

Abstract
We have combined immunogold labeling with the Miller spreading technique in order to localize proteins at the electron microscope (EM) level in whole mount nuclei from mouse and human fibroblasts. Anti-histone HI antibody labels nuclei uniformly, indicating that the nuclear interior is accessible to both antibodies and gold conjugates. Anti-topoisomerase I antibody labels nucleoli intensely, in agreement with previous immunofluorescent and biochemical data. Two different antibodies against the large subunit of RNA polymerase II (pol II) show preferential labeling of the nuclear periphery, as do antibodies against lamin, a known peripheral nuclear protein. Treatment of cells with a-amanitin results in loss of virtually all RNA polymerase II staining, supporting the specificity of labeling. Finally, when nuclei are incubated in the presence of biotin-UTP (bio-UTP) under run-off transcription conditions, incorporation is preferentially located at the nuclear periphery. These results support the conclusions that transcriptionally active pol II molecules are non-uniformly distributed in fibroblast nuclei, and that their differential distribution mirrors that of total pol II.
Data accumulated from a variety of approaches suggest that the interphase nucleus is an ordered structure (Cremer et al., 1982; Hancock and Hughes, 1982; Foe and Alberts, 1985). For example, the application of computer-assisted imaging of optical sections demonstrated a non-random arrangement of chromosomes in Drosophila melanogaster polytene nuclei (Agard and Sedat, 1983). Manuelidis and Borden (1988) employed in situ hybridization of chromosome-specific probes to show cell type-specific and distinct locations of chromosomes in central nervous system cells. In addition, specific DNA sequences, defined as attachment sites and thought to be involved in nuclear organization, have been identified around many genes (e.g., Mirkovitch et al., 1984; Cockerill and Garrard, 1986; Gasser and Laemmli, 1986), although their involvement in nuclear organization remains speculative. Taken together, these observations raise the question of whether or not there is a relationship between the expression of a sequence and its nuclear location.
Several experimental approaches have been used to determine where transcription occurs in nuclei. Electron microscope autoradiographs of sectioned cells after 3H-uridine pulse-labeling showed silver grains associated with so-called perichromatin fibers (Fakan et al., 1976). These fibers, thought to represent ribonucleoprotein complexes, are located at the periphery of condensed chromatin. However, recent experiments by Spector (1990) showed uridine incorporation primarily over nucleoli with diffuse low labeling of the nucleoplasm. Hutchison and Weintraub (1985) used a different approach to label putative transcription sites based on their preferential DNase I sensitivity (Weintraub and Groudine, 1976) and consequent preferential nick-translatability (Levitt et al., 1979). Cultured cell nuclei were nick-translated in the presence of biotinylated nucleotides and labeled regions located by immunofluorescence. Incorporation was clearly localized to the nuclear periphery in both chicken and mouse fibroblasts, whereas nuclei stained uniformly for DNA. Recently, Krystosek and Puck (1990) used nuclear nick-translation to demonstrate that normal and reverse-transformed CHO fibro-blast nuclei also show peripheral labeling. Although neither set of experiments located transcriptionally active chromatin directly, they both suggest nuclear compartmentalization in which transcriptionally active genes in fibroblasts are concentrated in the nuclear periphery. In contrast to these results, one highly transcribed viral gene has been localized to the nuclear interior by in situ hybridization (Lawrence et al., 1988).
We have developed an immunoelectron microscopic localization technique that can be quantitated easily and that permits a rapid assessment of nuclear antigen distribution. The technique was validated using antibodies with predictable distribution patterns based on previous work: anti-histone HI labeled nuclei uniformly, whereas anti-topoisomerase I preferentially labeled nucleoli. The intranuclear distribution of RNA polymerase II was then determined in a variety of experimental situations as an independent approach to investigate nuclear compartmentalization. The polyclonal antibodies used in most of these studies were directed against exon five of the large subunit of human pol II. The data obtained show that pol II is two- to three-fold more abundant in the nuclear periphery compared to the nuclear interior in both human and mouse fibroblasts. Labeling is specific, since treatment of cells with α-amanitin to destabilize pol II obliterates the signal. The pattern of bio-UTP incorporation in nuclear run-offs mirrors the differential distribution of pol II, supporting the hypothesis that many sequences transcribed by this enzyme are localized in this nuclear compartment.
Materials and methods
Immunoelectron microscopy and quantitation
Nuclei were obtained from mouse L929 or HeLa cells grown to 50% confluency in Joklik’s modified essential medium (Gibco) supplemented with 10% fetal calf serum (Irvine Scientific). Cells were pelleted at 800xg for 5 min., washed with 0.2 mM phosphate buffer (pH 7.5) in 0.1 M sucrose, lysed by incubation with 0.5% Nonidet-40 for 30 sec. at 25°C, and diluted with 2% volumes of PBS containing 0.1% Triton X-100. Nuclei were incubated in suspension with primary antibody at various dilutions for 2 h at 25°C, followed by deposition on parlodion-carbon-coated gold electron microscope (EM) grids by centrifugation through 1.0 M sucrose, essentially according to Rattner and Hamkalo (1978). Grids were rinsed in 0.4% Photoflo, air-dried, and transferred to 50 μl drops of goat anti-rabbit IgG conjugated to 15 nm colloidal gold particles (Janssen Pharmaceutica) diluted 1:10 in either 1% bovine serum albumin (BSA) buffer (20 mM Tris, pH 8.2, 0.15 M NaCl, 1% BSA, 0.14% Na-azide) or 0.5% gelatin buffer (20 mM Tris, pH 8.2, 0.15 M NaCl, 0.5% cold-water fish gelatin, 0.14% Na-azide). After incubation for 12–15 h at 25°C, grids were rinsed three times for 20 min. in 1% BSA buffer or 0.5% gelatin buffer followed by a 0.4% Photoflo rinse, and air dried. Electron microscopy was performed on a Zeiss 10CR electron microscope operated at 80 kV. Photographs are negatives so that colloidal gold particles appear as white particles. In order to obtain a quantitative estimate of antigen distribution, electron micrographs were analyzed as follows: Each micrograph was subdivided into one gm2 areas, the number of gold particles were counted in each of these areas, and averages were calculated for nuclear periphery, nuclear body, and background regions.
