4 and Table 3, indicate that CRAC336WT has the lowest affinity (6.86 × 10−2 M) for liposomes containing CC, which has a modified headgroup, followed by liposomes containing Desmo (2.39 × 10−4 M), which has a modified tail relative to that of Chol. Slightly reduced affinity, relative to Chol, was measured for liposomes containing DHC (2.53 × 10−7 M), which has a modified ring structure relative to that of Chol. These results suggest that the recognition of Chol by CRAC336WT occurs primarily at the hydroxyl and/or hydrocarbon groups of Chol.
Figure 3.
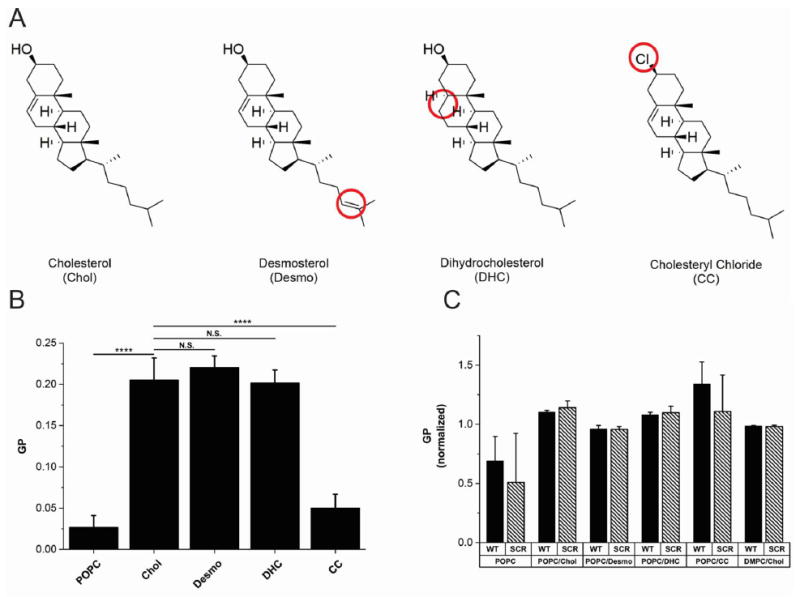
(A) Structures of the four sterols, Chol, DHC, Desmo, and CC, used in this work. The structural differences in each sterol relative to Chol are circled. (B) Membrane fluidity studies. Laurdan was incorporated into liposomes containing either 100% POPC or 80% POPC and 20% sterol at 23 °C. The fluidity of each membrane was quantified by the GP value of laurdan, with a lower GP indicating a more fluid membrane. This graph represents data averaged over six independent experiments. (C) Membrane disruption studies. Laurdan was incorporated into liposomes containing either 100% POPC, 80% POPC and 20% sterol, or 60% DMPC and 40% Chol at 23 °C. Disruption of bilayer packing after peptide incorporation was quantified by the GP value of laurdan, with a lower GP indicating the presence of water in the membrane core. Each GP value was normalized with respect to the GP value of the specified membrane in the absence of peptide. This graph represents data averaged over three independent experiments. The level of significance for both figures was determined using an unpaired two-sample t test. **** P ≤ 0.0001; *** P ≤ 0.001; ** P ≤ 0.01; NS, P > 0.05.
To verify that the observed inhibition of CRAC336WT binding in the presence of Desmo, CC, and DHC, relative to that with Chol, is due specifically to changes in sterol structure and not decreases in membrane fluidity that could prevent peptide association, a GP experiment was performed. Using POPC liposomes containing laurdan, a fluorescent molecule that is sensitive to the presence of water within the membrane,50 we measured the GP of membranes composed entirely of POPC or of POPC and one of the four sterols. As shown in Figure 3B, Chol significantly decreased fluidity in the membrane relative to that of 100% POPC, measured by an increase in GP, as expected. Both DHC and Desmo decreased fluidity similar to Chol. CC decreased the fluidity of the POPC membrane slightly, but much less so than Chol, DHC, or Desmo. This result indicates that the reduction in the level of binding observed in the ITC experiments is due to sterol structure and not overly tight packing of the membrane.
CRAC336WT Does Not Disrupt Membrane Packing
To determine if CRAC336WT perturbs bilayer packing in its interaction with the membrane, another laurdan fluorescence experiment was performed. Using laurdan-labeled liposomes of varying compositions, we calculated the GP before and after peptide addition, to measure the penetration of water into the membrane core, as a measure of bilayer disruption by the peptide. Figure 3C shows the GP of the membrane in the presence of CRAC336WT or CRAC336SCR, normalized by the membrane’s GP value in the absence of peptide. No statistical difference between the GP profiles of any of the membranes in the presence or absence of either CRAC336WT or CRAC336SCR was found. This results indicates that neither peptide induces penetration of water into the hydrophobic core of the membrane, suggesting that the peptides do not penetrate deeply into the membrane.
