

SUMMARY
Idiopathic pulmonary fibrosis (IPF) is characterized by aberrant accumulation of collagen-secreting myofibroblasts. Development of effective therapies is limited due to incomplete understanding of molecular mechanisms regulating myofibroblast expansion. FOXF1 transcription factor is expressed in resident lung fibroblasts, but its role in lung fibrosis remains unknown due to the lack of genetic mouse models. Through comprehensive analysis of human IPF genomics data, lung biopsies, and transgenic mice with fibroblast-specific inactivation of FOXF1, we show that FOXF1 inhibits pulmonary fibrosis. FOXF1 deletion increases myofibroblast invasion and collagen secretion and promotes a switch from N-cadherin (CDH2) to Cadherin-11 (CDH11), which is a critical step in the acquisition of the profibrotic phenotype. FOXF1 directly binds to Cdh2 and Cdh11 promoters and differentially regulates transcription of these genes. Re-expression of CDH2 or inhibition of CDH11 in FOXF1-deficient cells reduces myofibroblast invasion in vitro. FOXF1 inhibits pulmonary fibrosis by regulating a switch from CDH2 to CDH11 in lung myofibroblasts.
In Brief
Black et al. demonstrated that FOXF1 inhibits pulmonary fibrosis by preventing CDH2 to CDH11 cadherin switch in myofibroblasts.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is the most common and lethal interstitial lung disease, with a median survival rate of 3 years after diagnosis. Environmental, age-related, and genetic factors create an alveolar epithelium that is susceptible to injury from either endogenous or exogenous factors (Noble et al., 2012; Vaughan et al., 2015). Recurrent epithelial injury with deregulated repair of the epithelial barrier results in pulmonary fibrosis. IPF is characterized by scarring of pulmonary parenchyma due to accumulation of unrestrained myofibroblasts and excessive matrix deposition. Effective therapeutics for IPF is limited. Treatments targeting fibroblast functions have proved effective in slowing IPF progression but do not resolve fibrotic deposition (Selman et al., 2001; Ahluwalia et al., 2014; Chapman, 2012). Identifying molecular targets that regulate myofibroblast expansion and accumulation in the lung tissue is critical for development of novel therapeutic approaches for IPF.
The pathological feature of IPF is the formation of fibroblastic foci that reflect active myofibroblast differentiation and pathogenic processes. Differentiation of fibroblasts to myofibroblasts is associated with increased expression of alpha-smooth muscle actin (αSMA) stress fibers resulting in a pro-contractile, pro-secretory, pro-migratory, and hyper-proliferative phenotype (Hinz et al., 2007). Heterogeneous cell populations contribute to αSMA-expressing myofibroblasts, including resident fibro-blasts, mesenchymal stem cells, pericytes, circulating fibrocytes derived from bone marrow, and epithelial cells that differentiate into myofibroblasts via epithelial-to-mesenchymal transition (EMT) (Rock et al., 2011; Balli et al., 2013; Dulauroy et al., 2012; Hung et al., 2013). Resident lung stromal cells are progenitors for most myofibroblasts in fibrotic foci. Due to heterogeneity among stromal populations, identifying global fibroblast-specific markers remains elusive. Various fibroblast markers include PDGFRα, TBX4, DESMIN, VIMENTIN, and -COL1a1 (Hung et al., 2013; Rock et al., 2011; Hinz et al., 2007; Xie et al., 2016; Dulauroy et al., 2012; Lovgren et al., 2011). The differentiation of stromal fibroblasts into myofibroblasts is characterized by expression of αSMA. Molecular mechanisms causing their differentiation into myofibroblasts and transcriptional regulators of myofibroblast functions are not fully understood.
The Forkhead box (Fox) family of transcription factors regulates cell growth, tumor formation, tissue repair, organ development, and homeostasis (Costa et al., 2001; Kalin et al., 2011; Balli et al., 2012). Forkhead box F1 (FOXF1) is a mesenchymal-specific transcription factor expressed in multiple mesenchyme-derived cell types, including normal hepatic stellate cells, peri-bronchial smooth muscle cells, lung microvascular endothelial cells, and fibroblasts (Kalinichenko et al., 2003). FOXF1 haploin-sufficiency is linked to various developmental abnormalities and pathologies, including alveolar capillary dysplasia (Stankiewicz et al., 2009), lung edema (Cai et al., 2016), defects in gall bladder development (Kalinichenko et al., 2002a), and abnormal liver and lung repair after acute injury (Kalinichenko et al., 2002b, 2003). In addition, FOXF1 regulates lung mesenchymal stem cells (Walker et al., 2011; Malin et al., 2007). Although these reports establish FOXF1 as an important transcriptional regulator during organ development and acute injury, the role of FOXF1 in chronic lung diseases, such as pulmonary fibrosis, is not fully understood.
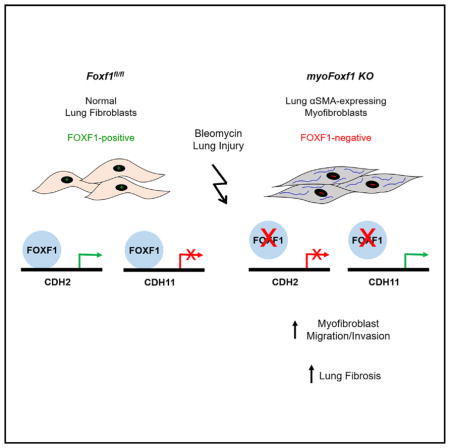
Through comprehensive analysis of human IPF genomics data, human normal lung and IPF biopsies, and transgenic mice with myofibroblast-specific deletion of Foxf1, the present study shows that loss of FOXF1 in myofibroblasts promotes pulmonary fibrosis. FOXF1-depleted myofibroblasts were more migratory, proliferative, and invasive and produced more collagens, all hallmarks of fibrotic pathogenesis. FOXF1 protein bound and transcriptionally regulated N-cadherin (Cdh2) and cadherin-11 (Cdh11) promoters. A switch between Cdh2 and Cdh11 expression caused myofibroblast activation, which was inhibited by FOXF1. Altogether, our data identify FOXF1 as an anti-fibrotic factor that regulates key myofibroblast functions driving fibrogenesis.
RESULTS
FOXF1 Expression Is Decreased in Human IPF Lung Fibroblasts and in Murine Bleomycin-Treated Fibrotic Lungs
To determine FOXF1 expression in fibroblasts from human normal and IPF lungs, we assessed gene expression profiles from clinical IPF datasets GEO: GSE44723 (Peng et al., 2013), GEO: GSE17978 (Emblom-Callahan et al., 2010), and GEO: GSE1724 (Renzoni et al., 2004). Collectively, these data show that FOXF1 mRNA expression was significantly decreased in human IPF fibroblasts compared to normal lung fibroblasts (Figure 1A). In normal human lungs, immunofluorescence staining showed FOXF1 colocalized with known fibroblast markers (αSMA, PDGFRa, and DESMIN) (Figure 1B). FOXF1 was absent in stromal cells within the fibroblastic foci of human IPF lesions (Figure 1B). Thus, FOXF1 mRNA and protein were decreased in human IPF.
Figure 1. Decreased FOXF1 in Human IPF and Mouse Bleomycin-Treated Lungs.

(A) FOXF1 mRNA is decreased in fibroblasts isolated from human IPF and normal lung tissues. Data obtained from GEO: GSE44723, GSE17978, and GSE1724. Data presented as mean ± SD.
(B) Decreased FOXF1 protein in myofibroblasts of IPF fibrotic foci. Lung sections from human IPF patients (n = 7) and normal lung donors (n = 6) stained with H&E or costained with antibodies against FOXF1 and fibroblast markers.
(C) Schematic of bleomycin-induced fibrosis in mice. Lungs from bleomycin-treated mice were harvested on day 14 for early fibrosis and day 28 for chronic fibrosis.
(D) Lung collagen was quantified by Sircol collagen assay.