Western blot of RNA polymerase II
Whole cell extracts of HeLa cells grown in Joklik medium (Gibco) with 7% horse serum (Gibco) were prepared according to Manley et al. (1980). Nuclear extracts were prepared as described by Dignam et al. (1983). Purified calf thymus RNA polymerase II (Hodo and Blatti, 1977), fusion protein, and cell extracts were electrophoresed on 10% SDS-polyacrylamide gels. Proteins were then electrotransferred from the gels onto nitrocellulose sheets (Schleicher and Schuell; 0.45 μm) using Biorad Trans-Blot cells for 30 min. (Towbin et al., 1979). After the blot was incubated with RB buffer (10 mM Tris-HCl, pH 7.9,150 mM NaCl, 1 mM EDTA, 0.1% Tween) plus 3% BSA for 3 hours at 4°C, antibody was added and incubated for 12–16 hours at 4°C. The blot was rinsed with RB buffer, followed by several washes with RB buffer plus 0.5% Tween 20 at room temperature. The nitrocellulose filter was then incubated at 4°C with RB buffer containing 3% BSA and 125I-goat anti-rabbit antibody (2 × 105 cpm/ml). The filter was washed with RB buffer plus 0.5% Tween 20, and exposed to X-ray film using Lightning Plus intensifying screens (DuPont) at −70°C.
Inhibition of RNA synthesis and destabilization of pol II
RNA polymerase II was inhibited and destabilized in mouse L929 cells by incubation in the presence of 5 μg/ml α-amanitin (Sigma) for 4 hours prior to nuclear isolation (Frederiksen et al., 1978). Polymerase inhibition was assessed by the reduction in 3HCTP as described below.
Nuclear run-off
Mouse L929 nuclei were prepared, and run-off reactions were carried out as previously described (Weber et al., 1977), except that both UTP and biotinylated UTP were added to the reaction. Cells were labeled for 15 min. in vitro in the presence of 0.2 mM ATP, 0.1 mM GTP and UTP, 0.09 mM CTP, 32.4 μCi 3H-CTP (1.0 mCi/ml, ICN), and 0.2 mM 11-C-bio UTP (BRL). The reaction was stopped by the addition of 4 pg/ml of actinomycin D and a 50-fold excess of UTP. Nuclei were washed five times in PBS to remove unincorporated nucleotides, and then resuspended in PBS containing 0.1% Triton X-100 prior to deposition onto EM grids. Incorporation was assayed after reaction with strep-tavidin conjugated to 20 nm colloidal gold particles (Janssen Pharmaceutica) diluted 1:10 in 0.5% gelatin buffer, followed by electron microscopy. A biochemical assay of incorporation was carried out by measuring the amount of acid-precipitable 3H in the washes and the nuclear pellet.
Results
The immunogold localization technique and tests of specificity
Figure 1 diagrams the steps employed for the immunoelectron microscopic localization of proteins in whole-mount nuclei. This protocol includes only minor modifications of that used to carry out in situ hybridization at the EM level with colloidal gold labeling (Hutchison et al., 1982; Narayanswami and Hamkalo, 1986). Briefly, interphase nuclei are incubated in suspension with primary antibody, deposited on EM grids by centrifugation, and the grids are incubated in secondary antibody conjugated to 15 nm gold particles. Specimens are not fixed, since unfixed nuclei retain good morphology, remain attached to the grid surface during incubations, and exhibit enhanced immunoreactivity compared to fixed specimens. However, despite reduced labeling, fixed material showed the same patterns of labeling. Time course experiments were carried out to determine optimal reaction conditions for maximum labeling. Although nuclei in these preparations swell and the nuclear envelope appears to be at least partially solubilized during specimen preparation, nucleoli remain well-defined, suggesting that gross nuclear reorganization did not occur.
Figure 1.
