

Abstract
The Conserved Oligomeric Golgi (COG) complex controls membrane trafficking and ensures Golgi homeostasis by orchestrating retrograde vesicle trafficking within the Golgi. Human COG defects lead to severe multi-systemic diseases known as COG-Congenital Disorders of Glycosylation (COG-CDG).
To gain better understanding of COG-CDGs we compared COG knockout cells to cells deficient to two key enzymes, Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase (MGAT1) and UDP-glucose 4-epimerase (GALE), which contribute to proper N- and O-glycosylation. While all knockout cells share similar defects in glycosylation, these defects only account for a small fraction of observed COG knockout phenotypes. Glycosylation deficiencies were not associated with the fragmented Golgi, abnormal endolysosomes, defective sorting and secretion or delayed retrograde trafficking indicating that these phenotypes are likely not due to hypoglycosylation, but to other specific interactions or roles of the COG complex.
Importantly, these COG deficiency specific phenotypes were also apparent in COG7-CDG patient fibroblasts proving the human disease relevance of our CRISPR knockout findings. The knowledge gained from this study has important implications, both for understanding the physiological role of COG complex in Golgi homeostasis in eukaryotic cells, and for better understanding human diseases associated with COG/Golgi impairment.
Keywords: COG complex, Golgi, glycosylation, CDG, COG-CDG, MGAT1, GALE, endolysosome, Lamp2, Cathepsin D
Graphical Abstract
Introduction
The conserved oligomeric Golgi (COG) complex is a master regulator of membrane trafficking at the Golgi 1–9. The COG complex influences a variety of processes through its many interactions with trafficking machinery. The most noted of effects of COG deficiency is misglycosylation10–13. In fact the deficiency/malfunction of the COG complex in humans is classified as a congenital disorder of glycosylation type-II (COG-CDG) or a congenital disorder of glycosylation affecting multiple pathways10,14–20. CDGs are a heterogeneous group of disorders with a wide variety of phenotypes dependent on the protein affected and the severity of the mutation in the affected protein. CDG patients are often identified due to an unexplained early onset of symptoms that have no traditional explanation, leading to genetic testing or to serum glycan profiling if a CDG is suspected. A congenital disorder of glycosylation is confirmed if serum proteins transferrin and apolipoprotein E (ApoE) (N- and O- glycosylated, respectively) are found to be underglycosylated. If transferrin alone is affected then a CDG-N-linked is diagnosed, if both are underglycosylated the patient has CDG-multiple pathway disorder. Patients with CDG-multiple pathway disorders can have misglycosylation of N- O- and lipid linked glycans 21. COG-CDG patients fall under this category. These patients suffer from severe multisystemic symptoms that affect the nervous system, liver, and skin (For review see: 22–24).
Though COG patients fit the diagnostic criteria for a CDG (having underglycosylated proteins/lipids) the COG complex is different from most proteins whose mutations cause CDGs in that it is primarily a vesicle trafficking molecule and not a glycosylation enzyme or sugar transporter, making its impact on glycosylation is secondary 25. We thus reasoned that COG deficient patients would not only have defects related to misglycosylation but also trafficking related defects that are independent of misglycosylation.
In order to elucidate COG’s role at the cellular level, we previously created CRISPR-mediated KOs of each COG subunit in human HEK293T cells and characterized these cells extensively26. We found that deficiency in any COG subunit caused severe misglycosylation, similar to that seen in patient samples. Additionally, trafficking disturbances as well as Golgi structure abnormalities were present in all COG subunit KOs. These changes overlapped well with findings in COG-CDG patient fibroblasts and COG deficient CHO cells 27–29.
In this study, we sought to uncover which of these COG KO phenotypes were downstream of hypoglycosylation and which where independent of it. To answer this question, we chose to knock-out two non-redundant genes, Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase (MGAT1) and UDP-glucose 4-epimerase (GALE) in the same model cell line. These KOs cause N- or N- and O-glycosylation defects, similar to COG KO glycosylation defects. We then characterized these new KOs alongside the COG KOs.
MGAT1 KO defects were first characterized in CHO cells (named Lec1 cells). These cells were obtained due to their resistance to various sugar-binding toxins as a result of defects in the N-glycosylation pathway. It was later discovered that the defective gene in Lec1 cells was MGAT1, a glycosylation enzyme that is responsible for adding the first GlcNAc to the growing N-glycan chain in the Golgi apparatus 30,31. Thus, without this enzyme only immature trimmed high mannose N-glycans are produced32. This gene was chosen due to being early in the N-glycosylation pathway in the Golgi meaning the effects of its deletion on N-glycosylation should be more severe than COG’s intermediate effects on glycosylation. Additionally, because it only affects N-glycosylation, this KO allows for the teasing apart of the contribution of various glycan classes (N- or O-glycans) to MGAT1, GALE, and COG KO cell phenotypes. There has yet to be a patient identified with an MGAT1 mutation, but MGAT1 KO in mice was found to be embryonic lethal, likely due to neural tube closure defects indicating the severity of the glycosylation defects produced when this enzyme is absent.33–35.
GALE deficiency was first characterized in CHO cells, where it was found to be an essential gene for LDL receptor stability, and was thus named ldlD 36. These cells were later shown to have a mutation in the gene encoding GALE, an epimerase that interconverts glucose and galactose as well as GalNAc and GlcNAc in the cytosol 37. In humans, mutations in GALE cause galactosemia type III, due to a buildup of galactose obtained from a regular diet that cannot be broken down due to the inability to convert this galactose to glucose. In standard culture conditions the primary sugar mammalian cells are supplemented with is glucose, so instead of a toxic buildup of galactose (as in the GALE null type III galactosemia patients) cells have little/no galactose/GalNAc causing defects in N- O- and lipid linked glycosylation, similar to COG mutants. In fact, COG1(LDLB) and COG2 (LDLC) were found in the same screen as GALE (LDLD) for all affecting the stability of the LDL receptor due to similar changes in glycosylation when mutated 38.
Using MGAT1 and GALE KOs along with COG KOs previously made in the same parental cell line (HEK293T) allowed for assessing which COG KO phenotypes resulted from misglycosylation due to mistrafficking and altered stability of glycosylation enzymes and which arose from other COG defects unrelated to glycosylation. Detailed analysis of HEK293T KO cells, using superresolution and electron microscopy, flow cytometry, lectin western blots (WB) and toxin retrograde trafficking assays, revealed that, on the cellular level, only a small subset of COG KO phenotypes are recapitulated in MGAT1 and GALE KOs, suggesting that COG dysfunction has significant influences that go beyond glycosylation defects.
Results
COG KO affects stability of key Golgi glycosylation enzymes in HEK293T cells
We and others 1,6,11–13,39–46 have proposed that COG deficiencies are affecting glycosylation via regulation of intra-Golgi trafficking of Golgi glycosylation machinery. Intracellular localization of MGAT1 13,40,41 and MAN2A1/Mann II 13,47 was previously shown to be severely affected in HeLa cells transiently knocked down for COG complex subunits. Previously we also reported that sialylation and fucosylation are decreased in COG KO cells while terminal mannose residues are increased 12. To gain a better understanding of which glycosylation enzymes are affected in COG KOs we probed knockout lysates for key glycosylation enzymes by western blot (WB). We found that nearly all tested Golgi enzymes are COG sensitive (Figure 1). In COG KO cells, MGAT1, B4GalT1 and ST6Gal1 all are dramatically decreased in abundance, while B4galT5 appears to be insensitive to COG malfunction (Figure 1A and B).
Figure 1. COG depletion severely affects protein levels of key Golgi glycosylation enzymes.

Western blot (A) and quantification (B) of WT and COG KO lysates probed for MGAT1, B4GalT1, B4GalT5, ST6GalT1 and actin (n=3). 10ug of total protein was loaded for each sample. * denotes significant difference from WT where p < 0.05. Protein levels are expressed in arbitrary units (AU) with WT being 100%. Actin was used as a loading control.
We then decided to select a pair of non-redundant glycosylation-related enzymes whose depletion would mimic COG KO misglycosylation patterns to elucidate which COG KO phenotypes would be recapitulated by hypoglycosylation alone. MGAT1, B4GalT1 and ST6Gal1 all operate in the N-glycosylation pathway and MGAT1 activity is necessary for subsequent addition of galactose and sialic acid, so MGAT1 was chosen to represent COG effects on N-glycosylation. In addition to defective N-glycosylation caused by reduced levels of MGAT1, B4GalT1, and ST6Gal1 COG deficiency also affects O- and lipid linked glycosylation, as previously stated. To model the effects of COG depletion on O-glycosylation as well as the effects of late stage N-glycosylation defects we chose GALE. GALE is a cytosolic enzyme, so its stability was not affected in COG KO cells (data not shown), but due to redundancy enzymes involved in O-glycosylation 48 and due to previous reports in CHO cells indicating its similar glycosylation changes to COG mutants 49 we selected GALE to affect both N- and O-glycosylation in the Golgi.
