

Abstract
Crabtree's catalyst was encapsulated inside the pores of the sulfonated MIL‐101(Cr) metal–organic framework (MOF) by cation exchange. This hybrid catalyst is active for the heterogeneous hydrogenation of non‐functionalized alkenes either in solution or in the gas phase. Moreover, encapsulation inside a well‐defined hydrophilic microenvironment enhances catalyst stability and selectivity to hydrogenation over isomerization for substrates bearing ligating functionalities. Accordingly, the encapsulated catalyst significantly outperforms its homogeneous counterpart in the hydrogenation of olefinic alcohols in terms of overall conversion and selectivity, with the chemical microenvironment of the MOF host favouring one out of two competing reaction pathways.
Keywords: allylic alcohols, Crabtree's catalyst, encapsulation, hydrogenation, metal–organic frameworks
Metal–organic frameworks (MOFs)1 are crystalline and permanently porous materials that have emerged as promising hosts for the immobilization of organometallic catalysts,2 since they allow control of the steric and chemical microenvironment around the encapsulated catalytically active species. This in turn could promote catalytic activity and selectivity through extended coordination sphere interactions. These concepts lie behind the exceptional reactivity and selectivity of metalloenzymes,3 however their transfer to the design and synthesis of artificial catalysts is challenging.4 Several examples of MOF‐supported catalysts showing exceptional overall catalytic activity have been reported.5, 6 Enhancement of selectivity between products of a single reaction pathway by control of the steric7 or the chemical8 microenvironment has also been demonstrated.
Crabtree's catalyst is one of the best commercially available homogeneous catalysts for hydrogenation of alkenes.9 However, it is deactivated in solution under hydrogenation conditions, forming catalytically inactive polymetallic hydride clusters.10 This self‐association reaction can be attenuated via modification of the coordination sphere of Ir11 or employment of larger weakly coordinating anions.12 Substrates bearing ligating functionalities such as olefinic alcohols show a more complicated behavior with Crabtree's catalyst since isomerization13 can also take place in parallel with hydrogenation.14
Here we use the Na+ salt of sulfonated MIL‐101(Cr) MOF (1‐SO3Na) to provide the anionic framework host for encapsulation of the cationic component of Crabtree's catalyst [Ir(cod)(PCy3)(py)][PF6] (2‐PF6) by cation exchange,15 forming 2@1‐SO3Na (Scheme 1). Encapsulation of cation 2 inside a well‐defined, anionic and hydrophilic microenvironment forms an efficient heterogeneous catalyst for the hydrogenation of non‐functionalized alkenes in solution, enables hydrogenation in the gas phase, and most importantly enhances the catalyst's activity and selectivity for the hydrogenation of olefinic alcohols by suppressing the competing isomerization reaction. The MOF chemical microenvironment directs substrates along one of two distinct reaction pathways.
Scheme 1.
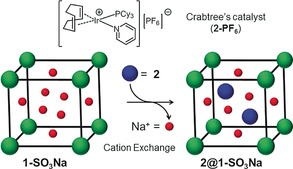
Encapsulation of the cationic component of Crabtree's catalyst (2, blue spheres) in sulfonated MIL‐101(Cr) (1‐SO3Na, cube) by exchange of the charge‐balancing Na+ cations (red spheres).
The sulfonated analogue of MIL‐101(Cr) (1‐SO3H)16 is a robust, readily synthesized anionic MOF. It is isostructural with pristine MIL‐101(Cr)17 with two charge‐balancing cations per formula unit, [HxNa2−x][Cr3(μ3‐O)(BDC‐SO3)3] (x=1.8±0.1, Figure S1, H2BDC‐SO3Na=2‐sulfoterephthalic acid sodium salt). Each cubic unit cell (a=87.63(3) Å) contains 8 bigger and 16 smaller mesopores, large enough to accommodate 2 (Figures S2 and S3). The cations within 1‐SO3H can be partially exchanged with Ag+[18] or [Rh(cod)(dppe)]+ [dppe=1,2‐bis(diphenylphosphino)ethane].19 To increase the number of exchangeable Na+ cations, 1‐SO3H was treated with AcONa/AcOH buffer solution (pH 4.7), forming [HyNa2−y][Cr3(μ3‐O)(BDC‐SO3)3] (1‐SO3Na, y=0.2±0.1, Table S1).
Compound 1‐SO3Na remains crystalline and mesoporous (Figures 1 a, b) with only a small change in the cubic unit cell parameter (a=87.99(4) Å) and a slight increase in the measured porosity (BET surface area=2005 m2 g−1, V P=0.91 cm3 g−1) and the pore size distribution, compared to 1‐SO3H (Figure S9).
Figure 1.