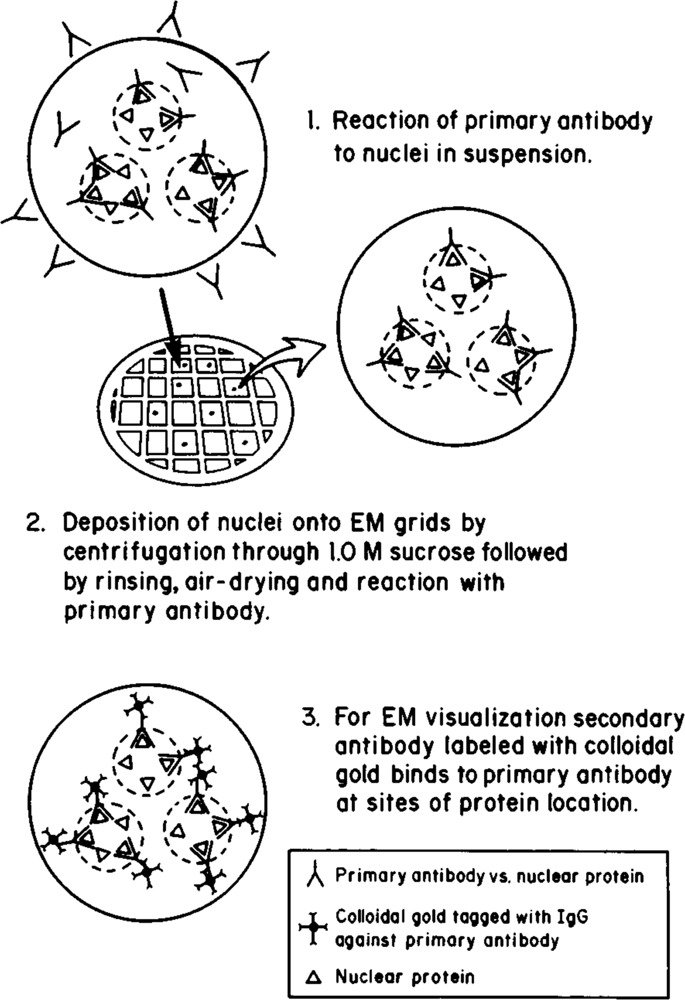
Immunoelectron microscopic localization technique for whole mount nuclei. Diagrammatic representation of the steps employed in the immunoelectron microscopic localization of nuclear antigens. Nuclei are reacted with primary antibody in suspension, deposited on EM grids by centrifugation, washed, and reacted with secondary antibody conjugated to colloidal gold particles. A nuclear protein is immunolocalized in situ by the visualization of colloidal gold particles under the EM.
In order to document the specificity of labeling, antibodies were localized, which detected antigens with predictable distributions. All the electron micrographs presented are negatives in order to permit better visualization of gold particles, which appear as white dots. In all cases, the labeling patterns illustrated are representative of at least 500 nuclei per experiment. Labeling is not due to nonspecific binding of the IgGs and/or gold to nuclei, since they are un-labeled after incubation with rabbit-anti-biotin (a “neutral” antibody) and secondary antibody-gold (Fig. 2a, Table 1). This control indicates that the nuclear labeling observed in subsequent experiments with antibodies directed against known nuclear antigens reflects bona fide reactions with the cognate antigens. In order to assess the accessibility of nuclei to antibodies and colloidal gold, HeLa nuclei were incubated with an antibody prepared against the major human HI subtype (H1A) (Clark et al., in preparation). Figure 2B illustrates a typical result with this antibody: gold particles are distributed relatively uniformly throughout the nucleus. Quantitation of these results (Table 1) supports this qualitative observation. Thus, nuclei prepared by nonionic detergent lysis of whole cells do not appear to exclude either IgGs or colloidal gold.
Figure 2.
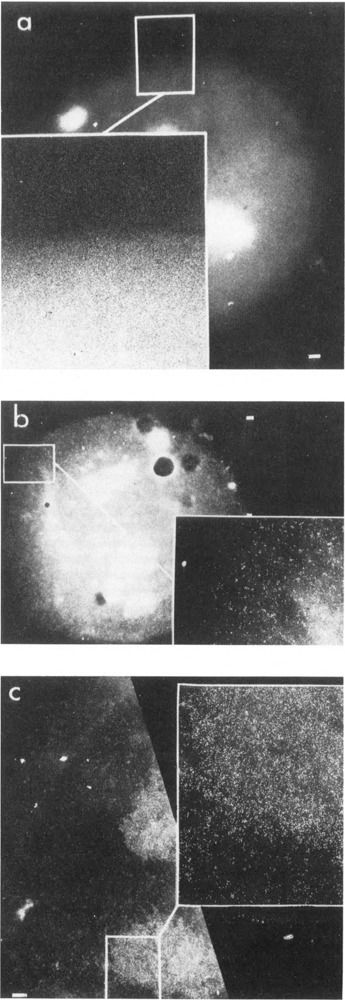
Immunoelectron microscopic labeling of control antigens. The reaction of a anti-biotin on mouse L929 cell nuclei, b anti-histone H1A on HeLa cell nuclei, and c anti-topoisomerase I on mouse L929 cell nuclei, followed by immunogold labeling (15 nm particles) as described in Methods, a. Rabbit anti-biotin antibody was used at a dilution of 1:50. b. Antibodies against human histone subtype HIA were obtained by injection of purified human placental histone H1A into rabbits by a modification of the Vaitukaitis method (Lewis et al., 1977). IgGs were purified by Affigel-blue (Bio-Rad) chromatography and concentration by ultrafiltration. A dilution of 1:50 was used for immunoelectron microscopic localization, c. Anti-topoisomerase I antibodies (the kind gift of Dr. Gerd Maul) were derived from a human scleroderma patient and purified by Affigel-blue (Bio-Rad) chromatography followed by ultrafiltration. A dilution of 1:20 to 1–50 was used for immunoelectron microscopic localization. Colloidal gold-secondary antibody complexes define antigen distributions. A single representative nucleus is shown in each case. Bar: 1 µm.
Table 1.
Distribution of antigens in cultured cell nuclei based on immunogold labeling.
| Antibody | Nuclear periphery | Nuclear body | Background | Peripheral/body ratio | Range |
|---|---|---|---|---|---|
| RNA polymerase II | |||||
| (-α-amanitin) | 341 ± 67 | 116 ± 35 | 5.1 ± 3.9 | 2.9 | (2.3–3.2) |
| (+α-amanitin) | 2.9 ± 1.9 | 5.8 ± 2.7 | 1.5 ± 1.4 | 0.5 | |
| Nuclear run-off | 46 ± 15 | 14 ± 6.3 | 1.8 + 1.8 | 3.2 | (3.2–4.0) |
| Lamins | 397 ± 85 | 146 + 59 | 1.1 +1.2 | 2.7 | |
| Histone H1A | 220 ± 33 | 262 ± 41 | 13 + 8.5 | 0.84 | |
| Biotin | 1.9 ± 1.4 | 1.5 ± 1.2 | 0.5 + 0.7 | 1.3 | |
Since the nucleolus is a structurally well-defined compact intranuclear compartment, we investigated the accessibility of an antigen known to be preferentially associated with ri-bosomal gene transcription. Topoisomerase I was chosen for this purpose, since specific antibodies are available, and there are extensive data in the literature documenting the presence of topoisomerase I at a high concentration on these genes (Fleischmann et al., 1984; Muller et al., 1985; Zhang et al., 1988; Culotta and Sollner-Webb, 1988; Rose et al., 1988). Incubations of nuclei with anti-topoisomerase I and colloidal gold results in a low level of labeling throughout most of the nucleus but intense labeling associated with nucleoli (Fig. 2C). This pattern is similar to that seen by immunofluorescence (Fleischmann et al., 1984; Muller et al., 1985; Rose et al., 1988). When Muller et al. (1985) localized topoisomerase I by imunogold staining of thin-sections only nucleoli were labeled, and those to a very low degree. In contrast to those results, the level of labeling we observe is so high that counting individual gold particles is not possible. Since EM immuno-localization to whole-mount nuclei affords a much higher level of labeling than observed with sections, the signal-to-noise ratio is high, and true sites and patterns of reaction are readily discerned.
Anti-RNA polymerase II localization
Based on the previous experiments, we concluded that the technique developed is capable of localizing antigens regardless of their nuclear location, even if they are buried in the center of the nucleus. The distribution of RNA polymerase II was determined with this technique in order to compare it with putative sites of transcription defined by in situ nick-translation studies (Hutchison and Weintraub, 1985; Krystosek and Puck, 1990). The antibody used in most of these experiments was made against the large subunit of RNA polymerase II (Cho et al., 1985). It is directed against amino acids 89–189 of the fifth exon of the large subunit (Rappaport et al., 1988), and its specificity is documented by both Western blot analysis (Fig. 3) and inhibition of transcription in whole cell extracts (data not shown).
Figure 3.
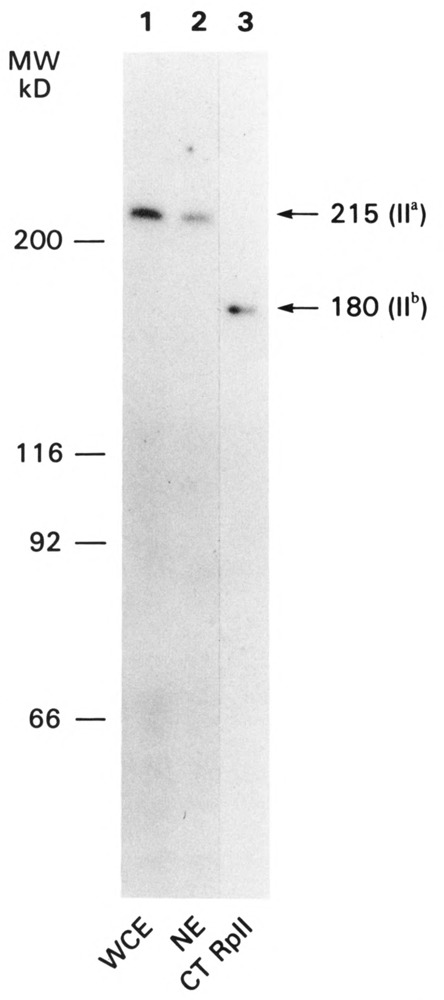
Electrophoretic analysis and immunodetection of the large subunit of RNA polymerase II. Whole cell extracts (40 µg of protein, lane 1), nuclear extracts (20 µg of protein, lane 2), or purified calf thymus RNA polymerase II (0.1 µg of protein, lane 3) were separated on 10% SDS-polyacrylamide gels. The proteins were transferred to nitrocellulose as described by Towbin et al. (1979) and reacted with affinity-purified anti-fusion protein antibody, followed by iodinated goat anti-rabbit IgG. An autoradiogram of the immunoreactive proteins is shown.
Figure 4a shows a typical mouse nucleus after immunogold labeling to localize pol II. Labeling is most apparent around the nuclear periphery. Chromatin at the nuclear periphery typically disperses slightly to form a halo around the body of the nucleus during cell lysis and centrifugation of the lysate onto the EM grid (see Fig. 1, Rattner and Hamkalo, 1979). Multiple experiments with both mouse and HeLa cells showed the same pattern with this antibody: preferential labeling of the nuclear periphery. In addition, a similar pattern was seen when primary antibody reactions were carried out after deposition of fixed or unfixed nuclei onto EM grids (data not shown). Immunofluorescent detection of this antibody also showed peripheral nuclear labeling (data not shown). A second antibody made against total calf thymus pol II (Kim and Dahmus, 1986) gave comparable results (Fig. 4b). Metaphase chromosomes were unlabeled (data not shown), indicating that the large subunit of RNA polymerase II disassociates or is inaccessible when chromosomes condense. Finally, no labeling was observed if the primary antibody was omitted from the reaction (data not shown).
Figure 4.
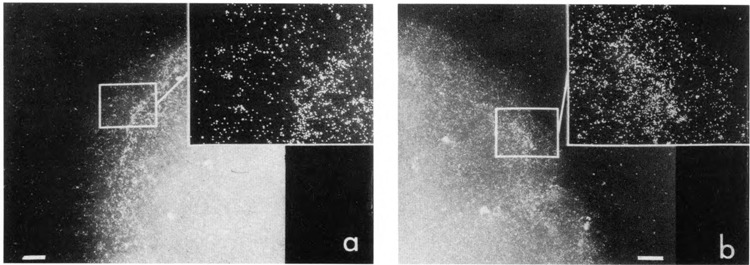
Immunoelectron microscopic localization of RNA polymerase II. Mouse L929 cell nuclei were reacted with one of two antibodies prepared against RNA polymerase II. Antibodies used in a were directed against a fusion protein containing a portion of β-galactosidase and the fifth exon of human RNA polymerase II (Cho et al., 1985; Rappaport et al., 1988). IgGs were prepared by ammonium sulfate precipitation and DE52 chromatography followed by a β-galactosidase affinity column to remove antibodies against the bacterial protein. A dilution of 1:40 was used. The antibodies used in b were prepared against total calf thymus RNA polymerase II (Kim and Dahmus, 1986) and were the generous gift of Dr. Michael Dahmus. They were purified by Affigel-blue (Bio-Rad) chromatography, concentrated by ultrafiltration, and used at a dilution of 1:40. In both cases, reaction sites were located with goat anti-rabbit colloidal gold. Bar: 1 μm.
A known peripheral nuclear antigen was localized as a control for the polymerase pattern. Figure 5 shows a nucleus after reaction with anti-nuclear lamina antibodies and colloidal gold. Although the nuclear lamina structure is not well-preserved in our preparations, labeling is concentrated at the edge of the nucleus as expected, often appearing as a band (Gerace et al., 1978; McKeon et al., 1983). Since anti-lamin gives the expected peripheral labeling pattern, we believe the pol II pattern accurately reflects the preferential peripheral location of pol II molecules.
Figure 5.
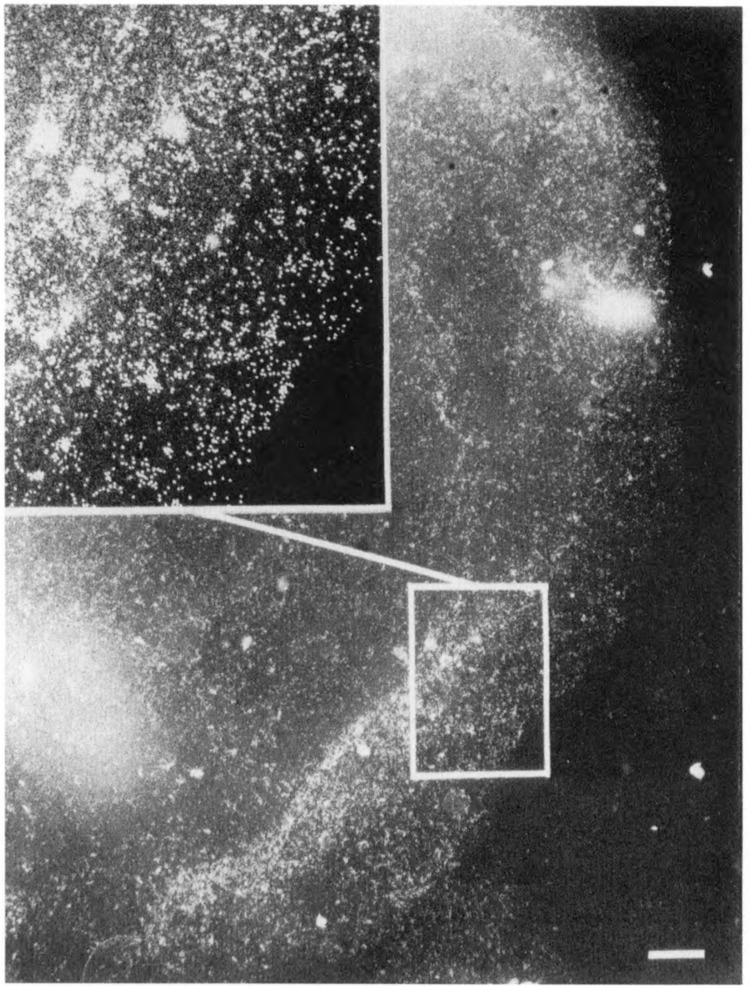
Immunoelectron microscopic localization of lamins. Mouse L929 cell nuclei were reacted with anti-lamin antibodies and secondary antibodies on colloidal gold. Antibodies derived from human autoimmune sera (McKeon et al., 1983) were the kind gift of Dr. Marc Kirschner. IgGs were purified by Affigel-blue (Bio-Rad) chromatography and concentrated by ultrafiltration. A dilution of 1:200 was used for immunoelectron microscopic localization. Bar: 1 μm.
Pol II-specific labeling was further validated by determining the effect on antibody labeling of pol II destabilization in the presence of α-amanitin. Guialis et al. (1979) showed a dramatic reduction in the half-life of the large sub-unit of Chinese hamster ovary pol II in the presence of this inhibitor: 70% of the subunit turned over after 5 hours in the presence of α-amanitin. Figure 6 shows a representative nucleus from cells grown in α-amanitin for 4 hours and then stained for pol II. Nuclear staining is virtually abolished, most likely due to the enhanced degradation of the reactive subunit.
Figure 6.
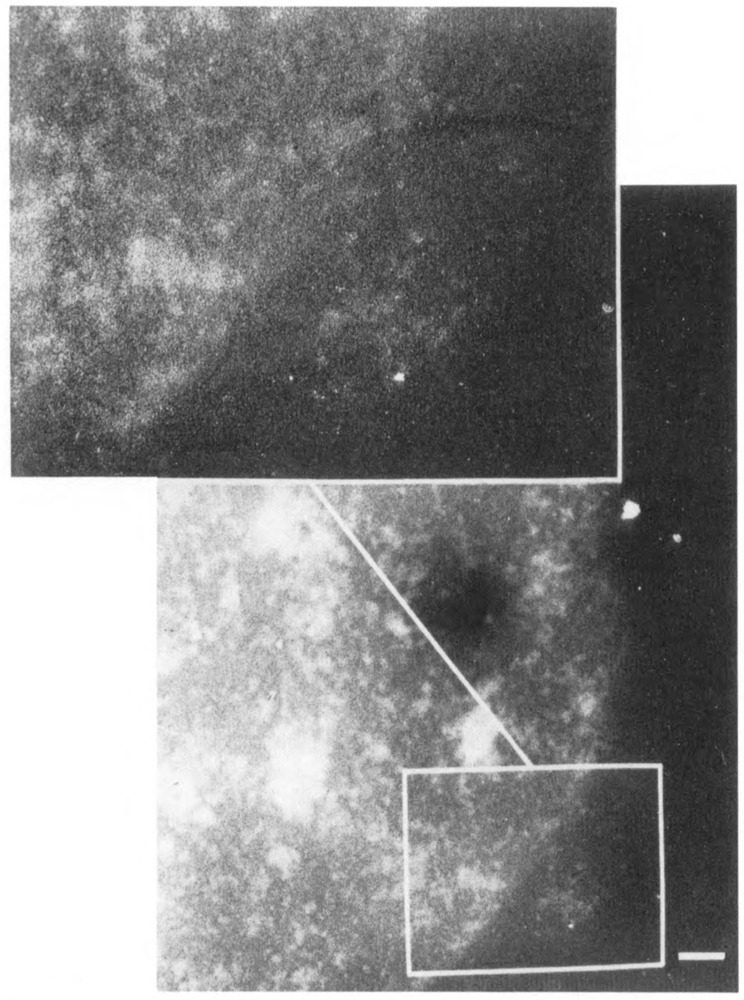
Immunolocalization of RNA polymerase II after α-amanitin treatment of cells. Mouse L929 cells were incubated in 5 μg/ml α-amanitin for 4 hours, and nuclei were released and incubated with anti-RNA polymerase II, followed by immunogold labeling as described in Methods. Bar: 1 μm.
Identification of sites of transcription in nuclear run-offs
The data presented indicate that the bulk of nuclear RNA polymerase II molecules are located at the periphery of fibroblast nuclei. Obviously these results do not distinguish between active and inactive enzymes. In order to locate actively transcribing polymerases, nuclear runoff reactions were carried out in the presence of bio-UTP followed by streptavidin staining. Figure 7 illustrates a typical nucleus after in vitro run-off transcription in the presence of bio-UTP and immunogold labeling. The level of labeling in these experiments is much lower because RNA polymerases incorporate bio-UTP only about 14% as well as unmodified nucleotides and do so only in the presence of UTP (Langer, 1986, data not shown). Nevertheless, the labeling pattern is similar to that observed for pol II with two to three times as many gold particles in the nuclear periphery. Nucleoli do not appear to label, probably because pol I incorporates bio-UTP even less well than pol II. When UTP was substituted for bio-UTP, no labeling was observed. In addition, labeling was abolished if transcription was inhibited by a-amanitin (data not shown). These two controls support the specificity of the reaction. A very rough estimate of the proportion of pol II molecules engaged in transcription can be made by comparing the density of labeling in run-offs with that for pol II (Table 1). Run-off labeling is about 15% that of pol II. This is probably a minimal estimate due to differences in the two types of experiments and the inefficient incorporation of bio-UTP.
Figure 7.
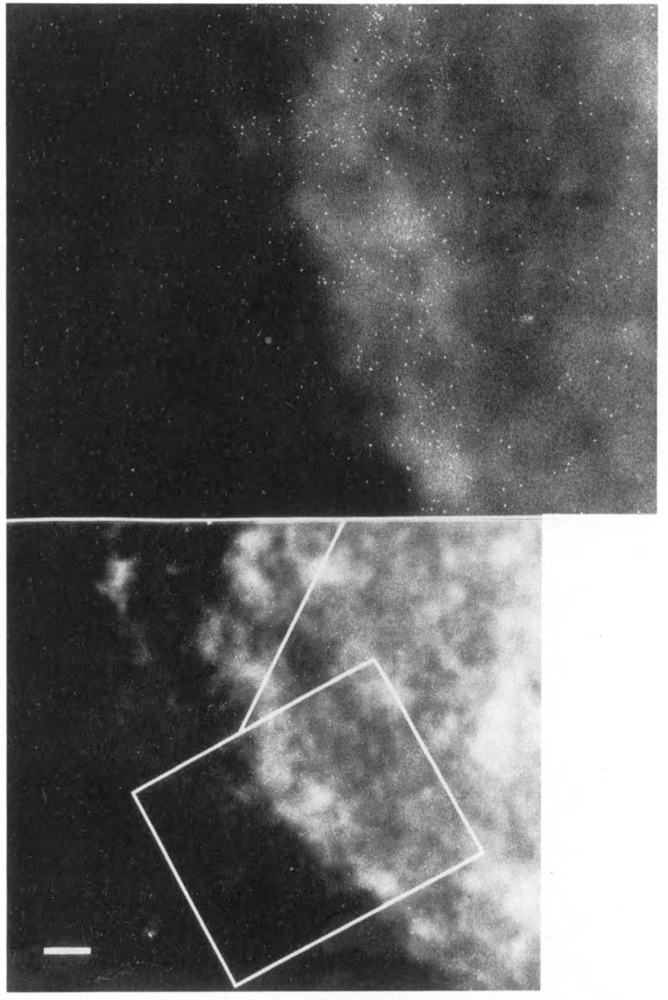
Immunolocalization of sites of transcription in a whole mount nucleus. Isolated nuclei were incubated under run-off transcription conditions (Weber et al., 1977) in the presence of biotinylated UTP in order to label sites active in transcription. Incorporated nucleotides were located with streptavidin-gold. Bar: 1 μm.
Quantitation of results and conclusions
Table 1 summarizes the results of a quantitative analysis of the data presented. Five nuclei from each localization were selected from different experiments for detailed analysis. The nuclear periphery was labeled with anti-RNA polymerase II antibody about three times more than the nuclear interior, and the peripheral/interior labeling ratio was similar for a known peripheral protein (lamin). In contrast, histone H1A is relatively uniformly distributed throughout the nucleus, while topoisomerase I is highly concentrated in nucleoli. When cells are treated with α-amanitin under conditions which result in pol II turnover, immunoreactivity disappears from the nucleus. The absolute number of gold particles varied among nuclei two- to three-fold. This variation could be due to variability in labeling or in the number of polymerases per nucleus. It is impossible to distinguish between these two alternatives without an independent determination of polymerase concentration per cell. Nevertheless, the striking similarity of peripheral/interior ratios, regardless of labeling intensity, supports a non-uniform polymerase distribution. Finally, transcribing polymerases, defined by nuclear runoff incorporation, also are about three-fold more abundant in the nuclear periphery.
Discussion
We have developed a simple immunoelectron microscopic technique which permits rapid quantitation of the relative amounts of nuclear antigens in different nuclear compartments. The specificity and potential to distinguish different patterns of protein distribution were validated with several control antigens. With these controls as a basis for comparison, the intranuclear distribution of RNA polymerase II molecules was determined in mouse and human fibroblasts. In both cases, using different antibodies, labeling of the nuclear periphery was several-fold greater than that of the body of the nucleus. A similar bias toward peripheral labeling was noted in nuclear run-off experiments, arguing that the bulk of transcriptionally active RNA polymerase II molecules are in this compartment. These results are in agreement with data from both Hutchison and Weintraub (1985) and Krystosek and Puck (1990), who showed that sequences which are preferentially nick-translated are peripheral in fibroblast nuclei.
Although topoisomerase I is associated with both active ribosomal and non-ribosomal transcription (Gilmour et al., 1986), it is not preferentially located in the nuclear periphery but is highly concentrated in nucleoli. One explanation for the lack of correspondence between topoisomerase I and RNA polymerase II distributions is based on the observation that the amounts of RNA polymerase II and topoisomerase I vary independently on different transcription units (Gilmour and Lis, 1985). In addition, the density of nascent transcripts on rRNA genes and their concentration in the nucleolus predicts the intense labeling observed.
The fact that the pattern of total nuclear RNA pol II labeling is very similar to the pattern of incorporation during nuclear run-offs suggests that the majority of enzyme molecules in the cells analyzed are sequestered in the nuclear periphery, regardless of their transcriptional state. It is possible that non-transcribing molecules are bound to chromatin at promoters of potentially active genes in pre-initiation complexes analogous to those described on uninduced heat-shock genes in Drosophila (Rougvie and Lis, 1988). However, all cells do not appear to sequester active sequences in the nuclear periphery. Chicken and newt erythrocytes exhibit channels of nick-translatable sequences which extend out to the nuclear envelope (Hutchison and Weintraub, 1985); nick-translatable sequences in human central nervous system cells appear to be in the nuclear interior (Manuelidis and Borden, 1988); and Lawrence et al. (1989) showed that integrated EBV genomes were restricted to the interior 0.8 radii of Namalwa cell nuclei. Nevertheless, most of the available data with fibroblasts support the differential distribution described here. If there are universal rules for interphase nuclear organization, they have yet to be discovered. It seems more likely that different organisms and different cell types will show profound differences in the location of a particular sequence based on the biology and physiology of the system. This view re-emphasizes a point previously made by Manuelidis and Borden (1988).
Although the similarity of labeling patterns for RNA polymerase II and the residual lamins may be coincidental, it may have some biological significance. Several years ago, Blobel (1985) proposed the “gene gating hypothesis,” which suggested that nuclear pore complexes, the nuclear lamins, and other components of the nuclear periphery are involved in nuclear organization, and that pore complexes interact specifically with transcribed regions of the genome. Although our studies do not resolve pore complexes, the finding that most of the RNA polymerase II molecules are preferentially located at the nuclear periphery does lend support to this hypothesis.
Acknowledgments
We thank Mrs. Agnes Demetrescu for excellent technical assistance, Dr. R. Zandomeni for the gift of purified calf thymus RNA polymerase II, Dr. M. Dahmus for anti-RNA polymerase II, and Dr. M. Kirschner for anti-lamin antibodies.
Research was supported by National Institutes of Health grant GM23241 to B. A. Hamkalo, and NIH grants AI13231 and CA10815 and American Cancer Society grant NP432 to R. Weinmann. R. F. Clark is supported by NIH Training Grant CA 09504-12.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Robert F. Clark is currently at the Department of Biology, Washington University, St. Louis, MO 63130. Ken W. Y. Cho is currently at the Department of Biological Chemistry, University of California, Los Angeles, CA 90024.
References
- Agard D. and Sedat J. (1983), Nature 302, 676–681. [DOI] [PubMed] [Google Scholar]
- Blobel G. (1985), Proc Natl Acad Sci USA 82, 8527–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K. W. Y., Khalili K., Zanndmeni R., and Weinmann R. (1985), J Biol Chem 260, 15204–15210. [PubMed] [Google Scholar]
- Cockerill P. N. and Garrard W. T. (1986), Cell 44, 273–282. [DOI] [PubMed] [Google Scholar]
- Cremer T., Cremer C., Baumann H., Leudtke E.-K., Sperling K., Teuber V., and Zorn C. (1982) Hum Genet 60, 40–56. [DOI] [PubMed] [Google Scholar]
- Culotta V. and Sollner-Webb B. (1988), Cell 52, 585–597. [DOI] [PubMed] [Google Scholar]
- Dignam J., Lebowitz R., and Roeder R. G. (1983), Nucl Acids Res 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakan S., Puvion E., and Spohr G. (1976) Exp Cell Res 99, 155–164. [DOI] [PubMed] [Google Scholar]
- Fleischmann G., Pflugfelder G., Steiner E., Javaherian K., Howard G., Wang J., and Elgin S. (1984), Proc Nad Acad Sci USA 81, 6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe V. and B. Alberts (1985), J Cell Biol 100, 1623–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen S., Hellung-Larsen P., and Gram Jensen E. (1978), FEBS Lett 87, 227–231. [DOI] [PubMed] [Google Scholar]
- Gasser S. M. and Laemmli U. K. (1986), Cell 46, 521–530. [DOI] [PubMed] [Google Scholar]
- Gerace L., Blum A., and Blobel G. (1978), J Cell Biol 79, 546–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour D. S. and Lis J. T. (1985), Mol Cell Biol 5, 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour D. S., Pflugfelder G., Wang J. C., and Lis J. T. (1986), Cell 44, 401–407. [DOI] [PubMed] [Google Scholar]
- Guialis A., Morrison K. E., and Ingles C. J. (1979), J Biol Chem 254, 4171–4176. [PubMed] [Google Scholar]
- Hancock R. and Hughes M. E. (1982), Cell 44, 201–212. [Google Scholar]
- Hodo H. G. II and Blatti S. P. (1977), Biochemistry 16, 2334–2343. [DOI] [PubMed] [Google Scholar]
- Hutchison N. J., Langer-Safer P. R., Ward D. C., and Hamkalo B. A. (1982), J Cell Biol 95, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison N. and Weintraub H. (1985), Cell 43, 471–482. [DOI] [PubMed] [Google Scholar]
- Kim W.-Y. and Dahmus M. E. (1986), J Biol Chem 261, 14219–14225. [PubMed] [Google Scholar]
- Krystosek A. and Puck T. T. (1990), Proc Natl Acad Sci USA 87, 6560–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer P. R. (1981), PhD Thesis, Yale University. [Google Scholar]
- Lawrence J. B., Singer R. H., and Marselle L. M. (1989), Cell 57, 493–502. [DOI] [PubMed] [Google Scholar]
- Levitt A., Axel R., and Cedar H. (1979), Dev Biol 69, 496–505. [DOI] [PubMed] [Google Scholar]
- Manley J. L., Fire A., Cano A., Sharp P. A., and Gefter M. L. (1980), Proc Natl Acad Sci USA 77, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. and Borden J. (1988), Chromosoma 96, 397–410. [DOI] [PubMed] [Google Scholar]
- McKeon F. D., Tuffanelli D. L., Fukujama K., and Kirschner M. W. (1983), Proc Natl Acad Sci USA 80, 4347–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkovitch J., Mirault M.-E., and Laemmli U. K. (1984), Cell 39, 223–232. [DOI] [PubMed] [Google Scholar]
- Muller M. T., Pfund W. P., Mehta V. B., and Trask D. K. (1985), EMBO J 4, 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanswami S. and Hamkalo B. (1986), Focus 8, 3–6. [Google Scholar]
- Rappaport J., Cho K., Saltzman A., Prenger J., Golomb M., and Weinmann R. (1988), Mol Cell Biol 8, 3136–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner J. B. and Hamkalo B. A. (1978), Chromosoma 69, 363–372. [DOI] [PubMed] [Google Scholar]
- Rattner J. B. and Hamkalo B. A. (1979), J Cell Biol 81, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K. M., Szopa J., Fan F.-S., Cheng Y.-C., Richter A., and Scheer U. (1988), Chromosoma 96,411–416. [DOI] [PubMed] [Google Scholar]
- Rougvie A. E. and Lis J. T. (1988), Cell 54, 795–804. [DOI] [PubMed] [Google Scholar]
- Spector D. L. (1990), Proc Natl Acad Sci USA 87, 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., and Gordon J. (1979), Proc Natl Acad Sci USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J., Jelinek W., and Darnell J. E. Jr. (1977), Cell 10, 611–616. [DOI] [PubMed] [Google Scholar]
- Weintraub H. and Groudine M. (1976), Science 193, 848–856. [DOI] [PubMed] [Google Scholar]
- Zhang H., Wang J. C., and Liu L. F. (1988), Proc Natl Acad Sci USA 85, 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]