CRAC336WT Peptides Inhibit LtxA Internalization
Previously, we showed that CRAC336WT is able to inhibit LtxA activity in target cells,23 and here, we have demonstrated that the peptide has a strong affinity for Chol. We therefore investigated the hypothesis that the peptide inhibits LtxA activity by preventing the toxin from binding to Chol and being subsequently internalized by the cell. To visually confirm the affinity of LtxA for Chol, we performed a confocal imaging experiment using DOPC/DPPC/Chol (1/1/1) GUVs. This lipid mixture phase segregates into a Chol-rich liquid ordered (lo) phase and a Chol-poor liquid disordered (ld) phase.51 The GUVs were labeled with the fluorescent probe NBD-PE, which has been shown to have a preferential affinity for the lo phase.52 As shown in Figure 4A, AF555-LtxA was able to bind to the DOPC/DPPC/Chol membranes, with a stronger affinity for the Chol-rich lo phase than for the Chol-poor ld phase. When the GUVs were preincubated with CRAC336WT (Figure 4B), LtxA was unable to bind to the membranes, indicating that the peptide blocks the ability of LtxA to bind Chol. The scrambled peptide, CRAC336SCR, which has no affinity for Chol, had no effect on the binding of LtxA to the membranes (Figure 4C). Quantification of the total LtxA intensity is shown in Figure 4D.
Figure 4.
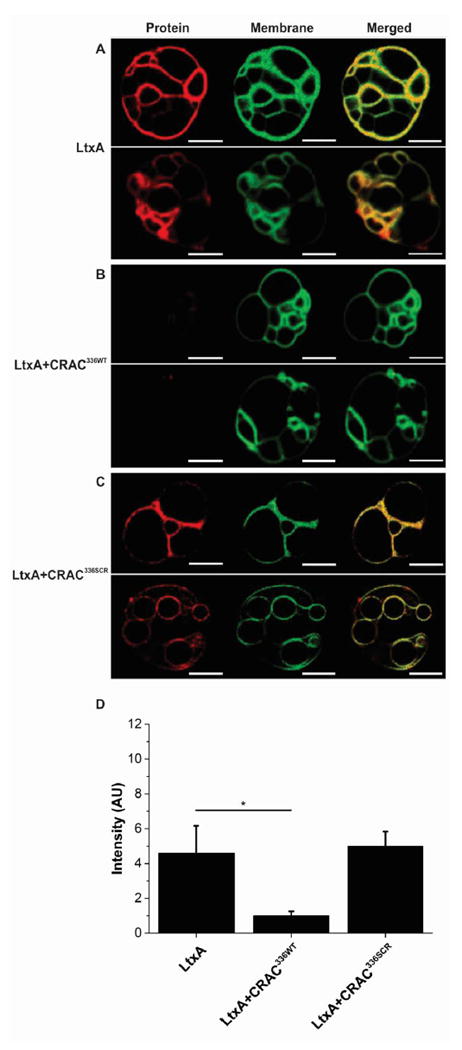
Peptide-mediated inhibition of binding of LtxA to Chol in GUVs. GUVs were composed of DOPC, DPPC, and Chol (1/1/1), labeled with NBD-PE (green), which partitions into the lo phase. (A) In the absence of peptide, AF555-LtxA (red) bound to the membranes where it colocalized with the Chol-rich lo phase. (B) In the presence of CRAC336WT at a peptide/toxin ratio of 100/1, LtxA was inhibited from binding to the membrane. (C) In the presence of CRAC336SCR at a peptide/toxin ratio of 100/1, LtxA was able to bind to the GUV membrane where it colocalized with the Chol-rich lo phase. Scale bars represent 10 μm. (D) Region of interest (ROI) analysis was performed to measure the fluorescence intensity of LtxA in the confocal images. The bar graph represents data averaged over five independent GUV image captures for each of the three conditions. The level of significance was determined using an unpaired two-sample t test. *P ≤ 0.05.
In addition to cholesterol binding, LtxA must also recognize and bind a cell surface receptor, LFA-1.21,53,54 We therefore investigated whether CRAC336WT-mediated inhibition of binding of LtxA to Chol is sufficient to inhibit internalization of LtxA into target cells. When AF555-LtxA was incubated for 30 min with THP-1 cells in the absence of the CRAC peptide, internalization of the toxin into THP-1 cells was observed, as shown in Figure 5A. In contrast, significantly less AF555-LtxA was detected inside the cells when the same amount of the AF555-LtxA was incubated with THP-1 cells pretreated with CRAC336WT, as shown in Figure 5B, demonstrating that CRAC336WT-mediated inhibition of binding of AF555-LtxA to Chol prevents the toxin from being internalized. THP-1 cells pretreated with CRAC336SCR, the control peptide, did not inhibit AF555-LtxA internalization, as shown in Figure 5C. Quantification of the total AF555-LtxA intensity is shown in Figure 5D.
Figure 5.