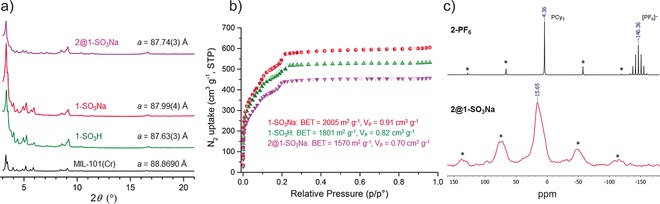
a) Comparison of PXRD patterns and unit cell parameter (Fd m space group) for 2@1‐SO3Na (magenta), 1‐SO3Na (red), 1‐SO3H (green) and MIL‐101(Cr) (calculated, black).17 Le Bail fits are included in the supporting information. b) N2 uptake of the desolvated materials at 77 K (BET=surface area, VP=pore volume). c) 31P{1H} MAS NMR spectrum of 2‐PF6 (black) and 2@1‐SO3Na (red). Spinning side bands are marked with an asterisk.
After establishing an appropriate cation exchange protocol using [Cp*2Co]+ as a cationic probe (Table S2 and Figures S6, S9–S12), as we have shown previously,15b 2‐PF6 was used as a cationic guest precursor. Since water can poison the catalytically active species,12 cation exchange was carried out using desolvated 1‐SO3Na as the anionic host in dry and degassed acetone, producing 2@1‐SO3Na. Crystallinity and particle morphology were retained after cation exchange with only a minor change in the cubic unit cell parameter (a=87.74(3) Å, Figure 1 a, see Le Bail fit in Figure S7 and SEM images in Figures S11 and S12), whereas BET surface area (1570 m2 g−1) and pore volume (0.70 cm3 g−1) were reduced, compared to 1‐SO3Na (Figure 1 b).
ICP‐OES after digestion of 2@1‐SO3Na gave an Ir content of 2.28 wt %, indicating that 7 % of the Na+ cations have been exchanged with 2 (Table S3), which is close to the upper limit of about 9 % calculated by accounting for the guest‐accessible space of the host MOF and the size of the cationic guest (Figures S1–S3). ICP‐OES also showed an equimolar Ir/P ratio, and only one broad peak was observed (δ P=15.65, fwhm≈15 ppm) in the 31P{1H} MAS NMR spectrum of 2@1‐SO3Na, assigned to the PCy3 ligand (Figure 1 c). Signals arising from the [PF6]− anion were not observed either in the 31P{1H} MAS or the 19F{1H} solution NMR spectra of 2@1‐SO3Na after digestion, in contrast with the respective spectra of 2‐PF6 (Figures 1 c and S13). The down‐field chemical shift and peak broadening observed for the signal due to the PCy3 ligand in the 31P{1H} MAS NMR spectrum of 2@1‐SO3Na, compared to 2‐PF6, likely originate from the different anionic environment surrounding 2.20
The 1H solution NMR spectrum of 2@1‐SO3Na after digestion showed three low intensity peaks at δ=8.22, 7.58 and 7.16 ppm, assigned to pyridine (Figure S14). Treatment of 2@1‐SO3Na with D2 gas resulted in deuteration of the cod ligand and formation of [D4]‐cyclooctane, as detected by 2H MAS NMR spectroscopy (Figure S15). These analytical and spectroscopic data are consistent with cation 2 being encapsulated intact inside the mesopores of 1‐SO3Na by a simple cation exchange process.
To explore the possible interaction of the sulfonate groups decorating the pore walls of 1‐SO3Na with the Ir center of 2 after encapsulation, the tosylate anion [OTs]− was selected to model the BDC‐SO3 linker. Two new complexes were synthesized, [Ir(cod)(PCy3)(py)][OTs] (2‐OTs) and [Ir(cod)(PCy3)(OTs)] (3), in which OTs− acts as a counter anion or as a ligand to Ir, respectively (Figures 2 a,b, Figures S16, S17, Table S4). 31P{1H} and 1H EXSY NMR spectroscopy in CD2Cl2 (Figures S18–S20) revealed that a dynamic reversible ligand exchange takes place between complexes 2‐OTs and 3, with OTs replacing pyridine in the coordination sphere of Ir (Figure 2 c). This suggests that the sulfonate groups in 2@1‐SO3Na may also play a non‐spectator role, with potential implications in catalysis, as discussed next.
Figure 2.
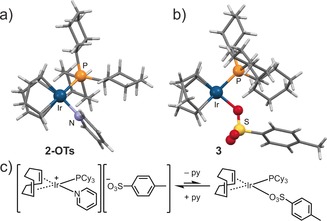
a) Single crystal structure of 2‐OTs (OTs− counter anion is not shown for clarity). b) Single crystal structure of 3. c) Reversible ligand exchange between 2‐OTs and 3 in CD2Cl2.
