

Abstract
Host glycans are paramount in regulating the symbiotic relationship between humans and their gut bacteria. The constant flux of host-secreted mucin at the mucosal layer creates a steady niche for bacterial colonization. Mucin degradation by keystone species subsequently shapes the microbial community. This study investigated the transcriptional response during mucin-driven trophic interaction between the specialised mucin-degrader Akkermansia muciniphila and a butyrogenic gut commensal Anaerostipes caccae. A. muciniphila monocultures and co-cultures with non-mucolytic A. caccae from the Lachnospiraceae family were grown anaerobically in minimal media supplemented with mucin. We analysed for growth, metabolites (HPLC analysis), microbial composition (quantitative reverse transcription PCR), and transcriptional response (RNA-seq). Mucin degradation by A. muciniphila supported the growth of A. caccae and concomitant butyrate production predominantly via the acetyl-CoA pathway. Differential expression analysis (DESeq 2) showed the presence of A. caccae induced changes in the A. muciniphila transcriptional response with increased expression of mucin degradation genes and reduced expression of ribosomal genes. Two putative operons that encode for uncharacterised proteins and an efflux system, and several two-component systems were also differentially regulated. This indicated A. muciniphila changed its transcriptional regulation in response to A. caccae. This study provides insight to understand the mucin-driven microbial ecology using metatranscriptomics. Our findings show that the expression of mucolytic enzymes by A. muciniphila increases upon the presence of a community member. This could indicate its role as a keystone species that supports the microbial community in the mucosal environment by increasing the availability of mucin sugars.
Electronic supplementary material
The online version of this article (10.1007/s10482-018-1040-x) contains supplementary material, which is available to authorized users.
Keywords: Butyrate, Cross feeding, Keystone species, Microbiome, Mucin, Transcriptional regulation, Verrucomicrobia
Introduction
The bacterial assembly at the mucosal layer of the human gastrointestinal tract is associated with gut health and disease (Ouwerkerk et al. 2013; Tailford et al. 2015). Although the microbial composition of the healthy mucosa has not been properly defined, it has been observed that strong deviations in the mucosal microbiota are associated with inflammatory bowel disease (IBD) (Kostic et al. 2014) and irritable bowel syndrome (IBS) (Lopez-Siles et al. 2014). At this mucosal site, host-produced mucin glycans and bioactive compounds collectively exert a selective pressure that enriches for a sub-population of mucosa-associated bacteria (Koropatkin et al. 2012; Ouwerkerk et al. 2013; Schluter and Foster 2012). Mucins are large and complex glycoproteins consisting of a protein core that is rich in proline, threonine and serine moieties, to which oligosaccharides are attached (Tailford et al. 2015). Mucins can function as an indigenous prebiotic in which only specialised members of intestinal microbiota are able to utilise it as the substrate for growth (Marcobal et al. 2013; Ouwehand et al. 2005; Tailford et al. 2015).
The intestinal symbiont, Akkermansia muciniphila is the sole human intestinal representative of the phylum Verrucomicrobia (de Vos 2017). A. muciniphila has adapted to mucosal environment in the gut (Derrien et al. 2008). The genome of A. muciniphila is equipped with an arsenal of mucin-degrading enzymes including proteases, glycosyl hydrolases (GH), and sulfatases (Derrien et al. 2016; van Passel et al. 2011). The mucin-degrading capacity and oxygen tolerance of A. muciniphila render it a key species in the mucosal niche (Ouwerkerk et al. 2016). This specialised mucin-degrading bacterium is detected at high prevalence (over 96%) in healthy Western adults (Collado et al. 2007; Derrien et al. 2008; Shetty et al. 2016). The abundance of A. muciniphila in the gut microbiota is inversely correlated with syndromes such as IBDs (both Crohn’s disease and ulcerative colitis) (Png et al. 2010), appendicitis (Swidsinski et al. 2011) and obesity (Everard et al. 2013). Furthermore, the potential therapeutic role of A. muciniphila has been demonstrated in mice by remedying symptoms of obesity and diabetes (Plovier et al. 2017) as well as alcoholic liver disease (Grander et al. 2017).
In addition to the health-promoting role of A. muciniphila via immune modulation, the extracellular mucin degradation by this bacterium could provide growth benefits to community members via trophic interactions (Belzer et al. 2017; Belzer and de Vos 2012; Derrien et al. 2016). Several in vitro studies have demonstrated the butyrogenic effect of complex carbohydrates via cross-feeding between glycan-degrading bifidobacteria and butyrogenic bacteria (Belenguer et al. 2006; De Vuyst and Leroy 2011; Falony et al. 2006; Rios-Covian et al. 2015; Riviere et al. 2015; Schwab et al. 2017). In the mucosal environment, mucolytic bacteria such as A. muciniphila, Bacteroides spp. and Ruminococcus spp. as well as butyrogenic members of the family Lachnospiraceae (also known as Clostridium cluster XIVa) and Ruminococcaceae (also known as Clostridium cluster IV) are enriched (Nava et al. 2011; Van den Abbeele et al. 2013). However, no mucolytic capacities of these butyrogenic bacteria are known, which suggested potential metabolic cross-feeding between the microbial groups. Butyrate production in the vicinity of epithelial cells is suggested to be important in maintaining gut health (Koh et al. 2016; Louis and Flint 2017).
In a previous study (Belzer et al. 2017), we showed that mucin degradation by A. muciniphila yields short chain fatty acids (SCFAs) and mucin-derived monosaccharides that support the growth and concomitant butyrate production of non-mucolytic butyrogens. In this paper, we used metatranscriptomics (RNA-seq) to study the molecular response of mucin-directed trophic interaction between A. muciniphila and an abutyrogenic bacterium from the family Lachnospiraceae (Anaerostipes caccae) which possesses metabolic capacity to convert acetate and lactate into butyrate (Duncan et al. 2004) and shows frequent occurrence at the mucosal niche (Nava et al. 2011; Van den Abbeele et al. 2013). We demonstrated the use of metatranscriptomics as an explorative approach to study the expressional changes of A. muciniphila in response to a community member. Notably, we showed that A. muciniphila increased its mucolytic activity to sustain the community.
Materials and methods
Bacterial strains and growth conditions
All bacteria were grown in anaerobic serum bottles sealed with butyl-rubber stoppers at 37 °C with N2:CO2 (80:20 ratio) in the headspace at 1.5 atm. Bacterial pre-cultures were prepared by overnight growth in: minimal media supplemented with type III hog gastric mucin (Sigma-Aldrich, St. Louis, USA) for A. muciniphila MucT (ATCC BAA-835)(Derrien et al. 2004), and peptone yeast glucose (PYG) medium for A. caccae L1-92 (DSM 14662) (Schwiertz et al. 2002). Growth was measured by spectrophotometer as optical density at 600 nm (OD600) (OD600 DiluPhotometer™, IMPLEN, Germany).
Co-culture experiment
Co-culture experiments were performed in minimal media (Plugge 2005) supplemented with purified hog gastric mucin (Miller and Hoskins 1981). Culture conditions were established as previously described (Belzer et al. 2017). A. muciniphila was inoculated at 1 × 106 cells to mucin media followed by 8 h of incubation to allow accumulation of metabolites. Subsequently, 1 × 106 cells of A. caccae (A.muc-A.cac co-cultures) were added to the A. muciniphila cultures. Cells were washed twice with phosphate-buffered saline (PBS) before addition to the co-cultures to prevent carryover of metabolites from the pre-cultures. Purified mucin (1.25 g l−1) was added to the media every 48 h. A schematic setup of the experiment is depicted in Fig. 1a. Cultures were sampled at 0, 1, 2, 4, 6, 8, 11, and 23 days for metabolites analysis. For transcriptomic analysis at day 8, bacteria pellets were preserved in Trizol® reagent (Invitrogen, Carlsbad, CA, USA) at − 20 °C storage till further RNA purification.
Fig. 1.
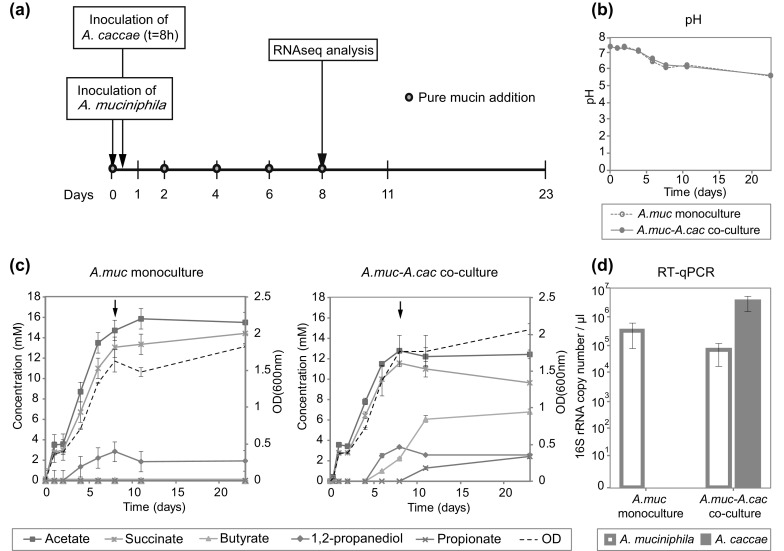
a Schematic overview of the interval-fed batch culture setup. A. muciniphila was inoculated at t = 0 h followed by A. caccae at t = 8 h to ensure substrate availability for butyrogen via extracellular mucin degradation by A. muciniphila. Limited amounts of pure mucin, 0.15% (v/v) were supplemented at 2 days intervals to maintain the abundance of A. muciniphila and to support the emergence of A. caccae. A sample for RNA-seq analysis was collected on day 8. b The pH and c metabolite profile of monocultures and co-cultures of the interval-fed batch culture, with arrow showing day 8. d Quantification of microbial composition on day 8 by RT-qPCR targeting 16S rRNA on total RNA. Error bars indicate the standard deviation of biological duplicates
High-performance liquid chromatography (HPLC)
For metabolites analysis, 1 ml of bacterial culture was centrifuged and the supernatant was stored at − 20 °C until HPLC analysis. Crotonate was used as the internal standard, and the external standards were lactate, formate, acetate, propionate, isobutyrate, butyrate, citrate, malate, succinate, fumarate, 1,2-propanediol, methanol, ethanol, 2-propanol, lactose, N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc),glucose, and galactose. Substrates conversion and products formation were measured with a Spectrasystem HPLC (Thermo Scientific, Breda, the Netherlands) equipped with a Hi-Plex-H column (Agilent, Amstelveen, the Netherlands) for the separation of organic acids and carbohydrates. A Hi-Plex-H column performs separation with diluted sulphuric acid on the basis of ion-exchange ligand-exchange chromatography. Measurements were conducted at a column temperature of 45 °C with an eluent flow of 0.8 ml min−1 flow of 0.01 N sulphuric acid. Metabolites were detected by refractive index (Spectrasystem RI 150, Thermo, Breda, the Netherlands).
RNA purification
Total RNA was isolated by a method combining the Trizol® reagent and the RNeasy Mini kit (QIAGEN GmbH, Hilden, Germany) as described previously (Chomczynski 1993; Zoetendal et al. 2006). Four microliter of p-mercaptoethanol and 0.4 ml of buffer RLT were added to 1 ml of Trizol® reagent containing the bacterial pellet. The mixture was transferred to a tube containing 0.8 g of glass beads (diameter 0.1 mm), followed by three times of bead beating for 1 min at 5.5 ms−1 with ice cooling steps in between. Subsequently, 0.2 ml of ice-cold chloroform was added. The solution was mixed gently followed by centrifugation at 12,000×g for 15 min at 4 °C. The RNA isolation was continued with the RNA clean-up according to the manufacturer’s instructions for the RNeasy Mini kit. Genomic DNA was removed by an on-column DNase digestion step during RNA purification (DNase I recombinant, RNase-free, Roche Diagnostics GmbH, Mannheim, Germany). Yield and RNA quality was assessed using the Experion™ RNA StdSens Analysis Kit in combination with the Experion™ System (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Quantitative reverse transcription PCR (RT-qPCR)
cDNA was synthesised using the ScriptSeq v2 RNA-Seq library preparation kit (Epicentre, Madison, WI, USA) according to the manufacturer’s instructions followed by purification using CleanPCR (CleanNA, the Netherlands). The cDNA was analysed by quantitative real-time PCR. Primers targeting 16S rRNA gene of A. muciniphila (AM1 5′-CAGCACGTGAAGGTGGGGAC-3′ and AM2 5′-CCTTGCGGTTGGCTTCAGAT-3′) (Collado et al. 2007), and A. caccae (OFF2555 5′-GCGTAGGTGGCATGGTAAGT-3′ and OFF2556 5′-CTGCACTCCAGCATGACAGT-3′) (Veiga et al. 2010) were used for quantification. Standard template DNA was prepared by 16S rRNA gene amplification of each bacterium with primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). Standard curves were prepared with nine standard concentrations from 100 to 108 gene copies μl−1. qPCR was performed in technical triplicate with iQ SYBR Green Supermix (Bio-Rad) in a total volume of 10 μl with primers at 500 nM in 384-well plates sealed with optical sealing tape. Amplification was performed with an iCycler (Bio-Rad) with the following protocol: one cycle of 95 °C for 10 min, 35 cycles of 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 30 s each, one cycle of 95 °C for 1 min, one cycle of 60 °C for 1 min, and a stepwise increase of the temperature from 60 to 95 °C (at 0.5 °C per 5 s) to obtain melt curve data. Data were analysed using the Bio-Rad CFX Manager 3.0.