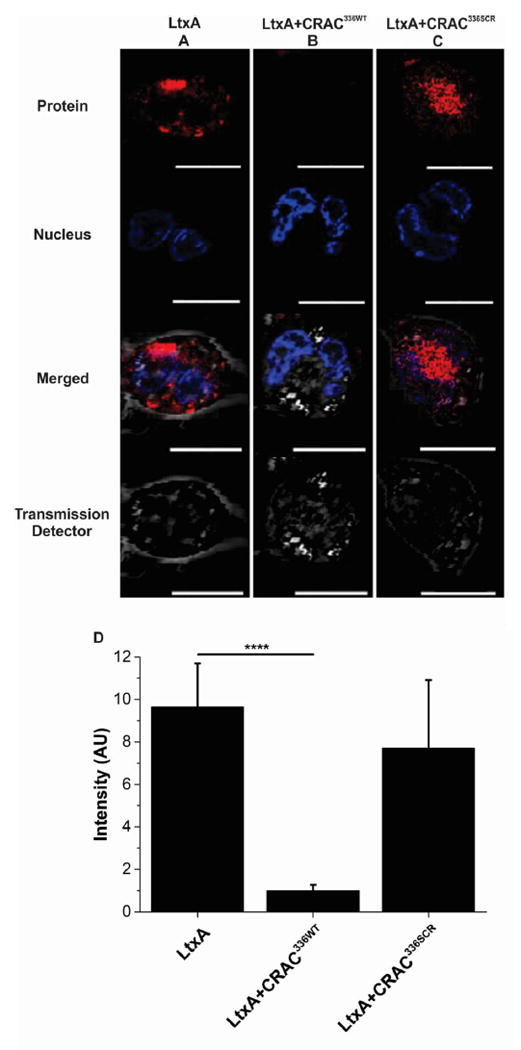
Peptide-mediated inhibition of LtxA internalization in THP-1 cells. THP-1 cells were differentiated into tissuelike macrophages, and the nuclei were labeled with NucBlue Live ReadyProbes reagent (Thermo Scientific, blue). (A) In the absence of peptide, AF555-LtxA (red) is located within THP-1 cells. (B) When the cells were preincubated with CRAC336WT for 30 min at a peptide/toxin ratio of 100/1, AF555-LtxA could not be internalized by the cells. (C) Preincubation of the cells with CRAC336SCR for 30 min at a peptide/toxin ratio of 100/1 did not inhibit AF555-LtxA internalization. Scale bars represent 10 μm. (D) Region of interest (ROI) analysis was performed to measure the fluorescence intensity of LtxA. The bar graph represents data averaged over every cell for each image captured, for each of the three conditions (images were enlarged for the sake of clarity, and not every cell measured is shown). The level of significance was determined using an unpaired two-sample t test. ****P ≤ 0.0001.
CRAC336WT Does Not Exhibit Long-Term Toxicity to Cells
To determine if the CRAC336WT peptide is toxic to cells over a period of time, which would prevent its future therapeutic use, we conducted a long-term viability study (Table 4). THP-1 cells were cultured in medium containing 40 μg/mL (15.9 μM) peptide of CRAC336WT alongside THP-1 cells grown in peptide-free medium. Over a period of 65 days, no peptide-mediated toxicity was observed in the cells, as shown in Table 3, suggesting that this peptide may represent a nontoxic method for blocking binding of the bacterial toxin and pathogen to Chol in host cells. The concentration used in the long-term toxicity assay is more than 2 times greater than the half-maximal inhibitory concentration (IC50), found previously for CRAC336WT inhibition of LtxA toxicity against THP-1 cells.23
Table 4.
Long-Term Effect of CRAC336WT on THP-1 Cell Viabilitya
| day | % viability
|
|
|---|---|---|
| untreated cells | cells with CRAC336WT | |
| 1 | 93 | 93 |
| 5 | 86 | 88 |
| 9 | 77 | 78 |
| 11 | 87 | 85 |
| 16 | 77 | 74 |
| 18 | 90 | 88 |
| 23 | 87 | 90 |
| 27 | 93 | 89 |
| 33 | 73 | 75 |
| 37 | 85 | 86 |
| 41 | 88 | 87 |
| 46 | 79 | 80 |
| 51 | 75 | 78 |
| 55 | 83 | 84 |
| 59 | 88 | 88 |
| 61 | 95 | 95 |
| 65 | 87 | 88 |
THP-1 cells were grown in medium supplemented with 40 μg/mL CRAC336WT or in the absence of peptide. Cell viability was measured using a Trypan blue assay.
Table 3.
Thermodynamic Parameters of the Interactions of CRAC336WT with Sterolsa
| KD (M) | ΔH (kJ/mol) | ΔS (kJ mol−1 K−1) | −TΔS (kJ/mol) | ΔG (kJ/mol) | |
|---|---|---|---|---|---|
| CRAC336WT and POPC | 3.81 × 10−3 | 32.58 | 0.155 | −46.8 | −14.2 |
| CRAC336WT and Chol | 5.05 × 10−8 | 2.81 | 0.152 | −46.2 | −43.4 |
| CRAC336WT and Desmo | 2.39 × 10−4 | 1.508 | 0.074 | −22.42 | −20.9 |
| CRAC336WT and DHC | 2.53 × 10−7 | 2.66 | 0.135 | −40.91 | −38.3 |
| CRAC336WT and CC | 6.86 × 10−2 | −0.80 | 0.019 | −5.76 | −4.96 |
The affinity and thermodynamic properties were obtained using the independent or two-site model, as described in Materials and Methods.
DISCUSSION
Treatment of bacterial illnesses has become increasingly difficult as the number of antibiotic-resistant organisms increases and the development of new antibiotics slows to record low numbers. The U.S. Centers for Disease Control and Prevention (CDC) recently estimated that more than 2 million people develop antibiotic-resistant infections annually in the United States, with more than 23000 dying as a result.55 Antibiotic resistance has been estimated to cost the U.S. health system $21–34 billion/year with an additional 8 million days in the hospital.56 In the past two years, both the CDC and World Health Organization (WHO) have issued recommendations for battling the issue of antibiotic resistance, which include preventing infections from occurring, improved tracking of resistant organisms, more conscientious use of current antibiotics, and the development of new antibiotic strategies.55,56
In this work, we have demonstrated the effective use of a Chol-binding peptide to inhibit the activity of a bacterial toxin, LtxA, a strategy that represents a novel antivirulence approach that has broad potential for the treatment of bacterial illnesses. We demonstrated that the CRAC336WT peptide has a strong affinity for Chol and resides near the membrane surface, where it blocks toxin recognition of Chol to inhibit membrane binding and subsequent internalization and activity of the toxin. Importantly, the peptide exhibits no long-term toxicity to the cells.
Chol, as a major component of animal cell plasma membranes, is a common target for pathogen entry, through a variety of direct and indirect interactions. Lipid rafts, which are enriched in Chol, are often targeted by pathogens because of the raft localization of a particular receptor.57-59 In addition, some pathogens, including intracellular bacteria,60 bacterial toxins such as the cholesterol-dependent cytolysins (CDCs),61,62 and viruses,63-66 recognize and directly bind to Chol on the plasma membrane. One method used by some viral and bacterial proteins to recognize Chol is the CRAC motif. The CRAC motif was first defined in the late 1990s,3 with a combination of a specific amino acid sequence and location at the end of a membrane-spanning α-helix, positioning the motif at the membrane interface.67 While the sequence definition is quite loose, the central tyrosine of this motif has been demonstrated to be essential in the recognition of Chol.4,25 Because of the flexibility of the CRAC definition, amino acid sequences that fit the definition do not necessarily bind to Chol, as we have shown with LtxA.12,25 In addition, an explanation for why this particular amino acid sequence binds Chol has not yet been discovered. Here, we investigated a specific CRAC motif known to bind to Chol to determine the mechanism by which it recognizes Chol and localizes in the membrane, thereby demonstrating its possible utility for therapeutic purposes.
In considering the use of this peptide to inhibit toxin activity, we considered two essential components: (1) localization at the membrane interface to weaken interactions of the toxin with essential membrane components, which may lead to cytotoxicity, and (2) strong binding energetics to inhibit binding by a toxin with a reported strong interaction with Chol.12 Our previous molecular dynamics study suggested that the CRAC336WT peptide interacts with the membrane interface but not the core,25 and investigation of the amino acid sequence suggested that the peptide would reside in a location where it could interact with both the aqueous solution and the membrane environment. In addition, we have previously demonstrated that a peptide corresponding to the CRAC motif in LtxA is able to inhibit activity,23 suggesting that the peptide itself has a strong affinity for Chol. We therefore undertook this work to demonstrate that our hypotheses were correct and to establish the potential use of this peptide in the inhibition of Chol binding by a range of pathogens.
To determine the membrane location of the CRAC336WT peptide, we used laurdan fluorescence to demonstrate that the peptide does not disrupt membrane packing, suggesting that the peptide sits near the interface of the membrane rather than deep in the hydrophobic core. In addition, we found that replacement of the hydroxyl group of Chol significantly weakens binding of the peptide, while substitutions to the B ring or hydrocarbon chains have weaker effects on binding, indicating that recognition of Chol by the peptide occurs near the membrane surface. These findings are consistent with previous findings regarding the CRAC motif found in the fusion protein, gp41, of HIV-1, LWYIK. A nearest neighbor recognition (NNR) study demonstrated that the peptide is sensitive to the packing of the bilayer, suggesting that the peptide must at least partially penetrate into the membrane.68 However, a molecular simulation experiment with the same peptide, along with some derivatives, demonstrated that the peptides prefer the membrane interface over the membrane core and interact with the hydroxyl group of Chol electrostatically.14 Magic-angle spinning nuclear magnetic resonance (MAS NMR) demonstrated in Chol-containing membranes that the peptide interacts with the A ring of Chol,69 near the membrane interface.
Recognition of the hydroxyl group of Chol may be conserved among pathogens, as it would be the first structural element of Chol they encounter upon interaction with a host cell membrane. For example, although they do not use a CRAC motif to recognize Chol, the CDCs produced by Gram-positive bacteria likewise require the presence of a sterol with an intact hydroxyl group.61 Variation in this region completely inhibits activity of the toxins; changes in the ring structure of the sterol reduce activity slightly, and changes to the hydrocarbon tail have no effect on toxin activity.