The catalytic performance of 2@1‐SO3Na was benchmarked against 2‐PF6 in the hydrogenation of non‐functionalized alkenes in CH2Cl2 under mild conditions (Table 1). Control experiments verified that 1‐SO3Na does not catalyze the hydrogenation of oct‐1‐ene (4). Introduction of 2@1‐SO3Na as the catalyst afforded complete hydrogenation of 4 to n‐octane, at loadings as low as 50 ppm (entries 1–3). When the loading was reduced to 10 ppm (entry 4), conversion of 4 to n‐octane reached 83 % (TON=8.3×104). Homogeneous catalyst 2‐PF6 under identical conditions produced comparable results, demonstrating that encapsulation is not detrimental to catalytic activity.
Table 1.
Hydrogenation of non‐functionalized alkenes with heterogeneous 2@1‐SO3Na and homogeneous 2‐PF6 catalysts.[a]
| Entry | Substrate | Loading | t | 2@1‐SO3Na | 2‐PF6 | |||
|---|---|---|---|---|---|---|---|---|
| [ppm] | [h] | Conv[b] | TON | Conv[b] | TON | |||
| [%] | [%] | |||||||
| 1 |
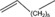
|
4 | 1000[c] | 3 | >99 | >990 | 100 | 1000 |
| 2 | 100[d] | 20 | 100 | 10 000 | 100 | 10 000 | ||
| 3 | 50[e] | 24 | 100 | 20 000 | – | – | ||
| 4 | 10[f] | 24 | 83 | 83 000 | 94 | 94 000 | ||
| 5 |
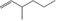
|
5 | 1000[c] | 3 | >99 | >990 | 100 | 1000 |
| 6 | 20 | 100 | 1000 | – | – | |||
| 7 |
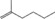
|
6 | 1000[c] | 3 | 10 | 100 | 12 | 120 |
| 8 | 20 | 26 | 260 | 37 | 370 | |||
| 9 |
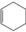
|
7 | 1000[c] | 3 | 69 | 690 | 100 | 1000 |
| 10 | 20 | 81 | 810 | – | – | |||
[a] CH2Cl2 solvent, T=20 °C. [b] Conversion (%) based on GC. [c] [alkene]=0.5 m, V=1 mL, 8 mmol of H2. [d] [alkene]=1.0 m, V=4 mL, 16 mmol of H2. [e] [alkene]=1.0 m, V=10 mL, 48 mmol of H2. [f] [alkene]=1.5 m, V=12 mL, 48 mmol of H2.
The branched, but unhindered, aliphatic alkene, 3‐methylhex‐1‐ene (5) was also completely hydrogenated using 2@1‐SO3Na at 1000 ppm loading (entries 5 and 6). The hindered aliphatic alkene, 2‐methylhex‐1‐ene (6) was only partially hydrogenated with either catalyst after 20 h (entries 7 and 8). Conversion did not increase any further after 72 h in either system, reflecting catalyst deactivation. When cyclohexene (7) was employed as a substrate, conversion reached 69 % in 3 h with 2@1‐SO3Na as the catalyst but increased only to 81 % after 20 h. On the contrary, 100 % conversion was observed with 2‐PF6 in 3 h (entries 9 and 10).
The different response observed for this bulkier substrate is consistent with hydrogenation taking place within the pores and not on the surface of 2@1‐SO3Na. The heterogeneity of the reaction was further established by carrying out a leaching test (Figure S21). Recycling of 2@1‐SO3Na was also possible with a small decrease in activity (82 % conversion) during the third cycle (Figure S22).
Compound 2@1‐SO3Na is a versatile catalyst which can also be employed in a gas/solid reaction,21 as demonstrated by the complete hydrogenation of but‐1‐ene over 2@1‐SO3Na in 2.5 h (4000 μmol of but‐1‐ene hydrogenated per 1 mg of Ir). Although finely ground solid 2‐PF6 was also active, dispersion of 2 in the porous anionic solid‐state support increases the number of accessible catalytic sites in 2@1‐SO3Na, resulting in a sixfold increase in activity compared to the non‐porous solid 2‐PF6 (Figure 3). Recycling of 2@1‐SO3Na was also successful upon exposure to fresh but‐1‐ene (Figure S23).
Figure 3.
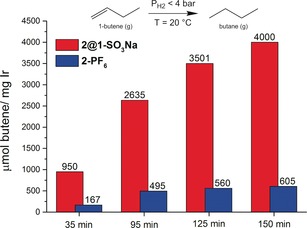
Conversion of but‐1‐ene into n‐butane in a gas/solid hydrogenation reaction over 2@1‐SO3Na (red) and 2‐PF6 (blue). Conditions: T=20 °C, P <4 bar, 0.5 mg of solid catalyst used.
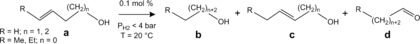
The mesopores of 2@1‐SO3Na are hydrophilic due to the presence of H‐bond accepting sulfonate groups as well as Lewis acidic CrIII sites and Na+ cations. Therefore, the reactivity of Crabtree's catalyst with substrates bearing functional groups that can interact with such an environment could significantly change due to encapsulation. We chose to explore this by using olefinic alcohols as substrates, whose fundamental characteristic is the competition between hydrogenation and isomerization upon turnover.22 Hydrogenation of a series of olefinic alcohols was carried out under a ≈20‐fold excess of H2 (Table 2).
Table 2.
Substrate conversion[a] and product selectivity[a,b] for hydrogenation of olefinic alcohols with heterogeneous 2@1‐SO3Na and homogeneous 2‐PF6 catalysts.[c]
| Entry | Substrate | t | 2@1‐SO3Na | 2‐PF6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [h] | Conv [%] |
b | c | d | Conv [%] |
b | c | d | |||
| 1 |

|
8 a | 3 | 34 | 100 | n.d.[d] | n.d. | 100 | 100 | n.d. | n.d. |
| 2 | 24 | 100 | 100 | n.d. | n.d. | – | – | – | – | ||
| 3 |
|
9 a | 3 | 22 | 100 | n.d. | n.d. | 100 | 100 | n.d. | n.d. |
| 4 | 24 | 100 | 100 | n.d. | n.d. | – | – | – | – | ||
| 5 |
|
10 a | 3 | 10 | 100 | n.d. | n.d. | 100 | 100 | n.d. | n.d. |
| 6 | 24 | 100 | 100 | n.d. | n.d. | – | – | – | – | ||
| 7 |
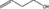
|
11 a | 3 | 33 | 95 | 5 | n.d. | 56 | 85 | 13 | 2 |
| 8 | 24 | 100 | 100 | n.d. | n.d. | 57 | 86 | 12 | 2 | ||
| 9 |
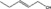
|
12 a | 3 | 41 | 93 | n.d. | 7 | 69[e] | 61 | n.d. | 35 |
| 10 | 24 | 96 | 92 | n.d. | 8 | 62[e] | 55 | n.d. | 19 | ||
| 11 |
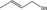
|
13 a | 3 | 26 | 92 | n.d. | 8 | 54[e] | 31 | n.d. | 54 |
| 12 | 24 | 82 | 90 | n.d. | 10 | 53[e] | 28 | n.d. | 26 | ||
[a] Based on 1H NMR using mesitylene as standard for verifying mass‐balance. [b] Yield of each product over total conversion. [c] 0.1 mol % loading, [substrate]=0.5 m in CH2Cl2, V=0.7 mL, ≈8 mmol of H2. [d] Not detected. [e] Formation of ill‐defined condensation products was also observed, especially in 24 h.
Complete hydrogenation of pent‐4‐en‐1‐ol (8 a), pent‐4‐en‐2‐ol (9 a), and 2‐methylbut‐3‐en‐1‐ol (10 a) to the respective alcohols 8 b–10 b was observed with 2‐PF6 in 3 h. Isomerization products were not detected (Figure S24), as reported for 2‐PF6 using similar substrates.23 Complete hydrogenation of 8 a–10 a to 8 b–10 b was also achieved with 2@1‐SO3Na, albeit in 24 h (Table 2, entries 1–6). Isomerization products were again not detected. Conversion in 3 h correlates well with the steric hindrance around the double bond of the substrate: 10 % for 10 a (more hindered), increasing to 22 % for 9 a (less hindered), and reaching 34 % for 8 a (linear). Olefinic alcohols 8 a–10 a were hydrogenated considerably slower with 2@1‐SO3Na, compared to the sterically comparable non‐functionalized alkenes 4 and 5 (Table 1). This is consistent with a strong interaction between the hydroxyl group of the olefinic alcohols and the chemical microenvironment of 2@1‐SO3Na.
Substrates which are intrinsically more susceptible to isomerization, such as the homoallylic (11 a) and allylic (12 a, 13 a) alcohols,23, 24 revealed a significant enhancement of reactivity and selectivity to hydrogenation with 2@1‐SO3Na, compared to its homogeneous counterpart. The homogeneous catalyst 2‐PF6 afforded 56 % conversion of 11 a in 3 h and 57 % in 24 h, indicative of catalyst deactivation (Table 2, entries 7 and 8, Figure S25). Moreover, isomerization of 11 a was also observed, producing a non‐negligible amount of the internal olefinic alcohol 11 c and traces of the aldehyde 11 d. As a result, selectivity to hydrogenation and formation of n‐butanol (11 b) was only 86 % for the homogeneous system.
By contrast, the heterogeneous catalyst 2@1‐SO3Na afforded complete conversion and 100 % selectivity to hydrogenation and formation of 11 b (Table 2, entries 7 and 8, Figure S26). Monitoring conversion over time for both systems (Figure S27) verified that 2‐PF6 is deactivated after 3 h, whereas 2@1‐SO3Na remained productive, affording full conversion in 6 h. Although traces of the internal olefin 11 c were detected in short reaction times, 11 c was subsequently also hydrogenated to 11 b. The encapsulated catalyst is thus more stable, more active with respect to overall conversion, and more selective.
The superior performance of 2@1‐SO3Na was even more pronounced in the hydrogenation of allylic alcohols that can isomerize directly to the respective aldehydes. Conversion under hydrogenation conditions for trans‐pent‐2‐en‐1‐ol (12 a, entries 9 and 10) and trans‐crotyl alcohol (13 a, entries 11 and 12) in 3 h with 2‐PF6 was 69 % and 54 %, respectively (Figure S28). Conversion did not increase after 24 h, indicating catalyst deactivation. Selectivity to hydrogenation was poor: 61 % for alcohol 12 b in 3 h with a substantial amount of the aldehyde 12 d formed (35 % selectivity), and 31 % for alcohol 13 b in 3 h with the aldehyde 13 d now being the main product (54 % selectivity). By contrast, overall conversion with 2@1‐SO3Na as the catalyst reached 96 % for 12 a and 82 % for 13 a in 24 h (Figure S29). Isomerization to the aldehydes 12 d and 13 d was significantly suppressed, resulting in ≥90 % selectivity for the alcohols 12 b and 13 b.
To probe the effect of the sulfonate group on stability and selectivity, we also investigated the homogeneous hydrogenation of crotyl alcohol using 2‐OTs and 3 as catalysts (Figure S30). Higher conversions were observed compared to 2‐PF6 (77 % for 2‐OTs and 83 % for 3 in 24 h) in accordance with OTs− being a more strongly coordinating anion, hence prolonging the catalyst's lifetime.25 By contrast, selectivity to hydrogenation did not significantly improve (39 % for 2‐OTs and 53 % for 3), remaining considerably lower than that of 2@1‐SO3Na (≥90 %).
The reaction pathways for the hydrogenation or isomerization of olefinic alcohols with the homogeneous catalyst 2‐PF6 likely share the same starting point, the formation of a cationic IrIII‐dihydride complex in which the hydroxyl group is also coordinated to Ir (Scheme 2, intermediate I), followed by migratory insertion (intermediate II).13, 14 Bifurcation into separate, competitive pathways then occurs: i) hydrogenation to the respective alcohol via reductive elimination (pathway A) or ii) isomerization to the internal olefin via β‐elimination, which requires an appropriately orientated vacant coordination site, followed by off‐cycle tautomerization to the aldehyde (pathway B).
Scheme 2.
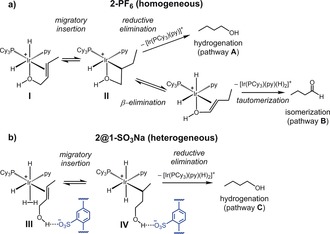
a) Competing pathways for hydrogenation (A) and isomerization (B) of olefinic alcohols with 2‐PF6. b) Proposed pathway for hydrogenation (C) and suppression of isomerization with 2@1‐SO3Na.
The significantly improved selectivity to hydrogenation observed with 2@1‐SO3Na suggests that isomerization is suppressed. We propose that this could take place due to extended coordination sphere interactions between the hydroxyl group of the olefinic alcohols and the chemical microenvironment around 2, such as H‐bonding to the sulfonate groups. This disfavors coordination of the hydroxyl group to Ir and enables formation of the dihydrogen complex III, in preference to I (pathway C). Productive hydrogenation occurs via an octahedral IrV‐trihydride species (IV), as proposed for non‐functionalized alkenes with Crabtree‐type catalysts26 and β‐elimination is suppressed since Ir is coordinatively saturated throughout.
Catalyst 2@1‐SO3Na also resulted in higher overall conversions for the hydrogenation of olefinic alcohols, compared to 2‐PF6. A series of selective poisoning experiments revealed that the isomerization products are not responsible for catalyst deactivation (Table S7). We thus suggest that 2@1‐SO3Na has a longer lifetime due to: i) spatial isolation of the positively charged catalytically active species inside the pores of the anionic MOF which hinders the formation of catalytically inactive clusters and/or ii) reversible coordination of the sulfonate anion, as shown with 2‐OTs and 3.
In summary, we demonstrate that the hybrid catalyst 2@1‐SO3Na is capable of hydrogenating non‐functionalized alkenes at low loadings in solution and in the gas phase under mild conditions. It outperforms its homogeneous counterpart in the hydrogenation of olefinic alcohols, showing significantly higher conversions under otherwise identical conditions. In addition, encapsulation results in a pronounced selectivity enhancement in favor of hydrogenation by suppressing the competing isomerization reaction due to extended coordination sphere interactions of the catalytic center with the chemically functionalized internal surface of the MOF. Capitalizing on such stability and selectivity enhancements is likely to be important in catalytic applications in continuous flow.27 In metalloenzymes, it is well‐established that well‐positioned amino acid residues around the active site control reactivity and selectivity.3 Here, the well‐defined, readily engineered MOF chemical microenvironment controls reactivity and selectivity of the encapsulated catalyst, allowing discrimination between two distinct reaction pathways.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The EPSRC is thanked for support for this work through the UK Catalysis Hub Consortium under grants EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1, and EP/M013219/1. A.S.W. also acknowledges EPSRC for funding (EP/M024210/1). M.J.R. is a Royal Society Research Professor.
A. Grigoropoulos, A. I. McKay, A. P. Katsoulidis, R. P. Davies, A. Haynes, L. Brammer, J. Xiao, A. S. Weller, M. J. Rosseinsky, Angew. Chem. Int. Ed. 2018, 57, 4532.
In memory of Gérard Férey
Contributor Information
Prof. Andrew S. Weller, Email: andrew.weller@chem.ox.ac.uk.
Prof. Matthew J. Rosseinsky, Email: M.J.Rosseinsky@liverpool.ac.uk.
References
- 1.
- 1a. Furukawa H., Cordova K. E., O'Keeffe M., Yaghi O. M., Science 2013, 341, 1230444; [DOI] [PubMed] [Google Scholar]
- 1b. Li B., Wen H.-M., Cui Y., Zhou W., Qian G., Chen B., Adv. Mater. 2016, 28, 8819–8860; [DOI] [PubMed] [Google Scholar]
- 1c. Zhou H.-C., Kitagawa S., Chem. Soc. Rev. 2014, 43, 5415–5418, and references therein. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Gascon J., Corma A., Kapteijn F., Llabrés i Xamena F. X., ACS Catal. 2014, 4, 361–378; [Google Scholar]
- 2b. Chughtai A. H., Ahmad N., Younus H. A., Laypkov A., Verpoort F., Chem. Soc. Rev. 2015, 44, 6804–6849; [DOI] [PubMed] [Google Scholar]
- 2c. Grigoropoulos A. in Modern Developments in Catalysis, World Scientific (Europe), London, 2016, pp. 123–158; [Google Scholar]
- 2d. Rogge S. M. J., Bavykina A., Hajek J., Garcia H., Olivos-Suarez A. I., Sepulveda-Escribano A., Vimont A., Clet G., Bazin P., Kapteijn F., Daturi M., Ramos-Fernandez E. V., Llabres i Xamena F. X., Van Speybroeck V., Gascon J., Chem. Soc. Rev. 2017, 46, 3134–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Ragsdale S. W., Chem. Rev. 2006, 106, 3317–3337; [DOI] [PubMed] [Google Scholar]
- 3b. Zhao M., Wang H.-B., Ji L.-N., Mao Z.-W., Chem. Soc. Rev. 2013, 42, 8360–8375; [DOI] [PubMed] [Google Scholar]
- 3c. Holm R. H., Solomon E. I., Chem. Rev. 2014, 114, 3367–3368, and references therein. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Jones C. W., Top. Catal. 2010, 53, 942–952; [Google Scholar]
- 4b. Raynal M., Ballester P., Vidal-Ferran A., van Leeuwen P. W. N. M., Chem. Soc. Rev. 2014, 43, 1660–1733 and 1734–1787; [DOI] [PubMed] [Google Scholar]
- 4c. Brown C. J., Toste F. D., Bergman R. G., Raymond K. N., Chem. Rev. 2015, 115, 3012–3035; [DOI] [PubMed] [Google Scholar]
- 4d. Hübner S., de Vries J. G., Farina V., Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar]
- 5.
- 5a. Fateeva A., Chater P. A., Ireland C. P., Tahir A. A., Khimyak Y. Z., Wiper P. V., Darwent J. R., Rosseinsky M. J., Angew. Chem. Int. Ed. 2012, 51, 7440–7444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7558–7562; [Google Scholar]
- 5b. Mitchell L., Williamson P., Ehrlichová B., Anderson A. E., Seymour V. R., Ashbrook S. E., Acerbi N., Daniels L. M., Walton R. I., Clarke M. L., Wright P. A., Chem. Eur. J. 2014, 20, 17185–17197; [DOI] [PubMed] [Google Scholar]
- 5c. Manna K., Zhang T., Carboni M., Abney C. W., Lin W., J. Am. Chem. Soc. 2014, 136, 13182–13185; [DOI] [PubMed] [Google Scholar]
- 5d. Fei H., Cohen S. M., J. Am. Chem. Soc. 2015, 137, 2191–2194; [DOI] [PubMed] [Google Scholar]
- 5e. Li Z., Xiao J.-D., Jiang H.-L., ACS Catal. 2016, 6, 5359–5365; [Google Scholar]
- 5f. Fracaroli A. M., Siman P., Nagib D. A., Suzuki M., Furukawa H., Toste F. D., Yaghi O. M., J. Am. Chem. Soc. 2016, 138, 8352–8355; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5g. Thacker N. C., Lin Z., Zhang T., Gilhula J. C., Abney C. W., Lin W., J. Am. Chem. Soc. 2016, 138, 3501–3509. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Canivet J., Aguado S., Schuurman Y., Farrusseng D., J. Am. Chem. Soc. 2013, 135, 4195–4198; [DOI] [PubMed] [Google Scholar]
- 6b. Rasero-Almansa A. M., Corma A., Iglesias M., Sanchez F., Green Chem. 2014, 16, 3522–3527; [Google Scholar]
- 6c. Burgess S. A., Kassie A., Baranowski S. A., Fritzsching K. J., Schmidt-Rohr K., Brown C. M., Wade C. R., J. Am. Chem. Soc. 2016, 138, 1780–1783; [DOI] [PubMed] [Google Scholar]
- 6d. Rimoldi M., Nakamura A., Vermeulen N. A., Henkelis J. J., Blackburn A. K., Hupp J. T., Stoddart J. F., Farha O. K., Chem. Sci. 2016, 7, 4980–4984; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Burgun A., Coghlan C. J., Huang D. M., Chen W., Horike S., Kitagawa S., Alvino J. F., Metha G. F., Sumby C. J., Doonan C. J., Angew. Chem. Int. Ed. 2017, 56, 8412–8416; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8532–8536. [Google Scholar]
- 7.
- 7a. Guo Z., Xiao C., Maligal-Ganesh R. V., Zhou L., Goh T. W., Li X., Tesfagaber D., Thiel A., Huang W., ACS Catal. 2014, 4, 1340–1348; [Google Scholar]
- 7b. Yuan S., Chen Y. P., Qin J. S., Lu W., Zou L., Zhang Q., Wang X., Sun X., Zhou H.-C., J. Am. Chem. Soc. 2016, 138, 8912–8919; [DOI] [PubMed] [Google Scholar]
- 7c. Liu L., Zhou T.-Y., Telfer S. G., J. Am. Chem. Soc. 2017, 139, 13936–13943. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Xiao D. J., Oktawiec J., Milner P. J., Long J. R., J. Am. Chem. Soc. 2016, 138, 14371–14379; [DOI] [PubMed] [Google Scholar]
- 8b. Shi W., Cao L., Zhang H., Zhou X., An B., Lin Z., Dai R., Li J., Wang C., Lin W., Angew. Chem. Int. Ed. 2017, 56, 9704–9709; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9836–9841; [Google Scholar]
- 8c. Li L., Yang Q., Chen S., Hou X., Liu B., Lu J., Jiang H.-L., Chem. Commun. 2017, 53, 10026–10029. [DOI] [PubMed] [Google Scholar]
- 9. Crabtree R., Acc. Chem. Res. 1979, 12, 331–337. [Google Scholar]
- 10.
- 10a. Chodosh D. F., Crabtree R. H., Felkin H., Morehouse S., Morris G. E., Inorg. Chem. 1982, 21, 1307–1311; [Google Scholar]
- 10b. Xu Y., Celik M. A., Thompson A. L., Cai H., Yurtsever M., Odell B., Green J. C., Mingos D. M. P., Brown J. M., Angew. Chem. Int. Ed. 2009, 48, 582–585; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 590–593. [Google Scholar]
- 11.
- 11a. Lee H. M., Jiang T., Stevens E. D., Nolan S. P., Organometallics 2001, 20, 1255–1258; [Google Scholar]
- 11b. Vazquez-Serrano L. D., Owens B. T., Buriak J. M., Inorg. Chim. Acta 2006, 359, 2786–2797; [Google Scholar]
- 11c. Kolychev E. L., Kronig S., Brandhorst K., Freytag M., Jones P. G., Tamm M., J. Am. Chem. Soc. 2013, 135, 12448–12459. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Smidt S. P., Zimmermann N., Studer M., Pfaltz A., Chem. Eur. J. 2004, 10, 4685–4693; [DOI] [PubMed] [Google Scholar]
- 12b. Moxham G. L., Douglas T. M., Brayshaw S. K., Kociok-Kohn G., Lowe J. P., Weller A. S., Dalton Trans. 2006, 5492–5505. [DOI] [PubMed] [Google Scholar]
- 13. Li H., Mazet C., Acc. Chem. Res. 2016, 49, 1232–1241. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Stork G., Kahne D. E., J. Am. Chem. Soc. 1983, 105, 1072–1073; [Google Scholar]
- 14b. Crabtree R. H., Davis M. W., J. Org. Chem. 1986, 51, 2655–2661. [Google Scholar]
- 15.
- 15a. Genna D. T., Wong-Foy A. G., Matzger A. J., Sanford M. S., J. Am. Chem. Soc. 2013, 135, 10586–10589; [DOI] [PubMed] [Google Scholar]
- 15b. Grigoropoulos A., Whitehead G. F. S., Perret N., Katsoulidis A. P., Chadwick F. M., Davies R. P., Haynes A., Brammer L., Weller A. S., Xiao J., Rosseinsky M. J., Chem. Sci. 2016, 7, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Akiyama G., Matsuda R., Sato H., Takata M., Kitagawa S., Adv. Mater. 2011, 23, 3294–3297; [DOI] [PubMed] [Google Scholar]
- 16b. Zhou Y.-X., Chen Y.-Z., Hu Y., Huang G., Yu S.-H., Jiang H.-L., Chem. Eur. J. 2014, 20, 14976–14980; [DOI] [PubMed] [Google Scholar]
- 16c. Juan-Alcañiz J., Gielisse R., Lago A. B., Ramos-Fernandez E. V., Serra-Crespo P., Devic T., Guillou N., Serre C., Kapteijn F., Gascon J., Catal. Sci. Technol. 2013, 3, 2311–2318. [Google Scholar]
- 17. Férey G., Mellot-Draznieks C., Serre C., Millange F., Dutour J., Surblé S., Margiolaki I., Science 2005, 309, 2040–2042. [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y., Li B., Krishna R., Wu Z., Ma D., Shi Z., Pham T., Forrest K., Space B., Ma S., Chem. Commun. 2015, 51, 2714–2717. [DOI] [PubMed] [Google Scholar]
- 19. Genna D. T., Pfund L. Y., Samblanet D. C., Wong-Foy A. G., Matzger A. J., Sanford M. S., ACS Catal. 2016, 6, 3569–3574. [Google Scholar]
- 20.Note: Although we cannot discount chemical shift changes in the 31P SS NMR spectrum due to the presence of paramagnetic CrIII centers, no significant changes were reported when molecular species were incorporated into pristine MIL-101(Cr). See reference [17].
- 21.
- 21a. Yang D., Odoh S. O., Borycz J., Wang T. C., Farha O. K., Hupp J. T., Cramer C. J., Gagliardi L., Gates B. C., ACS Catal. 2016, 6, 235–247; [Google Scholar]
- 21b. Chadwick F. M., McKay A. I., Martinez-Martinez A. J., Rees N. H., Kramer T., Macgregor S. A., Weller A. S., Chem. Sci. 2017, 8, 6014–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Moreno M., Kissell L. N., Jasinski J. B., Zamborini F. P., ACS Catal. 2012, 2, 2602–2613; [Google Scholar]
- 22b. Sadeghmoghaddam E., Gu H., Shon Y.-S., ACS Catal. 2012, 2, 1838–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Mantilli L., Mazet C., Tetrahedron Lett. 2009, 50, 4141–4144; [Google Scholar]
- 23b. Mantilli L., Gérard D., Torche S., Besnard C., Mazet C., Angew. Chem. Int. Ed. 2009, 48, 5143–5147; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5245–5249. [Google Scholar]
- 24. Uma R., Crevisy C., Gree R., Chem. Rev. 2003, 103, 27–51. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Rifat A., Kociok-Köhn G., Steed J. W., Weller A. S., Organometallics 2004, 23, 428–432; [Google Scholar]
- 25b. Piras E., Läng F., Rüegger H., Stein D., Wörle M., Grützmacher H., Chem. Eur. J. 2006, 12, 5849–5858. [DOI] [PubMed] [Google Scholar]
- 26. Verendel J. J., Pàmies O., Diéguez M., Andersson P. G., Chem. Rev. 2014, 114, 2130–2169. [DOI] [PubMed] [Google Scholar]
- 27. Hintermair U., Francio G., Leitner W., Chem. Commun. 2011, 47, 3691–3701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary