

Abstract
The self‐assembly of eight PdII cations and sixteen phenanthrene‐derived bridging ligands with 60° bite angles yielded a novel M8L16 metallosupramolecular architecture composed of two interlocked D 4h‐symmetric barrel‐shaped containers. Mass spectrometry, NMR spectroscopy, and X‐ray analysis revealed this self‐assembled structure to be a very large “Hopf link” catenane featuring channel‐like cavities, which are occupied by NO3 − anions. The importance of the anions as catenation templates became imminent when we observed the nitrate‐triggered structural rearrangement of a mixture of M3L6 and M4L8 assemblies formed in the presence of BF4 − anions into the same interlocked molecule. Furthermore, the densely packed structure of the M8L16 catenane was exploited in the preparation of a hexyloxy‐functionalized analogue, which further self‐assembled into vesicle‐like aggregates in a reversible manner.
Keywords: catenanes, interlocked structures, self-assembly, structural transformations, supramolecular chemistry
Mechanically interlocked structures continue to spark scientific interest and curiosity owing to their aesthetic appeal and the dynamic properties that are introduced by the mechanical bond.1 Although their preparation initially posed a synthetic challenge, the utilization of molecular templates2 and the implementation of reversible covalent bonds3 have resulted in high‐yielding and facile syntheses. In this regard, the highly directional coordinative bond has been the basis of a variety of interpenetrated structures, such as catenanes,4 rotaxanes,5 knots,6 Borromean rings,7 and more.8
Among the plethora of possible metal and ligand combinations, PdII cations together with N‐donor bridging ligands represent a robust and reliable combination widely utilized in the formation of hollow and interlocked metallosupramolecular structures.8b, 9 For assemblies obeying PdnL2n stoichiometry (n=6, 12, 24, 30 …), Fujita and co‐workers have demonstrated the topological dependence of the structure of hollow polyhedra on obtuse ligand bite angles.10 This work was recently expanded to the synthesis of a giant Pd48L96 Goldberg polyhedron.10e More acute ligand bite angles (≥60°) often yield Pd3L6 and Pd4L8 barrel‐shaped containers or Pd4L8 doubly bridged tetrahedra (Figure 1),11 whilst ligands with a parallel orientation of donors form lantern‐shaped Pd2L4 coordination cages.9 Catenation of the latter cages results in a larger family of Pd4L8 dimeric structures with a partitioned cavity capable of allosterically binding charged12 and neutral13 guest molecules. Under certain circumstances, these types of ligands also form a triply catenated link.14 More recently, the close proximity of electron donor/acceptor moieties in mixed‐ligand Pd4L8 interpenetrated cages has been exploited to study charge‐transfer phenomena,15 demonstrating that interlocked structures can also serve as platforms to examine interactions of densely packed functionalities.
Figure 1.
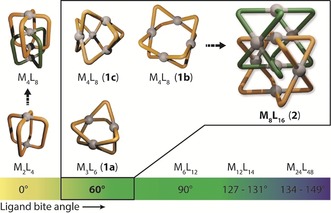
Comparison of the known MnL2n (M=PtII or PdII) structures according to the bite angle of their constituent ligands with the catenated M8L16 architecture 2. Metal‐mediated self‐assembly of a ligand with a 60° bite angle can give rise to M3L6 (1 a) or M4L8 structures (1 b or 1 c). In previous studies, only M2L4 cage structures were shown to undergo catenation to give M4L8 interlocked dimers.