(E) Foxf1 mRNA is decreased in isolated lung fibroblasts during lung fibrosis progression, shown by qRT-PCR. Data presented as mean ± SEM.
(F) Intensity of FOXF1 staining in lung fibroblasts is decreased during progression of fibrosis, as shown by flow cytometry. Mean fluorescence intensity of FOXF1 was determined for the CD326−CD45−CD31− lung fibroblasts.
(G) Percentage of FOXF1+ fibroblasts among CD326−CD45−CD31− lung fibroblasts in untreated and bleomycin-treated mice was determined by flow cytometry and presented as mean ± SD. See also Figure S1.
(H) Lung tissue sections from normal and bleomycin-treated mice were stained with H&E or Masson’s trichrome, or costained with antibodies against FOXF1 and fibroblast markers. n = 5 mice/group in (D)–(H).
All scale bars indicate 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S1.
We assessed FOXF1 expression in mouse fibroblasts during bleomycin-induced pulmonary fibrosis. Wild-type mice were administered two or four weekly doses of bleomycin. Lungs were examined 14 and 28 days after injury to evaluate early and chronic stages of fibrosis, respectively (Figure 1C) (Izbicki et al., 2002). Bleomycin caused severe pulmonary fibrosis with extensive collagen deposition (Figures 1C and 1D) and increased expression of profibrotic genes (Figure S1A). As lung fibrosis progressed, Foxf1 mRNA decreased in total lung lysates and in isolated lung fibroblasts (Figures 1E and S1A). Flow cytometry analysis of isolated lung fibroblasts at different time points during bleomycin-induced fibrosis showed a time-dependent decrease in the mean fluorescence intensity of FOXF1 staining and in the number of cells coexpressing FOXF1 and αSMA (Figures 1F, 1G, and S1B). Colocalization studies of normal, early fibrotic, and chronic fibrotic mouse lungs show that fibroblasts express FOXF1 in normal lungs but gradually decrease FOXF1 as fibrosis progresses (Figure 1H). Serial sections of fibrotic lungs show that smooth muscle cells coexpress αSMA and gamma-smooth muscle actin (γSMA) and are localized as thin sheaths around bronchial airway and blood vessels (Figures S1C and S1D). Smooth muscle cells were not found in fibrotic lesions that contain αSMA-expressing myofibroblasts (Figures S1C and S1D).
Genetic Deletion of Foxf1 in αSMA-Expressing Cells Exacerbates Bleomycin-Induced Pulmonary Fibrosis
Conditional inactivation of Foxf1 was carried out in αSMA-expressing myofibroblasts during pulmonary fibrosis. Mice carrying a Tamoxifen (Tam)-inducible αSMA-CREERT2 transgene (Wendling et al., 2009) were crossed with mice carrying Foxf1-floxed alleles (Ren et al., 2014) to generate αSMA-CREERT2; Foxf1fl/f (myoFoxf1 knockout [KO]) mice (Figure 2A). In the presence of Tam, Exon 1, encoding the FOXF1 DNA binding domain, was excised by Cre recombinase in αSMA-positive cells. mTmG reporter showed Cre-mediated recombination in ROSA26R(mTmG/+);αSMA-Cre-ERT2 (mTmG;αSMA-CreERT2) lungs and was increased after bleomycin treatment (Figures S2A and S2B). GFP was detected in 80% of αSMA-positive cells (Figure S2B). In uninjured mice, Tam treatment did not affect normal lung architecture of Foxf1fl/fl and myoFoxf1 KO mice (Figure S2C). To induce lung fibrosis, mice were treated with 1 U/kg bleomycin (Figure 2B). Compared to Figures 1C–1H, the lower dose of bleomycin was chosen to prevent mortality in the myoFoxf1 KO mice that developed severe fibrosis. After bleomycin injury, Tam-treated myoFoxf1 KO mice developed severe and persistent lung fibrosis, while few focal fibrotic lesions were found in Tam-treated Foxf1fl/fl mice (Figure 2C).
Figure 2. Deletion of Foxf1 in αSMA-Expressing Myofibroblasts Accelerates Bleomycin-Induced Pulmonary Fibrosis.

(A) Schematic of Foxf1 deletion in αSMA-CRE-ERT2;Foxf1fl/fl (myoFoxf1 KO) transgenic mice.
(B) Schematic of bleomycin treatment to induce lung fibrosis.
(C) Increased fibrosis and collagen deposition are shown with H&E, trichrome, and Sirius red and fast green stainings.
(D) Collagen deposition was quantitated with Sirius red, Sircol, and hydroxyproline assays using control Foxf1fl/fl (n = 9) and myoFoxf1 KO (n = 11) mouse lungs at day 21 after first bleomycin treatment.
(E) Increased number of cells in BALF from bleomycin-treated myoFoxf1 KO lungs.
(F) Increased number of macrophages in bleomycin-treated myoFoxf1 KO lungs. The number of MAC3+ cells was counted and quantified per 1,000 cells using 10 random microscope fields. Representative images are shown for n = 9–11 mice.
Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. All scale bars indicate 20 μm. See also Figure S2.
Collagen deposition was significantly increased in Tam-treated myoFoxf1 KO mice after bleomycin injury, as shown with trichrome or Sirius red collagen stainings and collagen protein assays (Sircol and hydroxyproline) (Figures 2C and 2D). Consistent with increased collagen deposition, total lung mRNAs for Col1a1, Col3a1, and αSMA were upregulated in myoFoxf1 KO lungs (Figure 3A). Depletion of FOXF1 in myoFoxf1 KO mice was associated with increased expression of proinflammatory genes IL1a and Tnfa (Figure 3A), inflammatory cells in bronchioalveolar lavage fluid (BALF), and increased numbers of MAC3-positive cells in lung parenchyma (Figures 2E and 2F). Depletion of FOXF1 in myoFoxf1 KO mice exacerbated bleomycin-induced pulmonary fibrosis, collagen deposition, and pulmonary inflammation, indicating that FOXF1 is an anti-fibrotic transcription factor.
Figure 3. Increased Myofibroblast Differentiation and Collagen Production in myoFoxf1 KO Lungs after Bleomycin.

(A) Foxf1 depletion increased mRNAs of collagens and inflammatory cytokines. n = 9–11 mice per group.
(B) Efficient FOXF1 deletion is shown by immunofluorescence costaining of αSMA and FOXF1 in fibrotic lesions of bleomycin-treated control and myoFoxf1 KO lungs. Percentage of αSMA+/FOXF1+ double-positive cells in control and myoFoxf1 KO mice was quantified and presented as mean ± SD. Scale bars indicate 20 μm. See also Figure S3.
(C and D) FOXF1 deletion increased activated myofibroblasts markers in isolated lung fibroblasts from bleomycin-treated myoFoxf1 KO (n = 6) and control (n = 5) mice, shown by (C) qRT-PCR and (D) western blot. Total lung was used to isolate RNA and protein. Western blot data were quantified using densitometry and presented as mean ± SD. See also Figure S3.
(E–G) FACS analysis shows an increase in the percentage of myofibroblasts (CD45−CD326−CD31−MHY11−αSMA+) and a decrease in the percentage of fibroblasts (CD45−CD326−CD31−MHY11−αSMA−) in bleomycin-treated myoFoxf1 KO lungs. (E) Representative dot plots and (F) quantification of the percentage of myofibroblasts and fibroblasts. Decreased percentage of FOXF1-positive lung myofibroblasts (CD45−CD326−CD31−MHY11−αSMA+) in myoFoxf1 KO mice (G) quantified by FACS analysis. n = 4 mice/group.
Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S4.
FOXF1 Deletion Induces Myofibroblast Differentiation
Immunofluorescence studies showed that the number of αSMA+/FOXF1+ cells was decreased in myofibroblastic foci of myoFoxf1 KO lungs compared to Foxf1fl/fl control lungs, indicating efficient Foxf1 deletion (Figures 3B and S3A). Decreased numbers of FOXF1+ myofibroblasts and diminished FOXF1 protein were detected in myoFoxf1 KO mice as early as day 3 after Tam administration (Figures S3A and S3B). To validate the efficiency of Foxf1 deletion, lung fibroblast fractions were isolated from uninjured, Tam- and bleomycin-treated control, and myoFoxf1 KO mice. Foxf1 mRNA was decreased in a time-dependent manner in fibroblast fractions isolated from bleomycin-treated myoFoxf1 KO lungs (Figures 3C and S3C). Decreased Foxf1 mRNA was associated with increased mRNAs of genes linked to activated myofibroblasts, including Acta2 (αSMA), vimentin (Vim), and Pdgfra, as well as extracellular matrix components fibronectin (Fn1), Col1a1, and Col3a1 (Figure 3C). Lung fibroblast fractions isolated from uninjured control and myoFoxf1 KO mice had similar levels of FOXF1, αSMA, VIM, and PDGFRa protein (Figure 3D). In contrast, fibroblast fractions isolated from bleomycin-treated Foxf1fl/fl lungs had reduced FOXF1 protein and increased αSMA, VIM, and PDGFRa protein levels compared to uninjured Foxf1fl/fl lungs, consistent with the development of lung fibrosis (Figure 3D). Deletion of Foxf1 in myoFoxf1 KO myofibroblasts further increased αSMA, VIM, and PDGFRa protein levels (Figure 3D), consistent with exacerbated pulmonary fibrosis in myoFoxf1 KO mice (Figures 2C and 2D). Because isolated lung fibroblast fractions consist of a mix of fibroblasts and myofibroblasts, we used fluorescence-activated cell sorting (FACS) analysis to determine the proportions of each of these cell types in response to Foxf1 deletion and bleomycin treatment (Figure S4A). Uninjured Foxf1fl/fl and myoFoxf1 KO lung had similar percentages of fibroblast (CD31−CD45−CD326−MHY11−αSMA−) and myofibroblast (CD31−CD45−CD326−MHY11−αSMA+) populations (Figures 3E, 3F, and S4A). However, after bleomycin treatment, the percentage of myofibroblasts increased, while fibroblast populations decreased in myoFoxf1 KO mice (Figures 3E and 3F). The percentage of αSMA+/FOXF1+ myofibroblasts was reduced in myoFoxf1 KO mice compared to Foxf1fl/fl controls (Figures 3E and 3G). This is consistent with efficient deletion of Foxf1 by the αSMA-CreERT2 transgene. The percentage of smooth muscle cells (CD31−CD45−CD326−αSMA+MHY11+) did not change in myoFoxf1 KO lungs (Figures S4B–S4E). Collectively, these data demonstrate that genetic deletion of Foxf1 in αSMA-expressing myofibroblasts caused myofibroblast differentiation and collagen production, associated with increased expression of αSMA, Vim, Pdgfrα, Fn1, Col1a1, and Col3a1, known markers of activated myofibroblasts.
Deletion of Foxf1 Increased Myofibroblast Migration and Invasion
To determine whether FOXF1 regulates functions of myofibroblasts during pulmonary fibrosis, we assessed proliferation, migration, and invasion of primary lung fibroblasts isolated from Foxf1fl/fl control and myoFoxf1 KO mice. Bleomycin-treated myoFoxf1 KO mice had ~2-fold higher fibroblast cell numbers compared to Foxf1fl/fl mice (Figure 4A). mRNAs of proliferation-specific Foxm1, Ccnb1, Cdc25b, and Aurkb genes were increased in total lung and myofibroblasts isolated from myo-Foxf1 KO lungs (Figure 4B). Colocalization studies demonstrated increased numbers of Ki-67- and FOXM1-positive cells in myoFoxf1 KO lungs (Figure 4C). Using transwell migration and Matrigel invasion assays, we measured the migration and invasion of fibroblasts isolated from bleomycin-treated Foxf1fl/fl and myoFoxf1 KO mice. Migration and invasion of myoFoxf1 KO fibroblasts increased ~30% compared to Foxf1fl/fl fibro-blasts (Figures 4D–4G). No differences in migration or invasion were detected in uninjured, Tam-treated Foxf1fl/fl, and myoFoxf1 KO fibroblasts (Figures 4D–4G). Depletion of Foxf1 increased myofibroblast migration and invasion, exacerbating pulmonary fibrosis in bleomycin-treated myoFoxf1 KO mice.
Figure 4. Deletion of Foxf1 Increases Fibroblast Migration and Invasion.

(A) Increased numbers of fibroblast isolated from bleomycin-treated myoFoxf1 KO lungs.
(B) Increased mRNAs of proliferation-specific genes in fibroblasts isolated from myoFoxf1 KO lungs are shown by qRT-PCR. n = 7–8 mice per group.
(C) Ki-67 and αSMA costaining in bleomycin-treated lungs. The number of Ki-67+ cells was counted and quantified per image using 10 random microscope fields. n = 7–8 mice per group.
(D) Increased migration of fibroblasts isolated from bleomycin-treated myoFoxf1 KO lungs was shown using transwell inserts.
(E) Migrated cells were stained with crystal violet and counted.
(F) Increased invasion of FOXF1-deficient fibroblasts was assessed using Matrigel-coated transwell inserts.
(G) Invaded cells were stained with crystal violet and counted. n = 3 mice/group.
Experiments were seeded in triplicates. Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. All scale bars indicate 20 μm.
FOXF1 Regulates Genes Essential for Myofibroblast Differentiation and Function
RNA sequencing (RNA-seq) analysis was performed to compare genome-wide mRNA expression profiles of fibroblasts isolated from bleomycin-treated myoFoxf1 KO and Foxf1fl/fl lungs. Purity of isolated fibroblasts was assessed by the absence of markers for endothelial, epithelial, and immune cells (Figure S5). RNA-seq analysis identified 2,803 transcripts differentially expressed in myoFoxf1 KO fibroblasts using the following criteria: absolute value of fold change > 2 and false discovery rate (FDR)-adjusted p value < 0.01 (Figure 5A). The most enriched functional categories in myoFoxf1 KO lung fibroblasts were related to response to wound healing, extracellular matrix organization, cell migration, focal adhesion, and cell adhesion (Figures 5B and 5C), consistent with the increased invasive phenotype of myoFoxf1 KO lung fibroblasts (Figures 4D–4G). Foxf1 mRNA was decreased 2.4-fold in myoFoxf1 KO lung fibroblasts (Figure 5D), a finding confirmed by qRT-PCR (Figure 5E). Expression of several genes from each functional category was validated by qRT-PCR, including Jup, Adam10, Rock1, Ccl2, Cdh2, and Cdh11 and genes involved in cell junctions, extracellular matrix organization, inflammatory response, and migration (Figures 5D and 5E). Cdh2 was not detected in myoFoxf1 KO lung fibroblasts; however, Cdh11 mRNA was increased (Figure 5E). Thus, FOXF1 regulates fibroblast genes associated with pulmonary fibrosis.
Figure 5. FOXF1 Regulates Genes Essential for Myofibroblast Differentiation and Functions.

(A) Differentially expressed genes in bleomycin-treated myoFoxf1 KO lungs were identified using RNA-seq analysis. Heatmap of RNA-seq results showing 2,809 coding genes significantly upregulated (red) or downregulated (green) between isolated fibroblasts from bleomycin-treated Foxf1fl/fl and those from myoFoxf1 KO lungs. Samples from three mice per genotype were pooled for RNA-seq analysis.
(B and C) Increased (B) and decreased (C) functional pathways influenced by Foxf1 deletion were identified using the ToppGene Suite (https://toppgene.cchmc.org/). The statistical significance of each functional pathway was presented using negative log2 transformation of the p value.
(D) Fold changes in mRNA for several genes from the RNA-seq mapped in (A).
(E) qRT-PCR validation of several genes found to be differentially expressed by RNA-seq analysis. n = 7–8 mice per genotype. See also Figure S5.
(F) FOXF1 regulates CDH2-CDH11 cadherin switching in mouse lung fibroblasts. Representative images of western blot analysis of CDH2 and CDH11 levels in isolated lung fibroblast fractions from Foxf1fl/fl and myoFoxf1 KO lungs. n = 7–8 mice per group.
(G and H) Immunofluorescence costaining of (G) αSMA and CDH2 or (H) CDH11 in fibrotic lesions of Foxf1fl/fl and myoFoxf1 KO lungs. Representative images of the staining are shown for n = 9–11 mice. Scale bars indicate 20 μm.
(I) FACS shows an increase in the percentage of CDH11+CDH2− and CDH2+CDH11+ myofibroblasts (CD45−CD326−CD31−MHY11−αSMA+) in bleomycin-treated myoFoxf1 KO lungs. Representative dot plots and quantification of the percentage of CDH2+CDH11−, CDH11+CDH2−, and CDH2+CDH11+ myofibroblasts are shown. n = 4 mice/group.
Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S6.
FOXF1 Regulates CDH2-CDH11 Cadherin Switch in Mouse and Human Lung Fibroblasts
CDH2 and CDH11 are homophilic adhesion molecules identified as mesenchymal-specific cadherins (Alimperti and Andreadis, 2015; Agarwal et al., 2008). Because cadherin signaling is associated with pathogenic fibroblast functions (Hinz et al., 2004; Chang et al., 2011; Schneider et al., 2012), we focused our next studies on the FOXF1-dependent regulation of CDH2 and CDH11. Western blot analysis of isolated myofibroblasts and colocalization studies showed decreased CDH2 and increased CDH11 in myofibroblasts isolated from bleomycin-treated myoFoxf1 KO lungs (Figures 5F–5H). In addition, FACS analysis showed myoFoxf1 KO mice had increased percentages of CDH11+CDH2− cells among myofibroblast and fibroblast populations (Figures 5I and S6A–S6C). In vitro studies using NIH 3T3 fibroblasts with stable expression or inhibition of FOXF1 showed that FOXF1 expression positively correlated with CDH2 and inversely correlated with CDH11 protein and mRNA levels (Figures S7A and S7B).
Next, we assessed CDH2 and CDH11 expression in human IPF lungs using clinical datasets wherein FOXF1 mRNA was decreased (Figure 1A). Analysis revealed that CDH2 mRNA was significantly decreased (Figure 6A) but CDH11 mRNA was increased in human IPF fibroblasts (Figure 6B). Consistent with mRNA data, CDH2 protein levels were decreased but CDH11 protein levels were increased in human IPF lungs, as shown by immunostaining (Figures 6C and 6D) and western blot (Figure 6E). These data demonstrated that FOXF1 regulates a switch between CDH2 and CDH11 expression in myofibroblasts that may contribute to fibrosis in human and mouse lungs.
Figure 6. FOXF1 Regulates CDH2-CDH11 Cadherin Switching in Human IPF Lung Fibroblasts.

(A and B) CDH2 (A) and CDH11 (B) mRNA data using fibroblasts isolated from human IPF and normal lung samples obtained from GEO: GSE44723, GSE17978, and GSE1724. Data presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test.
(C and D) Lung sections from normal human lung donors and IPF patients costained with αSMA and CDH2 (C) or CDH11 (D). Representative images are shown. IPF samples are n = 7, and normal lung samples are n = 6. Scale bars indicate 20 μm.
(E) Western blot analysis demonstrated decreased FOXF1 and CDH2 but increased CDH11 protein in human IPF compared to normal lungs.
FOXF1 Directly Binds to and Regulates Transcriptional Activity of Cdh2 and Cdh11 Promoters
Because inactivation of FOXF1 in myofibroblasts was associated with decreased CDH2 and increased CDH11 expression in vivo and in vitro, we tested whether FOXF1 directly binds to and regulates transcription of Cdh2 and Cdh11 genes. Using chromatin immunoprecipitation sequencing (ChIP-seq) analysis with FOXF1 antibodies, FOXF1 binding peaks were identified within the −5 kb promoter region of Cdh2 (Figure S7C), indicating that FOXF1 binds to the Cdh2 promoter region. FOXF1 binding to the Cdh2 promoter was confirmed by a conventional ChIP assay, in which FOXF1 and FLAG antibodies were used to immunoprecipitate FOXF1 in mouse NIH 3T3 fibroblasts stably expressing a His-FLAG-tagged Foxf1 construct (Figures 7A and S7D). Thirteen potential FOXF1 binding sites were identified in the −5 kb Cdh2 promoter region (Figure 7A; Table S2). FOXF1 physically bound to the −1,925/−1,776 base pair (bp) region of the Cdh2 promoter, which contains two FOXF1-binding motifs (Figure 7A). FOXF1 binding was also detected in the promoter of Pdgfb, a known transcriptional target of FOXF1 (Bolte et al., 2015; Ren et al., 2014) used as a positive control (Figure 7A). FOXF1 did not bind to Gapdh DNA, which lacked FOXF1 binding sites (Figure 7A). To test whether FOXF1 directly regulated Cdh2 transcription, we cloned the −3,087/+71 bp Cdh2 promoter region, which contains high-affinity FOXF1 binding sites, into the pGL2-Basic luciferase (LUC) vector to generate the pGL2-Cdh2-Luc reporter (Figure 7B). This reporter was cotransfected with CMV-Empty or CMV-Foxf1 expression plasmids into HEK293T cells. CMV-Foxf1 plasmid induced transcriptional activity of the Cdh2 promoter region (Figure 7B). In addition, the FOXF1 binding region had mono- and tri-methylation of histone 3 lysine 4 (H3K4) (Figure S7C), which was shown to be associated with active transcription (Barski et al., 2007). This is consistent with the findings that FOXF1 increased Cdh2 mRNA and protein (Figures 5G–5I). Thus, FOXF1 transcriptionally activates the Cdh2 gene.
Figure 7. FOXF1 Regulates Myofibroblast Invasion through Transcriptional Activation of CDH2 and Repression of CDH11.

(A) Schematic drawing of the mouse Cdh2 promoter region with potential FOXF1 DNA binding sites (black boxes). See also Table S2. ChIP assays in NIH 3T3 cells showed direct binding of FOXF1 to the indicated regions in Cdh2 promoter. ChIP assay of FOXF1 binding to the −1,435/−1,428 bp mouse Pdgfb promoter region was used as a positive control. Gapdh DNA, which lacks potential FOXF1 binding motifs, was used as a negative control. FOXF1 binding is shown relative to immunoglobulin G (IgG). Data represent one of two independent experiments.
(B) Schematic drawing of the pGL2-mCdh2-Luc construct containing the Cdh2 promoter region. The mouse Cdh2 promoter (−3,087/+71 bp) was cloned into the pGL2-Basic LUC vector and cotransfected with CMV-Empty vector (CMV-EV) or CMV-Foxf1. LUC assay shows FOXF1 transcriptionally activates the Cdh2 promoter. Data are representative of three independent experiments.
(C) Schematic drawings of the mouse Cadherin-11 (Cdh11) promoter. ChIP assays showed direct binding of FOXF1 to the indicated regions in the Cdh11 promoter. See also Figure S7 and Table S2.
(D) Schematic drawing of the pGL2-mCdh11-Luc construct containing the Cdh11 promoter region. The mouse Cdh11 promoter (−2,497/−137 bp) was cloned into the pGL2-Basic LUC vector and cotransfection with CMV-EV or CMV-Foxf1. LUC assay shows FOXF1 transcriptionally inhibits the Cdh11 promoter.
(E) Re-expression of CDH2 in FOXF1-deficient human CCL-210 lung fibroblasts decreased the invasion of fibroblasts through the basement membrane matrix (upper panels). CCL-210 lung fibroblasts were transfected with control siRNA, FOXF1 siRNA, control empty vector, or CDH2 OE, or cotransfected with siFOXF1 and CDH2 OE constructs. The efficiency of transfection is shown using qRT-PCR and western blot (bottom panels). Representative images and quantification of CCL-210 invasion are shown. Data presented as mean ± SEM.
(F) Depletion of CDH11 in FOXF1-deficient human lung fibroblasts decreased the invasion of fibroblasts through the basement membrane matrix (upper panels). CCL-210 fibroblasts were transfected with control siRNA, FOXF1 siRNA, or CDH11 siRNA or cotransfected with siFOXF1 and siCDH11. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S7.
(G) Schematic drawing shows loss of FOXF1 during myofibroblast differentiation is associated with the loss of CDH2 and gain of CDH11. FOXF1 directly regulates the CDH2-CDH11 cadherin switch by transcriptionally activating CDH2 and repressing CDH11. The cadherin switch is essential for myofibroblast differentiation, migration, and invasion.
ChIP-seq analysis was also used to assess whether FOXF1 bound to the −5 kb promoter region of the Cdh11 gene (Figure S7E). In contrast to Cdh2, methylation marks on Cdh11 demonstrated significant tri-methylation of histone 3 lysine 27 (H3K27) in the same binding region of Cdh11 (Figure S7E), indicating that FOXF1 may repress transcription from this site. Four potential FOXF1 binding sites were found in the Cdh11 promoter region (Figure 7C; Table S2). FOXF1 directly bound to the −2,401/−2,146 bp mouse Cdh11 promoter region (which contain two FOXF1-binding motifs), as shown by ChIP (Figure 7C; Table S2). To test whether FOXF1 directly inhibits Cdh11 transcription, we cloned the −2,497/−137 bp Cdh11 promoter region, which contains high-affinity FOXF1 binding sites, into the pGL2-Basic LUC vector to generate the pGL2-Cdh11-Luc reporter (Figure 7D; Table S2). FOXF1 decreased transcriptional activity of the Cdh11 promoter region, indicating that FOXF1 is a repressor of the Cdh11 gene (Figure 7D). Altogether, these data demonstrate that FOXF1 activates Cdh2 but inhibits Cdh11 transcription, regulating a switch between these cadherins during pulmonary fibrogenesis.
FOXF1 Regulates Fibroblast Invasion through Activation of CDH2 and Repression of CDH11
Because FOXF1 activates CDH2 and represses CDH11 during lung fibrogenesis in mice and in IPF patients, we assessed whether increased invasion of FOXF1-deficient myofibroblasts depends on CDH2 and CDH11. FOXF1 was depleted from human CCL-210 lung fibroblasts, and migration was assessed in vitro. Inhibition of FOXF1 increased fibroblast invasion through basement membrane matrixes (Figure 7E). Re-expression of CDH2 or inhibition of CDH11 reduced invasion of FOXF1-defi-cient lung fibroblasts, indicating that CDH2 and CDH11 function downstream of FOXF1 to regulate fibroblast invasion (Figures 7E and 7F). In reciprocal experiments, overexpression of FOXF1 decreased fibroblast invasion through basement membrane matrix (Figures S7F and S7G). Inhibition of CDH2 or overexpression of CDH11 in CCL-210 fibroblasts expressing exogenous FOXF1 was sufficient to restore fibroblast invasion to the levels found in control cells (Figures S7F and S7G). These rescue experiments identified CDH2 and CDH11 as key targets of FOXF1 in human lung fibroblasts. Collectively, our mouse and human lung data suggest that FOXF1 inhibits pulmonary fibrosis by decreasing myofibroblast differentiation, migration, and invasion through transcriptional regulation of CDH2 and CDH11 (Figure 7G).
DISCUSSION
IPF is a lethal fibrotic lung disease with limited therapies. Treatments that target fibroblast populations and fibrogenic pathways have proved most effective to halt IPF expansion (Canestaro et al., 2016). Identifying molecular mechanisms that regulate stromal cell activation are critical to provide novel therapeutics for IPF (Scotton and Chambers, 2007). In the present study, we identified FOXF1 protein as an anti-fibrotic factor that regulates fibroblast functions in vitro and in vivo. Deletion of FOXF1 during fibroblast-to-myofibroblast transition exacerbates bleomycin-induced lung fibrosis by regulating Cdh2 to Cdh11 cadherin switching and increasing migration and invasion of myofibroblasts.
IPF is triggered by recurrent epithelial cell injury that induces unrestrained differentiation of myofibroblasts, the key effector cells in fibrosis. Myofibroblasts express αSMA stress fibers, deposit collagen, and localize to fibrotic foci, which are the active sites of IPF (Selman et al., 2001; Ahluwalia et al., 2014). We showed that FOXF1 is expressed in normal lung fibroblasts but decreased during myofibroblast activation and fibrogenesis. At late stages of lung fibrosis, FOXF1 was absent in myofibroblasts within fibrotic foci, a finding consistent with previous studies (Melboucy-Belkhir et al., 2014). However, the same group reported that IPF fibroblasts expressed FOXF1 in vitro, which is inconsistent with their findings in IPF fibrotic foci in vivo. These inconsistencies in FOXF1 expression levels may reflect different biological conditions in fibrotic lungs in vivo compared to cell cultures in vitro or different protocols used for isolation and culture of lung fibroblasts. The data presented in this manuscript using conditional KO mice clearly show that FOXF1 acts as an anti-fibrotic factor in vivo and in vitro.
To understand the role of FOXF1 in lung fibrogenesis, we specifically deleted Foxf1 in αSMA-expressing myofibroblasts during bleomycin-induced lung fibrosis. Our current data show that FOXF1 expression in fibroblasts controls key pathogenic functions that contribute to IPF. FOXF1-deficient fibroblasts isolated from fibrotic lung had a greater capacity to migrate to and invade the matrix. The importance of fibroblast migration and invasion in the pathogenesis of IPF has been highlighted in histological observations (Selman et al., 2001). In IPF, fibroblastic foci are usually located distal to alveolar basement membranes and under poorly adherent alveolar epithelium. The locations of these lesions suggest that fibroblasts and myofibroblasts must migrate and invade through damaged epithelial basement membranes to enter alveolar spaces, where they accumulate and form fibroblastic foci.
Cadherins are adhesion molecules that mediate calcium-dependent, homophilic cell-to-cell adhesion and regulate a variety of processes in a tissue-type- and cell-type-specific context. CDH2 and CDH11 have been described as mesenchymal-specific cadherins (Chang et al., 2011; Agarwal et al., 2008) and are critical regulators of mesenchymal stem cell differentiation and fate decisions (Alimperti and Andreadis, 2015). Primary fibroblasts changed cadherin expression from CDH2 to CDH11 in αSMA-positive myofibroblasts during dermal wound healing. Myofibroblast differentiation was associated with increased CDH11 and decreased CDH2 levels, as well as changes in functional phenotypes of pathogenic myofibroblasts (Pittet et al., 2008; Hinz et al., 2004). After epithelial injury, profibrotic mediators induce EMT and regulate CDH2 to promote fibrosis (Tanjore et al., 2009; Balli et al., 2013). However, it is not clear whether changes in CDH2 levels are essential for the development of fibrosis. CDH11 contributes to pulmonary fibrosis by regulating transforming growth factor β (TGF-β) production in alveolar macrophages and EMT in alveolar epithelial cells (Schneider et al., 2012). However, the role of CDH11-expressing fibroblasts in fibrosis is unknown. In the present study, we demonstrated that FOXF1 is directly bound to Cdh2 and Cdh11 promoters and differentially regulates transcription of these genes. We also showed that FOXF1 controls fibroblast invasion in vitro via CDH2 and CDH11. The action of cadherins involves both homophilic cell-to-cell adhesion and actin cytoskeleton organization critical for cell motility and invasion. Both CDH2 and CDH11 bind to β-catenin and regulate the Wnt/β-catenin pathway. This is in line with several reports that CDH2 and CDH11 mediate migration and invasion in a variety of cell types (Valencia et al., 2004; Kaur et al., 2012). Thus, on the basis of our findings, FOXF1 and its downstream targets, CDH2 and CDH11, may represent promising targets for anti-fibrotic therapy in progressive fibrotic diseases, including IPF.
In normal lung, αSMA marks perivascular and peribronchiolar smooth muscle cells, which are localized as sheaths around bronchi and blood vessels, as well as myofibroblasts that are discontinuously located at the alveolar entrance ring. Because αSMA-CreERT2 also targets smooth muscle cells, it is possible that Foxf1 deletion from smooth muscle cells can contribute to increased lung fibrosis in FOXF1-deficient mice, possibly through release of paracrine inflammatory mediators (Kendall and Feghali-Bostwick, 2014). However, we did not observe changes in the percentage of smooth muscle cells in FOXF1-deficient lungs, indicating that FOXF1 may have distinct functions in myofibroblasts and smooth muscle cells. We also observed increased inflammation in bleomycin-treated myoFoxf1 KO lungs. Inflammatory responses are triggered during fibrogenesis. An increase in bronchoalveolar (BAL) immune cells number has been identified as a predictive biomarker of progressive lung diseases, including IPF (Wynn and Ramalingam, 2012; Shen et al., 1988). Therefore, increased inflammation may contribute to profibrotic responses in FOXF1-deficient mice.
FOX transcription factors regulate several biological processes including cell proliferation, tumor development, tissue repair, organ development, and homeostasis (Costa et al., 2001; Kalin et al., 2011). FOXM1 is a well-known transcriptional activator of cell-cycle regulatory genes, such as Cdc25B, Cyclin B1, Plk1, Topo2, and Aurora B kinase (Wang et al., 2005). FOXM1 is overexpressed in many human cancers and solid tumors (Cai et al., 2013; Balli et al., 2011; Kalin et al., 2011). FOXM1 promotes lung fibrosis by inducing EMT and stimulating pulmonary inflammation through increased expression of pro-inflammatory mediators (Balli et al., 2013). In the current study, FOXM1 was increased in FOXF1-deficient fibroblast fractions, a finding consistent with increased cell proliferation in myoFoxf1 KO fibrotic foci. We found that myoFoxf1 KO mice had decreased fibroblast but increased myofibroblast populations. Because proliferation and the percentage of myofibroblasts increased in bleomycin-injured myoFoxf1 KO mice, it is possible that the change in the ratios of fibroblasts to myofibroblasts can contribute to increased migration and invasion of myoFoxf1 KO fibroblasts in vitro. This suggests a potential role of FOXM1-dependent proliferation in the progression of lung fibrosis in FOXF1-deficient mice.
Because of the lack of common markers that label heterogeneous fibroblast populations that contribute to pulmonaryfibrosis, it is technically challenging to target fibroblast populations before myofibroblast differentiation (Rock et al., 2011; Balli et al., 2013; Dulauroy et al., 2012; Hung et al., 2013). Future studies identifying markers that are shared by fibroblast subsets are critical to better define fibroblast populations important for fibrosis.
In summary, FOXF1 inhibits pulmonary fibrosis by preventing CDH2-CDH11 cadherin switching and regulating myofibroblast migration and invasion. On the basis of these findings, FOXF1 and its downstream effectors may represent promising targets for anti-fibrotic therapy in IPF.
EXPERIMENTAL PROCEDURES
Transgenic Mice and Bleomycin-Induced Pulmonary Fibrosis Models
The Foxf1fl/fl mouse line was generated as 129/C57BL/6 hybrids and bred into the C57BL/6 mouse background (Ren et al., 2014). Foxf1fl/fl mice were bred with αSMA-Cre-ERT2 mice (Wendling et al., 2009) to generate αSMA-Cre-ERT2; Foxf1fl/fl (abbreviated as myoFoxf1 KO) mice. Single transgenic Foxf1fl/fl litter-mates were used as controls. Both male and female mice aged 8–12 weeks were used for all experiments. ROSA26R(mTmG/mTmG) mice (Jackson Laboratory) were crossed with αSMA-Cre-ERT2 mice to generate mTmG;αSMA-CreERT2 mice. For bleomycin-induced fibrosis studies, mice were administered 1 or 2 U/kg of bleomycin sulfate (EMD Biosciences) intratracheally one time per week for 2, 3, or 4 weeks. Tam was given (40 mg/kg; Sigma) by intraperitoneal injection on days 2, 3, and 4 after bleomycin treatment. Mice were harvested 1 week after final bleomycin treatment. Quantification of lung collagen content was performed using the Sircol collagen assay (Biocolor, Carrickfergus, UK) and the HydroxyprolineAssay Kit (Sigma) following the manufacturers’ protocols.
Human Lung Samples
Lung samples were obtained at the University of Vienna and the University of Giessen Lung Center from patients with IPF (n = 9), and control organ donors (n = 6) were obtained anonymously following lung transplantation. Diagnosis was done according to the American Thoracic Society/European Respiratory Society (ATS/ERS) criteria for IPF. Utilization of human lung samples was reviewed and approved by the Ethics Committee of Justus-Liebig-University of Giessen.
Immunohistochemistry, Immunofluorescence Staining, and Real-Time qRT-PCR
Lung paraffin sections were stained with H&E, and immunofluorescence staining was performed as previously described (Ren et al., 2010). Images were obtained using a Zeiss AxioPlan 2 microscope. Antibody information is shown in Table S1. The RNeasy Mini Kit (QIAGEN) was used to isolate total RNA from total lung lysates or isolated fibroblasts. qRT-PCR was performed using the StepOnePlus Real-Time PCR system (Applied Biosystems) as previously described (Cai et al., 2016). TaqMan probes used are listed in Table S1.
Isolation of Mouse Fibroblasts
Mouse lung fibroblasts were isolated from mouse lungs as previously described (Tager et al., 2004; Jiang et al., 2010).
Flow Cytometry
Flow cytometry experiments were conducted as previously described (Ren et al., 2010; Kalin et al., 2008). Table S1 lists antibodies used. Fibroblast sub-populations were identified as CD45−CD326−CD31−MHY11−PDGFRa+. Myofibroblasts were defined as CD45−CD326−CD31−MHY11−αSMA+. Smooth muscle cell populations were defined as CD45−CD326−CD31−MHY11+αSMA+. Cells were permeabilized and stained with fluorescence-labeled antibodies (Table S1). 7AAD (eBioscience) or fixed viability dye (Thermo Fisher Scientific) was used to label dead cells. Flow cytometry data were acquired using FACSCanto II or LSR II (BD Biosciences).
Transwell Migration and Invasion Assays
Migration and invasion of human or mouse cells were performed as described previously (Jiang et al., 2010; Li et al., 2011). 100,000 fibroblasts were seeded in 0.5% fetal bovine serum (FBS) media onto an 8 μm pore permeable transwell insert or BioCoat Matrigel Invasion Chamber (BD), and cell migration or invasion was performed in the presence of 10% FBS complete medium. After 48 hr, media were removed, and the polycarbonate filters with the migrated or invaded cells were stained with crystal violet. The migrating or invading cells of each sample were counted in ten randomly selected fields.
Western Blot
Western blot analysis was done as described previously (Cheng et al., 2014). The full primary antibody list is shown in Table S1. β-actin was used as loading control. The signals from the primary antibody were amplified by horseradish peroxidase (HRP)-conjugated immunoglobulin G (IgG) (Bio-Rad) and detected with Pierce ECL western blotting substrate (Thermo Fisher Scientific) followed by autoradiography.
RNA-Seq Analysis
RNAfrom fibroblasts isolated from bleomycin-treated Foxf1fl/fl and myoFoxf1KO mouse lungs was used for RNA-seq analysis. Genes were prefiltered by counts > 20 and reads per kilobase million (RPKM) > 1 in at least one sample. Differentially expressed genes were identified using DESeq with fold change > 2 and an adjusted p value (FDR) < 0.01, and the p value was calculated using Z test (Pearson’s chi-square test) in the Agilent GeneSpring GX suite (Anders et al., 2012). Differentiated expressed genes were analyzed using the ToppGene Suite (Chen et al., 2009). The statistical significance of each bioprocess was calculated using a negative logarithm (−log10) of the p value. The heatmap was constructed on differentially expressed genes using JMP Genomics 6.0.
Constructs and Transfection Studies
NIH 3T3 and normal human lung fibroblast CCL-210 (ATCC) cells were cultured as described previously (Wang et al., 2013). Plasmid and small interfering RNA (siRNA) transfections were performed using Lipofectamine 2000 (Invitrogen). For transient knockdown, siRNAs targeting either open reading frame (ORF) (mouse, 5′-GAAAGGAGUUUGUCUUCU C-3′; human, 5′-GGAA AUGCCAGGCGCUCAAUU-3′) or siFoxF1-3′UTR (mouse, 5′-CCAGAUACGU GGAAA GAAUUU-3′; human, 5′-GCAGAAAGGUUAAGG CACUUU-3′) were purchased from Dharmacon. For retroviral-mediated stable knockdown of Foxf1, short hairpin RNA (shRNA) targeting the 3′ UTR of mouse Foxf1 was used (5′-AAATGTTAGTGGTGGGTCTGA-3′). Stable cell lines were generated using lentiviruses carrying either pLKO.1:Puro-shControl or pLKO.1:Puro-shFoxf1-3′UTR followed by puromycin selection (Pradhan et al., 2016; Milewski et al., 2017b). Murine Foxf1 cDNAs were PCR amplified, and N-terminal FLAG and C-terminal His tags were introduced by using PCR as previously described (Milewski et al., 2017a). PCR was performed using AccuPrime Pfx DNA Polymerase according to the manufacturer’s protocol (Invitrogen). The retroviral and lentiviral particles were made in Cincinnati Children’s Viral Vector Core facility, and the generation of stable cell lines was done as described previously (Milewski et al., 2017a).
ChIP and LUC Assays
ChIP was performed as previously described (Cai et al., 2016). Stable NIH 3T3 cells overexpressing FOXF1 were crosslinked and sonicated to create DNA fragments between 300 and 500 bp. Protein/DNA complexes were used for immunoprecipitation with FOXF1 antibody (R&D Systems) or control goat IgG (Vector Laboratories). Reverse crosslinked ChIP DNA samples were subjected to real-time PCR, using specified primer sets outlined in Table S2. The −3,087/+71 kb region of the mouse Cdh2 promoter (NC_000084.6) was PCR amplified and cloned into the pGL2-Basic LUC vector using the following primers: 5′-TGCAGACTGAAGTTAACTGCAGAG-3′ and 5′-TGCTCGAGGCC GTGCGCGTT-3′. The −2,497/−137 kb region of the mouse Cdh11 promoter (NC_000074.6) was cloned using the following primers: 5′-CAAGTGTGCA GCGCCACTTGA-3′ and 5′-GCTAGTGGCAGGAATGAG AAAC-3′. LUC constructs were previously described (Milewski et al., 2017b). HEK293T cells were transfected with CMV-Foxf1 or CMV-empty plasmids, as well as with LUC constructs. CMV-Renilla was used as an internal control to normalize transfection efficiency.
Statistics
Statistical significance differences in measured variables between experimental and control groups were assessed by Student’s t test (two-tailed) or one-way ANOVA with Bonferroni test. p < 0.05 was considered significant. Values for all measurements were expressed as mean ± SD or as mean ± SEM. Statistical analysis was performed and data were graphically displayed using GraphPad Prism v.5.0 for Windows (GraphPad).
Study Approval
All animal studies were approved by Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee and covered under our animal protocol (IACUC2016-0070). Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee is an AAALAC- and NIH-accredited institution (NIH Insurance No. 8310801).
Supplementary Material
Highlights.
FOXF1 is decreased in human IPF and mouse myofibroblasts from fibrotic lung
Loss of FOXF1 increases collagen production and invasion of lung myofibroblasts
FOXF1 differentially regulates transcription of CDH2 and CDH11 cadherins
Myofibroblast invasion depends on FOXF1-regulated switch from CDH2 to CDH11
Acknowledgments
We thank Pierre Chambon and Anna-Mae Diehl for αSMA-CREERT2 mice, the CCHMC Gene Expression Core for technical assistance with RNA-seq, and Yina Du for bioinformatics analysis. We appreciate Andreas Günther and co-workers of Giessen University, who provided IPF tissues. This work was supported by NIH grants T32-HR007752 (to M.B.), R01HL132849 (to T.V.K.), R56HL126660 (to T.V.K.), RO1HL84151 (to V.V.K.), and RO1HL123490 (to V.V.K.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE110408.
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.067.
AUTHOR CONTRIBUTIONS
M.B. and T.V.K. conceived and designed the study. M.B. and T.L. performed the in vivo experiments. M.B., D.M., and X.R. participated in in vitro and culture experiments. Y.X. designed and performed the RNA-seq bioinformatics analysis. M.B., D.M., and T.V.K. analyzed data. M.B., D.M., X.R., Y.X., V.V.K., and T.V.K. interpreted the data. V.V.K. provided critical reagents and intellectual discussions. M.B. and T.V.K. wrote the paper. All authors discussed the data. T.V.K. approved the submission of the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Agarwal SK, Lee DM, Kiener HP, Brenner MB. Coexpression of two mesenchymal cadherins, cadherin 11 and N-cadherin, on murine fibroblast-like synoviocytes. Arthritis Rheum. 2008;58:1044–1054. doi: 10.1002/art.23369. [DOI] [PubMed] [Google Scholar]
- Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190:867–878. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimperti S, Andreadis ST. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem Cell Res. 2015;14:270–282. doi: 10.1016/j.scr.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balli D, Zhang Y, Snyder J, Kalinichenko VV, Kalin TV. Endothelial cell-specific deletion of transcription factor FoxM1 increases urethane-induced lung carcinogenesis. Cancer Res. 2011;71:40–50. doi: 10.1158/0008-5472.CAN-10-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balli D, Ren X, Chou FS, Cross E, Zhang Y, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene. 2012;31:3875–3888. doi: 10.1038/onc.2011.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32:231–244. doi: 10.1038/emboj.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bolte C, Ren X, Tomley T, Ustiyan V, Pradhan A, Hoggatt A, Kalin TV, Herring BP, Kalinichenko VV. Forkhead box F2 regulation of platelet-derived growth factor and myocardin/serum response factor signaling is essential for intestinal development. J Biol Chem. 2015;290:7563–7575. doi: 10.1074/jbc.M114.609487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Balli D, Ustiyan V, Fulford L, Hiller A, Misetic V, Zhang Y, Paluch AM, Waltz SE, Kasper S, Kalin TV. Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. J Biol Chem. 2013;288:22527–22541. doi: 10.1074/jbc.M113.455089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Bolte C, Le T, Goda C, Xu Y, Kalin TV, Kalinichenko VV. FOXF1 maintains endothelial barrier function and prevents edema after lung injury. Sci Signal. 2016;9:ra40. doi: 10.1126/scisignal.aad1899. [DOI] [PubMed] [Google Scholar]
- Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug Treatment of Idiopathic Pulmonary Fibrosis: Systematic Review and Network Meta-Analysis. Chest. 2016;149:756–766. doi: 10.1016/j.chest.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci USA. 2011;108:8402–8407. doi: 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman HA. Epithelial responses to lung injury: role of the extracellular matrix. Proc Am Thorac Soc. 2012;9:89–95. doi: 10.1513/pats.201112-053AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XH, Black M, Ustiyan V, Le T, Fulford L, Sridharan A, Medvedovic M, Kalinichenko VV, Whitsett JA, Kalin TV. SPDEF inhibits prostate carcinogenesis by disrupting a positive feedback loop in regulation of the Foxm1 oncogene. PLoS Genet. 2014;10:e1004656. doi: 10.1371/journal.pgen.1004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RH, Kalinichenko VV, Lim L. Transcription factors in mouse lung development and function. Am J Physiol Lung Cell Mol Physiol. 2001;280:L823–L838. doi: 10.1152/ajplung.2001.280.5.L823. [DOI] [PubMed] [Google Scholar]
- Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18:1262–1270. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- Emblom-Callahan MC, Chhina MK, Shlobin OA, Ahmad S, Reese ES, Iyer EPR, Cox DN, Brenner R, Burton NA, Grant GM, Nathan SD. Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics. 2010;96:134–145. doi: 10.1016/j.ygeno.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol Biol Cell. 2004;15:4310–4320. doi: 10.1091/mbc.E04-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM, Duffield JS. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188:820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002;83:111–119. doi: 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Liang J, Campanella GS, Guo R, Yu S, Xie T, Liu N, Jung Y, Homer R, Meltzer EB, et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J Clin Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin TV, Meliton L, Meliton AY, Zhu X, Whitsett JA, Kalinichenko VV. Pulmonary mastocytosis and enhanced lung inflammation in mice heterozygous null for the Foxf1 gene. Am J Respir Cell Mol Biol. 2008;39:390–399. doi: 10.1165/rcmb.2008-0044OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinichenko VV, Zhou Y, Bhattacharyya D, Kim W, Shin B, Bambal K, Costa RH. Haploinsufficiency of the mouse Forkhead Box f1 gene causes defects in gall bladder development. J Biol Chem. 2002a;277:12369–12374. doi: 10.1074/jbc.M112162200. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Zhou Y, Shin B, Stolz DB, Watkins SC, Whitsett JA, Costa RH. Wild-type levels of the mouse Forkhead Box f1 gene are essential for lung repair. Am J Physiol Lung Cell Mol Physiol. 2002b;282:L1253–L1265. doi: 10.1152/ajplung.00463.2001. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Bhattacharyya D, Zhou Y, Gusarova GA, Kim W, Shin B, Costa RH. Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–117. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- Kaur H, Phillips-Mason PJ, Burden-Gulley SM, Kerstetter-Fogle AE, Basilion JP, Sloan AE, Brady-Kalnay SM. Cadherin-11, a marker of the mesenchymal phenotype, regulates glioblastoma cell migration and survival in vivo. Mol Cancer Res. 2012;10:293–304. doi: 10.1158/1541-7786.MCR-11-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, Noble PW. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208:1459–1471. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovgren AK, Kovacs JJ, Xie T, Potts EN, Li Y, Foster WM, Liang J, Meltzer EB, Jiang D, Lefkowitz RJ, Noble PW. β-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci Transl Med. 2011;3:74ra23. doi: 10.1126/scitranslmed.3001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin D, Kim IM, Boetticher E, Kalin TV, Ramakrishna S, Meliton L, Ustiyan V, Zhu X, Kalinichenko VV. Forkhead box F1 is essential for migration of mesenchymal cells and directly induces integrin-beta3 expression. Mol Cell Biol. 2007;27:2486–2498. doi: 10.1128/MCB.01736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melboucy-Belkhir S, Pradère P, Tadbiri S, Habib S, Bacrot A, Brayer S, Mari B, Besnard V, Mailleux A, Guenther A, et al. Forkhead Box F1 represses cell growth and inhibits COL1 and ARPC2 expression in lung fibro-blasts in vitro. Am J Physiol Lung Cell Mol Physiol. 2014;307:L838–L847. doi: 10.1152/ajplung.00012.2014. [DOI] [PubMed] [Google Scholar]
- Milewski D, Balli D, Ustiyan V, Le T, Dienemann H, Warth A, Breuhahn K, Whitsett JA, Kalinichenko VV, Kalin TV. FOXM1 activates AGR2 and causes progression of lung adenomas into invasive mucinous adenocarcinomas. PLoS Genet. 2017a;13:e1007097. doi: 10.1371/journal.pgen.1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewski D, Pradhan A, Wang X, Cai Y, Le T, Turpin B, Kalinichenko VV, Kalin TV. FoxF1 and FoxF2 transcription factors synergistically promote rhabdomyosarcoma carcinogenesis by repressing transcription of p21Cip1CDK inhibitor. Oncogene. 2017b;36:850–862. doi: 10.1038/onc.2016.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R, Sridhar S, Tyagi G, Phillips JE, Garrido R, Harris P, Burns L, Renteria L, Woods J, Chen L, et al. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS ONE. 2013;8:e59348. doi: 10.1371/journal.pone.0059348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet P, Lee K, Kulik AJ, Meister JJ, Hinz B. Fibrogenic fibroblasts increase intercellular adhesion strength by reinforcing individual OB-cadherin bonds. J Cell Sci. 2008;121:877–886. doi: 10.1242/jcs.024877. [DOI] [PubMed] [Google Scholar]
- Pradhan A, Ustiyan V, Zhang Y, Kalin TV, Kalinichenko VV. Forkhead transcription factor FoxF1 interacts with Fanconi anemia protein complexes to promote DNA damage response. Oncotarget. 2016;7:1912–1926. doi: 10.18632/oncotarget.6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Zhang Y, Snyder J, Cross ER, Shah TA, Kalin TV, Kalinichenko VV. Forkhead box M1 transcription factor is required for macrophage recruitment during liver repair. Mol Cell Biol. 2010;30:5381–5393. doi: 10.1128/MCB.00876-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Ustiyan V, Pradhan A, Cai Y, Havrilak JA, Bolte CS, Shannon JM, Kalin TV, Kalinichenko VV. FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circ Res. 2014;115:709–720. doi: 10.1161/CIRCRESAHA.115.304382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzoni EA, Abraham DJ, Howat S, Shi-Wen X, Sestini P, Bou-Gharios G, Wells AU, Veeraraghavan S, Nicholson AG, Denton CP, et al. Gene expression profiling reveals novel TGFbeta targets in adult lung fibroblasts. Respir Res. 2004;5:24. doi: 10.1186/1465-9921-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DJ, Wu M, Le TT, Cho SH, Brenner MB, Blackburn MR, Agarwal SK. Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J. 2012;26:503–512. doi: 10.1096/fj.11-186098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- Selman M, King TE, Jr, Pardo A American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- Shen AS, Haslett C, Feldsien DC, Henson PM, Cherniack RM. The intensity of chronic lung inflammation and fibrosis after bleomycin is directly related to the severity of acute injury. Am Rev Respir Dis. 1988;137:564–571. doi: 10.1164/ajrccm/137.3.564. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, Polosukhin V, Zhao LH, Sakamoto H, Blackwell TS, Luster AD. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia X, Higgins JMG, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, Watts GFM, Mizoguchi E, Simmons B, Patel DD, et al. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004;200:1673–1679. doi: 10.1084/jem.20041545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker N, Badri L, Wettlaufer S, Flint A, Sajjan U, Krebsbach PH, Keshamouni VG, Peters-Golden M, Lama VN. Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am J Pathol. 2011;178:2461–2469. doi: 10.1016/j.ajpath.2011.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IC, Ustiyan V, Zhang Y, Cai Y, Kalin TV, Kalinichenko VV. Foxm1 transcription factor is required for the initiation of lung tumorigenesis by oncogenic KrasG12D. Oncogene. 2013;33:5391–5396. doi: 10.1038/onc.2013.475. [DOI] [PubMed] [Google Scholar]
- Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. doi: 10.1002/dvg.20448. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W, Kumar M, Xiao R, D’Armiento J, Metzger D, et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest. 2016;126:3063–3079. doi: 10.1172/JCI85328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.