MGAT1 and GALE are efficiently knocked out in HEK293T cells and cause hypoglycosylation
MGAT1 and GALE KOs were obtained using CRISPR/Cas9 strategy (see Materials and Methods section) to create gene-disrupting frame shift mutations resulting in the absence of the targeted protein (Figure 2A). MGAT1 and GALE KO appeared to grow at a similar rate to the parental cell line, as was previously observed for the COG KOs (26 and data not shown). In addition, both MGAT1 and GALE KOs caused the hypermobility of lysosomal Lamp2 protein on SDS-PAGE indicating protein misglycosyation. The shift for Lamp2 was more pronounced in the MGAT1 KO than the GALE KO and similar to the COG KOs, indicating that N-glycosylation contributes more to Lamp2 mobility than O-glycosylation. Inversely, Lamp2 protein stability appeared to be affected more in GALE KOs that MGAT1 indicating that O-glycosylation contributes to Lamp2 protein stability. Lamp2 stability was also affected to varying degrees in the COG KOs (again, possibly due to defects in O-glycosylation) (Figure 2B). Because Lamp2 has both N-and O-glycosylation sites and showed shifts in mobility for all KOs we next sought to further characterize changes in N- and O- glycosylation in these KO cells compared to COG KOs.
Figure 2. CRISPR/Cas9-induced KO of MGAT1 and GALE results in severe Lamp2 glycosylation and stability defects.

(A) WB analysis of HEK293T cells depleted for MGAT1 and GALE. (B) MGAT1, GALE and COG KO cell lysates were analyzed for Lamp2 glycosylation and stability. All KOs show increased Lamp2 gel mobility indicating hypoglycosylation. (C) Quantification of Lamp2 abundance in KO cells vs control (n=3). * denotes significant difference from WT levels where p < 0.05. Protein levels are expressed in arbitrary units (AU) with WT being 100%. Actin was used as a loading control.
COG KOs shares some misglycosylation patterns with MGAT1 and GALE KOs and differs on others
To further assess glycosylation defects in MGAT1, GALE, and COG KO cells we used sugar-specific lectins to probe both total cellular and plasma membrane expressed glycoconjugates. For these assays, a subset of COG KOs was chosen, including subunits from lobe A (COG2 and COG4) and B (COG7) of the COG complex. We previously showed that these knockouts all have similar glycosylation and trafficking phenotypes, so these subsets are representative of whole complex malfunction 12. Lectins chosen for this characterization were: Galanthus nivalis lectin(GNL), Griffonia simplicifolia lectin II (GS-II) and Helix pomatia agglutinin (HPA) (Figure 3A–D). GNL binds terminal 1,3 and 1,6 linked mannose residues. These residues should only be accumulated in cells where Golgi glycosylation is compromised in its early Golgi stages. As shown in Figure 3A and D, GNL binds minimally to total glycoproteins isolated from control HEK293T and GALE KO cell lysates as these glycans are processed past the high mannose stage. In contrast, GNL binds to glycoproteins in MGAT1 and COG KO lysates at much higher levels indicating that even the early stages of glycosylation at the Golgi are impaired in these cells. This MGAT1 KO phenotype is in agreement with previously published data obtained in CHO cells 31 and confirms MGAT1 impairment in both MGAT1 KO and COG KO cells, as the N-glycan structures that are synthesized in the absence of MGAT1 have 2 binding sites for GNL each. GS-II binds to terminal GlcNAc residues 50. GSII was used to assess the level of terminal GlcNAc residues in MGAT1, GALE, and COG KO cell lysates (Figure 3B and D). These residues should only be left exposed in cells that have defects in later Golgi glycosylation reactions involving the addition of galactose and sialic acid residues. GS-II had a higher affinity for glycoproteins in GALE KO cell lysates alone, as the N-glycans in these cells are not able to add galactose or sialic acid residues to their N-glycans. Though COG KOs have reduced B4GalT1 protein levels, glycoproteins isolated from all COG KO lysates did not demonstrate elevated affinity to GS-II lectin by WB. Instead, COG KO lysates have increased specific binding to HPA (Figure 3C and D). HPA binds to terminal N-acetylgalactosaminyl residues in O-glycan structures, which are partially modified O-glycans, and has been shown to bind at a high affinity to multiple kinds of cancer cells51–55. These specific O-glycan structures were absent in GALE cells (due to the absence of the initiating sugar for O-glycosylation) and buried under further processing in WT and MGAT1 KO cells.
Figure 3. Both MGAT1 and GALE KOs result in severe global glycosylation defects.

WT HEK293T cells along with COG, GALE, and MGAT1 KO cells were analyzed by lectin blot (A–C) and flow cytometry analysis (D–F) for GNL (A, D), GS-II (B, E) and HPA (C, F) binding. COG and MGAT1 KO cell lysates show increased affinity to terminal high mannose binding lectin GNL. GALE KO cell lysates show increased affinity for GlcNAc binding GS-II lectin. COG KO lysates show increased affinity to GalNAc binding HPA. (D–F) Top: Flow cytometry of fluorescent lectin binding to non-permeabilized cells. 30,000 cells were counted for each condition. Bottom: an example glycan that binds to each lectin analyzed.
The lectin blots analyze all glycoproteins in the cells regardless of localization. To complement this data, we performed flow cytometry with fluorescently-tagged lectins (Figure 3D). In this assay non-permeabilized cells were treated with lectins on ice and then analyzed for fluorescence (i.e. lectin binding levels). Using this procedure allowed us to only look at glycoproteins which had reached the cell surface, meaning that there would be no background signal from glycoprotein still undergoing modifications. Our results from this experiment were in agreement with the data obtained from lectin blots. GNL bound strongest to MGAT1 KO cells, with the COG KO cells binding highly as well, presumably due to MGAT1 instability. GSII bound the strongest to GALE KO cells as in the lectin blots, though in this assay increased binding can also be seen in COG KOs. This increase was likely below the detection level for the lectin blot assay but shows the heterogeneous nature of misglycosylation in COG KOs. HPA bound to COG KOs with a much higher affinity than any of the other cells, as seen in the lectin blot assay, indicating that not only are these O-glycan modifications increased in COG KO cells, but that abnormally modified glycoconjugates were delivered to the cell surface.
Obtained lectin WB and flow cytometry binding patterns serve as confirmation of KO of the targeted gene and show that MGAT1 and GALE have similar or more severe glycosylation defects compared to COG KOs.
COG KO specifically affects Golgi structure
We next sought to test if dramatic altering of glycosylation observed in MGAT1 and GALE knock-outs would alter Golgi structure, as the COG subunit KOs do. For this assessment, we employed superresolution fluorescent microscopy which allowed us to distinguish the fragmented Golgi structures in COG KO cells from control HEK293T Golgi ribbon structures (Figure 4) that we have previously observed by electron microscopy 26. We used GM130 (a cis-Golgi marker) and Rab6 (a trans-Golgi marker), both COG-insensitive peripheral membrane proteins 56, to analyze Golgi organization in MGAT1 and GALE KO cells compared to COG KO cells. As expected, we observed severe fragmentation of Golgi apparatus in COG2 KO cells, but we saw no visible alterations in the Golgi structure of MGAT1 or GALE KOs (Figure 4). This indicates that misglycosylation itself or its secondary effects do not cause the fragmentation of the Golgi that is seen in COG KO cells.
Figure 4. COG KO Golgi fragmentation phenotype is not related to glycosylation defects.

Airyscan super resolution microscopy of HEK293T and GALE, MGAT1 and COG4 KO cells was used to determine if the Golgi structure (marked by GM130(cis, green) and Rab6 (trans, red)) is affected in MGAT1 and GALE cells. Golgi of WT, MGAT1 and GALE KO cells are not fragmented, while COG4 KO cells demonstrate intense Golgi fragmentation. Nuclear DNA was stained with Hoechst (blue). Scale bar 10 μm. Insert 3× zoom.
To continue our analysis of the relationship between MGAT1 and GALE KO cell phenotypes to COG KO cell phenotypes we wanted to ensure that COG complex localization itself was not disturbed by MGAT1 or GALE depletion. As shown in Figure 5 the COG complex (both lobe A (COG3, green) and lobe B (COG8, red)) is still normally localized in the perinuclear area (Figure 5 arrows) and of the same abundance in MGAT1 and GALE KO cells as WT cells, while COG2 KO alters the perinuclear localization (COG8) and protein abundance (COG3) of its COG subunit partners.
Figure 5. Golgi glycosylation defects do not affect localization of the COG complex.

MGAT1, GALE, and COG KOs were fixed and stained for COG8 (red) and COG3 (green) and Hoechst was used to mark nuclear DNA (merged). All cell lines but the COG2 KO showed normal perinuclear localization of COG proteins (arrows). Scale bar 10μm.
COG KO specifically affects endolysosomal structure
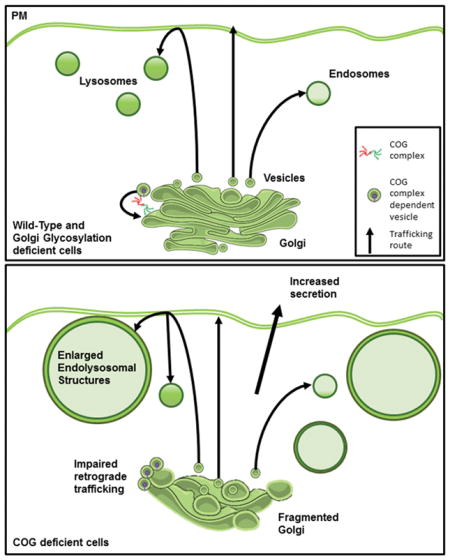
Surprisingly in COG KO cells, in addition to Golgi fragmentation, we also observed dramatic alterations of the endosomal/lysosome system. Upon KO of any COG subunit, enlarged vacuolar structures, some being in excess of 5μm across were readily visible by differential interference contrast (DIC) microscopy (Figure 6A). Upon further characterization we discovered that these structures immunostained with endogenous lysosomal protein Lamp2 (Figure 6B), as well as with Rab7 and CD63 (JBB, VL, unpublished observation), indicating they are late-endosomal and/or lysosomal in origin. We next sought demonstrate that the enlarged structures visible by DIC microscopy were the same Lamp2 positive structures in fixed and stained cells. To answer this, we transiently transfected COG KO cells with a Lamp2-GFP plasmid and then imaged cells using fluorescence and DIC microscopy simultaneously. As shown in Figure 6C the all vacuoles seen in DIC were also labeled with Lamp2-GFP in transfected cells (Figure 6C). We will further refer to these COG KO-induced compartments as enlarged endolysosomal structures (EELSs). These EELSs were not seen in MGAT1 or GALE cells (Figure 6A and B), indicating that this is likely an effect of defective endosomal/Golgi trafficking due to COG specific malfunction and not a byproduct of hypoglycosylation of lysosomal, Golgi, or trafficking proteins.