Despite the vast structural diversity of reported PdnL2n polyhedral structures, interpenetrated assemblies larger than the Pd4L8 dimeric cages remained hitherto undiscovered. Herein, we report the assembly of an interlocked metallosupramolecule with the formula M8L16 (M=PdII), which is the largest PdnL2n catenane reported to date. Palladium‐mediated self‐assembly of the dimeric structure was achieved with Pd(NO3)2 and a rigid bis(monodentate) ligand L with a bite angle of 60°. The presence of NO3 − anions is crucial for the formation of the large catenane, and can also trigger the structural rearrangement of a mixture of Pd3L6 and Pd4L8 assemblies formed in the presence of BF4 − anions into the same interpenetrated structure. Through alkyl functionalization of the ligand backbone, an amphiphilic catenane was prepared that further self‐assembled into vesicle‐like aggregates, demonstrating that the densely packed nature of the Pd8L16 architecture can be utilized as a platform for higher‐order supramolecular aggregation.
Phenanthrene‐based ligand L1 (Figure 2) readily underwent self‐assembly with [Pd(CH3CN)4](BF4)2 to form a D 4h‐symmetric Pd4L8 (1 b) container in DMSO as the only product.11c In CD3CN, however, a 1:2:0.2 mixture of a D 3h‐symmetric Pd3L6 (1 a) container, 1 b, and a D 2d‐symmetric tetrahedron (1 c) was obtained (Figure 1).16 As both solvent and anion are known to dramatically affect the assembly of coordination cage structures,9d, 11 we carried out further investigations, including the reaction of L1 with Pd(NO3)2. Self‐assembly in the presence of nitrate anions again yielded 1 b as the major product in DMSO (Figure S10); however, in CD3CN, we observed an entirely different outcome.
Figure 2.
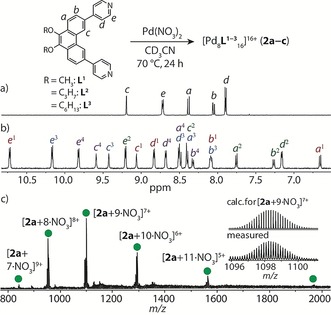
Top: Reaction equation representing the formation of Pd8L16 (2 a–2 c). Bottom: 1H NMR spectra (500 MHz, CD3CN, 25 °C) of a) ligand L1 and b) 2 a. c) ESI‐MS spectrum of 2 a with the measured and calculated isotope patterns of [2 a+9 NO3]7+ shown in the inset.
Heating a 2:1 mixture of L1 and Pd(NO3)2 in CD3CN at 70 °C for 24 h resulted in the quantitative formation of a new species (2 a), as indicated by 1H NMR spectroscopy and ESI mass spectrometry. In the 1H NMR spectrum, a total of 20 aromatic signals were identified in the range of δ=6.6–10.7 ppm (compared with 5 aromatic signals for L1), along with four signals in the region of the methoxy protons (Figures 2 and S11). ESI‐MS analysis gave a spectrum with several prominent peaks consistent with the formula [Pd8L1 16+n NO3](16−n)+ (n=7–12; Figure 2 c). By COSY NMR analysis, we identified four distinct sets of aromatic signals of equal ratios for L1. Further analysis by 1H‐1H NOESY revealed numerous cross‐peaks (Figure S14), including notable through‐space contacts between the Hc protons of sets 1/2 and 3/4. We therefore postulated that the fourfold splitting is due to L1 being in two different chemical environments and losing its twofold symmetry in 2 a. The exclusive formation of 2 a was further confirmed by DOSY NMR analysis (Figure S16), which revealed that all of the assigned proton signals correspond to the same diffusion coefficient, with the derived hydrodynamic radius (1.24 nm) pointing towards a rather compact structure.
According to the empirical predictions for PdnL2n assemblies,10 a structure with a composition of Pd8L16 is incompatible with a spherical, hollow topology, unless it is a transient intermediate towards larger Pd12L24 polyhedra.10d In light of the fourfold signal splitting observed in the 1H NMR spectrum of 2 a, our Pd8L1 16 assembly is therefore likely to be composed of two interpenetrating Pd4L1 8 subunits. To gain insight into whether the possible monomeric Pd4L8 assemblies (1 b or 1 c) are transient intermediates preceding 2 a, time‐resolved 1H NMR experiments were performed on a 2:1 mixture of L1 and Pd(NO3)2 at 70 °C in MeCN (Figure S17). Only the proton resonances of 2 a evolved gradually over 20 h, and no other species were detected, suggesting that any intermediates involved in the self‐assembly process are polymeric or short‐lived in solution.