In addition to localization at the membrane interface, we hypothesized that strong affinity for Chol would be required for the peptide to efficiently inhibit binding of pathogens to Chol with reported affinity for Chol. For this reason, we investigated the CRAC336WT peptide of LtxA. Previously, we showed that LtxA has a very strong affinity, on the order of 10−12 M, for liposomes containing 40% Chol.12 Here, we have shown that much of the affinity of LtxA for Chol is driven by the CRAC336 motif, as the peptide alone has an affinity for Chol comparable to that of the full-length protein. The affinity of the CRAC336WT peptide is several orders of magnitude stronger than reported affinities of pneumolysin (4 × 10−7 M),70 the cytolethal distending toxin (Cdt) produced by A. actinomycetemcomitans (2 × 10−6),71 α-hemolysin (HlyA) produced by Escherichia coli (1.6 × 10−5),72 and the invasion plasmid antigen B (ipaB) produced by Shigella flexneri (1.8 × 10−5),73 suggesting that the peptide may be able to inhibit binding by these and other pathogenic proteins.
We predicted that a CRAC peptide that binds Chol at the membrane surface would best be able to block the pathogen’s recognition of Chol. Our results here represent a proof of concept of this idea; experiments are currently underway to improve Chol recognition at the interface by CRAC peptides and thus more efficiently block toxin and pathogen activity. Because many pathogens recognize Chol as an initial step in their activity against host cells, we expect that this peptide will have broad applications for the treatment of viral and bacterial diseases, as there are currently no viable approaches to inhibit this interaction. Previously proposed strategies include cyclodextrins, which have a strong affinity for Chol and are able to extract the sterol from the membrane.74 This molecule has been shown to extract Chol from HIV-1 and SIV-1 virions, resulting in decreased infectivity, and was therefore proposed as a possible topical microbicide.75 In addition, β-cyclodextrins have been proposed for the treatment of intracellular Leishmania infections;76 however, because of cyclodextrins’ alteration of cell processes and viability, its therapeutic use is limited. Here, we have demonstrated that the CRAC336WT peptide, perhaps because its interaction with Chol occurs only near the membrane interface, does not induce cytotoxicity in host cells. The ability of the CRAC336WT peptide to inhibit toxin activity by blocking its interaction with Chol in the host cell plasma membrane is a novel concept that has wide-ranging applications in bacterial and viral pathogenesis.
Supplementary Material
Acknowledgments
The authors thank Drs. Damien Thevenin, Marcos Pires, and Lesley Chow for their technical assistance in peptide purification.
Funding
This work was supported by the National Institutes of Health (R00DE022795) and Sigma Xi, the Scientific Research Society (E. Koufos).
ABBREVIATIONS
- CD
circular dichroism
- Chol
cholesterol
- CRAC
cholesterol recognition amino acid consensus
- CDCs
cholesterol-dependent cytolysins
- CC
cholesteryl chloride
- Cdt
cytolethal distending toxin
- Desmo
desmosterol
- DCM
dichloromethane
- DHC
dihydrocholesterol
- DMF
dimethylformamide
- Fmoc
9-fluorenylmethyloxycarbonyl
- GPCR
G protein-coupled receptor
- GP
generalized polarization
- GUV
giant unilamellar vesicle
- HSV-1
herpes simplex virus type 1
- HPLC
high-performance liquid chromatography
- ITO
indium tin oxide
- ITC
isothermal titration calorimetry
- LUV
large unilamellar vesicle
- LtxA
leukotoxin A
- ld
liquid disordered
- lo
liquid ordered
- LFA-1
lymphocyte function-associated antigen-1
- MAS NMR
magic-angle spinning nuclear magnetic resonance
- MALDI-TOF MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- MRE
mean residue ellipticity
- MeOH
methanol
- MLVs
multilamellar vesicles
- NBD-PE
N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine
- DIEA
N,N-diisopropylethylamine
- PMA
phorbol 12-myristate 13-acetate
- PDMS
polydimethylsiloxane
- HBTU
tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- TFA
trifluoroacetic acid
- CDC
U.S. Centers for Disease Control and Prevention
- VSV
vesicular stomatitis virus
- WHO
World Health Organization
- HOBt
1-hydroxybenzotriazole monohydrate
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DMPC
1,2-ditetradecanoyl-sn-glycero-3-phosphocholine
- laurdan
6-dodecanoyl-2-dimethylaminonaphthalene
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00430.
Peptide synthesis and purification (Figure S1), ITC models and raw data for LtxA, CRAC336WT, and CRAC336SCR with POPC and POPC/Chol (Figure S2), raw CD data (Figure S3), and ITC raw data for CRAC336WT with POPC/sterol (Figure S4). (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Yeagle PL. Modulation of membrane function by cholesterol. Biochimie. 1991;73:1303–1310. doi: 10.1016/0300-9084(91)90093-g. [DOI] [PubMed] [Google Scholar]
- 2.Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 4.Jamin N, Neumann JM, Ostuni MA, Vu TK, Yao ZX, Murail S, Robert JC, Giatzakis C, Papadopoulos V, Lacapere JJ. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol Endocrinol. 2005;19:588–594. doi: 10.1210/me.2004-0308. [DOI] [PubMed] [Google Scholar]
- 5.Fantini J, Carlus D, Yahi N. The fusogenic tilted peptide (67−78) of α-synuclein is a cholesterol binding domain. Biochim Biophys Acta, Biomembr. 2011;1808:2343–2351. doi: 10.1016/j.bbamem.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 6.Wood WG, Schroeder F, Igbavboa U, Avdulov NA, Chochina SV. Brain membrane cholesterol domains, aging and amyloid β-peptides. Neurobiol Aging. 2002;23:685–694. doi: 10.1016/s0197-4580(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 7.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. β-amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 9.Jafurulla M, Tiwari S, Chattopadhyay A. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem Biophys Res Commun. 2011;404:569–573. doi: 10.1016/j.bbrc.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boesze-Battaglia K, Brown A, Walker L, Besack D, Zekavat A, Wrenn S, Krummenacher C, Shenker BJ. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J Biol Chem. 2009;284:10650–10658. doi: 10.1074/jbc.M809094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown AC, Balashova NV, Epand RM, Epand RF, Bragin A, Kachlany SC, Walters MJ, Du Y, Boesze-Battaglia K, Lally ET. Aggregatibacter actinomycetemcomitans leukotoxin utilizes a cholesterol recognition/amino acid consensus site for membrane association. J Biol Chem. 2013;288:23607–23621. doi: 10.1074/jbc.M113.486654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenwood AI, Pan J, Mills TT, Nagle JF, Epand RM, Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim Biophys Acta, Biomembr. 2008;1778:1120–1130. doi: 10.1016/j.bbamem.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vishwanathan SA, Thomas A, Brasseur R, Epand RF, Hunter E, Epand RM. Hydrophobic substitutions in the first residue of the CRAC segment of the gp41 protein of HIV. Biochemistry. 2008;47:124–130. doi: 10.1021/bi7018892. [DOI] [PubMed] [Google Scholar]
- 15.Thaa B, Levental I, Herrmann A, Veit M. Intrinsic membrane association of the cytoplasmic tail of influenza virus M2 protein and lateral membrane sorting regulated by cholesterol binding and palmitoylation. Biochem J. 2011;437:389–397. doi: 10.1042/BJ20110706. [DOI] [PubMed] [Google Scholar]
- 16.Luz-Madrigal A, Asanov A, Camacho-Zarco AR, Sampieri A, Vaca L. A cholesterol recognition amino acid consensus domain in GP64 fusion protein facilitates anchoring of baculovirus to mammalian cells. J Virol. 2013;87:11894–11907. doi: 10.1128/JVI.01356-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch RA. Pore-forming cytolysins of gram-negative bacteria. Mol Microbiol. 1991;5:521–528. doi: 10.1111/j.1365-2958.1991.tb00723.x. [DOI] [PubMed] [Google Scholar]
- 18.Taichman NS, Dean RT, Sanderson CJ. Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect Immun. 1980;28:258–268. doi: 10.1128/iai.28.1.258-268.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taichman NS, Shenker BJ, Tsai CC, Glickman LT, Baehni PC, Stevens R, Hammond BF. Cytopathic effects of Actinobacillus actinomycetemcomitans on monkey blood leukocytes. J Periodontal Res. 1984;19:133–145. doi: 10.1111/j.1600-0765.1984.tb00802.x. [DOI] [PubMed] [Google Scholar]
- 20.Taichman NS, Simpson DL, Sakurada S, Cranfield M, DiRienzo J, Slots J. Comparative studies on the biology of Actinobacillus actinomycetemcomitans leukotoxin in primates. Oral Microbiol Immunol. 1987;2:97–104. doi: 10.1111/j.1399-302x.1987.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 21.Kieba IR, Fong KP, Tang HY, Hoffman KE, Speicher DW, Klickstein LB, Lally ET. Aggregatibacter actinomycetemcomitans leukotoxin requires β-sheets 1 and 2 of the human CD11a β-propeller for cytotoxicity. Cell Microbiol. 2007;9:2689–2699. doi: 10.1111/j.1462-5822.2007.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lally ET, Kieba IR, Sato A, Green CL, Rosenbloom J, Korostoff J, Wang JF, Shenker BJ, Ortlepp S, Robinson MK, Billings PC. RTX toxins recognize a β2 integrin on the surface of human target cells. J Biol Chem. 1997;272:30463–30469. doi: 10.1074/jbc.272.48.30463. [DOI] [PubMed] [Google Scholar]
- 23.Brown AC, Koufos E, Balashova NV, Boesze-Battaglia K, Lally ET. Inhibition of LtxA toxicity by blocking cholesterol binding with peptides. Mol Oral Microbiol. 2016;31:94–105. doi: 10.1111/omi.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong KP, Pacheco CM, Otis LL, Baranwal S, Kieba IR, Harrison G, Hersh EV, Boesze-Battaglia K, Lally ET. Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cell Microbiol. 2006;8:1753–1767. doi: 10.1111/j.1462-5822.2006.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller CM, Brown AC, Mittal J. Disorder in cholesterol-binding functionality of CRAC peptides: a molecular dynamics study. J Phys Chem B. 2014;118:13169–13174. doi: 10.1021/jp5106423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fine DH, Furgang D, Schreiner HC, Goncharoff P, Charlesworth J, Ghazwan G, Fitzgerald-Bocarsly P, Figurski DH. Phenotypic variation in Actinobacillus actinomycetemcomitans during laboratory growth: implications for virulence. Microbiology. 1999;145(Part 6):1335–1347. doi: 10.1099/13500872-145-6-1335. [DOI] [PubMed] [Google Scholar]
- 27.Forman MS, Nishikubo JB, Han RK, Le A, Balashova NV, Kachlany SC. Gangliosides block Aggregatibacter actinomycetemcomitans leukotoxin (LtxA)-mediated hemolysis. Toxins. 2010;2:2824–2836. doi: 10.3390/toxins2122824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bangham AD, Horne RW. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J Mol Biol. 1964;8:660–668. doi: 10.1016/s0022-2836(64)80115-7. [DOI] [PubMed] [Google Scholar]
- 29.MacDonald RC, MacDonald RI, Menco BP, Takeshita K, Subbarao NK, Hu LR. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim Biophys Acta, Biomembr. 1991;1061:297–303. doi: 10.1016/0005-2736(91)90295-j. [DOI] [PubMed] [Google Scholar]
- 30.Estes DJ, Mayer M. Electroformation of giant liposomes from spin-coated films of lipids. Colloids Surf, B. 2005;42:115–123. doi: 10.1016/j.colsurfb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Angelova MI, Soléau S, Méléard P, Faucon F, Bothorel P. Preparation of giant vesicles by external AC electric fields. Kinetics and applications. Trends in Colloid and Interface Science VI. 1992;89:127–131. [Google Scholar]
- 32.Freire E, Mayorga OL, Straume M. Isothermal Titration Calorimetry. Anal Chem. 1990;62:950A–959A. [Google Scholar]
- 33.Voglino L, McIntosh TJ, Simon SA. Modulation of the binding of signal peptides to lipid bilayers by dipoles near the hydrocarbon-water interface. Biochemistry. 1998;37:12241–12252. doi: 10.1021/bi9805792. [DOI] [PubMed] [Google Scholar]
- 34.Voglino L, Simon SA, McIntosh TJ. Orientation of LamB signal peptides in bilayers: influence of lipid probes on peptide binding and interpretation of fluorescence quenching data. Biochemistry. 1999;38:7509–7516. doi: 10.1021/bi990099q. [DOI] [PubMed] [Google Scholar]
- 35.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 37.Lees JG, Miles AJ, Wien F, Wallace BA. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics. 2006;22:1955–1962. doi: 10.1093/bioinformatics/btl327. [DOI] [PubMed] [Google Scholar]
- 38.Abdul-Gader A, Miles AJ, Wallace BA. A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy. Bioinformatics. 2011;27:1630–1636. doi: 10.1093/bioinformatics/btr234. [DOI] [PubMed] [Google Scholar]
- 39.Provencher SW, Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 40.van Stokkum IH, Spoelder HJ, Bloemendal M, van Grondelle R, Groen FC. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 41.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biologicalimage analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pokorny A, Yandek LE, Elegbede AI, Hinderliter A, Almeida PF. Temperature and composition dependence of the interaction of delta-lysin with ternary mixtures of sphingomyelin/cholesterol/POPC. Biophys J. 2006;91:2184–2197. doi: 10.1529/biophysj.106.085027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veatch SL, Keller SL. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys Rev Lett. 2005;94:148101. doi: 10.1103/PhysRevLett.94.148101. [DOI] [PubMed] [Google Scholar]
- 44.Brown AC, Towles KB, Wrenn SP. Measuring raft size as a function of membrane composition in PC-based systems: Part II–ternary systems. Langmuir. 2007;23:11188–11196. doi: 10.1021/la7006342. [DOI] [PubMed] [Google Scholar]
- 45.Seelig J, Ganz P. Nonclassical hydrophobic effect in membrane binding equilibria. Biochemistry. 1991;30:9354–9359. doi: 10.1021/bi00102a031. [DOI] [PubMed] [Google Scholar]
- 46.Bronowska AK. Thermodynamics-Interaction Studies-Solids, Liquids and Gases. InTech; Rijeka, Croatia: 2011. Thermodynamics of Ligand-Protein Interactions: Implications for Molecular Design; pp. 1–49. [Google Scholar]
- 47.Luque I, Freire E. Structure-based prediction of binding affinities and molecular design of peptide ligands. Methods Enzymol. 1998;295:100–127. doi: 10.1016/s0076-6879(98)95037-6. [DOI] [PubMed] [Google Scholar]
- 48.Ladbury JE, Klebe G, Freire E. Adding calorimetric data to decision making in lead discovery: a hot tip. Nat Rev Drug Discovery. 2010;9:23–27. doi: 10.1038/nrd3054. [DOI] [PubMed] [Google Scholar]
- 49.Nunez S, Venhorst J, Kruse CG. Target-drug interactions: first principles and their application to drug discovery. Drug Discovery Today. 2012;17:10–22. doi: 10.1016/j.drudis.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 50.Parasassi T, De Stasio G, d’Ubaldo A, Gratton E. Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys J. 1990;57:1179–1186. doi: 10.1016/S0006-3495(90)82637-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veatch SL, Keller SL. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys J. 2003;85:3074–3083. doi: 10.1016/S0006-3495(03)74726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dietrich C, Bagatolli LA, Volovyk ZN, Thompson NL, Levi M, Jacobson K, Gratton E. Lipid rafts reconstituted in model membranes. Biophys J. 2001;80:1417–1428. doi: 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dileepan T, Kachlany SC, Balashova NV, Patel J, Maheswaran SK. Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infect Immun. 2007;75:4851–4856. doi: 10.1128/IAI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DiFranco KM, Gupta A, Galusha LE, Perez J, Nguyen TV, Fineza CD, Kachlany SC. Leukotoxin (Leukothera(R)) targets active leukocyte function antigen-1 (LFA-1) protein and triggers a lysosomal mediated cell death pathway. J Biol Chem. 2012;287:17618–17627. doi: 10.1074/jbc.M111.314674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demuth D, James D, Kowashi Y, Kato S. Interaction of Actinobacillus actinomycetemcomitans outer membrane vesicles with HL60 cells does not require leukotoxin. Cell Microbiol. 2003;5:111–121. doi: 10.1046/j.1462-5822.2003.00259.x. [DOI] [PubMed] [Google Scholar]
- 56.Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004;23:4538–4549. doi: 10.1038/sj.emboj.7600471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Goot FG, Harder T. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin Immunol. 2001;13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- 58.Manes S, del Real G, Martinez-A C. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3:557–568. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberger CM, Brumell JH, Finlay BB. Microbial pathogenesis: lipid rafts as pathogen portals. Curr Biol. 2000;10:R823–R825. doi: 10.1016/s0960-9822(00)00788-0. [DOI] [PubMed] [Google Scholar]
- 60.Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1651. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 61.Palmer M. Cholesterol and the activity of bacterial toxins. FEMS Microbiol Lett. 2004;238:281–289. doi: 10.1016/j.femsle.2004.07.059. [DOI] [PubMed] [Google Scholar]
- 62.Zitzer A, Zitzer O, Bhakdi S, Palmer M. Oligomerization of Vibrio cholerae cytolysin yields a pentameric pore and has a dual specificity for cholesterol and sphingolipids in the target membrane. J Biol Chem. 1999;274:1375–1380. doi: 10.1074/jbc.274.3.1375. [DOI] [PubMed] [Google Scholar]
- 63.Phalen T, Kielian M. Cholesterol is required for infection by Semliki Forest virus. J Cell Biol. 1991;112:615–623. doi: 10.1083/jcb.112.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kielian MC, Helenius A. Role of cholesterol in fusion of Semliki Forest virus with membranes. J Virol. 1984;52:281–283. doi: 10.1128/jvi.52.1.281-283.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses. 2001;17:1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- 66.Scheiffele P, Roth MG, Simons K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997;16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Epand RM. Proteins and cholesterol-rich domains. Biochim Biophys Acta, Biomembr. 2008;1778:1576–1582. doi: 10.1016/j.bbamem.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 68.Mukai M, Krause MR, Regen SL. Peptide Recognition of Cholesterol in Fluid Phospholipid Bilayers. J Am Chem Soc. 2015;137:12518–12520. doi: 10.1021/jacs.5b08132. [DOI] [PubMed] [Google Scholar]
- 69.Epand RM, Sayer BG, Epand RF. Peptideinduced formation of cholesterol-rich domains. Biochemistry. 2003;42:14677–14689. doi: 10.1021/bi035587j. [DOI] [PubMed] [Google Scholar]
- 70.Nollmann M, Gilbert R, Mitchell T, Sferrazza M, Byron O. The role of cholesterol in the activity of pneumolysin, a bacterial protein toxin. Biophys J. 2004;86:3141–3151. doi: 10.1016/S0006-3495(04)74362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boesze-Battaglia K, Walker LP, Zekavat A, Dlakic M, Scuron MD, Nygren P, Shenker BJ. The Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Active Subunit CdtB Contains a Cholesterol Recognition Sequence Required for Toxin Binding and Subunit Internalization. Infect Immun. 2015;83:4042–4055. doi: 10.1128/IAI.00788-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vazquez RF, Mate SM, Bakas LS, Fernandez MM, Malchiodi EL, Herlax VS. Novel evidence for the specific interaction between cholesterol and α-haemolysin of Escherichia coli. Biochem J. 2014;458:481–489. doi: 10.1042/BJ20131432. [DOI] [PubMed] [Google Scholar]
- 73.Hayward RD, Cain RJ, McGhie EJ, Phillips N, Garner MJ, Koronakis V. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol Microbiol. 2005;56:590–603. doi: 10.1111/j.1365-2958.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- 74.Szejtli J. Introduction and General Overview of Cyclodextrin Chemistry. Chem Rev. 1998;98:1743–1754. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 75.Graham DR, Chertova E, Hilburn JM, Arthur LO, Hildreth JE. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with β-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J Virol. 2003;77:8237–8248. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pucadyil TJ, Chattopadhyay A. Cholesterol: a potential therapeutic target in Leishmania infection? Trends Parasitol. 2007;23:49–53. doi: 10.1016/j.pt.2006.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.