Figure 6. Golgi glycosylation defects are not related to COG-KO-specific accumulation of enlarged endolysosomal structures (EELS).

(A) DICIII of KO cells vs WT HEK293T cells reveled large vacuoles in COG KO cells (arrows). (B) Immunostaining of lysosomal protein Lamp2 in control and KO cells demonstrate specific accumulation of Lamp2 (red)-positive vacuolar EELS compartment in COG KO cells. Nuclear DNA was stained with Hoechst (blue). Insert is 2× zoom of original image (C) To show correlation between the vacuoles seen in live cell (A) and the structures visualized by immunostaining of Lamp2 (B) COG2 KO cells were transfected with Lamp2-GFP and live cell imaged. Arrows point to EELS. Scale bar 10 μm in A and B 5 μm in C.
COG KO affects retrograde trafficking while MGAT1 and GALE KOs do not
After verifying that MGAT1 and GALE glycosylation mutants didn’t have any unintended/unexpected effects on Golgi structure or on the COG complex that could interfere with our comparisons, we next sought to determine if impaired glycosylation results in trafficking disturbances. It has previously been extensively reported that the COG complex is involved in retrograde trafficking at the Golgi 40,57–59. This has been demonstrated both by resistance of Golgi collapse upon Brefeldin A treatment in COG deficient cells 14,60,61, and by defects in Shiga toxin and Subtilase cytotoxin (SubAB) trafficking 40,57. We chose to use the SubAB toxin trafficking assay, as previously performed with COG KO cells, with MGAT1 and GALE KO cells to assess the role of glycosylation in the retrograde trafficking of this toxin. SubAB toxin binds to the plasma membrane and then traffics through the Golgi to the ER where it cleaves target protein GRP78. In this assay, SubAB toxin is added to cells for different time points then cell lysates are assessed for cleavage of GRP78, indicating that the toxin has reached ER via retrograde trafficking (Figure 7A). GRP78 undergoes 50% cleavage before 30 minutes in GALE and MGAT1 KO cells (at 30 minutes GRP78 is 59% cleaved in MGAT1 KO cells and 62%cleaved in GALE KO cells, Figure 7B) while in COG2 KO cells it takes an additional hour to obtain the same degree of cleavage (58% cleavage at 90 minutes, Figure 7B). This suggests that COG’s effect on retrograde trafficking extends outside of its effects on glycosylation.
Figure 7. Retrograde trafficking is impaired in COG KO cells but not MGAT1 or GALE.

Cells were treated with Subtilase cytotoxin (SubAB) for the indicated time points at 37°C then collected and lysed. Samples were analysed by WB (A, FL= Full length GRP78) and quantified using fluorescent densitometry for cleavage of SubAB target protein GRP78/Bip (B). SubAB takes ~60 minutes longer to reach the ER and cleave GRP78 in COG KO cells than MGAT1 or GALE KO cells. (n=2) * denotes significance from MGAT1 values # denotes significance from GALE values. MGAT1 and GALE were not statistically significant from one another. P < 0.05. Error bars represent standard deviation.
COG KOs alone cause perturbations in sorting and overall secretion
To further assess if the COG KO effects on the efficiency of intracellular trafficking were related to hypoglycosylation we next tested protein sorting in MGAT1 and GALE KO cells as well as COG KO cells using secretion of lysosomal protease cathepsin D. At the cellular level, secretion of immature cathepsin D has been associated with defective protein sorting and trafficking 62,63. Previously we showed that immature cathepsin D is secreted in significant amounts in COG KO cells but not the in parental cell line. We repeated this experiment with MGAT1 and GALE KO cells to assess the role of hypoglycosylation in this phenotype (Figure 8). COG KOs secreted 5–10 times more pro-cathepsin D than WT cells (Figure 8B). In contrast, neither MGAT1 KO nor GALE KO recapitulated the pro-cathepsin D secretion phenotype seen in COG KO cells (Figure 8A and B). This indicates that missorting and secretion of cathepsin D is not due to misglycosylation of a protein or lipid involved in this transport process, or misglycosylation of cathepsin D itself, but rather the actual transport process of this enzyme is altered in COG-deficient cells. Interestingly, mature cathepsin D was readily visible in COG KO cell lysates (Figure 8A), despite the significant amount of unprocessed enzyme that was secreted, indicating that COG dysfunction only partially affected Golgi sorting machinery and allowing some cathepsin D to reach the lysosomes and become fully processed. This mature intracellular amount was not different from WT, GALE KO, or MGAT1 KO. Interestingly, an additional ~40 kDa band of underprocessed Cathepsin D was detected in GALE KO lysates, indicating a novel role of O-glycosylation in Cathepsin D intracellular processing.
Figure 8. Golgi glycosylation defects do not affect Cathepsin D sorting.

WT cells along with MGAT1, GALE, and COG KOs were placed in serum free media for 36 hours. Media was then collected and concentrated 5x, and cells were collected and lysed. 10μL each of media (Secreted, A) and cell lysates (Intracellular, A) were analyzed for the presence of cathepsin D (A). Amount of secreted Cathepsin D compared to intracellular was quantified by densitometry (B). COG KOs have significant amounts of immature cathepsin D secreted that MGAT1 or GALE KOs do not (A and B). (n=4 (COG2 n=2)) * denotes significant difference from WT, p< 0.05. AUs are standardized to the highest secretor in each experiment. Error bars represent standard deviation.
To further understand the impact that COG KO was having on Golgi sorting and secretion compared to MGAT1 and GALE KOs we collected and analyzed total proteins secreted into the conditioned media compared to proteins contained in cell lysates (Figure 9A). While GALE KO cells did demonstrate slightly enhanced secretion compared to WT cells (~1.7x) COG KOs secreted 4–6 times more total protein and protein species than WT cells and MGAT1 and GALE KO cells (Figure 9B). This was not due to cell lysis in COG KO cells, which was minimal (Figure 9C), but is likely due to defective sorting of multiple proteins at the Golgi/TGN.
Figure 9. Protein secretion is stimulated in COG KO cells.

MGAT1, GALE, and COG KOs were placed in serum free media for 36 hours. Media was then collected and concentrated 5x, and cells were collected and lysed. 10μL of media (A, Med) and cell lysates (A, CL) were analyzed for protein abundance via Coomassie Blue stain (A) and GNL lectin and tubulin blots (C). Tubulin blot was used as a loading control and to insure minimal lysis was present in conditioned media. (B)Total protein amount secreted was quantified and standardized to WT (n=4, COG2 n=2). * denotes significance from WT, p< 0.05. Error bars represent standard deviation.
We next probed these samples by lectin blot to see if their secreted glycan profile was different than the intracellular one, due to under glycosylated product not being sorted properly. For this assay we utilized GNL, as this lectin binds to the most severely under glycosylated N-glycans. We found that though COG and MGAT1 KO cell lysates bind GNL similarly, the GNL-positive bands in the COG KO secretome have different intensities and banding profiles from MGAT1 KO secretome further indicating that altered secretion in COG KO cells is mostly due to COG trafficking defects and not due to hypoglycosylation of secretory proteins or trafficking receptors alone.
GALE and MGAT1 double KO cells do not phenocopy COGs effects on sorting, secretion, endosomal/lysosomal morphology and Golgi morphology
MGAT1 depletion affects N-glycosylation at the earliest stages of Golgi processing. GALE depletion affects medial/trans Golgi N- glycosylation and the initial step of mucin type O- glycosylation completely inhibiting this pathway. Though we expected the single KOs to cause similar defects to the COG complex, MGAT1 KO cells affect N-glycosylation most severely and GALE mutations affect mucin type O-glycosylation most severely. To induce even more severe Golgi glycosylation defects, we created compound MGAT1 and GALE double KOs to hamper essentially all glycosylation modifications at the Golgi. These cells were then studied to assess if this severe block in Golgi glycosylation could better phenocopy COG KOs. GALE and MGAT1 double KO cells (G/M DKOs) have the same viability of the single GALE KOs (data not shown) and lack both MGAT1 and GALE proteins (Figure 10A). G/M DKOs were then assessed for Lamp2 stability and mobility (Figure 10A). In G/M DKO cells, Lamp2 showed a similar mobility to the MGAT1 single KO, and increased mobility when compared to the GALE single KO. However, similar to the GALE single KO the stability of Lamp2 was reduced in the G/M DKO cells, confirming our hypothesis that the hypermobility of Lamp2 is mostly affected by N-glycosylation, but the stability is mostly affected by O-glycosylation.
Figure 10. GALE/MGAT1 DKO cells show glycosylation defects but normal Golgi and lysosomal structure.

GALE/MGAT1 DKO (G/M DKO) characterization. (A) Potential DKOs were screened for the absence of MGAT1 protein. Clones were also screened for hypermobility of Lamp2. DKO clones showed Lamp2 shifts similar to MGAT1 single KO, but with stability issues similar to GALE single KOs. (B) DKO cells were analyzed for Golgi structure abnormalities using GM130 (green) as a cis Golgi marker and Rab6 (red) as a trans-Golgi marker. G/M DKO cells still retained a ribbon structure (inserts 3x, B). (C) G/M DKO cells were imaged by electron microscopy to determine the ultrastructure of the Golgi. Scale bar 1 μm (D) DKOs were screened for abnormal EELS using Lamp2-GFP as a lysosomal and EELS marker. No enlarged Lamp2 positive structures were visible. EELS in COG2 KO shown with arrows. Scale bar is 10 μm.
We then utilized the G/M DKO cells in a subset of the assays performed above to assess the effects of severe Golgi glycosylation impairment on Golgi and lysosomal morphology (Figure 10B–D) as well as sorting and secretion (Figure 11). The Golgi structure was assessed by superresolution microscopy using GM130 as a cis-Golgi marker and Rab6 as a trans-Golgi marker (Figure 10B) as was done in the single KOs (Figure 4). We found that, like the single knockouts, G/M DKO cells retained a ribbon-like Golgi morphology and proper segregation of cis/trans Golgi markers, unlike the fragmented Golgi observed in COG KO cells. To confirm that the Golgi structure was not altered in G/M DKO cells we performed electron microscopy on these cells (Figure 10C). The Golgi structure in G/M DKO cells was indistinguishable from that of WT cells, showing stacked Golgi structures clustered near each other in both cell types. In contrast, COG KO cells showed a severely dysmorphic Golgi, as we have previously observed in these cells 26. This suggest that maintenance of Golgi structure is not dependent on Golgi glycosylation.
Figure 11. Protein secretion and sorting of Cathepsin D in MGAT1/GALE DKO cells does not phenocopy COG defects.

MGAT1/GALE DKO cells were placed in serum free media for 36 hours alongside with MGAT1, GALE, and COG KOs. Media was then collected and concentrated 10× and cells were collected and lysed. 10μl each of media (A) and cell lysates (A) were analyzed for protein abundance via Coomassie Blue stain (A) and for cathepsin D by WB (C). (B)Total protein amount secreted was quantified and standardized to WT (n=4, n=2 for G/M DKO). (D) Total Cathepsin D secreted was quantified and standardized to intracellular protein abundance and then to the highest secretor (A.U.). * denoted significant change from WT, p< 0.05. Error bars represent standard deviation.
G/M DKO cells were then screened for the presence of Lamp2 positive EELSs by transfection with Lamp2-GFP, along with COG KO cells, then cells were analyzed by DIC and fluorescence confocal microscopy simultaneously, as was done for Figure 6. COG KO cells showed EELSs that were Lamp2 positive. In contrast, G/M DKO cells did not contain enlarged vacuoles and Lamp2-GFP positive compartments appeared to resemble normal lysosome size and distribution, suggesting that even the most severe Golgi hypoglycosylation does not affect lysosomal morphology.
While no Golgi or lysosomal morphology changes were observed in these cells it is possible that altered sorting or enhanced secretion observed in COG KO cells could be mimicked in G/M DKO cells. To test this possibility, we collected conditioned media and cells from G/M DKO cells, single MGAT1 and GALE KO cells, WT cells, and COG KO cells. Conditioned media was first analyzed for total protein abundance (relative to intracellular protein abundance, Figure 11A and B). G/M DKO cells performed similarly to GALE single KO cells, with slightly enhanced protein secretion (~ 1.7× that of WT), but not to the same extent as that of COG KOs (~ 4× that of WT). This suggests that COG KOs effects on trafficking as well as glycosylation are contributing to this phenotype. We next assessed the conditioned media from the G/M DKOs for enhanced secretion of pro-cathepsin D, indicative of missorting at the TGN (Figure 11C and D). G/M DKO cells did not secrete a significant amount of cathepsin D, 8–10× less than that of COG KO cells, again indicating that COG-dependent missorting of cathepsin D is independent of Golgi glycosylation.
COG7-CDG patient fibroblasts have altered endolysosomes and defects in sorting/secretion
To further investigate confirm these COG KO specific changes in another COG deficient cell line, we characterized COG7-CDG fibroblasts obtained from a COG7-CDG patient that were previously shown to have far less COG7 protein than control fibroblasts 64. Similar to their CRISPR KO counterparts, large 1–5+μm intracellular vacuole-like structures were visible by phase contrast microscopy in COG7-CDG cells (~30% all cells), but absent in control fibroblast (Figure 12A). We then analyzed these structures by electron microscopy and saw multiple electron scarce vacuoles in COG7-CDG fibroblasts but not in control cells. These cells were then immunostained for Lamp2 and confirmed that there are enlarged Lamp2 positive in these cells similar to our COG KO cells (Figure 12C). These findings indicate that the altered EELSs in COG KO HEK293T cells are not a cell type specific response, but an attribute of COG-deficient cells.
Figure 12. COG7-CDG patient fibroblasts phenocopy glycosylation-independent defects observed in HEK293T Cog KO cells.

(A) Phase contrast images of COG7-CDG patient fibroblasts reveled that ~30% of cells contain vacuoles (arrows). (B) EM analysis of patient fibroblasts indicates abundant vacuole-like EELS structures throughout the cell, though smaller than those in HEK293T cells (arrows). (C) Immunostaining of COG7-CDG patient fibroblasts and control fibroblasts reveal enlarged Lamp2 positive EELS in patient fibroblasts but not in control. (D and E) COG7-CDG patient fibroblasts and control fibroblasts lysates and concentrated conditioned media were analyzed for Cathepsin D (D), Col6A1, GPP130, and α-tubulin (E). Col6A1 and cathepsin D were secreted 3× more in COG7-CDG patient cells than control (n=2, calculated via densitometry) while transmembrane protein GPP130 was not secreted in either. α-tubulin was used as a control for lysis. Scale bars are 5μm.
We next looked for COG-specific protein sorting and secretion abnormalities in these cells. Conditioned media was collected along with cells for control and COG7-CDG fibroblasts and analyzed by western blot. As in the HEK COG KO cells (Figure 8 and 11C and D), COG deficient fibroblasts secreted more of cathepsin D than their control cell counterparts (~3× more as assessed by densitometry, Figure 12D). We also analyzed the conditioned media for COL6A1, a normal secretory product of fibroblasts. We found that its secretion was also increased in the COG7-CDG cells around 3× that of control fibroblasts (Figure 10D). This was not due to cell lysis, as there was little/no alpha tubulin in the concentrated media. Additionally TGN transmembrane protein GPP130 was not found in the conditioned media, even though it is COG sensitive, as previously reported56 showing that the altered secretion and sorting is specific for a subset of proteins.
Taken together this strongly suggests that COG deficiency causes COG specific, glycosylation-independent defects in Golgi sorting, lysosomal morphology, and altered secretion; not only in our KO model, but also in COG7-CDG patient derived cells.
Discussion
In this study, we sought to determine which phenotypic changes in COG-deficient cells are an effect of hypoglycosylation of transmembrane and secretory proteins and which are more specific to other COG functions.
In all studied eukaryotic systems COG complex malfunction affects N- and O- glycosylation through mistrafficking of glycosylation enzymes 39,41,65–67, but in addition to this wide-spread defect, several trafficking regulators (v-SNAREs Bet1L/GS15 and GOSR1/GS28) as well as Golgi-localized cytoplasmic enzymes are also affected 40,68–70. Thus, we reasoned that some of the phenotypes we observe must be related to COG-specific trafficking functions that aren’t related to hypo-glycosylation. We then postulated that, if this were true, it would have important implications for broadening our understanding of how defects in intra-Golgi trafficking specifically related to COG function impacts the cell. Additionally, uncovering COG-dependent phenotypes unrelated to glycosylation defects would broaden our understanding of COG-CDGs, as these cellular phenotypes could cause symptoms that are atypical of other CDGs and therefore require different treatments.
To test this hypothesis, we created mutants that show Golgi-based defects in glycosylation similar to or more severe than the ones observed in COG KO cells. MGAT1 and GALE were chosen for this purpose as they created early and late blocks in N-glycosylation, respectively. GALE KO also prevented O-glycosylation by removing available GalNAc. Then to induce an even more dramatic Golgi hypoglycosylation scenario we created GALE and MGAT1 DKO cells, effectively halting most glycosylation at the Golgi. We reasoned that these KO cells would affect the same glycosylated intracellular and secreted proteins as COG KOs and therefore should allow for the deduction of which COG KO phenotypes were actually downstream effects of hypo-glycosylation.
What we discovered was that only a subset of COG KO cell phenotypes are mimicked by hypo-glycosylation alone. Only phenotypes that were clearly assessing glycosylation (Lamp2 altered mobility and stability, altered lectin binding) showed similarities between MGAT1 KOs, GALE KOs, GALE/MGAT1 DKOs and COG KOs. Phenotypes not phenocopied by MGAT1 KOs, GALE KOs, or GALE/MGAT1 DKOs include: a severely fragmented Golgi structure, delayed plasma membrane-Golgi-ER retrograde trafficking, altered TGN sorting and secretion, and accumulation of EELS.
A subset of glycosylation-independent COG KO phenotypes could arise as a secondary manifestation of permanent Golgi fragmentation. Golgi fragmentation has an important physiological role in mitosis 71. Golgi fragmentation has also been observed to proceed apoptosis, alter cell migration, and impair polarized secretion72. Persistent fragmentation has been noted pathological states, such as cancers (e.g. GOLPH3 is a well-known oncogene and its overexpression in cancer cells has been linked to Golgi fragmentation and enhanced cell proliferation73), pathogenic infection (e.g. Chlamydia remodels the Golgi in later stages on infection 74), and neurological diseases (Golgi fragmentation has been observed in Alzheimer’s disease, ALS, Parkinsons, and Creutzfeldt-Jacob disease 72,75,76). Fragmentation of the Golgi in cancer has been theorized to proceed cancer cells becoming more metastatic 77. Golgi fragmentation in neurodegenerative disorders contributes to the pathology of these disease by causing defective protein processing and sorting and eventually leads to neuronal death 78. Persistent Golgi fragmentation was seen in all COG KO cells. COG KO Golgi were swollen and vesiculated and far less stacked/organized than control or MGAT1/GALE DKO cells (Figure 10 and 26). This is potentially due to a disbalance between anterograde and retrograde trafficking, but could also be due to changes in lipid compositions affecting Golgi membrane curvature or altered levels of pH or calcium at the Golgi affecting the lumen and inducing swelling 27,79–81. What specific alterations in COG KO cells are causing this persistent fragmentation, and how this fragmentation contributes to other phenotypes observed in COG KO cells will be an important avenue for future investigation. Interestingly, our recent experiments with permanent attachment of COG subunits to the Golgi membrane indicate that the expression of Golgi-restricted COG4 and COG7 was sufficient to block EELS accumulation but failed to rescue Golgi fragmentation in COG KO cells (Climer et al, in press)). At the same time Golgi attached COG subunits were sufficient for restoring normal Cathepsin D sorting, indicating that some glycosylation-independent phenotypes in COG KO cells may not be directly related to Golgi fragmentation.
Strikingly, COG-dependent morphological changes were always evident downstream of the Golgi. EELS were present in COG KOs as well as COG7 deficient fibroblasts. These structures were massive and very acidic (JB and VL, unpublished observation), yet normal sized lysosomes were also present in these cells, potentially explaining how some cathepsin D is processed into its mature form while the rest is secreted. Interestingly, previous knockdown studies of the Golgi-associated retrograde protein (GARP) complex, a related multisubunit tethering complex, also found enlarged lysosomes82. COG and GARP have shared interactions with the STX16 SNARE complex at the TGN83, further supporting the idea that altered trafficking/sorting at the TGN is causing the enlarged lysosomal phenotype. Further investigation of EELSs origin, molecular composition and their relationship to trafficking/sorting at the Golgi will be an important future study.
Protein sorting defects, common to all COG deficient cells, were likely related to EELS accumulation. Cathepsin D is known to be secreted in cases of altered sorting at the TGN 63,82. In this study we found that this COG KO phenotype is also not shared with other glycosylation-deficient cells, ‘further supporting a role for the COG complex in TGN cargo sorting. By what mechanism is this missorting happening? While it is possible that partially impaired mannose-6-phosphate (M6P) tagging or M6P receptor stability/localization could cause Cathepsin D missorting as it does in GARP KDs82 and select lysosomal storage disorders84 we do not believe this to be the case in COG deficiency. COG KO cells demonstrate normal distribution of the mannose-6-phosphate cation independent receptor 26 and we have also seen no change in the cation dependent M6P receptor localization (data not shown). In agreement with this conclusion, previous studies of COG7-CDG patients demonstrated increase in secreted lysosomal enzyme activity with a normal level of mannose-6-phosphate 85. This study also showed no effect on M6PR-CI distribution in patient fibroblasts indicating that this missorting exists in COG KO cells as well as patient cells and appears to be independent of M6P tagging or receptor localization for both. So, what is causing the secretion of cathepsin D in COG deficient cells? It is possible that sortilin is involved 86 or another yet unidentified pathway. Elucidating what M6P independent missorting mechanism of lysosomal enzymes in COG deficient cells will be an interesting future study.
In addition to altered lysosomal enzyme targeting, COG-specific increase in protein bulk secretion was also observed. When extracellular proteins were assessed we found that COG KOs tended to secrete more protein and protein species over all, compared to WT, MGAT1 KO, GALE KO or MGAT1/GALE DKO cells. Changes in the secretome have the potential to cause multi-systemic changes in a COG deficient organism. Discovering what other proteins are contained in the COG KO secretome will likely give a better understanding of what sorting process(es) is/are defective at the Golgi in COG deficient cells.
What could be causing these COG specific changes? We hypothesize that the COG complex is not only affecting the sorting and retention of Golgi enzymes, but also of packaging and sorting at the TGN for outbound cargo, such as cathepsin D. This altered packaging and sorting is not only affecting the Golgi but post-Golgi compartments and secretion at the plasma membrane. The COG complex could exert these effects on the TGN through a variety of different mechanisms including: impaired lipid homeostasis, altered ion concentrations, mislocalization of trafficking molecules at the TGN or, most likely, a combination these mechanisms. Additionally, most of these proposed mechanisms could also contribute to the Golgi fragmentation phenotype observed in COG KO cells.
COG deficiency is known to affect glycolipids such as GM3 70, but what about other, non-glycosylated lipids? COG2 mutant CHO cells have reduced levels of sphingomyelin (~25% of WT cells) due to mislocalized sphingomyelin synthase27. These cells also showed increase in ceramide levels (3× that of WT) and mislocalization of the ceramide transfer protein, CERT. These changes could cause destabilization of lipid microdomains containing cholesterol and SM at the Golgi which could then disperse proteins that normally localize to these lipid domains (transmembrane sorting proteins or ion transporters like the v-ATPase, which could in turn affect Golgi pH)87.
COG deficiency has been shown to affect localization and stability of several ion transporters like ATP7A and SLC31A1 88) thus changing the ion concentrations at the Golgi, which are known to be important for retention of sorting molecules like GPP130 (COG sensitive)56, TMEM165, and Cab45 63.
The COG complex is a master coordinator of trafficking components and functionally interacts with several trafficking components at the TGN including members of the GARP/EARP complex, the STX6/STX16/Vamp4/Vti1a SNARE complex, and GOLPH3 4,67. Deficiency of the COG complex could either cause these components to be mislocalized and/or destabilized, as is the case of VAMP4 in COG8 deficient patient cells89. COG deficiency could also make these trafficking interactions less efficient as was the case for STX16 SNARE complex formation in COG deficient fibroblasts of COG knock-down in HeLa cells. These changes then lead to altered cycling between the TGN and endosomes89, which could affect the steady state protein composition at the Golgi thus altering sorting.
In summary, we have found that the majority of COG KO phenotypes previously reported by our lab were not due to Golgi-based glycosylation defects mimicked by GALE and MGAT1 deficiency, but rather due to other more COG specific trafficking roles. We hope that these cellular insights will shed light, not only on COG facilitated membrane trafficking, but also on the heterogeneity of COG-CDG patient symptoms and perhaps eventually lead to different or better ways to manage these symptoms.
Materials and Methods
Cell culture
HEK293T cells (either CRISPR KOs or WT (ATCC)) were used for all experiments except those involving COG7-CDG female patient fibroblasts in Figure 12 (these fibroblasts were obtained as a gift from Dr. Hudson Freeze and initially described in 64. Cells were cultured using Dulbecco’s Modified Eagle Medium (DMEM)/F12 50/50 (Corning) with 10% (v/v) Fetal Bovine Serum (FBS) (Thermo Fisher) added. Cells were kept at 37°C 5% CO2 in a humidity-controlled incubator (90%). HEK cells were passed via gentle resuspension. COG7-CDG fibroblasts were trypsinized to passage.
Antibodies and reagents
Antibodies used in this study include: mouse [anti-cathepsin D (WB 1:500, Sigma), b-actin (1:1000, Sigma), Lamp2 (WB 1:100 DSHB), GM130 (IF 1:500, BD), mCOG3 (1:500)] ; goat [anti-GRP78 (WB 1:1000, Santa Cruz), B4GalT1 (1:500, R&D), ST6GalT1 (1;200, R&D)]; rabbit [anti-MGAT1 (1:300, Abcam), GALE (1:500, Abcam), B4GalT5 (1:300, Abcam), TMEM165 (1:500, Sigma), GAPDH (1:1000, Cell Signaling), COG8 (1:500, Sigma)]; sheep [anti-TGN46(1:500, Bio-Rad)]
Lectins used: Helix pomatia agglutinin (HPA)-Alexa 647 (Thermo Fisher), Galanthus nivalis lectin(GNL)-647 90, Griffonia simplicifolia (GS-II)-647 90(Thermo Fisher).
Other Reagents include: Subtilase cytotoxin (1.49 mg/ml), a gift from Dr. J. Paton 91
Creating CRISPR KOs
COG KOs were created as previously described26,92. MGAT1 and GALE KOs were generated in a similar fashion using HEK293T cells. Cells were co-transfected with plasmid DNA encoding Cas9 (Genecopoeia, Cat. No. CP-C9NU-01) and 2 sgRNAs (Genecopoeia, For GALE: Cat No. HCP206766-SG01–3-B-a Target sequence: TGGAAGTTATCGATGACCAC and Cat No. HCP206766-SG01–3-B-c Target sequence CCTTTTTGAAGAGACGCTGT; For MGAT1: Cat No. HCP265906-SG01–3-B-a Target sequence CACCCGGGAAGTGATTCGCC and Cat No. HCP265906-SG01–3-B-c Target sequence GTGGGGCGCTATCCTCTTTG) using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s protocol. MGAT1 KO cells were then stained with GNL one week after transfection with CRISPR plasmids and single cell sorted to obtain clonal populations. GALE cells were selected with Hygromycin (200μg/mL) for 2–4 days post transfection. Surviving cells were then single cell sorted into clonal populations. Clonal populations of both MGAT1 and GALE were screened by western blot for absence of targeted protein.
GALE/MGAT1 DKO cells were created by treating GALE single KO cells with the same MGAT1 gRNAs used to create the single MGAT1 KOs. GALE/MGAT1 DKO cells were selected with Hygromycin (200μg/mL) for 3 days post transfection then individual colonies were collected and assessed for increased GNL binding and the absence of MGAT1 protein. Cells lacking MGAT1 and displaying increased GNL binding were used for all G/M DKO experiments.
Cell lysis and western blot analysis
Cells were lysed in hot 2% SDS and heated for 5 minutes then 6× SDS sample buffer with βME was added to samples then samples were mixed and frozen until needed. For western blot (WB) analysis samples were loaded into Bio-Rad 4–15% gels or 7 or 9% lab made gels. Gels were then transferred to nitrocellulose paper using the Thermo Scientific Pierce G2 Fast Blotter. Membranes were rinsed in PBS then blocked using Odyssey blocking buffer for 20 minutes then primary antibody was added and incubated overnight at 4°C. Membrane was then washed with PBS then incubated with Licor IRDye secondary antibodies in 680CW and 800CW in 5% milk in PBS for 45–60 minutes. Blot was then washed and imaged using the Odyssey Imaging System. Images were processed using the Licor Image Studio software.
Lectin blot
Lectins (described under antibodies and reagents) were conjugated to Alexa647 fluorophore which can be detected by the Odyssey. Membranes were blocked with blocking buffer or 3%BSA then lectin was added, and the membrane was incubated for 1 hour at room temp or overnight at 4°C then washed and imaged.
Immunofluorescence
Cells were plated on 12 mm glass coverslips (#1, 0.17 mm thickness) that has been coated with collagen I (final concentration 50μg/mL, Corning) and washed. Cells were allowed to grow to 50–90% confluency before fixation and processing. Once proper confluency was reached media was removed and cells were then fixed with 4% paraformaldehyde (freshly made from 16% stock solution) diluted in Dulbecco’s phosphate-buffered saline (DPBS); Electron Microscopy Sciences) for 15 minutes. Cells were then permeabilized with 0.1% Triton X-100 for one minute. Cells were then treated with 50 mM ammonium chloride for 5 minutes and then either treated with 6M urea for 2 minutes when necessary for the antibody (COG3 staining) then washed with DPBS or just washed with DPBS. Following washes cells were blocked twice with 1% BSA, 0.1% saponin in DPBS for 10 minutes each then removed and primary antibody (diluted in 1% cold fish gelatin, 0.1% saponin in DPBS) was added and cells were incubated for 1 hour. Cells were then washed with DPBS and incubated with fluorescently conjugates secondary antibodies (diluted in 1% cold fish gelatin, 0.1% saponin in DPBS) for 45 minutes. Cells were then washed two times with PBS then coverslips with cells were dunked in dPBS 10 times then water 10 times. Coverslips containing cells were mounted on glass microscope slides using Prolong® Gold antifade reagent (Life Technologies). Samples were allowed to cure at room temp in the dark for one to two days before imaging. Cells were imaged with the 63× oil 1.4 numerical aperture (NA) objective of a LSM880 Zeiss Laser inverted microscope outfitted with confocal optics and Airyscan or with the Elyra Zeiss microscope using the 63× or 100× objective with the 3D SIM mode. All images are maximum intensity projections of 8 slices created in the Zen2 software unless noted.
Secretion assay
Cells were plated in 6-well plates and grown to 90–100% confluency. Cells were then rinsed carefully with DPBS 5× then plated in 1 ml of serum-free, chemically-defined media (BioWhittaker Pro293a-CDM, Lonza) with 1× GlutaMAX (100× stock, Gibco) added per well. Cells were kept in media 24–36 hrs then the media was collected and spun down to remove floating cells. Supernatant was transferred to a new tube and then concentrated using a 10k concentrator (Amicon® Ultra 10k, Millipore), final concentration was 5–10× that of cell lysates. Cells were collected separately in PBS and spun down. Supernatant was removed and cells were lysed in hot 2% SDS. Samples were analyzed by western blot as detailed above.
Subtilase cytotoxin (SubAB) trafficking assay
SubAB assay was performed essentially as previously described 93 with minor changes. Each cell line was plated in 1 well of a 6-well plate and allowed to grow to 70–80% confluency. Media was removed and replace with 500μL of fresh media (as described above, with no antibiotics or antimycotics). A 6× stock of SubAB in media was then made (39.3 μg/mL). 100μL of stock solution was added to each well at the appropriate time point for a final concentration of 6.5 μg/mL. Time points were 180, 120, 90, 60, 30, and 0 minutes with the toxin. Cells were incubated with the toxin for the appropriate amount of time at 37°C in 5%CO2 with 90% humidity. At the end media with toxin was collected and disposed of in bleach and cells were collected in DPBS and spun down at 600xg for 3 minutes. Supernatant was removed, and cells were lysed in 2%SDS. Lysates were analyzed for GRP78 using the western blot protocol detailed above. Actin was used as a loading control for quantification.
High Pressure Freezing, Freeze Substitution, and Electron Microscopy
Electron microscopy for Figure 10 was performed as described in 94. Briefly, cells were fixed in 0.1M sodium cacodylate buffer (pH 6.8) with 2.5% glutaraldehyde and 0.05% malachite green for 20 minutes on ice. Samples were then treated with 0.5% osmium tetroxide and 0.8% potassium ferricyanide in 0.1 M sodium cacodylate for 30 minutes at room temperature, and 1% tannic acid on ice for 20 minutes. Finally, samples were stained with uranyl acetate at room temperature for one hour. Samples were then dehydrated with ethanol in increasing concentrations prior to embedding in Araldite 502/Embed 812 resin (EMS).
Electron microscopy for Figure 12 was performed as previously described26. Briefly, cells were plated on sapphire disk to be at 100% confluency at the time of freezing. Disk were placed in cryoprotectant (PBS with 2% agarose, 100 mM D-mannitol, and 2% FBS) then subjected to high pressure freezing using a Leica EM PACT2 with rapid transfer system. Samples were placed in a staining cocktail (acetone with 2% osmium tetroxide, 0.1% Glutaraldehyde, and 1% ddH2O) in liquid nitrogen then transferred to a freeze substitution chamber at −90°C. Cells were slowly warmed to 0°C using the following protocol: −90°C for 22 hours, warm 3°C/hour to −60°C, −60°C for 8 hours, warm 3°C/hour to −30°C, −30°C for 8 hours, warm 3°C/hour to 0°C. Tubes containing samples were placed on ice and were stained using the following protocol: samples were washed with acetone 3×, stained with a 1% Tannic acid/1% ddH2O solution in acetone for 1 hour, washed 3 more times with acetone, then stained with a 1% osmium tetroxide/1% ddH2O solution in acetone for 1 hour followed by 3 more acetone washes. Samples were then embedded in Araldite 502/Embed 812 resin (EMS) with DMP-30 activator added and processed in a Biowave at 70°C under vacuum for 3 minutes per embedding step. Samples were then baked at 60°C for 48 hours before holders were removed and samples were cut. Post cutting staining was done with uranyl acetate and lead citrate prior to imaging.
For both Figure 10 and 12 Ultrathin sections were imaged at 80 kV on a FEI Technai G2 TF20 transmission electron microscope. Images were taken with a FEI Eagle 4kX USB Digital Camera.
Statistical analysis
All results are representative of at least 3 independent experiments unless noted. WB are representative images from 3 repeats. Western blots were quantified via densitometry using the Licor Image Studio software. Error bars for all graphs calculated based on standard deviation. P values calculated using a 2-tailed students t-test. Significance was defined as p < 0.05. AUs are defined in each figure.
Synopsis.
The Conserved Oligomeric Golgi (COG) complex controls membrane trafficking and Golgi homeostasis by orchestrating retrograde vesicle trafficking. COG deletions results in defects in glycosylation, trafficking, and sorting as well as morphological abnormalities in the Golgi and endolysosomal compartments. A block in Golgi glycosylation does not recapitulate the majority of COG deficient phenotypes indicating that these defects are due to interruption of specific roles of the COG complex independent of its role in the maintenance of Golgi glycosylation machinery.
Acknowledgments
We are thankful to Santiago Di Pietro, Hudson Freeze, James Paton, Brian Storrie and Daniel Ungar as well as others who provided reagents and advice. We would like to thank Leslie Climer and Wei Wang for helpful discussion. We would also like to thank the DNA Sequencing and Flow Cytometry cores (NIH grant P20GM103625), and the Digital Microscopy core facilities for the use of their equipment and expertise. This work was supported by the NIH grants GM083144 and U54 GM105814. The authors declare no competing financial interests.
Footnotes
Author contributions
J.B.B. wrote the article and made substantial contributions to conception and design, acquisition of data, analysis, and interpretation of data. T.K. participated in drafting the article, performed experiments for portions of 1–4 and interpreted the data. I.P. performed experiments for portions of Figure 10 and 12 and interpreted the data. V.V.L. wrote the article and made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data.
References
- 1.Ungar D, Oka T, Brittle EE, et al. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. Journal of Cell Biology. 2002 Apr 29;157(3):405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lupashin VV, Suvorova ES, Duden R. Yeast COG complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNARES and with COPI vesicle coat proteins. FEMS Congress of European Microbiologists Abstract Book. 2003;2003(1) [Google Scholar]
- 3.Zolov S, Lupashin V. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. Molecular Biology of the Cell. 2004 Nov;15:460A–460A. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willett R, Ungar D, Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem Cell Biol. 2013 Sep 01;140(3):271–283. doi: 10.1007/s00418-013-1117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willett R, Pokrovskaya I, Kudlyk T, Lupashin V. Multipronged interaction of the COG complex with intracellular membranes. Cellular logistics. 2014 Jan 1;4(1):e27888. doi: 10.4161/cl.27888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willett R, Blackburn JB, Climer L, et al. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Scientific reports. 2016 Jul 07;6:29139. doi: 10.1038/srep29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whyte JRC, Munro S. The SeC34/35 golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Developmental cell. 2001 Oct;1(4):527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- 8.Ram RJ, Li BJ, Kaiser CA. Identification of Sec36p, Sec37p, and Sec38p: Components of yeast complex that contains Sec34p and Sec35p. Molecular Biology of the Cell. 2002 May;13(5):1484–1500. doi: 10.1091/mbc.01-10-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter DM, Paul KS, Waters MG. Purification and characterization of a novel 13 S hetero-oligomeric protein complex that stimulates in vitro Golgi transport. J Biol Chem. 1998;273(45):29565–29576. doi: 10.1074/jbc.273.45.29565. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Steet RA, Bohorov O, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004 May;10(5):518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 11.Foulquier F. COG defects, birth and rise! Biochim Biophys Acta. 2009 Sep;1792(9):896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 12.Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin VV. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Front Cell Dev Biol. 2016;4:23. doi: 10.3389/fcell.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011 Dec;21(12):1554–1569. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kranz C, Ng BG, Sun L, et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Human molecular genetics. 2007 Apr 1;16(7):731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 15.Paesold-Burda P, Maag C, Troxler H, et al. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Human molecular genetics. 2009 Nov 15;18(22):4350–4356. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- 16.Reynders E, Foulquier F, Leao Teles E, et al. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Human molecular genetics. 2009 Sep 1;18(17):3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lubbehusen J, Thiel C, Rind N, et al. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Human molecular genetics. 2010 Sep 15;19(18):3623–3633. doi: 10.1093/hmg/ddq278. [DOI] [PubMed] [Google Scholar]
- 18.Kodera H, Ando N, Yuasa I, et al. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clinical genetics. 2015 May;87(5):455–460. doi: 10.1111/cge.12417. [DOI] [PubMed] [Google Scholar]
- 19.Foulquier F, Vasile E, Schollen E, et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci U S A. 2006 Mar 7;103(10):3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peanne R, de Lonlay P, Foulquier F, et al. Congenital disorders of glycosylation (CDG): Quo vadis? European journal of medical genetics. 2017 Oct 24; doi: 10.1016/j.ejmg.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Sparks SE, Krasnewich DM. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 22.Climer LK, Dobretsov M, Lupashin V. Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. Frontiers in Neuroscience. 2015;9:405. doi: 10.3389/fnins.2015.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foulquier F. COG defects, birth and rise! Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792(9):896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 24.Climer LK, Hendrix RD, Lupashin VV. Conserved Oligomeric Golgi and Neuronal Vesicular Trafficking. Handbook of experimental pharmacology. 2017 Oct 21; doi: 10.1007/164_2017_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Molecular Genetics and Metabolism. 2008;93(1):15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- 26.Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin V. COG Complex complexities: Detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Frontiers in Cell and Developmental Biology. 2016 Mar 30;:4. doi: 10.3389/fcell.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spessott W, Uliana A, Maccioni HJ. Cog2 null mutant CHO cells show defective sphingomyelin synthesis. J Biol Chem. 2010 Dec 31;285(53):41472–41482. doi: 10.1074/jbc.M110.150011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podos SD, Reddy P, Ashkenas J, Krieger M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. The Journal of cell biology. 1994;127(3):679–691. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steet R, Kornfeld S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Mol Biol Cell. 2006 May;17(5):2312–2321. doi: 10.1091/mbc.E05-08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R, Stanley P. Transfection of a human gene that corrects the Lec1 glycosylation defect: evidence for transfer of the structural gene for N-acetylglucosaminyltransferase I. Molecular and cellular biology. 1989 Dec 1;9(12):5713–5717. doi: 10.1128/mcb.9.12.5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Stanley P. Five Lec1 CHO cell mutants have distinct Mgat1 gene mutations that encode truncated N-acetylglucosaminyltransferase I. Glycobiology. 2003 Jan;13(1):43–50. doi: 10.1093/glycob/cwg003. [DOI] [PubMed] [Google Scholar]
- 32.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A. 2002 Oct 15;99(21):13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi S, Williams SA, Seppo A, et al. Inactivation of the Mgat1 gene in oocytes impairs oogenesis, but embryos lacking complex and hybrid N-glycans develop and implant. Molecular and cellular biology. 2004 Nov;24(22):9920–9929. doi: 10.1128/MCB.24.22.9920-9929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38) Biochem J. 1990;270(1):97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye Z, Marth JD. N-glycan branching requirement in neuronal and postnatal viability. Glycobiology. 2004;14(6):547–558. doi: 10.1093/glycob/cwh069. [DOI] [PubMed] [Google Scholar]
- 36.Kingsley DM, Krieger M. Receptor-mediated endocytosis of low density lipoprotein: somatic cell mutants define multiple genes required for expression of surface- receptor activity. Proc Natl Acad Sci U S A. 1984;81(17):5454–5458. doi: 10.1073/pnas.81.17.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kingsley DM, Kozarsky KF, Hobbie L, Krieger M. Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell. 1986;44(5):749–759. doi: 10.1016/0092-8674(86)90841-x. [DOI] [PubMed] [Google Scholar]
- 38.Kingsley DM, Kozarsky KF, Segal M, Krieger M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. The Journal of cell biology. 1986;102(5):1576–1585. doi: 10.1083/jcb.102.5.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suvorova ES, Duden R, Lupashin VV. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. Journal of Cell Biology. 2002 May 13;157(4):631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. Journal of Cell Biology. 2005 Feb 28;168(5):747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shestakova A, Zolov S, Lupashin V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic. 2006 Feb;7(2):191–204. doi: 10.1111/j.1600-0854.2005.00376.x. [DOI] [PubMed] [Google Scholar]
- 42.Smith RD, Lupashin VV. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydrate Research. 2008 Aug 11;343(12):2024–2031. doi: 10.1016/j.carres.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson BC, Smith RD, Ungar D, et al. Structural basis for a human glycosylation disorder caused by mutation of the COG4 gene. Proc Natl Acad Sci U S A. 2009 Aug 11;106(32):13329–13334. doi: 10.1073/pnas.0901966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fisher P, Ungar D. Bridging the Gap between Glycosylation and Vesicle Traffic. Front Cell Dev Biol. 2016;4:15. doi: 10.3389/fcell.2016.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynders E, Foulquier F, Annaert W, Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology. 2011 Jul;21(7):853–863. doi: 10.1093/glycob/cwq179. [DOI] [PubMed] [Google Scholar]
- 46.Rosnoblet C, Peanne R, Legrand D, Foulquier F. Glycosylation disorders of membrane trafficking. Glycoconjugate journal. 2013 Jan;30(1):23–31. doi: 10.1007/s10719-012-9389-y. [DOI] [PubMed] [Google Scholar]
- 47.Oka T, Vasile E, Penman M, et al. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5- and COG7-deficient mammalian cells. J Biol Chem. 2005 Sep 23;280(38):32736–32745. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- 48.Tran DT, Ten Hagen KG. Mucin-type O-glycosylation during development. J Biol Chem. 2013 Mar 08;288(10):6921–6929. doi: 10.1074/jbc.R112.418558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kingsley DM, Kozarsky KF, Segal M, Krieger M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. The Journal of Cell Biology. 1986;102(5):1576–1585. doi: 10.1083/jcb.102.5.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamb JE, Shibata S, Goldstein IJ. Purification and Characterization of Griffonia simplicifolia Leaf Lectins. Plant Physiology. 1983;71(4):879–887. doi: 10.1104/pp.71.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pour PM, Burnett D, Uchida E. Lectin binding affinities of induced pancreatic lesions in the hamster model. Carcinogenesis. 1985 Dec;6(12):1775–1780. doi: 10.1093/carcin/6.12.1775. [DOI] [PubMed] [Google Scholar]
- 52.Fenlon S, Ellis IO, Bell J, Todd JH, Elston CW, Blamey RW. Helix pomatia and Ulex europeus lectin binding in human breast carcinoma. The Journal of pathology. 1987 Jul;152(3):169–176. doi: 10.1002/path.1711520305. [DOI] [PubMed] [Google Scholar]
- 53.Kakeji Y, Tsujitani S, Mori M, Maehara Y, Sugimachi K. Helix pomatia agglutinin binding activity is a predictor of survival time for patients with gastric carcinoma. Cancer. 1991 Dec 01;68(11):2438–2442. doi: 10.1002/1097-0142(19911201)68:11<2438::aid-cncr2820681119>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 54.Schumacher U, Higgs D, Loizidou M, Pickering R, Leathem A, Taylor I. Helix pomatia agglutinin binding is a useful prognostic indicator in colorectal carcinoma. Cancer. 1994 Dec 15;74(12):3104–3107. doi: 10.1002/1097-0142(19941215)74:12<3104::aid-cncr2820741207>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 55.Lescar J, Sanchez JF, Audfray A, et al. Structural basis for recognition of breast and colon cancer epitopes Tn antigen and Forssman disaccharide by Helix pomatia lectin. Glycobiology. 2007 Oct;17(10):1077–1083. doi: 10.1093/glycob/cwm077. [DOI] [PubMed] [Google Scholar]
- 56.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI Complexes Interact to Control the Abundance of GEARs, a Subset of Golgi Integral Membrane Proteins. Molecular Biology of the Cell. 2004 May 1;15(5):2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith RD, Willett R, Kudlyk T, et al. The COG Complex, Rab6 and COPI Define a Novel Golgi Retrograde Trafficking Pathway that is Exploited by SubAB Toxin. Traffic. 2009 Oct;10(10):1502–1517. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laufman O, Hong W, Lev S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. The Journal of cell biology. 2011 Aug 8;194(3):459–472. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kudlyk T, Willett R, Pokrovskaya ID, Lupashin V. COG6 Interacts with a Subset of the Golgi SNAREs and Is Important for the Golgi Complex Integrity. Traffic. 2013 Feb;14(2):194–204. doi: 10.1111/tra.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foulquier F, Ungar D, Reynders E, et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Human molecular genetics. 2007 Apr 1;16(7):717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 61.Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol Genet Metab. 2008 Jan;93(1):15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- 62.Perez-Victoria FJ, Mardones GA, Bonifacino JS. Requirement of the Human GARP Complex for Mannose 6-phosphate-receptor-dependent Sorting of Cathepsin D to Lysosomes. Mol Biol Cell. 2008 Mar 26; doi: 10.1091/mbc.E07-11-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crevenna AH, Blank B, Maiser A, et al. Secretory cargo sorting by Ca2+-dependent Cab45 oligomerization at the trans-Golgi network. The Journal of cell biology. 2016;213(3):305–314. doi: 10.1083/jcb.201601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ng BG, Kranz C, Hagebeuk EE, et al. Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Mol Genet Metab. 2007 Jun;91(2):201–204. doi: 10.1016/j.ymgme.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004 May;15(5):2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan X, Cao K, Liu F, et al. Arabidopsis COG Complex Subunits COG3 and COG8 Modulate Golgi Morphology, Vesicle Trafficking Homeostasis and Are Essential for Pollen Tube Growth. PLoS genetics. 2016 Jul;12(7):e1006140. doi: 10.1371/journal.pgen.1006140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frappaolo A, Sechi S, Kumagai T, et al. COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J Cell Sci. 2017 Nov 01;130(21):3637–3649. doi: 10.1242/jcs.209049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sohda M, Misumi Y, Yamamoto A, et al. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic. 2010 Dec;11(12):1552–1566. doi: 10.1111/j.1600-0854.2010.01123.x. [DOI] [PubMed] [Google Scholar]
- 69.Sohda M, Misumi Y, Yoshimura S, et al. The interaction of two tethering factors, p115 and COG complex, is required for Golgi integrity. Traffic. 2007 Mar;8(3):270–284. doi: 10.1111/j.1600-0854.2006.00530.x. [DOI] [PubMed] [Google Scholar]
- 70.Spessott W, Uliana A, Maccioni HJ. Defective GM3 synthesis in Cog2 null mutant CHO cells associates to mislocalization of lactosylceramide sialyltransferase in the Golgi complex. Neurochemical research. 2010 Dec;35(12):2161–2167. doi: 10.1007/s11064-010-0319-8. [DOI] [PubMed] [Google Scholar]
- 71.Lowe M, Rabouille C, Nakamura N, et al. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94(6):783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- 72.Gosavi P, Gleeson PA. The Function of the Golgi Ribbon Structure – An Enduring Mystery Unfolds! BioEssays. 2017;39(11):1700063. doi: 10.1002/bies.201700063. n/a. [DOI] [PubMed] [Google Scholar]
- 73.Buschman MD, Xing M, Field SJ. The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Frontiers in Neuroscience. 2015;9:362. doi: 10.3389/fnins.2015.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heuer D, Lipinski AR, Machuy N, et al. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2008;457:731. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- 75.Gonatas NK, Stieber A, Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. Journal of the Neurological Sciences. 2006;246(1):21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 76.Wei J-H, Seemann J. Golgi ribbon disassembly during mitosis, differentiation and disease progression. Current Opinion in Cell Biology. 2017 Aug 01;47:43–51. doi: 10.1016/j.ceb.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrosyan A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochemistry & molecular biology journal. 2015;1(1):16. doi: 10.21767/2471-8084.100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haase G, Rabouille C. Golgi Fragmentation in ALS Motor Neurons. New Mechanisms Targeting Microtubules, Tethers, and Transport Vesicles. Front Neurosci. 2015;9:448. doi: 10.3389/fnins.2015.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kellokumpu S, Sormunen R, Kellokumpu I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS Letters. 2002;516(1–3):217–224. doi: 10.1016/s0014-5793(02)02535-8. [DOI] [PubMed] [Google Scholar]
- 80.Micaroni M, Perinetti G, Berrie CP, Mironov AA. The SPCA1 Ca2+ Pump and Intracellular Membrane Trafficking. Traffic. 2010;11(10):1315–1333. doi: 10.1111/j.1600-0854.2010.01096.x. [DOI] [PubMed] [Google Scholar]
- 81.Sepúlveda MR, Wuytack F, Mata AM. High levels of Mn2+ inhibit secretory pathway Ca2+/Mn2+-ATPase (SPCA) activity and cause Golgi fragmentation in neurons and glia. Journal of Neurochemistry. 2012;123(5):824–836. doi: 10.1111/j.1471-4159.2012.07888.x. [DOI] [PubMed] [Google Scholar]
- 82.Pérez-Victoria FJ, Mardones GA, Bonifacino JS. Requirement of the Human GARP Complex for Mannose 6-phosphate-receptor-dependent Sorting of Cathepsin D to Lysosomes. Molecular Biology of the Cell. 2008 Jun 1;19(6):2350–2362. doi: 10.1091/mbc.E07-11-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hong W, Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014 Jan;24(1):35–43. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 84.Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Molecular Genetics and Metabolism. 2012 Apr 01;105(4):542–550. doi: 10.1016/j.ymgme.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 85.Spaapen LJM, Bakker JA, Meer SBvd, et al. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. Journal of inherited metabolic disease. 2005;28(5):707–714. doi: 10.1007/s10545-005-0015-z. [DOI] [PubMed] [Google Scholar]
- 86.Canuel M, Korkidakis A, Konnyu K, Morales CR. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochemical and Biophysical Research Communications. 2008 Aug 22;373(2):292–297. doi: 10.1016/j.bbrc.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 87.Gkantiragas I, Brügger B, Stüven E, et al. Sphingomyelin-enriched Microdomains at the Golgi Complex. Molecular Biology of the Cell. 2001 Jun 1;12(6):1819–1833. doi: 10.1091/mbc.12.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Comstra HS, McArthy J, Rudin-Rush S, et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. eLife. 2017 Mar 29;:6. doi: 10.7554/eLife.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laufman O, Freeze HH, Hong W, Lev S. Deficiency of the Cog8 Subunit in Normal and CDG-Derived Cells Impairs the Assembly of the COG and Golgi SNARE Complexes. Traffic (Copenhagen, Denmark) 2013;14(10):1065–1077. doi: 10.1111/tra.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Willett RA, Pokrovskaya ID, Lupashin VV. Fluorescent microscopy as a tool to elucidate dysfunction and mislocalization of Golgi glycosyltransferases in COG complex depleted mammalian cells. Methods in molecular biology (Clifton, NJ) 2013;1022:61–72. doi: 10.1007/978-1-62703-465-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. The Journal of experimental medicine. 2004 Jul 05;200(1):35–46. doi: 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Blackburn JB, Lupashin VV. Creating Knockouts of Conserved Oligomeric Golgi Complex Subunits Using CRISPR-Mediated Gene Editing Paired with a Selection Strategy Based on Glycosylation Defects Associated with Impaired COG Complex Function. In: Brown WJ, editor. The Golgi Complex: Methods and Protocols. New York, NY: Springer New York; 2016. pp. 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith RD, Willett R, Kudlyk T, et al. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic (Copenhagen, Denmark) 2009;10(10):1502–1517. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pokrovskaya ID, Szwedo JW, Goodwin A, Lupashina TV, Nagarajan UM, Lupashin VV. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cellular Microbiology. 2012 May;14(5):656–668. doi: 10.1111/j.1462-5822.2012.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]