Owing to difficulties encountered with obtaining crystals of 2 a that are suitable for X‐ray analysis, we focused on modifying the solubility of the catenane by utilizing a propoxy‐functionalized ligand L2. Under the same reaction conditions as for 2 a, the reaction of L2 with Pd(NO3)2 gave an analogous Pd8L16 interlocked structure (2 b), as confirmed by 1H NMR spectroscopy and ESI‐MS analysis (Figures S18–S21). Single crystals (diffracting up to 1.28 Å with synchrotron radiation)17 were grown by slow vapor diffusion of diisopropyl ether into a concentrated CD3CN solution of 2 b, enabling unambiguous structure elucidation by X‐ray crystallography. 2 b crystallized in the monoclinic space group P21/n, with one Pd8L16 molecule in the asymmetric unit. The large interpenetrated structure, which is composed of two interlocked D 4h‐symmetric containers,18 can be described as a “Hopf link” (Figure 3) with D 2d symmetry.
Figure 3.
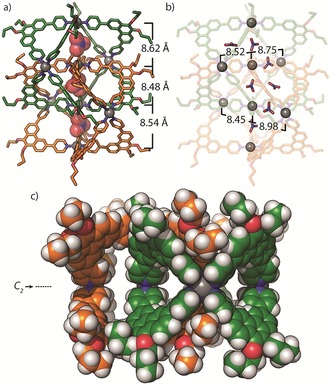
X‐ray structure of 2 b:24 a) Showing Pd⋅⋅⋅Pd distances and encapsulated NO3 − anions along the major C2 axis, b) highlighting the Pd⋅⋅⋅Pd distances of cations orthogonal to the major C2 axis, c) a space‐filling representation of the [2]catenane with NO3 − anions removed for clarity.
In the structure of 2 b, the Pd4L8 monomers are geometrically related to one another by a 90° rotation along the major C 2 axis, creating an even partition of three channel‐like cavities, each accommodating a NO3 − counterion (Figure 3 a). The central cavity is enclosed by phenanthrene moieties of L2, which participate in offset π‐stacking with an average separation of 3.7 Å. In contrast to the Pd4L8 interpenetrated cages,8b 2 b appears to be sterically restricted from mechanical movement owing to the respective tilt of the ligands between adjacent Pd4L8 units. In addition to the three NO3 − anions found along the C2 axis, four more (of a total of 13 located in the Fourier difference map) were found within the interpenetrated structure; however, not all of them occupied defined cavities. For example, 2 b contains four smaller cavities located perpendicular to the major C 2 axis along the Pd3 planes (Figure 3 b), with only two of the four cavities being occupied. Accordingly, the Pd⋅⋅⋅Pd separations for the empty cavities are 0.2–0.5 Å shorter than those of the filled cavities.19
Next, we examined whether NO3 − anions trigger a transformative rearrangement of the mixture of 1 a, 1 b, and 1 c that is formed when L1 is reacted with [Pd(CH3CN)4](BF4)2 16 in CD3CN (during the course of our studies, a single crystal of 1 c was isolated and analyzed by X‐ray crystallography, further supporting 1 c as one of the Pd‐mediated assemblies of L1; Figures 4 b and S38–S40). The mixture of 1 a, 1 b, and 1 c was therefore heated at 70 °C for 24 h with 4, 8, and 12 equivalents of NO3 −. In each case, 1H NMR spectroscopy revealed that 2 a was formed exclusively (Figure 4); however, the reaction proceeded with the highest yield when 8 equivalents of NO3 − were used (Figure S27). ESI‐MS of this sample confirmed the structural conversion of 1 a–1 c into 2 a as it revealed signals corresponding to [Pd8L1 16+n X](16−n)+ (n=8–11, X=Cl−, NO3 −, and BF4 −), with each signal distribution comprising a bias towards X=8 NO3 − and (8−n) BF4 − (Figure S28). To gain further insight into the role of the nitrate anions in the system, we performed the same transformation with a 15N‐labeled NO3 − source. Inverse‐gated 15N NMR analysis revealed a single 15N signal, which corresponds to the free 15NO3 − signal of the tetrabutylammonium salt used in the transformation (Figure S29). This indicates that the NO3 − anions are not tightly bound and are free to exchange with free NO3 − in the solvent, which is consistent with the channel‐like cavities observed in the X‐ray structure of 2 b. The reason for why catenation is observed only in the presence of NO3 − anions could therefore be related to their optimal size as templates for this dimer.12b, 14, 20 Indeed, 1 a–1 c were not transformed into the catenated product upon extended heating (70 °C, 24 h) or in the presence of PF6 − anions (Figures S9 and S31).
Figure 4.
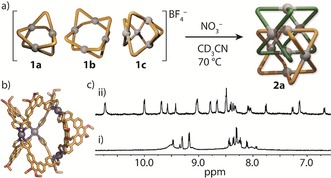
a) The NO3 −‐mediated transformation of 1 a–1 c into 2 a. b) X‐ray structure of 1 c.24 c) 1H NMR spectra (500 MHz, CD3CN) of i) 1 a–1 c and ii) the same mixture after heating for 24 h in the presence of NO3 − anions.
Finally, we examined whether the structurally dense nature of the Pd8L16 [2]catenane can be utilized as a platform for further functionalization and supramolecular aggregation. It is worth noting that hierarchical aggregates of nanocages have scarcely been reported, and the underlying mechanisms of such systems primarily rely upon counterion‐mediated interactions and π‐stacking.21 To promote a different aggregation pathway, we prepared a dihexyloxy‐functionalized ligand (L3) for the palladium‐mediated self‐assembly of amphiphilic catenane 2 c (Figures S22–S26). Remarkably, in MeCN, 2 c rapidly and spontaneously further self‐assembled into a colloid of nanoscalar vesicular aggregates by virtue of the dense hydrophobic interactions between molecules of 2 c (Figure 5). This is in contrast to the behavior of 2 a and 2 b, which remain in solution owing to the shorter alkyl chains of their respective ligands. At 25 °C, a 1H NMR spectrum of a cloudy solution of 2 c revealed the complete absence of proton resonances. At elevated temperatures (50–70 °C), however, the solution of 2 c became completely transparent, and the characteristic NMR spectroscopic signature of the interlocked molecule was observed (Figures S22 and S23). Dynamic light scattering (DLS) analysis of 2 c revealed particles with diameters of 150±45 nm (PDI=0.09) at 20 °C, which were observed to incrementally swell upon dilution (Figure S32), which is indicative of a vesicular‐type assembly.22 Variable temperature (VT) DLS revealed that this aggregate begins to disassemble between 40 and 50 °C, at which point a smaller aggregate (36.2±20.4 nm) is formed along with particles measuring 2.3±0.7 nm in diameter, the latter coinciding with the approximate dimensions of a single molecule of 2 c. Interestingly, cooling the sample back to 20 °C resulted in the recovery of the original, approximately 150 nm large aggregate (Figure 5 a), with the MeCN solution becoming cloudy once again. The reversibility of the temperature‐dependent supramolecular aggregation was demonstrated over three cycles by DLS with no detectable degradation (Figure S33). High‐resolution TEM analysis of 2 c (Figures 5 b and S34) confirmed the presence of spherical particles, albeit with a slightly broader distribution (80–200 nm).23 This is in contrast to the TEM analysis of 2 a, which revealed small particles with diameters of approximately 3 nm, which correspond to non‐aggregated molecules of 2 a (Figure S36). This observation further supports our hypothesis that the hydrophobic hexyloxy chains in 2 c promote hierarchical aggregation, which is facilitated by the structurally dense nature of this type of interlocked architecture.
Figure 5.
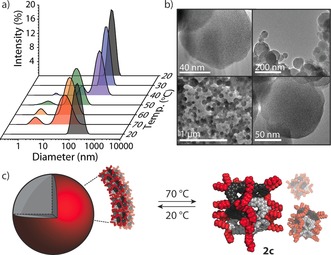
a) Variable‐temperature DLS. b) TEM images of 2 c. c) Temperature‐dependent aggregation of 2 c (plausible model based on the X‐ray structure of 2 b).
In conclusion, we have reported the self‐assembly of a 24‐component Pd8L16 [2]catenane composed of two interlocked D 4h‐symmetric Pd4L8 barrel‐shaped containers. The formation of the large catenane was shown to depend on the presence of NO3 − counterions by NMR, ESI‐MS, and crystallographic analysis. We also showed that NO3 − anions can trigger the structural rearrangement of a mixture of Pd3L6 and Pd4L8 assemblies into the same interpenetrated product. Indeed, the X‐ray crystal structure of 2 b revealed that the channel‐like cavities are occupied by NO3 − anions, which not only charge‐balance the structure but also facilitate the interpenetration of the Pd4L8 barrel‐shaped monomers. Exohedral functionalization of the structurally dense catenane with hexyloxy chains resulted in the hierarchical assembly of vesicle‐like aggregates, which could be reversibly assembled and disassembled by a change in temperature. We are currently investigating the use of this aggregate as a molecular delivery and release vessel.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Andreas Brockmeyer and Dr. Petra Janning (Max‐Planck Institute for Molecular Physiology, Dortmund) and Dr. Holm Frauendorf (Georg‐August University Göttingen) for mass spectrometry, Dr. Ashley Slattery (Adelaide Microscopy) for assistance with TEM measurements, and Björn Holzapfel (TU Dortmund) for assistance with DLS measurements. W.M.B. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship and acknowledges support through an Adelaide University Ramsay Fellowship. This work has been financially supported by an ERC Consolidator grant 683083 (RAMSES) and the DFG‐funded Cluster of Excellence RESOLV (EXC 1069). The crystallographic experiments for 1 c were performed at the PXII (X06SA) beamline of the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland. Diffraction data for 2 b were collected at PETRA III at DESY, a member of the Helmholtz Association (HGF). We thank Saravanan Panneerselvam for assistance in using synchrotron beamline P11 (I‐20170404).17
W. M. Bloch, J. J. Holstein, B. Dittrich, W. Hiller, G. H. Clever, Angew. Chem. Int. Ed. 2018, 57, 5534.
Contributor Information
Dr. Witold M. Bloch, Email: witold.bloch@adelaide.edu.au.
Prof. Dr. Guido H. Clever, Email: guido.clever@tu-dortmund.de.
References
- 1.
- 1a. Stoddart J. F., Chem. Soc. Rev. 2009, 38, 1802–1820; [DOI] [PubMed] [Google Scholar]
- 1b. Vickers M. S., Beer P. D., Chem. Soc. Rev. 2007, 36, 211–225; [DOI] [PubMed] [Google Scholar]
- 1c. Janke M., Rudzevich Y., Molokanova O., Metzroth T., Mey I., Diezemann G., Marszalek P. E., Gauss J., Bohmer V., Janshoff A., Nat. Nanotechnol. 2009, 4, 225–229; [DOI] [PubMed] [Google Scholar]
- 1d. Collin J.-P., Dietrich-Buchecker C., Gaviña P., Jimenez-Molero M. C., Sauvage J.-P., Acc. Chem. Res. 2001, 34, 477–487; [DOI] [PubMed] [Google Scholar]
- 1e. Angew. Chem. Int. Ed 2016, 55, 13925; Angew. Chem 2016, 128, 14129.