Transcriptome sequencing (RNA-seq)
Total RNA samples were further processed by Baseclear for RNA-seq (Leiden, the Netherlands). Depletion of ribosomal RNA was performed using the Ribo-Zero™ Kit for bacteria (Epicentre, Madison, WI, USA) followed by quality monitoring using the Agilent 2100 BioAnalyzer system. Library construction for whole transcriptome sequencing was done using the TruSeq Stranded mRNA Library Prep Kit (Illumina, USA). The barcoded cDNA libraries were analysed using BioAnalyzer and were subsequently pooled and sequenced. Single read 50 bp sequencing was performed on two lanes using the Illumina HiSeq 2500 platform.
Transcriptome analysis
The RNA-seq data was pre-processed for quality control. Ribosomal RNA was removed with SortMeRNA v2.0 (Kopylova et al. 2012) followed by all TruSeq adapters removal with Cutadapt v1.1.a (Martin 2011). Next, quality trimming was performed using Sickle v1.33 (Joshi and Fass 2011) with a score of 30 for threshold indicating a base calling confidence of 99.9%. Reads trimmed to a length < 50 bp were removed. Reads were subsequently mapped to the relevant bacterial genomes with Bowtie2 v0.6 (Langmead and Salzberg 2012) using default settings. HTSeq v0.6.1p1 was used to determine the read count for each protein coding region (Anders et al. 2015). All these steps were performed within a local Galaxy environment (Afgan et al. 2016). More detailed information about the data analysis can be found in Table S1. Non-mapping reads of the two samples with the lowest mapping rate (both of the A. muciniphila monocultures) were collapsed to unique reads with the fastx toolkit version 0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/). A blast search (with standard parameters, except for an e-value of 0.0001) of these unique reads was performed against the NCBI NT database (download 22.01.2014), against the human microbiome (download 08.05.2014), the NCBI bacterial draft genomes (download 23.01.2014), and the human genome (download 30.12.2013, release 08.08.2013, NCBI Homo sapiens annotation release 105). Taxonomy was estimated with a custom version of the LCA algorithm as implemented in MEGAN (Huson et al. 2011). Default parameters were used with the customization that only hits exceeding a bitscore of 50 and a length of more than 25 nucleotides were considered. 98% of the non-mapping reads were not classified, with Akkermansia accounting for 1.15% of the classified reads (Table S2). Differential gene expression was assessed using DESeq2 (Love et al. 2015). Raw RNA-seq sequence files can be accessed at the European Nucleotide Archive under accession numbers ERR1907419, ERR1907420, ERR1907423, and ERR1907424.
Carbohydrate-active enzymes (CAZymes) prediction
CAZymes were predicted with dbCAN version 3.0 (Yin et al. 2012), transmembrane domains with TMHMM version 2.0c (Krogh et al. 2001) and signal peptides with signalP 4.1 (Petersen et al. 2011).
Results
Metabolite profile of A. muciniphila monocultures and co-cultures with A. caccae
Co-culturing of A. muciniphila and A. caccae was performed followed by RT-qPCR, HPLC and metatranscriptomic analysis. The metabolites detected in the cultures were comparable with previous findings (Belzer et al. 2017). A. muciniphila grown as monoculture produced acetate, succinate and 1,2-propanediol as the major metabolites from pure mucin degradation (Fig. 1c). On day 8 the A.muc-A.cac co-cultures yielded around 2 mM butyrate and a low amount of propionate was detected (Fig. 1c). The mucin sugars (galactose, GalNAc, and GlcNAc) were below the detection limit of 0.5 mM.
The transcriptomes of A. muciniphila monocultures and co-cultures with A. caccae
Transcriptomic samples were analysed on day 8 of the interval-fed batch cultures, when the major metabolites were accumulated (Fig. 1c) and a stable bacterial composition was established (Belzer et al. 2017). On average 27 million reads were generated per sample, which is above the recommended sequence depth of 5–10 million reads for a single bacterial transcriptome (Haas et al. 2012). The detailed information about the data analysis can be found in Table S1. The RT-qPCR targeting 16S rRNA on total RNA showed an A. muciniphila to A. caccae ratio of 1:50 (Fig. 1d). On the other hand, the ratio of sequenced transcripts mapped to the genome of A. muciniphila versus A. caccae was 1:1 (Table S1).
Differential expression between A. muciniphila in monocultures and co-cultures with A. caccae
The genome of A. muciniphila possesses a total of 2176 predicted protein-coding sequences (CDSs) (van Passel et al. 2011) of which 2137 (98%) were found to be expressed in this study (Table S3). Differential expression analysis (DESeq2) was performed to compare the gene expression of A. muciniphila in mono- and co-culture conditions. The overall transcriptional response differentiated between the mono- and co-cultures (Pearson’s correlation = 0.88 ± 0.02) (Fig. 2).
Fig. 2.

Hierarchical clustering showing the Pearson’s correlation of the transcriptome samples as calculated from A. muciniphila CDS count performed with Python 2.7.12 and SciPy version 0.17.1 (van der Walt et al. 2011)
We used cut-offs of q < 0.05 and fold change > 2 for significantly regulated genes (Schurch et al. 2016). A total of 12% A. muciniphila genes were differentially regulated between mono- and co-cultures, with 148 upregulated genes and 132 downregulated genes (Table S3). Interestingly, two groups of contiguous genes were differentially regulated at high fold change (Fig. 3a). In the co-cultures, the upregulation of the annotated response regulator Amuc_1010 was coupled with the upregulation of a putative operon containing the genes Amuc_1011, Amuc_1012, Amuc_1013, and Amuc_1014 (Fig. 3b). Whereas, the putative operon consisting of Amuc_2041, Amuc_2042 and Amuc_2043 was downregulated in the co-cultures (Fig. 3c). Furthermore, several putative two-component systems were differentially expressed (Table 1).
Fig. 3.
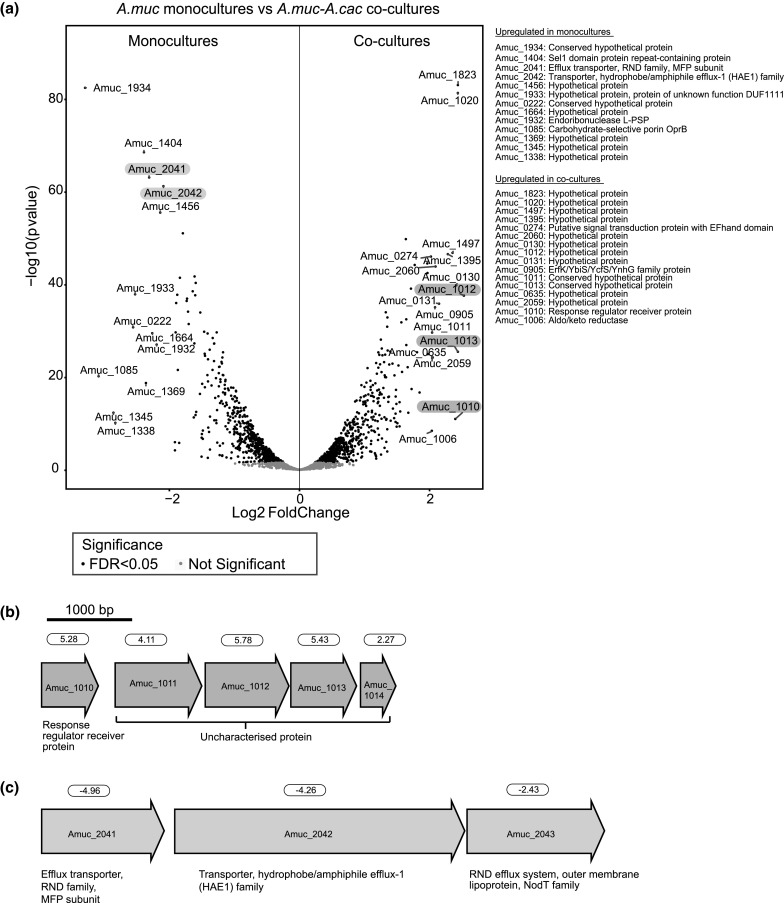
a Volcano plots showing p-values correlated to fold changes in gene expression of A. muciniphila observed in monocultures versus co-cultures with A. caccae. Positive fold changes indicate upregulation in co-cultures, and negative fold changes indicate upregulation in monocultures. Locus tags for genes with Log2 fold change > 2 (or fold change > 4) are labelled. b Response regulator and putative operon upregulated in the co-cultures. c Putative operon upregulated in the monocultures. Fold changes are listed above the respective genes
Table 1.
The differential expression of putative two-component systems in A. muciniphila
| Locus tag | A.muc-A.cac co-culture | Function | |
|---|---|---|---|
| q value | Fold change | ||
| Amuc_0311 | < 0.05 | 1.96 | Signal transduction histidine kinase, nitrogenspecific, NtrB |
| Amuc_0312 | < 0.05 | 2.19 | Two-component, sigma54 specific, transcriptional regulator, Fis family |
| Amuc_0827 | < 0.05 | 1.44 | Osmo-sensitive K+ channel signal transduction histidine kinase |
| Amuc_0828 | < 0.05 | 1.74 | Two-component transcriptional regulator, winged helix family |
| Amuc_1109 | < 0.05 | − 1.89 | Histidine kinase |
| Amuc_1110 | 0.53 | − 1.07 | Two-component transcriptional regulator, winged helix family |
| Amuc_1727 | 0.63 | 1.06 | Integral membrane sensor signal transduction histidine kinase |
| Amuc_1728 | 0.25 | 1.13 | Two-component transcriptional regulator, winged helix family |
| Amuc_1010 | < 0.05 | 5.28 | Response regulator receiver protein |
Negative values indicate upregulation in monocultures and positive values indicate upregulation in co-cultures
Gene ontology analysis (Table 2) showed overall increase expression of hydrolase activity, DNA recombination enzymes, and sulphuric ester hydrolase activity in the co-cultures whereas ribosome, structural constituent of ribosome and translation were downregulated. The list of A. muciniphila CAZymes is summarised in Table S4. The overall expression of glycosyl hydrolases was upregulated in the co-cultures. Signal peptides and transmembrane domains prediction showed putative extracellular activity for glycosyl hydrolases required for the degradation of mucin O-glycan chains including GH2, GH20, GH29, GH33, GH84, GH89, and GH98.
Table 2.
Gene ontology (GO) analysis of the differentially regulated A. muciniphila genes (q < 0.05) in co-cultures
| GO term | Total count in A.muc genome | Percentage upregulated | Percentage downregulated |
|---|---|---|---|
| A.muc-A.cac co-culture | |||
| GO:hydrolase activity, hydrolyzing O-glycosyl compounds | 30 | 0.60 | 0.03 |
| GO:DNA recombination | 17 | 0.53 | 0.06 |
| GO:sulphuric ester hydrolase activity | 12 | 0.50 | 0.17 |
| GO:transporter activity | 27 | 0.22 | 0.52 |
| GO:magnesium ion binding | 16 | 0.19 | 0.44 |
| GO:tRNA processing | 11 | 0.18 | 0.55 |
| GO:cytoplasm | 66 | 0.17 | 0.48 |
| GO:pyridoxal phosphate binding | 20 | 0.15 | 0.45 |
| GO:RNA binding | 37 | 0.14 | 0.46 |
| GO:GTP binding | 20 | 0.10 | 0.55 |
| GO:transferase activity | 21 | 0.10 | 0.43 |
| GO:tRNA aminoacylation for protein translation | 24 | 0.08 | 0.71 |
| GO:cellular amino acid metabolic process | 12 | 0.08 | 0.50 |
| GO:aminoacyl-tRNA ligase activity | 25 | 0.08 | 0.72 |
| GO:nucleotide binding | 40 | 0.08 | 0.58 |
| GO:intracellular | 42 | 0.07 | 0.79 |
| GO:NAD binding | 15 | 0.07 | 0.33 |
| GO:ribosome | 50 | 0.02 | 0.88 |
| GO:structural constituent of ribosome | 55 | 0.02 | 0.89 |
| GO:translation | 57 | 0.02 | 0.88 |
The list contains GO with total count in genome higher than 10 and absolute percentage difference higher than average value. GO with overall expression upregulated or downregulated in co-cultures are marked in bold and italic respectively
Genes expression in relation to the metabolites production
We examined the transcripts of the co-cultures to reconcile the metabolite findings. The transcripts for A. caccae showed median of relative abundance around 0.005% and maximum value of 2.07%. The list of A. caccae genes is displayed in Table S5. It is reported that A. caccae metabolises acetate to butyrate by employing the most prevalent butyrate production pathway via acetyl-coenzyme A (CoA) (Vital et al. 2014). The relative abundances of all transcripts involved in the metabolism pathways are summarised in Table 3. Our data indicated that the majority of enzymes involved in the acetyl-CoA pathway were expressed at a relative abundance higher than 0.1%, with over 2% of total transcripts accounted for butyrate production. In addition, A. caccae possesses genomic capacity to synthesis butyrate by using 4-aminobutyrate or succinate as the precursor. However, the expression of this pathway was low, with the relative abundance of transcripts lower than 0.01%, indicating that acetyl-CoA was the dominant pathway.
Table 3.
The relative abundance (%) of A. caccae transcripts for genes involved in butyrate synthesis pathway
| Enzyme | Locus tag | Dup1 | Dup2 |
|---|---|---|---|
| Interconversion of pyruvate to acetyl-CoA | |||
| Pyruvate dehyrogenase complex | ANACAC_01488 | < 0.00 | < 0.00 |
| ANACAC_01489 | < 0.00 | < 0.00 | |
| ANACAC_01490 | < 0.00 | < 0.00 | |
| ANACAC_01491 | < 0.00 | < 0.00 | |
| ANACAC_01492 | < 0.00 | < 0.00 | |
| Formate C-acetyltransferase | ANACAC_01621 | < 0.00 | < 0.00 |
| ANACAC_00664 | < 0.00 | < 0.00 | |
| Pyruvate synthase | ANACAC_00834 | 1.83 | 1.85 |
| Interconversion of pyruvate to lactate | |||
| l-Lactate dehydrogenase | ANACAC_01148 | 0.01 | 0.01 |
| ANACAC_03769 | 0.02 | 0.02 | |
| Acetyl-CoA pathway | |||
| Acetyl-CoA C-acetyltransferase | ANACAC_00256 | 0.34 | 0.37 |
| Acetoacetyl-CoA reductase | ANACAC_00254 | 0.35 | 0.39 |
| 3-Hydroxybutyryl-CoA dehydratase | ANACAC_03496 | 0.01 | 0.02 |
| ANACAC_00255 | 0.21 | 0.23 | |
| Butyryl-CoA dehydrogenase | ANACAC_00252 | 0.50 | 0.50 |
| ANACAC_00253 | 0.54 | 0.56 | |
| ANACAC_03492 | 0.00 | 0.00 | |
| Phosphate acetyltransferase | ANACAC_00344 | 0.13 | 0.15 |
| Acetate kinase | ANACAC_00343 | 0.17 | 0.18 |
| Butyryl-CoA:acetate CoA-transferase | ANACAC_01149 | 0.16 | 0.17 |
| 4-Aminobutyrate/succinate pathway | |||
| Hydroxybutyrate dehydrogenase | ANACAC_00166 | < 0.00 | < 0.00 |
| 4-Hydroxybutyrate coenzyme A transferase | ANACAC_00165 | < 0.00 | < 0.00 |
| 4-Hydroxybutanoyl-CoA dehydratase | ANACAC_00167 | < 0.00 | < 0.00 |
| ANACAC_02698 | < 0.00 | < 0.00 | |
Nutrients interdependency between A. muciniphila and A. caccae
The genomes of A. muciniphila and A. caccae were inspected for B vitamins and amino acids auxotrophy to investigate potential nutrient interdependency. A. muciniphila lacked the upstream genes required for vitamin B12 biosynthesis including CbiL, CobG, CbiGF, CobF, CbiECA and CobAT. Complementarily, A. caccae was predicted to possess a complete vitamin B12 biosynthesis pathway (Table 4). However, no vitamin B12 transporter was found in the A. caccae genome. We found indications for aspartate auxotrophy of A. caccae (Table S6) however the bacterium was reported to grow in minimal defined media supplemented with glucose without additional nitrogen source (Belzer et al. 2017). Furthermore, A. caccae lacks the genes to synthesise the cofactor lipoate required for dehydrolipoate dehydrogenase, EC 1.8.1.4. The different enzyme complexes containing this enzyme are involved in citrate cycle, glycine, serine, and threonine metabolism, and valine, leucine, and isoleucine degradation. Nevertheless, A. caccae could acquire lipoate via salvage pathway and we observed the upregulation of lipoate biosynthesis by A. muciniphila in co-cultures.
Table 4.
Genomic prediction of B vitamins biosynthesis (presence = 1 and absence = 0) based on the combination of essential functional roles by Magnusdottir et al. (2015)
| B7 | B12 | B9 | B3 | B5 | B6 | B2 | B1 | |
|---|---|---|---|---|---|---|---|---|
| Biotin | Cobalamin | Folate | Niacin | Pantothenate | Pyridoxin | Riboflavin | Thiamin | |
| Akkermansia muciniphila MucT | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| Anaerostipes caccae L1-92 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
Discussion
In this study, we demonstrated the use of metatranscriptomics as an explorative approach to decipher bacterial interaction in the mucosal environment. Two representative mucosa-associated species, namely A. muciniphila and A. caccae, were used to show the ecological dependency between a mucin degrader and a butyrate producer. Importantly, this study revealed changes in the expression of genes involved in host-glycan catabolism and trophic interactions between the gut commensals. This interplay leads to the formation of butyrate in the mucosal layer that is proposed to be beneficial to the host (Koh et al. 2016; Louis and Flint 2017).
In the presence of A. caccae, A. muciniphila upregulated mucin-degrading genes involved in hydrolase and sulphuric ester hydrolase activity. The majority of these mucin-degrading enzymes were predicted to function in the extracellular compartment (Ottman et al. 2016), which could lead to the degradation of oligosaccharide chains consisting of GalNAc, GlcNAc, mannose, galactose, fucose and sialic acid (Moran et al. 2011). Previous work demonstrated that A. caccae as well as Eubacterium hallii and Faecalibacterium prausnitzii could utilise the mucin-derived sugars including galactose, mannose and GlcNAc for growth (Belzer et al. 2017; Lopez-Siles et al. 2012). The fermentation of these monosaccharides results in butyrate production. Since both A. muciniphila and the butyrate-producer rely on the uptake of mucin-derived sugars for growth in our model, a higher extracellular concentration of A. muciniphila-derived mucolytic enzymes could contribute to substrate availability in the community. Concurrently, A. muciniphila showed downregulation of ribosomal genes in the co-cultures, which implied a lower growth rate of A. muciniphila. The qPCR results of genomic 16S rRNA gene ratio from a previous publication on extracted DNA showed a A.muciniphila to A. caccae ratio of 100:1 (Belzer et al. 2017). In this study, the ratio of 16SrRNA in total RNA samples quantified by RT-qPCR showed a A. muciniphila to A. caccae ratio of 1:50, whereas, the sequenced transcripts ratio was 1:1. The discrepancy could be the result of differential expression between ribosomal and messenger RNA. Note that total RNA could contain 95–99% of ribosomal RNA (Zoetendal et al. 2006) and that the number of ribosomes per cell correlates with the growth rate (Fegatella et al. 1998). In addition, A. muciniphila and A. caccae contain 3 and 12 copies of the rRNA operon, respectively. Taken together, these results indicate that A. muciniphila dominated in terms of cells number but A. caccae showed proportionally higher growth rate and transcriptional activity.
The co-culturing of two representative mucosa-associated bacteria has demonstrated the major pathways for intestinal SCFAs biosynthesis. The overview of this mucin-directed trophic interaction is shown in Fig. 4. A. caccae cross-fed on a part of the mucin sugars liberated by A. muciniphila for central metabolism. In addition, A. caccae can incorporate A. muciniphila-derived acetate for butyrate production via butyryl-CoA:acetate CoA-transferase enzyme(Duncan et al. 2004; Louis and Flint 2009; Louis and Flint 2017). Moreover, A. muciniphila could benefit from the corrinoids released by A. caccae (Degnan et al. 2014). Pseudo-vitamin B12 from E. hallii could activate the propionate production by A. muciniphila via the succinate pathway (Belzer et al. 2017). A low level of propionate was detected after day 8 in A.muc-A.cac co-cultures (Belzer et al. 2017). Propionate is likely produced by A. muciniphila because A. caccae is not known to produce propionate and it does not possess the genes involved in the known propionate biosynthesis pathways i.e. the succinate, acrylate, and propanediol pathways (Louis and Flint 2017). Nevertheless, A. caccae is predicted to synthesise vitamin B12 but lacked a vitamin B12 transporter. Upon cell lysis, the release of cellular vitamin B12 by A. caccae could facilitate methylmalonyl-CoA mutase enzymes (Amuc_1983 and Amuc_1984) of A. muciniphila to produce propionate (Degnan et al. 2014). The upregulation of cobalamin-dependent methylmalonyl-CoA mutase genes in monocultures indicated an attempt by the organism to activate the propionate production pathway in the absence of the essential cofactor (Fig. S1), as the conversion of methylmalonyl-CoA to propionyl-CoA is thermodynamically favourable (Dimroth and Schink 1998). The exergonic decarboxylation of methylmalonyl-CoA could be coupled to sodium ion export to extracellular space for the establishment of a proton gradient via a sodium-proton antiporter to generate ATP (Ottman et al. 2017a).
Fig. 4.
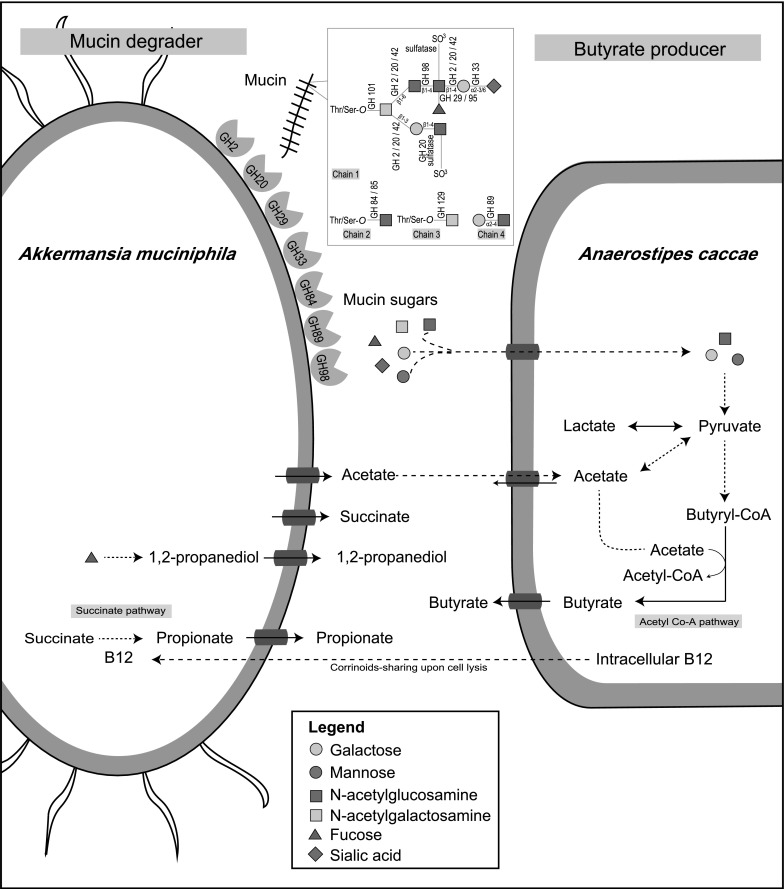
Schematic representation of mucin-driven trophic interaction between A. muciniphila and A. caccae. A. muciniphila degrades oligosaccharides chain of mucin by extracellular glycosyl hydrolases. The structure for O-linked glycan chains and CAZymes action sites are adapted from Tailford et al. (2015). Chain 1 is a hypothetical mucin glycan chain, chain 2 is O-GlcNAc often found on other glycoproteins, chain 3 (Tn antigen) and chain 4 are found in gastro-duodenal mucin. In addition, mannose could be released from degradation of N-linked glycan chains. A. caccae utilises some of the mucin-derived sugars (galactose, mannose and GlcNAc) and acetate released by A. muciniphila for growth and concomitant butyrate production
Interestingly, two putative operons and several two-component systems were differentially regulated, indicating the mode of transcriptional regulation by A. muciniphila in response to A. caccae. A previous study has demonstrated that the presence of one organism is often associated with transcriptional changes in the other (Plichta et al. 2016). In the co-culture with A. caccae, A. muciniphila downregulated a putative operon consisting of Amuc_2041 (efflux transporter, RND family, MFP subunit), Amuc_2042 (transporter, hydrophobe/amphiphile efflux-1 (HAE1) family) and Amuc_2043 (RND efflux system, outer membrane lipoprotein, NodT family). The membrane fusion protein (MFP) is described as a component of drug resistance, nodulation, and the cell division (RND) family involved in the transportation of drug molecules (Anes et al. 2015). HAE1 is involved in toxin production and resistance processes (Anes et al. 2015). The outer membrane lipoproteins from the NodT family are predicted to primarily export small molecules rather than proteins. This efflux system was reported to play a role in multidrug resistance of Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa (Nikaido and Takatsuka 2009). A similar resistance mechanism could be employed by the Gram-negative A. muciniphila, and this study suggested the down-tuning of the efflux pump expression in the presence of a community member.
The annotated response regulator Amuc_1010 and the adjacent predicted operon consisting of Amuc_1011, Amuc_1012, Amuc_1013, and Amuc_1014, were upregulated in the co-cultures. Amuc_1010 is likely not a two-component system as it encoded only for the LytTR DNA-binding domain without the CheY-like receiver domain. Amuc_1010 could be autoregulatory as cis-acting regulatory elements were predicted at its upstream region using MEME (Bailey et al. 2009) (data not shown). Amuc_1011, Amuc_1012, Amuc_1013, and Amuc_1014 were annotated as uncharacterised proteins, and Amuc_1011 was predicted as an outer membrane protein (Ottman et al. 2016). Further research is needed to investigate this interesting gene cluster with unidirectional arrangement and a short intercistronic region that could likely be co-transcribed. The upregulation of the outer membrane protein could be associated with host colonization, persistence and immunomodulation (Galdiero et al. 2012). A recent study showed that an immune-stimulatory outer membrane protein of A. muciniphila (Amuc_1100) (Ottman et al. 2017b) is able to ameliorate the metabolic symptoms of obese and diabetic mice (Plovier et al. 2017). However, Amuc_1100 was not found to be differentially regulated in this study.
In addition, A. muciniphila upregulated several two-component systems in the co-cultures. Two-component systems consist of a membrane bound sensor histidine kinase and a cytoplasmic response regulator, which are often encoded by adjacent genes, enable bacteria to response to changing environment by altering gene expression (Monedero et al. 2017). However, the roles of two-component systems in A. muciniphila grown in the co-cultures were not yet identified. Studies showed that they could be involved in the regulation of physiological processes in commensal bacteria, such as stress responses, regulation of metabolism, and resistance to antimicrobial peptides (Monedero et al. 2017). The gastrointestinal pathogen, enterohemorrhagic E. coli (EHEC), was reported to encode the two-component system FusKR. This system provides a growth advantage and modulates the expression of virulence genes upon sensing of fucose liberated by Bacteroides thetaiotaomicron during growth in media containing mucin (Pacheco et al. 2012). The metabolism of mucin-derived fucose by A. muciniphila yielded 1,2-propanediol (Ottman et al. 2017a). As such, fucose metabolism by A. muciniphila could confer colonization resistance against the fucose-dependent enteric pathogens (Pickard and Chervonsky 2015).
In conclusion, we demonstrated the use of metatranscriptomics to provide in-depth mechanistic understanding of bacterial interaction. The trophic interaction between mucosal keystone species A. muciniphila and A. caccae could result in beneficial butyrate production at close proximity to the host epithelium. We revealed the expressional changes of A. muciniphila in response to A. caccae and demonstrated the provider role of A. muciniphila by upregulating the mucolytic activity to sustain the community at the mucosa niche.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Thi Phuong Nam Bui, Kees C.H. van der Ark, Maria Suarez-Diez, Teunke van Rossum and Fons Stams for their constructive comments.Furthermore, we would like to thank Bart Nijsse for technical support. This work was partly supported by a special grant from Nutricia Research, the SIAM Gravitation Grant 024.002.002 of the Netherlands Organization for Scientific Research, and the IP/OP program Systems Biology (project KB-17-003.02-023).
Author contributions
LWC, WMdV, JK and CB contributed to study conception. LWC, SA and CB contributed to experimental design. LWC performed the experiments. LWC and BH analysed data. LWC, BH, SA, PS, WMdV, JK and CB interpreted data and revised the manuscript. LWC and BH wrote the manuscript.
Compliance with ethical standards
Conflict of interest
JK is the employee of Nutricia Research. LWC and CB are financially supported by Nutricia Research. There was no involvement of the company in the content of this work.
Footnotes
Electronic supplementary material
The online version of this article (10.1007/s10482-018-1040-x) contains supplementary material, which is available to authorized users.
References
- Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anes J, McCusker MP, Fanning S, Martins M. The ins and outs of RND efflux pumps in E coli. Front Microbiol. 2015;6:587. doi: 10.3389/fmicb.2015.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, Flint HJ. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbol. 2006;72:3593–3599. doi: 10.1128/AEM.72.5.3593-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer C, de Vos WM. Microbes inside–from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer C, Chia LW, Aalvink S, Chamlagain B, Piironen V, Knol J, de Vos WM. Microbial metabolic networks at the mucus layer lead to diet-independent butyrate and vitamin B12 production by intestinal symbionts. Mbio. 2017 doi: 10.1128/mBio.00770-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, et al. The Metacyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques. 1993;15(532–534):536–537. [PubMed] [Google Scholar]
- Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol. 2007;73:7767–7770. doi: 10.1128/AEM.01477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos WM. Microbe Profile: akkermansia muciniphila: a conserved intestinal symbiont that acts as the gatekeeper of our mucosa. Microbiology. 2017 doi: 10.1099/mic.0.000444. [DOI] [PubMed] [Google Scholar]
- De Vuyst L, Leroy F. Cross-feeding between bifidobacteria and butyrate-producing colon bacteria explains bifdobacterial competitiveness, butyrate production, and gas production. Int J Food Microbiol. 2011;149:73–80. doi: 10.1016/j.ijfoodmicro.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Degnan PH, Taga ME, Goodman AL. Vitamin B12 as a modulator of gut microbial ecology. Cell Metab. 2014;20:769–778. doi: 10.1016/j.cmet.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microb. 2008;74:1646–1648. doi: 10.1128/AEM.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microbial Pathogenesis. 2016 doi: 10.1016/j.micpath.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Dimroth P, Schink B. Energy conservation in the decarboxylation of dicarboxylic acids by fermenting bacteria. Arch Microbiol. 1998;170:69–77. doi: 10.1007/s002030050616. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. 2004;70:5810–5817. doi: 10.1128/AEM.70.10.5810-5817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. P Natl Acad Sci. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Vlachou A, Verbrugghe K, De Vuyst L. Cross-feeding between Bifidobacterium longum BB536 and acetate-converting, butyrate-producing colon bacteria during growth on oligofructose. Appl Environ Microb. 2006;72:7835–7841. doi: 10.1128/AEM.01296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegatella F, Lim J, Kjelleberg S, Cavicchioli R. Implications of rRNA operon copy number and ribosome content in the marine oligotrophic ultramicrobacterium Sphingomonas sp. strain RB2256. Appl Environ Microbiol. 1998;64:4433–4438. doi: 10.1128/aem.64.11.4433-4438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdiero S, Falanga A, Cantisani M, Tarallo R, Della Pepa ME, D’Oriano V, Galdiero M. Microbe-host interactions: structure and role of Gram-negative bacterial porins. Curr Protein Peptide Sci. 2012;13:843–854. doi: 10.2174/138920312804871120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grander C, et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2017 doi: 10.1136/gutjnl-2016-313432. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? Bmc Genomics. 2012 doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Fass J (2011) Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]
- Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Siles M, Khan TM, Duncan SH, Harmsen HJM, Garcia-Gil LJ, Flint HJ. Cultured representatives of two major phylogroups of human colonic faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl Environ Microb. 2012;78:420–428. doi: 10.1128/AEM.06858-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Siles M, et al. Mucosa-associated faecalibacterium prausnitzii and escherichia coli co-abundance can distinguish irritable bowel syndrome and inflammatory bowel disease phenotypes. Int J Med Microbiol. 2014;304:464–475. doi: 10.1016/j.ijmm.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- Love M, Anders S, Kim V, Huber W (2015) RNA-Seq workflow: gene-level exploratory analysis and differential expression [version 1; referees: 2 approved] vol 4. [DOI] [PMC free article] [PubMed]
- Magnusdottir S, Ravcheev D, de Crecy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. 2015;6:148. doi: 10.3389/fgene.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A, Southwick AM, Earle KA, Sonnenburg JL. A refined palate: bacterial consumption of host glycans in the gut. Glycobiology. 2013;23:1038–1046. doi: 10.1093/glycob/cwt040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011 [Google Scholar]
- Miller RS, Hoskins LC. Mucin degradation in human colon ecosystems. fecal population densities of mucin-degrading bacteria estimated by a “most probable number” method. Gastroenterology. 1981;81:759–765. [PubMed] [Google Scholar]
- Monedero V, Revilla-Guarinos A, Zúñiga M. Physiological role of two-component signal transduction systems in food-associated lactic acid bacteria. Advances Appl Microbiol. 2017;99:1–51. doi: 10.1016/bs.aambs.2016.12.002. [DOI] [PubMed] [Google Scholar]
- Moran AP, Gupta A, Joshi L. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut. 2011;60:1412–1425. doi: 10.1136/gut.2010.212704. [DOI] [PubMed] [Google Scholar]
- Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5:627–638. doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2009;1794:769–781. doi: 10.1016/j.bbapap.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman N, et al. Characterization of outer membrane proteome of akkermansia muciniphila reveals sets of novel proteins exposed to the human intestine. Front Microbiol. 2016;7:1157. doi: 10.3389/fmicb.2016.01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman N, et al. Genome-scale model and omics analysis of metabolic capacities of Akkermansia muciniphila reveal a preferential mucin-degrading lifestyle. Appl Environ Microbiol. 2017 doi: 10.1128/AEM.01014-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman N, et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE. 2017;12:e0173004. doi: 10.1371/journal.pone.0173004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouwehand AC, Derrien M, de Vos W, Tiihonen K, Rautonen N. Prebiotics and other microbial substrates for gut functionality. Curr Opin Biotechnol. 2005;16:212–217. doi: 10.1016/j.copbio.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Ouwerkerk JP, de Vos WM, Belzer C. Glycobiome: bacteria and mucus at the epithelial interface. Best Pract Res Clin Gastroenterol. 2013;27:25–38. doi: 10.1016/j.bpg.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Ouwerkerk JP, van der Ark KC, Davids M, Claassens NJ, Robert Finestra T, de Vos WM, Belzer C. Adaptation of Akkermansia muciniphila to the oxic-anoxic interface of the mucus layer. Appl Environ Microbiol. 2016 doi: 10.1128/AEM.01641-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492:113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Pickard JM, Chervonsky AV. Intestinal Fucose as a Mediator of Host-Microbe Symbiosis. J Immunol. 2015;194:5588–5593. doi: 10.4049/jimmunol.1500395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plichta DR, et al. Transcriptional interactions suggest niche segregation among microorganisms in the human gut. Nat Microbiol. 2016 doi: 10.1038/nmicrobiol.2016.152. [DOI] [PubMed] [Google Scholar]
- Plovier H, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23:107–113. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- Plugge CM. Anoxic media design, preparation, and considerations. Methods Enzymol. 2005;397:3–16. doi: 10.1016/S0076-6879(05)97001-8. [DOI] [PubMed] [Google Scholar]
- Png CW, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- Rios-Covian D, Gueimonde M, Duncan SH, Flint HJ, de los Reyes-Gavilan CG. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol Lett. 2015 doi: 10.1093/femsle/fnv176. [DOI] [PubMed] [Google Scholar]
- Riviere A, Gagnon M, Weckx S, Roy D, De Vuyst L. Mutual Cross-Feeding Interactions between Bifidobacterium longum subsp longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl Environ Microb. 2015;81:7767–7781. doi: 10.1128/AEM.02089-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter J, Foster KR. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012;10:e1001424. doi: 10.1371/journal.pbio.1001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch NJ, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA. 2016;22:839. doi: 10.1261/rna.053959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Ruscheweyh HJ, Bunesova V, Pham VT, Beerenwinkel N, Lacroix C. Trophic interactions of infant bifidobacteria and eubacterium hallii during l-fucose and fucosyllactose degradation. Front Microbiol. 2017;8:95. doi: 10.3389/fmicb.2017.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiertz A, et al. Anaerostipes caccae gen. nov., sp. nov., a new saccharolytic, acetate-utilising, butyrate-producing bacterium from human faeces. Syst Appl Microbiol. 2002;25:46–51. doi: 10.1078/0723-2020-00096. [DOI] [PubMed] [Google Scholar]
- Shetty SA, Hugenholtz F, Lahti L, Smidt H, Vos WMd (2016) Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies FEMS Microbiology Reviews [DOI] [PMC free article] [PubMed]
- Swidsinski A, et al. Acute appendicitis is characterised by local invasion with Fusobacterium nucleatum/necrophorum. Gut. 2011;60:34–40. doi: 10.1136/gut.2009.191320. [DOI] [PubMed] [Google Scholar]
- Tailford LE, Crost EH, Kavanaugh D, Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet. 2015;6:81. doi: 10.3389/fgene.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Abbeele P, et al. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013;7:949–961. doi: 10.1038/ismej.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Colbert SC, Varoquaux G. The numpy array: a structure for efficient numerical computation. Comput Sci Eng. 2011;13:22–30. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- van Passel MWJ, et al. The genome of akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS ONE. 2011 doi: 10.1371/journal.pone.0016876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga P, et al. Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. Proc Natl Acad Sci. 2010;107:18132–18137. doi: 10.1073/pnas.1011737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio. 2014 doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucl Acids Res. 2012;40:W445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoetendal EG, Booijink CC, Klaassens ES, Heilig HG, Kleerebezem M, Smidt H, de Vos WM. Isolation of RNA from bacterial samples of the human gastrointestinal tract. Nat Protoc. 2006;1:954–959. doi: 10.1038/nprot.2006.143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.