- 2.
- 2a. Meyer C. D., Joiner C. S., Stoddart J. F., Chem. Soc. Rev. 2007, 36, 1705–1723; [DOI] [PubMed] [Google Scholar]
- 2b. Mullen K. M., Beer P. D., Chem. Soc. Rev. 2009, 38, 1701–1713; [DOI] [PubMed] [Google Scholar]
- 2c. Ayme J.-F., Beves J. E., Campbell C. J., Leigh D. A., Chem. Soc. Rev. 2013, 42, 1700–1712; [DOI] [PubMed] [Google Scholar]
- 2d. Denis M., Goldup S. M., Nat. Rev. Chem. 2017, 1, 61. [Google Scholar]
- 3.
- 3a. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 5126–5130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5226–5230; [Google Scholar]
- 3b. Hasell T., Wu X., Jones J. T., Bacsa J., Steiner A., Mitra T., Trewin A., Adams D. J., Cooper A. I., Nat. Chem. 2010, 2, 750–755. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Fujita M., Acc. Chem. Res. 1999, 32, 53–61; [Google Scholar]
- 4b. Prusty S., Krishnaswamy S., Bandi S., Chandrika B., Luo J., McIndoe J. S., Hanan G. S., Chand D. K., Chem. Eur. J. 2015, 21, 15174–15187; [DOI] [PubMed] [Google Scholar]
- 4c. Wood C. S., Ronson T. K., Belenguer A. M., Holstein J. J., Nitschke J. R., Nat. Chem. 2015, 7, 354–358. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Sauvage J.-P., Acc. Chem. Res. 1998, 31, 611–619; [Google Scholar]
- 5b. Beves J. E., Blight B. A., Campbell C. J., Leigh D. A., McBurney R. T., Angew. Chem. Int. Ed. 2011, 50, 9260–9327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9428–9499. [Google Scholar]
- 6.
- 6a. Engelhard D. M., Freye S., Grohe K., John M., Clever G. H., Angew. Chem. Int. Ed. 2012, 51, 4747–4750; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4828–4832; [Google Scholar]
- 6b. Guo J., Mayers P. C., Breault G. A., Hunter C. A., Nat. Chem. 2010, 2, 218–222; [DOI] [PubMed] [Google Scholar]
- 6c. Barran P. E., Cole H. L., Goldup S. M., Leigh D. A., McGonigal P. R., Symes M. D., Wu J., Zengerle M., Angew. Chem. Int. Ed. 2011, 50, 12280–12284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12488–12492. [Google Scholar]
- 7.
- 7a. Huang S.-L., Lin Y.-J., Li Z.-H., Jin G.-X., Angew. Chem. Int. Ed. 2014, 53, 11218–11222; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11400–11404; [Google Scholar]
- 7b. Chichak K. S., Cantrill S. J., Pease A. R., Chiu S.-H., Cave G. W. V., Atwood J. L., Stoddart J. F., Science 2004, 304, 1308–1312. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Fielden S. D. P., Leigh D. A., Woltering S. L., Angew. Chem. Int. Ed. 2017, 56, 11166–11194; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11318–11347; [Google Scholar]
- 8b. Frank M., Johnstone M. D., Clever G. H., Chem. Eur. J. 2016, 22, 14104–14125; [DOI] [PubMed] [Google Scholar]
- 8c. Horner K. E., Miller M. A., Steed J. W., Sutcliffe P. M., Chem. Soc. Rev. 2016, 45, 6432–6448; [DOI] [PubMed] [Google Scholar]
- 8d. Lewis J. E. M., Beer P. D., Loeb S. J., Goldup S. M., Chem. Soc. Rev. 2017, 46, 2577–2591; [DOI] [PubMed] [Google Scholar]
- 8e. Forgan R. S., Sauvage J.-P., Stoddart J. F., Chem. Rev. 2011, 111, 5434–5464; [DOI] [PubMed] [Google Scholar]
- 8f. Huang S.-L., Hor T. S. A., Jin G.-X., Coord. Chem. Rev. 2017, 333, 1–26. [Google Scholar]
- 9.
- 9a. Clever G. H., Punt P., Acc. Chem. Res. 2017, 50, 2233–2243; [DOI] [PubMed] [Google Scholar]
- 9b. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 9c. Bloch W. M., Clever G. H., Chem. Commun. 2017, 53, 8506–8516; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Sun Q.-F., Iwasa J., Ogawa D., Ishido Y., Sato S., Ozeki T., Sei Y., Yamaguchi K., Fujita M., Science 2010, 328, 1144–1147; [DOI] [PubMed] [Google Scholar]
- 10b. Bunzen J., Iwasa J., Bonakdarzadeh P., Numata E., Rissanen K., Sato S., Fujita M., Angew. Chem. Int. Ed. 2012, 51, 3161–3163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3215–3217; [Google Scholar]
- 10c. Yokoyama H., Ueda Y., Fujita D., Sato S., Fujita M., Chem. Asian J. 2015, 10, 2292–2295; [DOI] [PubMed] [Google Scholar]
- 10d. Fujita D., Yokoyama H., Ueda Y., Sato S., Fujita M., Angew. Chem. Int. Ed. 2015, 54, 155–158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 157–160; [Google Scholar]
- 10e. Fujita D., Ueda Y., Sato S., Mizuno N., Kumasaka T., Fujita M., Nature 2016, 540, 563–566. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Suzuki K., Kawano M., Fujita M., Angew. Chem. Int. Ed. 2007, 46, 2819–2822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2877–2880; [Google Scholar]
- 11b. Ganta S., Chand D. K., Dalton Trans. 2015, 44, 15181–15188; [DOI] [PubMed] [Google Scholar]
- 11c. Bloch W. M., Abe Y., Holstein J. J., Wandtke C. M., Dittrich B., Clever G. H., J. Am. Chem. Soc. 2016, 138, 13750–13755; [DOI] [PubMed] [Google Scholar]
- 11d. Klein C., Gütz C., Bogner M., Topić F., Rissanen K., Lützen A., Angew. Chem. Int. Ed. 2014, 53, 3739–3742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3814–3817; [Google Scholar]
- 11e. Samanta D., Chowdhury A., Mukherjee P. S., Inorg. Chem. 2016, 55, 1562–1568; [DOI] [PubMed] [Google Scholar]
- 11f. Jansze S. M., Cecot G., Wise M. D., Zhurov K. O., Ronson T. K., Castilla A. M., Finelli A., Pattison P., Solari E., Scopelliti R., Zelinskii G. E., Vologzhanina A. V., Voloshin Y. Z., Nitschke J. R., Severin K., J. Am. Chem. Soc. 2016, 138, 2046–2054; [DOI] [PubMed] [Google Scholar]
- 11g. Chand D. K., Biradha K., Kawano M., Sakamoto S., Yamaguchi K., Fujita M., Chem. Asian J. 2006, 1, 82–90. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Freye S., Hey J., Torras-Galán A., Stalke D., Herbst-Irmer R., John M., Clever G. H., Angew. Chem. Int. Ed. 2012, 51, 2191–2194; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2233–2237; [Google Scholar]
- 12b. Freye S., Michel R., Stalke D., Pawliczek M., Frauendorf H., Clever G. H., J. Am. Chem. Soc. 2013, 135, 8476–8479; [DOI] [PubMed] [Google Scholar]
- 12c. Sekiya R., Fukuda M., Kuroda R., J. Am. Chem. Soc. 2012, 134, 10987–10997; [DOI] [PubMed] [Google Scholar]
- 12d. Li Y.-H., Jiang J.-J., Fan Y.-Z., Wei Z.-W., Chen C.-X., Yu H.-J., Zheng S.-P., Fenske D., Su C.-Y., Barboiu M., Chem. Commun. 2016, 52, 8745–8748. [DOI] [PubMed] [Google Scholar]
- 13. Löffler S., Lübben J., Krause L., Stalke D., Dittrich B., Clever G. H., J. Am. Chem. Soc. 2015, 137, 1060–1063. [DOI] [PubMed] [Google Scholar]
- 14. Zhu R., Lübben J., Dittrich B., Clever G. H., Angew. Chem. Int. Ed. 2015, 54, 2796–2800; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2838–2842. [Google Scholar]
- 15.
- 15a. Frank M., Ahrens J., Bejenke I., Krick M., Schwarzer D., Clever G. H., J. Am. Chem. Soc. 2016, 138, 8279–8287; [DOI] [PubMed] [Google Scholar]
- 15b. Ahrens J., Frank M., Clever G. H., Schwarzer D., Phys. Chem. Chem. Phys. 2017, 19, 13596–13603. [DOI] [PubMed] [Google Scholar]
- 16. Bloch W. M., Holstein J. J., Hiller W., Clever G. H., Angew. Chem. Int. Ed. 2017, 56, 8285–8289; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8399–8404. [Google Scholar]
- 17. Burkhardt A., Pakendorf T., Reime B., Meyer J., Fischer P., Stübe N., Panneerselvam S., Lorbeer O., Stachnik K., Warmer M., Rödig P., Göries D., Meents A., Eur. Phys. J. Plus 2016, 131, 56. [Google Scholar]
- 18.We use the term “container” to describe the general shape of the Pd4L8 D 4h-symmetric monomers.
- 19.To obey the D 2d symmetry of the Pd8L16 catenane, the NO3 − anions are expected to rapidly exchange with free NO3 − anions in solution.
- 20.We note that catenation of this system is also dependent on CH3CN as the solvent. The fact that catenation is not observed in DMSO may be due to DMSO solvent molecules stabilizing or packing more efficiently in the cavity of the D 4h-symmetric Pd4L8 momomer than CH3CN; see Ref. [11a].
- 21.
- 21a. Li D., Zhang J., Landskron K., Liu T., J. Am. Chem. Soc. 2008, 130, 4226–4227; [DOI] [PubMed] [Google Scholar]
- 21b. Li D., Zhou W., Landskron K., Sato S., Kiely C. J., Fujita M., Liu T., Angew. Chem. Int. Ed. 2011, 50, 5182–5187; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5288–5293; [Google Scholar]
- 21c. Li H., Luo J., Liu T., Chem. Eur. J. 2016, 22, 17949–17952; [DOI] [PubMed] [Google Scholar]
- 21d. Zhang Y., Zhou Q.-F., Huo G.-F., Yin G.-Q., Zhao X.-L., Jiang B., Tan H., Li X., Yang H.-B., Inorg. Chem. 2017, https://doi.org/10.1021/acs.inorgchem.7b02777; [DOI] [PubMed] [Google Scholar]
- 21e. Clegg J. K. et al., Angew. Chem. Int. Ed. 2010, 49, 1075–1078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1093–1096. [Google Scholar]
- 22.
- 22a. Discher D. E., Eisenberg A., Science 2002, 297, 967–973; [DOI] [PubMed] [Google Scholar]
- 22b. Marques E. F., Regev O., Khan A., Lindman B., Adv. Colloid Interface Sci. 2003, 100–102, 83–104; [Google Scholar]
- 22c. Sauer M., Meier W., Chem. Commun. 2001, 55–56. [DOI] [PubMed] [Google Scholar]
- 23.The broader distribution may be due to partial desolvation and deformation of the vesicle-like aggregates because of solvent loss.
- 24. https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201800490 1811308 (1 c) and 1811309 (2 b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary