

ABSTRACT
Antimicrobial peptides (AMPs), including chemokines, are produced during infections to kill pathogenic bacteria. To fill in gaps in knowledge about the sensitivities of Streptococcus pneumoniae and related Streptococcus species to chemokines and AMPs, we performed a systematic, quantitative study of inhibition by chemokine CXCL10 and the AMPs LL-37 and nisin. In a standard Tris-glucose buffer (TGS), all strains assayed lacked metabolic activity, as determined by resazurin (alamarBlue) reduction, and were extremely sensitive to CXCL10 and AMPs (50% inhibitory concentration [IC50], ∼0.04 μM). In TGS, changes in sensitivities caused by mutations were undetectable. In contrast, strains that retained reductive metabolic activity in a different assay buffer (NPB [10 mM sodium phosphate {pH 7.4}, 1% {vol/vol} brain heart infusion {BHI} broth]) were less sensitive to CXCL10 and AMPs than in TGS. In NPB, mutants known to respond to AMPs, such as Δdlt mutants lacking d-alanylation of teichoic acids, exhibited the expected increased sensitivity. S. pneumoniae serotype 2 strain D39 was much (∼10-fold) less sensitive to CXCL10 killing in NPB than serotype 4 strain TIGR4, and the sensitivity of TIGR4 was unaffected by the absence of capsule. Candidate screening of strain D39 revealed that mutants lacking Opp (ΔamiACDEF) oligopeptide permease were significantly more resistant to CXCL10 than the wild-type strain. This increased resistance could indicate that Opp is a target for CXCL10 binding or that it transports CXCL10 into cells. Finally, ΔftsX or ΔftsE mutants of Bacillus subtilis or amino acid changes that interfere with FtsX function in S. pneumoniae did not impart resistance to CXCL10, in contrast to previous results for Bacillus anthracis, indicating that FtsX is not a general target for CXCL10 binding.
IMPORTANCE S. pneumoniae (pneumococcus) is a human commensal bacterium and major opportunistic respiratory pathogen that causes serious invasive diseases, killing millions of people worldwide annually. Because of its increasing antibiotic resistance, S. pneumoniae is now listed as a “superbug” for which new antibiotics are urgently needed. This report fills in knowledge gaps and resolves inconsistencies in the scientific literature about the sensitivity of S. pneumoniae and related Streptococcus pathogens to chemokines and AMPs. It also reveals a new mechanism by which S. pneumoniae can acquire resistance to chemokine CXCL10. This mechanism involves the Opp (AmiACDEF) oligopeptide transporter, which plays additional pleiotropic roles in pneumococcal physiology, quorum sensing, and virulence. Taking the results together, this work provides new information about the way chemokines kill pneumococcal cells.
KEYWORDS: chemokine antimicrobial activity, chemokine resistance mechanisms, CXCL10 family chemokines, LL-37, nisin, FtsEX functions
INTRODUCTION
Chemokines are small (7- to 12-kDa), cationic chemotactic cytokines that bind to cell surface receptors on leukocytes, such as macrophages, neutrophils, and T lymphocytes, and that attract them to sites of host infection (reviewed in references 1 and 2). Chemokines have highly conserved structures similar to those of members of the defensin class of antimicrobial peptides (AMPs), consisting of a three-stranded beta-sheet, a C-terminal alpha-helix that tends to be amphipathic, and two intramolecular disulfide bonds (reviewed in references 3, 4, and 5). Many chemokines, including CXCL10, form dimers and higher-order oligomers which may have functional implications (2–4, 6). Synergism with other chemokines through heterocomplex formation may also be important for function (reviewed in reference 7).
CXCL9, CXCL10, and CXCL11 are inflammatory CXC family chemokines that interact with the chemokine receptor CXCR3 (8, 9) and are produced by pharyngeal epithelial cells in response to gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) (10). Numerous studies show CXCL10 and CXCL9 to be highly induced in respiratory cells by bacterial pathogens, including the major human respiratory pathogen Streptococcus pneumoniae (11–14).
Besides their chemotactic function, many chemokines exhibit direct antimicrobial activity against a broad range of bacteria (15, 16). Chemokines share many structural and functional features with AMPs, a group of low-molecular-weight, cationic peptides produced by host cells to kill invading bacteria (5, 17, 18). By electron microscopy criteria, antimicrobial chemokines appear to induce lysis of the bacterial membrane (19, 20). Since bacterial membranes are highly anionic, the attraction of positively charged chemokines to the anionic bacterial membrane is thought to mediate initial binding (21, 22). Studies measuring membrane depolarization and nucleic acid staining in real time show that some AMPs, such as the human cathelicidin LL-37, interact with the membrane nonspecifically to cause cell lysis (23, 24). In addition, LL-37 has also been shown to enter the cell and bind cytoplasmic targets, such as acyl carrier protein (25). Other AMPs interact with specific components of the cell membrane or intracellular targets (22). The lantibiotic nisin shows high affinity for the cell wall precursor external lipid II, and this interaction is thought to facilitate insertion of nisin into the cell membrane, leading to pore formation and lysis (26, 27). In addition, nisin and other lantibiotics displace lipid II from its functional localization in Gram-positive bacteria, thus blocking cell wall synthesis (28). While some chemokines and AMPs are constitutively expressed at distinct sites in the host (16), many are induced upon bacterial infection (11, 14, 29).
Due to the electrostatic interactions between cationic chemokines or AMPs and the anionic bacterial cell membrane, antimicrobial activity of many chemokines or AMPs is decreased or abolished at salt concentrations above 50 to 150 mM in in vitro assays (30, 31). An increase in the salt concentration in the airway surface liquid of the lower respiratory tract in cystic fibrosis patients has been implicated in decreasing the antimicrobial activity of AMPs, leading to bacterial infection (30, 32). In this respect, sweat and mucosal secretions, such as saliva, are produced under conditions of low ionic concentrations that would favor antimicrobial activities (16). Standard MIC assays can be inapplicable for determining the antimicrobial activity of AMPs and chemokines against some bacteria whose growth is impeded by low-salt conditions. One readout format for AMP and chemokine killing is bacterial survival determined by analysis of CFU counts, which can be performed in a variety of media or buffers. A commonly used buffer for this assay is hypo-osmotic 10 mM Tris-HCl (pH 7.4) containing 5 mM glucose (TGS; see Materials and Methods) (11, 31, 33). Another readout format for AMP or chemokine killing is the fluorescence signal of indicator dyes, such as resazurin (alamarBlue), which provides a measurement of bacterial reductive metabolism (34–38). This resazurin-based assay requires cells to be metabolically active in the incubation buffer.
Bacterial pathogens employ numerous strategies to evade chemokine- and AMP-mediated killing (39, 40). Gram-positive bacteria repel AMPs by increasing the positive charge of the cell surface, most often through the addition of d-alanine or phosphorylcholine to teichoic acids (41–43). It has also been suggested that the d-alanylation of teichoic acids decreases the permeability of the cell wall, thus limiting access of chemokines and AMPs to the bacterial cell membrane (44). Anionic exopolysaccharide capsules, a virulence factor of many bacterial pathogens, can sensitize bacteria to AMPs, possibly by masking protective positive-charge modifications (45, 46). However, some pathogens actively shed their capsule upon interaction with AMPs, and the released capsule fragments may bind AMPs, providing a sink imparting resistance to the unencapsulated cells (47, 48). Cell surface proteases have also been shown to degrade chemokines and AMPs (20, 40). Recently, ABC family transporters have been proposed to provide AMP resistance, possibly by removing AMPs from the cell surface, where membrane permeabilization can occur, and transporting them into the cytoplasm for proteolytic degradation (49). Most, if not all, of these defense mechanisms are induced when bacteria sense chemokine and AMP stress, often through the activity of bacterial two-component systems (50–52).
Recent work has shown that a ΔftsX mutant exhibits resistance to chemokines CXCL9 and CXCL10 in Bacillus anthracis (53, 54). FtsX is a conserved bacterial cell division protein that is essential in some bacteria, including S. pneumoniae (55–58). FtsX is an integral membrane protein with two extracellular loops (ECL1 and ECL2) that dimerizes in phospholipid nanodiscs (59). In Escherichia coli and likely other bacteria, FtsX plays a role in regulating penicillin-binding protein (PBP) activity in the divisome (60). In S. pneumoniae and several other bacterial species, FtsX is a component of a peptidoglycan (PG) hydrolase complex, along with cytoplasmic FtsE ATPase and an extracellular peptidoglycan hydrolase, such as PcsB in S. pneumoniae (57, 61). In its role as a subunit of PG hydrolases, the extracellular loops of FtsX interact with and regulate the activity of PG hydrolases in PG remodeling (57, 61, 62). It has been proposed that CXCL10 kills B. anthracis by interacting with FtsX, thereby misregulating PG hydrolysis, which leads to cell lysis (53, 54, 63).
Antimicrobial activity of some chemokines, including CXCL10, has been reported previously for several Streptococcus species other than S. pneumoniae using different assay conditions and readout formats (11, 31, 64–66). While the sensitivities of S. pneumoniae serotype 4 strain TIGR4 to CXCL9 (11) and of S. pneumoniae serotype 19F to CXCL14 (33) have been published, limited information is available about the sensitivity to or mechanism of killing of different S. pneumoniae serotype strains by chemokines, despite the upregulation of CXCL10-family chemokines during pneumococcal infection (11, 33, 67). More has been reported about the antimicrobial effects of various AMPs, including LL-37, nisin (52), α-defensin human neutrophil proteins 1 to 3 (HNP1 to -3) (45), and human β-defensins (68), against S. pneumoniae, again using different assay formats.
In this paper, we report a quantitative comparison of the antimicrobial properties of CXCL10 and other chemokines and AMPs against wild-type (WT) and mutant S. pneumoniae strains in media in which cells are metabolically static or active. To this end, we evaluated different incubation buffers for production of metabolically active cells in which the fluorescence signal in a resazurin-based assay was correlated with cell viability by CFU counting. Optimal results were obtained in an assay buffer that retained reductive metabolic activity (NPB) and that contained 10 mM sodium phosphate (pH 7.4) and 1% (vol/vol) brain heart infusion (BHI) broth. We focused on chemokine CXCL10 because of its possible interaction with FtsX in B. anthracis to cause cell lysis (53, 54, 63) and because of our interest in FtsX function in S. pneumoniae (57, 59, 61). Previously studied AMP LL-37 and bacteriocin nisin were included for comparison.
Our results show that CXCL10 has direct antimicrobial activity against S. pneumoniae and other Streptococcus species and that the sensitivity of S. pneumoniae to CXCL10 varies for cells in different media and metabolic conditions and between S. pneumoniae serotypes. Several candidate mutants were surveyed for possible CXCL10 and AMP resistance. We found that mutants deficient for the Opp (AmiACDEF [AmiA-F]) general oligopeptide transporter showed increased resistance to CXCL10, whereas dlt mutants deficient for d-alanylation of teichoic acids were more sensitive to CXCL10. However, the results obtained with deletion mutants of ftsX and ftsE in Bacillus subtilis and amino acid changes that interfere with FtsX function in S. pneumoniae did not show resistance to CXCL10, in contrast to ftsEX mutants of B. anthracis (53, 54). Together, these results indicate that different Gram-positive species are susceptible to CXCL10 antimicrobial activity via several distinct mechanisms.
RESULTS
S. pneumoniae metabolic activity varies in different media used for chemokine and AMP assays.
Many bacteria, including S. pneumoniae, mount a significant response to AMP stress in growth medium, including transcriptional regulation of several genes as well as dynamic capsule shedding (47, 52). Previous reports determined the antimicrobial activity of CXC family chemokines against Streptococcus species suspended in low-cation and low-salt buffered hypo-osmotic solutions, such as TGS (10 mM Tris-HCl, 5 mM glucose, pH 7.4), that minimize salt inhibition of chemokine and AMP antimicrobial activity (30–32, 69). However, it was unclear whether S. pneumoniae cells are metabolically active in assay buffers such as TGS. Therefore, we used a resazurin (alamarBlue)-based fluorescence assay (34–38) to determine the metabolic state of S. pneumoniae serotype 2 (strain D39) cells in TGS and in other buffered solutions used previously to determine chemokine and AMP antimicrobial activity (see Materials and Methods). In this assay, nonfluorescent resazurin enters bacterial cells and is reduced to fluorescent resorufin by bacterial metabolites, such as NADPH (34). During active metabolism, these molecules are continuously recycled to their reduced state and produce more resorufin, generating increasing fluorescence signal over time (Fig. 1A). Starting with equal cell masses, the fluorescence signal at a given time point provides a measurement of bacterial reductive metabolism (Fig. 1A), even in nonproliferating cells (Fig. 1B). Additionally, killing of bacterial cells by chemokines or AMPs can be monitored as a decrease in fluorescence in this assay.
FIG 1.
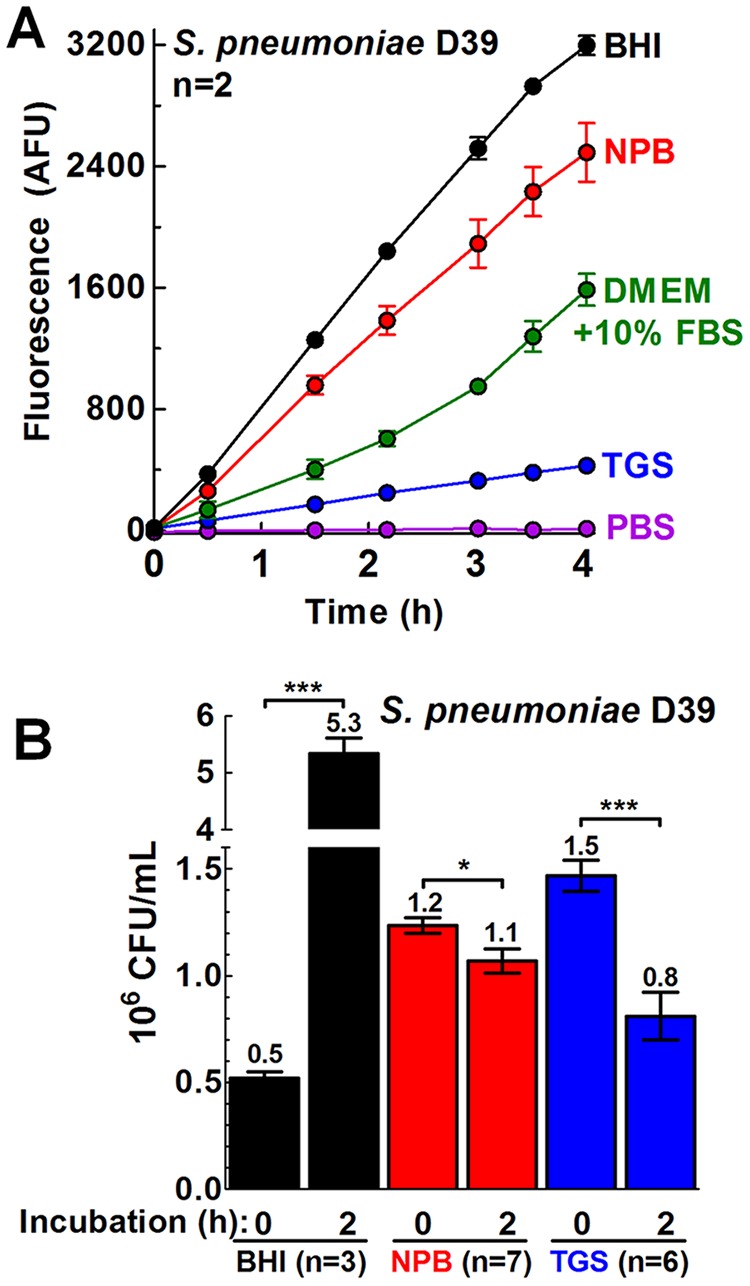
S. pneumoniae metabolic activity varies in different media used for chemokine and AMP assays. S. pneumoniae D39 (IU1690) was grown in BHI broth until the early log phase (OD620 of 0.1 to 0.13) and then washed and diluted in BHI broth, in 10 mM sodium phosphate (pH 7.4)–1% (vol/vol) BHI broth (NPB), in DMEM–10% (vol/vol) FBS, in 10 mM Tris-HCl–5 mM glucose (pH 7.4) (TGS), or in PBS, all containing resazurin (alamarBlue) dye (10% total volume). The increase of fluorescence signal with time was due to the accumulation of fluorescent resorufin produced from the reduction of cell-permeative resazurin. Fluorescence is used as an indication of bacterial reductive metabolism. (A) Plots of fluorescence signal over time (AFU, arbitrary fluorescence units). Cells were incubated at 37°C in 5% CO2, and fluorescence was measured periodically from h 0 to h 4. Each point represents the mean ± standard error of the mean (SEM) of results of two independent experiments, each with duplicate reactions. (B) CFU counts per milliliter of S. pneumoniae D39 (IU1690) before or after incubation in BHI broth, TGS, or NPB. Bars represent mean 106 CFU/ml ± SEM (n = number of independent experiments). The statistical significance of the results of comparisons between the 0-h and 2-h time points for each buffer was determined using the Mann-Whitney U test. *, P < 0.05; ***, P < 0.001.
Brain heart infusion (BHI) broth is a rich medium used routinely to grow S. pneumoniae in liquid culture and was included here as a positive control for both metabolic activity and cell proliferation (Fig. 1A and B, respectively). NPB (10 mM sodium phosphate [pH 7.4] containing 1% [vol/vol] BHI broth) was used previously to investigate the antibacterial activity of human β-defensin 3 (hBD-3) and LL-37 against nontypeable Haemophilus influenzae (70). S. pneumoniae D39 cells in NPB in the resazurin-based assay showed 78% of the metabolic activity seen with cells incubated in BHI broth over 4 h (Fig. 1A), even though there was no increase in cell mass over 6 h in NPB (as measured by culture turbidity at an optical density of 620 nm [OD620]) (not shown) or in cell proliferation over 2 h (assayed by CFU) (Fig. 1B). Dulbecco's modified Eagle's medium (DMEM), a defined cell culture medium, supplemented with 10% (vol/vol) fetal bovine serum (FBS), was used previously to demonstrate the antimicrobial activity of CXCL9 and CXCL10 against Bacillus anthracis (53, 54), despite the relatively high (∼160 mM) cation concentration in DMEM and the fact that serum decreases the antimicrobial activity of some AMPs (71). S. pneumoniae D39 cells exhibited an ∼50% decrease in reductive metabolic activity in DMEM–10% (vol/vol) FBS compared to BHI broth (Fig. 1A). S. pneumoniae D39 cells resuspended in TGS or phosphate-buffered saline (PBS) (pH 7.4) displayed 13% or <1% metabolic activity, respectively, compared to cells incubated in BHI broth over 4 h, suggesting a minimally reductive, static metabolism (Fig. 1A).
After 2 h of incubation, CFU assays indicated that the level of S. pneumoniae D39 cells in BHI broth increased ∼10-fold (Fig. 1B), while the cells in NPB and TGS were at ∼87% and ∼55% of their initial CFU, respectively (Fig. 1B). CFU numbers were not corrected for cell chaining, which would reduce the apparent CFU level per milliliter. Live-dead staining assays performed using a LIVE/DEAD BacLight bacterial viability kit (see Materials and Methods) showed that ∼75% of the cells were viable after dilution in NPB and that a similar proportion of viable cells (64%) was present after 2 h of incubation (data not shown), consistent with the CFU data. Together, these results indicate that the levels of S. pneumoniae D39 cellular reductive metabolism varied widely under the assay conditions used in previous studies that were performed to determine chemokine and AMP antimicrobial activity against a variety of bacteria. S. pneumoniae reductive metabolism was minimal in assay buffers, such as TGS, and remained robust in NPB, despite a lack of overt cell growth (Fig. 1). In this paper, S. pneumoniae cells assayed in TGS or NPB are referred to as metabolically static or metabolically active, respectively. As shown below, the metabolically static S. pneumoniae cells assayed in TGS were considerably more sensitive to chemokines and AMPs than the metabolically active cells assayed in NPB.
Metabolically static S. pneumoniae cells in TGS are sensitive to CXCL10 family chemokines at concentrations in the 0.03 to 0.06 μM range.
Previous studies demonstrated antimicrobial activity of the three CXCL10 family chemokines against several Streptococcus species, including S. pneumoniae TIGR4 sensitivity to CXCL9, for cells in TGS (11, 20, 31). We recapitulated these experiments for CXCL10 (Materials and Methods). CXCL10 exhibited potent antimicrobial activity against S. pneumoniae in a CFU assay, regardless of the serotype or the presence of capsule, with 50% inhibitory concentrations (IC50s) ranging between 0.033 and 0.058 μM for encapsulated and unencapsulated variants of D39 (serotype 2) and TIGR4 (serotype 4) (Table 1, rows 1, 2, 4, and 5; see also Fig. S1 in the supplemental material). The level of killing of S. pneumoniae D39 recorded for CXCL9 was similar to that seen with CXCL10 (Table 2, rows 1 and 4), and these IC50s are comparable to the estimated IC50s for killing by CXCL9 of S. pyogenes (0.06 μM) or S. pneumoniae TIGR4 (∼0.03 μM) reported previously (11, 31). The IC50 value for killing by CXCL14 of S. pneumoniae D39 was 0.08 μM, which is ∼2-fold higher than that for CXCL9 or CXCL10 for S. pneumoniae D39 (Table 2, rows 1 and 5) but is ∼4-fold lower than the IC50 (∼0.3 μM) reported for CXCL14 killing of an S. pneumoniae serotype 19F isolate under the same conditions (33), demonstrating the applicability of this assay to chemokines outside the CXCL10 family. Finally, in TGS, CXCL10 exhibited similar levels of antimicrobial activity against S. pneumoniae D39, S. pneumoniae TIGR4, Streptococcus mitis, Streptococcus sanguinis, and Streptococcus mutans strains (Table 1, rows 1, 4, and 6 to 8), which contrasts with the lack of susceptibility (MIC >23 μM) to CXCL10 reported for S. mitis, S. sanguinis, and S. mutans in radial-diffusion assays (65, 66).
TABLE 1.
IC50 values determined for CXCL10-treated cells in TGS using a CFU survival assaya
| Strain or speciesb | IC50 (μM)c | 95% CI (μM)d |
|---|---|---|
| S. pneumoniae | ||
| D39 | 0.039 | 0.034–0.045 |
| D39 Δcps | 0.058 | —e |
| D39 ΔOPT | 0.054 | — |
| TIGR4 | 0.040 | 0.028–0.056 |
| TIGR4 Δcps | 0.033 | 0.009–0.126 |
| S. mitis | 0.062 | 0.046–0.084 |
| S. sanguinis | 0.057 | — |
| S. mutans | 0.055 | 0.034–0.088 |
Bacterial strains were cultured in BHI broth, resuspended in TGS buffer (10 mM Tris-HCl with 5 mM glucose, pH 7.4), and assayed for the number of CFU after treatment with CXCL10 for 2 h as described in Materials and Methods.
Strain numbers and the ranges of CXCL10 concentrations used for the assays were as follows: for D39, IU1690, 0.004 to 0.231 μM; for D39 Δcps, IU1945, 0.004 to 0.116 μM; for D39 ΔOPT (defined as ΔamiA-F Δ[spd_1167-spd_1170] ΔaliA ΔaliB), IU11919, 0.004 to 0.116 μM; for TIGR4, IU11966, 0.004 to 0.116 μM; for TIGR4 Δcps, IU12001, 0.004 to 0.116 μM; for S. mitis, IU11303, 0.004 to 0.116 μM; for S. sanguinis, IU11305, 0.007 to 0.231 μM; for S. mutans, IU11309, 0.007 to 0.463 μM.
IC50 values were obtained from pooled data from at least two independent experiments and fitted to a dose-response curve (log of inhibitor versus response-variable slope) using GraphPad Prism.
Data represent 95% confidence intervals of the IC50.
—, 95% CI could not be calculated due to a high Hill slope value.
TABLE 2.
IC50 values determined for chemokine-treated S. pneumoniae cells in TGS using a CFU survival assaya
| Chemokine | D39 |
D39 Δcps |
||
|---|---|---|---|---|
| IC50 (μM)b | 95% CI (μM)c | IC50 (μM)b | 95% CI (μM)c | |
| CXCL10 (aa 1–77) | 0.039 | 0.034–0.045 | 0.058 | — |
| N-terminal CXCL10 (aa 1–54) | 0.14 | 0.12–0.16 | 0.23 | — |
| C-terminal CXCL10 (aa 55–77) | 1.1 | 0.76–1.6 | 1.5 | 0.82–2.9 |
| CXCL9 | 0.047 | 0.032–0.068 | Nd | |
| CXCL14 | 0.080 | 0.055–0.116 | Nd | |
D39 (IU1690) and D39 Δcps (IU1945) strains were cultured in BHI broth, resuspended in TGS buffer (10 mM Tris-HCl with 5 mM glucose, pH 7.4), and assayed for the number of CFU after treatment with chemokines for 2 h as described in Materials and Methods. The ranges of chemokine concentrations used for the assays were as follows: for D39, 0.004 to 0.231 μM (CXCL10), 0.014 to 1.85 μM (N-terminal CXCL10) (aa 1 to 54), 0.058 to 18.5 μM (C-terminal CXCL10) (aa 55 to 77), 0.004 to 0.116 μM (CXCL9), and 0.007 to 0.231 μM (CXCL14); for D39 Δcps, 0.004 to 0.116 μM (CXCL10), 0.014 to 0.98 μM (N-terminal CXCL10) (aa 1 to 54), and 0.116 to 3.7 μM (C-terminal CXCL10) (aa 55 to 77).
IC50 values were obtained from pooled data from at least two independent experiments and fitted to a dose-response curve (log of inhibitor versus response-variable slope) using GraphPad Prism.
Data represent 95% confidence intervals of the IC50 values. —, 95% CI data could not be calculated due to a high Hill slope value.
Metabolically active S. pneumoniae cells in NPB are significantly less sensitive to CXCL10 and AMPs than S. pneumoniae cells in TGS.
To compare levels of CXCL10 killing of S. pneumoniae strains under different buffer conditions, a CFU survival assay was performed for bacteria treated with CXCL10 in NPB or TGS. S. pneumoniae D39 in NPB was 50-fold to 100-fold less sensitive to CXCL10 than S. pneumoniae D39 in TGS, with an IC50 of 1.9 μM compared to 0.016 μM (Fig. 2A; see also Table S2 [rows 1 and 2] in the supplemental material). The IC50 of 0.016 μM for S. pneumoniae D39 in TGS (Fig. 2A; see also Table S2) differs somewhat from the value of 0.039 μM shown in Tables 1 and 2 and Fig. S1 due to differences in the number of CFU added in each set of experiments (see Materials and Methods). Nevertheless, it is clear that cells are killed by CXCL10 at a higher concentration in NPB than in TGS. The antimicrobial activities of human cathelicidin LL-37 and the lantibiotic nisin were also determined in a CFU assay against S. pneumoniae D39 cells in TGS or NPB (Fig. 2B and C; see also Table S2, rows 1 to 3). S. pneumoniae D39 in NPB was 10-fold to 25-fold or 3-fold less sensitive to LL-37 or nisin, respectively, than S. pneumoniae D39 in TGS. Furthermore, metabolically active S. pneumoniae D39 in NPB was less sensitive to CXCL10 than to LL-37 or nisin, whereas in metabolically static S. pneumoniae D39 in TGS, the sensitivity trend was reversed (Fig. 2; see also Table S2).
FIG 2.
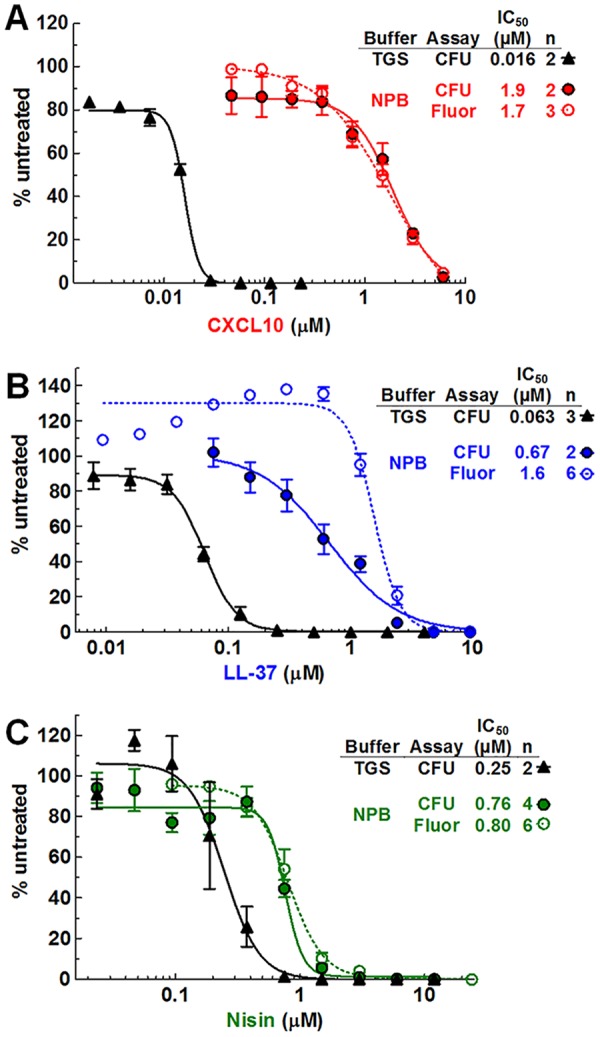
S. pneumoniae D39 cells in NPB are significantly less sensitive to CXCL10 and AMPs than S. pneumoniae cells in TGS. The sensitivity of S. pneumoniae D39 (IU1690) to CXCL10 (A), LL-37 (B), and nisin (C) was determined using a CFU survival assay (closed symbols) under both the TGS conditions and the NPB conditions described in Materials and Methods. In addition, S. pneumoniae D39 sensitivity to CXCL10, LL-37, and nisin in NPB was determined using a fluorescence-based antimicrobial assay (open circles). “n” indicates the number of independent experiments, each with duplicate reactions. Each data point represents the mean ± SEM (where not visible, error bars are smaller than the symbol). Dose-response curves were fitted to pooled data in GraphPad Prism, using the “log of inhibitor versus response-variable slope” function. See Table S2 for 95% CI values.
Previous work has shown that the results obtained with a resazurin-based fluorescence assay correlate well with those of a standard CFU assay under various bacterial and buffer conditions and can be used to determine the antimicrobial activity of chemokines and AMPs (34, 35, 53, 54). We performed parallel resazurin-based fluorescence assays and CFU assays with CXCL10-, LL-37-, or nisin-treated S. pneumoniae D39 incubated in NPB. Fluorescence assays of CXCL10 and nisin killing of S. pneumoniae D39 gave IC50s indistinguishable from those given by the CFU assays (Fig. 2A and C; see also Table S2, lines 2 and 3). However, for LL-37, the IC50 was ∼2-fold lower for the CFU assay than for the fluorescence assay (Fig. 2B), possibly due to an increase in metabolic activity at subinhibitory concentrations of LL-37 (50, 52, 72). This effect was reproducible in 6 of 6 independent experiments, with the fluorescence readout increasing to ∼140% in samples containing 0.3 μM LL-37 compared to the untreated control (Fig. 2B).
Benefits of this fluorescence assay include the ability to measure changes in metabolic activity over time and the elimination of inconsistencies in CFU measurements due to cell chaining. However, unencapsulated S. pneumoniae D39 Δcps strains were not assayed in NPB, because, unexpectedly, only about 25% of the S. pneumoniae D39 Δcps cells survived incubation in NPB, as indicated by decreased CFU levels after incubation (Fig. S2A) and decreased fluorescence (Fig. S2B). In comparison, encapsulated S. pneumoniae D39 does not actively increase in cell mass and number (Fig. 1B; see also Fig. S2A) during incubation in NPB, but the cells continue to exhibit reductive metabolism that reduces the indicator dye with time (Fig. 1A; see also Fig. S2B). While the rates of reductive metabolism of untreated cells differed among the strains tested (Fig. S3A and B), no strict correlation was obvious between endpoint fluorescence and sensitivity to CXCL10 (Fig. S3C). While loss of capsule and/or the biosynthesis of capsule profoundly affected the viability of serotype 2 S. pneumoniae D39 in NPB medium (Fig. S2), it had little effect on the endpoint reductive metabolism of serotype 4 S. pneumoniae TIGR4 (Fig. S3A and C).
S. pneumoniae D39 ΔamiA-F (oligopeptide permease) mutants show resistance to CXCL10 in NPB.
An ABC transporter system designated Sap (sensitivity to antimicrobial peptides) in nontypeable H. influenzae has been shown to provide AMP resistance by importing AMPs, such as LL-37, for degradation in the cytoplasm, while sap mutants show increased sensitivity to AMPs (49, 70, 73). Since the sap operon has homology to the well-characterized oligopeptide import ABC transport system Opp (73), we investigated whether disruption of similar transport systems affected the sensitivity of S. pneumoniae to CXCL10. We constructed a deletion mutant of the operon encoding Opp in S. pneumoniae (AmiACDEF) (74–77) (ΔamiA-F, IU11759), a deletion mutant of another operon encoding Opp-like system spd_1167 to spd_1170 (Δ[spd_1167-spd_1170], IU11778), and deletion mutants of aliA (ΔaliA, IU11848) and aliB (ΔaliB, IU11850), encoding two additional oligopeptide substrate-binding proteins of the AmiA-F uptake system (74). Additionally, a deletion mutant of amiD, corresponding to one of two required transmembrane permeases in the amiA-F operon, was constructed (ΔamiD, IU14488), along with a merodiploid strain complementing ΔamiD by the presence of a wild-type copy of amiD+ expressed ectopically at the bgaA locus under the control of the constitutive ftsA promoter (ΔamiD ΔbgaA::PftsA-amiD+, IU14510). Finally, a quadruple mutant (designated “ΔOPT”; IU11919) containing deletions of amiA-F, spd_1167-spd_1170, aliA, and aliB was constructed.
ΔamiA-F and ΔamiD mutants were more resistant to CXCL10 than the amiA-F+ parent S. pneumoniae D39 strain. In a dose-response experiment, the ΔamiA-F mutant exhibited increased resistance to CXCL10 in NPB, with an IC50 of 1.4 μM (95% confidence interval [CI], 1.1 to 1.8) compared to 0.87 μM (95% CI, 0.8 to 0.96) for S. pneumoniae D39 (Fig. 3A). In additional experiments performed with single doses of 3 μM CXCL10 (Fig. 3B) or 6 μM CXCL10 (Fig. S4B), the ΔamiA-F mutant displayed ∼3-fold- or 5-fold-increased fluorescence, respectively, relative to S. pneumoniae D39. The single gene deletion ΔamiD mutant displayed ∼6-fold-increased fluorescence relative to S. pneumoniae D39 in testing performed with 3 μM CXCL10 (Fig. 3B), whereas a ΔamiD strain complemented by ectopic amiD+ showed CXCL10 sensitivity indistinguishable from that seen with S. pneumoniae D39 (Fig. 3B). A mutant containing a nonpolar deletion of ΔamiA-F (IU13780) also demonstrated a >4-fold increase in fluorescence signal compared to the amiA-F+ parent, ruling out the possibility of polar effects in the ΔamiA-F mutant (Fig. S4A). Similarly to experiments performed with S. pneumoniae D39 (Fig. 2A), an experiment measuring killing of the S. pneumoniae ΔamiA-F mutant with 6 μM CXCL10 using CFU and fluorescence analyses in parallel showed agreement between the assays (Fig. S4C). Notably, the CXCL10 resistance of these ABC transporter mutants is dependent upon incubation in NPB buffer, as a ΔOPT mutant with deletions of amiA-F, spd_1167-spd_1170, aliA, and aliB showed CXCL10 resistance relative to the S. pneumoniae D39 OPT+ parent in NPB (Fig. 3 and 4, left) but not in TGS (Table 1, line 3). We conclude from these multiple data sets that S. pneumoniae ΔamiA-F and ΔamiD mutant strains exhibit increased resistance to CXCL10 relative to S. pneumoniae D39 and that this resistance is correlated with metabolic activity in NPB.
FIG 3.
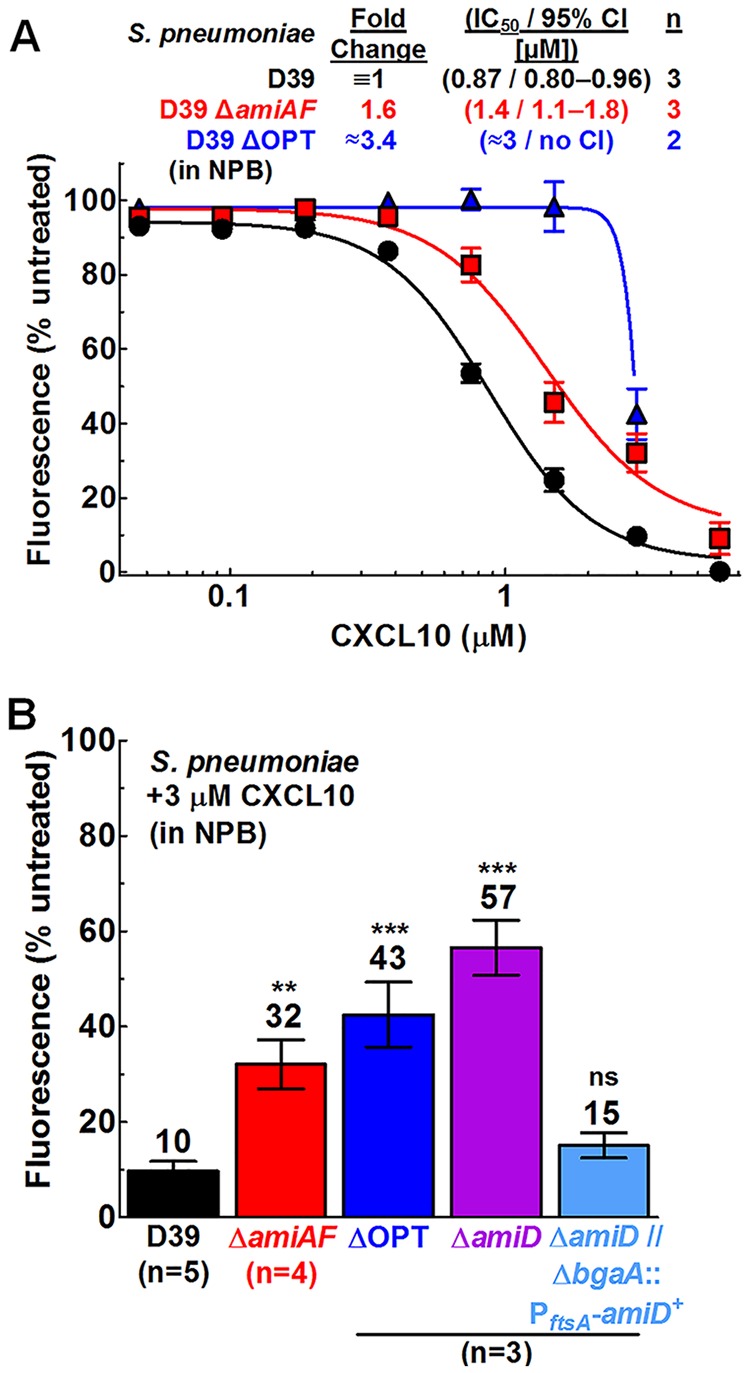
S. pneumoniae D39 ΔamiA-F, ΔamiD, and ΔOPT mutants show resistance to CXCL10 in NPB relative to the wild-type strain, and complementation of the ΔamiD mutant by an ectopic amiD+ strain restores CXCL10 sensitivity to wild-type levels. The sensitivity to CXCL10 in NPB was determined using a fluorescence-based antimicrobial assay (described in Materials and Methods). (A) S. pneumoniae D39 (IU1690), ΔamiA-F (ΔamiAF) (IU11759), and ΔOPT (IU11919). (B) The strains described above as well as the ΔamiD mutant (IU14488) and the ΔamiD ΔbgaA::PftsA-amiD+ mutant (IU14510) were treated with 3 μM CXCL10. Each data point or bar represents the mean ± SEM (where not visible, error bars are smaller than the symbol). “n” indicates the number of biologically independent replicates, each with duplicate reactions. For the titration experiments described for panel A, dose-response curves were fitted to pooled data in GraphPad Prism, using the “log of inhibitor versus response-variable slope” function. For the complementation experiments described for panel B, statistical significance relative to S. pneumoniae D39 was determined using the Mann-Whitney U test. **, P < 0.01; ***, P < 0.001; ns, not significant.
FIG 4.
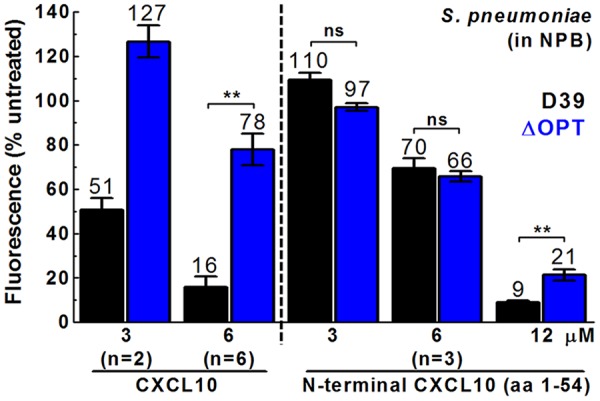
S. pneumoniae D39 ΔOPT does not show increased resistance to N-terminal CXCL10 relative to S. pneumoniae D39, and N-terminal CXCL10 exhibits decreased antimicrobial activity compared to full-length CXCL10 for S. pneumoniae D39. A fluorescence-based antimicrobial assay was performed with full-length CXCL10 (left 4 bars) or N-terminal CXCL10 (aa 1 to 54) (right 6 bars) with S. pneumoniae D39 (IU1690) or S. pneumoniae ΔOPT (IU11919) in NPB. Each bar represents the mean ± SEM of results of 3 independent experiments (unless otherwise noted). The statistical significance of the results of comparisons between strains at the same concentration was determined using the Mann-Whitney U test (except for 3 μM CXCL10, where n = 2). Asterisks denote difference between strains at the same concentration. **, P < 0.01; ns, not significant.
In contrast to the increased CXCL10 resistance of the ΔamiA-F mutants relative to wild-type S. pneumoniae D39, single mutants with deletions of ΔaliA, ΔaliB, and Δ[spd_1167-spd_1170] showed fluorescence values similar to the values seen with the wild-type strain at a high concentration of CXCL10 (6 μM) (Fig. S4B). Additionally, in two independent sets of experiments using 3 or 6 μM CXCL10 (Fig. 3; see also Fig. S4B), the ΔOPT mutant exhibited fluorescence values similar to those exhibited by the ΔamiA-F mutant (∼4-fold to 5-fold increase relative to S. pneumoniae D39). Taken together, these data suggest that inactivation of the AmiA-F oligopeptide transporter is the main factor involved in the increased CXCL10 resistance relative to S. pneumoniae D39 at 3 or 6 μM CXCL10. However, titration experiments showed that the ΔOPT mutant may be more resistant to CXCL10 than the ΔamiA-F mutant at intermediate concentrations, particularly at 1.5 μM, where the ΔOPT mutant showed almost no decrease in fluorescence relative to untreated cells (Fig. 3A). These data suggest that the cumulative ΔaliA, ΔaliB, and/or Δ[spd_1167-spd_1170] mutations may have contributed to the increased CXCL10 resistance of the ΔOPT mutant relative to the ΔamiA-F mutant at intermediate CXCL10 concentrations.
We observed variability in the absolute fluorescence values of CXCL10-treated cells between the sets of experiments, likely due to differences between batches of commercially available CXCL10. For example, the IC50s for S. pneumoniae D39 were 1.7 and 0.87 μM for CXCL10 from 2 different batches (Fig. 2A and 3A), while the IC50s for the ΔamiA-F mutant were >6 and 1.4 μM, respectively (Fig. 3A; see also Fig. S3C and S4B and C). Within each batch, the variability of fluorescence values is low, leading to relatively small standard error values (Fig. 2A and 3A). This variability of different lots of commercial CXCL10 limits comparisons of absolute IC50 and fluorescence values among sets of experiments. However, the mutants with a deletion in the amiA-F operon (the ΔamiA-F, ΔamiD, and ΔOPT mutants) consistently demonstrated ∼3-fold- to 5-fold-increased fluorescence relative to S. pneumoniae D39 after CXCL10 treatment.
Previous work in B. anthracis established that the N-terminal domain of CXCL10 (N-CXCL10; amino acids [aa] 1 to 54) showed reduced antimicrobial activity against wild-type B. anthracis and no antimicrobial activity against B. anthracis mutants lacking an active FtsEX complex (54). We tested whether N-CXCL10 retained antimicrobial activity against an oligopeptide transporter mutant of S. pneumoniae. The IC50 of S. pneumoniae D39 treated with N-CXCL10 in NPB was estimated to be ∼7 μM (Fig. 4); therefore, N-CXCL10 was ∼4-fold less effective than full-length CXCL10 at killing S. pneumoniae D39 (IC50 of 1.7 μM) (Table 3). Notably, the S. pneumoniae D39 ΔOPT mutant did not show increased resistance to N-CXCL10 at 3 or 6 μM compared to the S. pneumoniae D39 parent (Fig. 4). Similarly, N-CXCL10 was ∼3-fold to 4-fold less effective than full-length CXCL10 at killing encapsulated and unencapsulated S. pneumoniae D39 cells in TGS (Table 2, rows 1 and 2).
TABLE 3.
Relative fold change and IC50 values determined for CXCL10-treated, LL-37-treated, or nisin-treated Streptococcus cells with a fluorescence-based antimicrobial assay in NPB buffera
| Strain | CXCL10 |
LL-37b |
Nisinc |
|||
|---|---|---|---|---|---|---|
| Fold changed | IC50/95% CI (μM)e | Fold changed | IC50/95% CI (μM)e | Fold changed | IC50/95% CI (μM)e | |
| S. pneumoniae | ||||||
| D39 | ≡1f,g | 1.7/1.1–2.7f | ≡1 | 1.6/1.5–1.7 | ≡1 | 0.80/0.71–0.90 |
| D39 ΔamiA-F | 1.6g | See Fig. 3Ag | 0.69 | 1.1/1.0–1.2 | 2.3 | 1.8/1.6–2.1 |
| D39 ΔOPT | 3.4g | See Fig. 3Ag | 0.81 | 1.3/1.2–1.4 | 2.0 | 1.6/1.2–2.0 |
| D39 ΔdltA | 0.28f | 0.47/0.43–0.50f | 0.29 | 0.46/0.38–0.54 | 0.70 | 0.56/0.40–0.77 |
| TIGR4 | 0.08f | 0.14/0.12–0.16f | 0.21 | 0.33/0.13–0.84 | 0.19 | 0.15/0.07–0.31 |
| TIGR4 Δcps | 0.10f | 0.17/0.16–0.18f | 0.29 | 0.46/0.07–2.8 | 0.23 | 0.18/0.16–0.20 |
| S. mitis | See Fig. S6Ah | 1.3i | 2.1/1.7–2.8i | Ndk | ||
| S. sanguinis | See Fig. S6Ah | 0.36i | 0.57/0.56–0.59i | Nd | ||
| S. mutans | See Fig. S6Ah | ∼3.1i | ∼5i,j | Nd | ||
IC50 and 95% CI values for S. pneumoniae D39 in response to treatment with CXCL10, LL-37, and nisin were determined with a fluorescence-based antimicrobial assay in NPB buffer as described in Materials and Methods. The strains used were S. pneumoniae D39 (IU1690), S. pneumoniae D39 ΔamiA-F (IU11759), S. pneumoniae D39 ΔOPT (IU11919, ΔOPT ≡ ΔamiA-F Δ[spd_1167-spd_1170] ΔaliA ΔaliB), S. pneumoniae D39 ΔdltA (IU12470), S. pneumoniae TIGR4 (IU11966), S. pneumoniae TIGR4 Δcps (IU12001), S. mitis (IU11303), S. sanguinis (IU11305), and S. mutans (IU11309).
Unless otherwise noted, the inhibition curves from which fold change, IC50, and 95% CI values were obtained are shown in Fig. S5A and B.
Unless otherwise noted, the inhibition curves from which fold change, IC50, and 95% CI values were obtained are shown in Fig. S5C and D.
Fold change values were determined by dividing the IC50 for the mutant by the IC50 for S. pneumoniae D39.
IC50 values were obtained from pooled data from at least two independent experiments and fitted to a dose-response curve (log of inhibitor versus response-variable slope) using GraphPad Prism. 95% CI data represent the 95% confidence interval of the IC50.
The inhibition curves from which fold change, IC50, and 95% CI values were obtained are shown in Fig. 5A and B.
A separate set of experiments was performed to obtain inhibition curves for the ΔamiA-F and ΔOPT mutants (see Fig. 3A for curves, IC50 values, and 95% CIs). While variability in the absolute IC50 values is seen between batches of commercially available CXCL10, the relative levels of resistance of the ΔamiA-F and ΔOPT mutants to S. pneumoniae D39 remained consistent, so fold change values relative to S. pneumoniae D39 are included in this table.
Full titration curves were not completed, but single-point assays for 0.2 and 1 μM CXCL10 are shown in Fig. S6A.
Inhibition curves from which fold change, IC50, and 95% CI values were obtained are shown in Fig. S6B.
The 95% confidence interval could not be calculated due to the approximate nature of the IC50 value.
Nd, not determined.
S. pneumoniae D39 ΔamiA-F mutants are more resistant to nisin in NPB whereas ΔdltA mutants are more sensitive to CXCL10, LL-37, and nisin than the D39 parent.
ΔamiA-F and ΔOPT mutants showed slightly higher sensitivity to LL-37 in NPB than the S. pneumoniae D39 strain (Table 3, rows 1 to 3; see also Fig. S5A), whereas those mutants were more resistant to nisin (Table 3, rows 1 to 3; see also Fig. S5C). In contrast, a mutant of the dlt operon (ΔdltA, IU12470) which lacks the ability to add d-alanine residues to teichoic acids (41, 42, 78) showed ∼3.5-fold-lower IC50s than S. pneumoniae D39 after CXCL10 or LL-37 treatment (Fig. 5A; see also Fig. S5A) (Table 3, rows 1 and 4) but was only slightly more sensitive to nisin (Table 3, rows 1 and 4; see also Fig. S5C). Taken together, these results extend to CXCL10 the previous conclusion that dlt operon mutants of S. pneumoniae and other Streptococcus species are more sensitive than their parent strains to AMPs such as LL-37 and nisin (41, 78).
FIG 5.
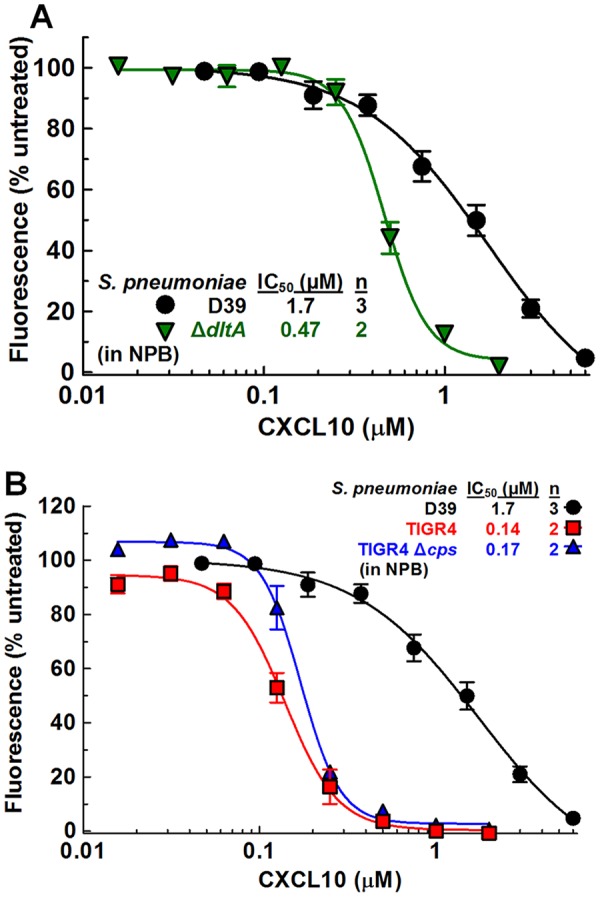
S. pneumoniae D39 ΔdltA mutant and S. pneumoniae TIGR4 strains show greater CXCL10 sensitivity in NPB than S. pneumoniae D39. The sensitivity to CXCL10 in NPB was determined using a fluorescence-based antimicrobial assay, as described in Materials and Methods. (A) S. pneumoniae D39 (IU1690; data from Fig. 2A) and S. pneumoniae D39 ΔdltA (IU12470). (B) S. pneumoniae D39 (IU1690; data from Fig. 2A), S. pneumoniae TIGR4 (IU11966), and S. pneumoniae TIGR4 Δcps (IU12001). “n” indicates the number of independent experiments, each with duplicate reactions. Each data point represents the mean ± SEM (where not visible, error bars are smaller than the symbol). Dose-response curves were fitted to pooled data in GraphPad Prism, using the “log of inhibitor versus response-variable slope” function. See Table 3 for 95% CI values.
S. pneumoniae TIGR4 and S. pneumoniae TIGR4 Δcps are more sensitive to CXCL10, LL-37, and nisin than S. pneumoniae D39.
We directly compared the sensitivities of serotype 2 S. pneumoniae D39 and serotype 4 S. pneumoniae TIGR4 to inhibition by CXCL10, LL-37, and nisin. S. pneumoniae D39 and S. pneumoniae TIGR4 produce different extracellular polysaccharide capsules, and their genomes differ by as much as 10% (79, 80). Encapsulated and unencapsulated variants of S. pneumoniae TIGR4 were considerably more sensitive than S. pneumoniae D39 to CXCL10, LL-37, and nisin in NPB (Fig. 5B; see also Fig. S5B and D) (Table 3, rows 1, 5, and 6). For CXCL10, the IC50s were >10-fold lower for S. pneumoniae TIGR4 than for S. pneumoniae D39, which contrasts with the similar low IC50s of S. pneumoniae TIGR4 and S. pneumoniae D39 in TGS (Fig. 5B; see also Fig. S1) (Table 1, rows 1, 2, 4 and 5). After LL-37 or nisin treatment, S. pneumoniae TIGR4 IC50s were 3-fold to 5-fold lower than those calculated for S. pneumoniae D39 (Table 3, rows 1, 5, and 6; see also Fig. S5B and D). Notably, the dose-response curves of encapsulated and unencapsulated S. pneumoniae TIGR4 strains for LL-37 in NPB are abnormally shaped, resulting in wide 95% confidence intervals (Table 3, lines 5 and 6; see also Fig. S5B).
These differences in sensitivity to CXCL10, LL-37, and nisin suggest enhanced killing of S. pneumoniae TIGR4 by AMPs or, conversely, enhanced resistance mechanisms of S. pneumoniae D39. Additionally, unencapsulated S. pneumoniae D39 cells lost viability in NPB, while unencapsulated S. pneumoniae TIGR4 cells maintained viability (Fig. S2 and S3A). Whether these responses can be attributed simply to capsule chemical differences between the two S. pneumoniae serotype strains or to other mechanisms, such as differences in capsule shedding (81), remains to be determined. Comparisons of the CXCL10 sensitivities of S. pneumoniae D39 and TIGR4 with those of S. mitis, S. mutans, and S. sanguinis did not reveal a correlation with reductive metabolic activity in NPB (Fig. S6A and S3C, top). S. mitis and S. mutans exhibited the greatest metabolic activity in NPB but showed sensitivity to CXCL10 similar to that seen with S. pneumoniae D39 (Fig. S6A and S3C, top). For LL-37, S. mutans was the most resistant species, S. pneumoniae D39 and S. mitis showed intermediate resistance, and S. pneumoniae TIGR4 and S. sanguinis were the most sensitive (Fig. S6B). These differences among Streptococcus species again likely reflect multiple mechanisms of killing, resistance, or both.
ΔftsX and ΔftsE mutants of B. subtilis and S. pneumoniae ftsX mutants show the same sensitivity to CXCL10 or N-CXCL10 as the ftsX+ ftsE+ parents.
Previous work showed that ΔftsX and nonfunctional ftsE mutants of B. anthracis are 2-fold- to 3-fold more resistant to CXCL10 than the wild-type strain and were completely resistant to N-CXCL10 at the highest concentration tested (2.8 μM), suggesting an FtsEX-dependent mode of killing mediated by N-CXCL10, while the C-terminal α-helix of CXCL10 may kill B. anthracis independently of FtsEX (53, 54). Because ftsX and ftsE are essential in S. pneumoniae (57), we first tested whether ΔftsX (markerless) and ΔftsE mutations of another Bacillus species, B. subtilis, imparted resistance to CXCL10 killing. In contrast to B. anthracis, B. subtilis ΔftsX and ΔftsE mutants showed sensitivity to CXCL10 or N-CXCL10 similar to that seen with the ftsX+ ftsE+ parent in TGS or NPB (Fig. 6; see also Fig. S7A to C and Table S3). Previous studies of B. anthracis assayed for CXCL10 killing in DMEM–10% (vol/vol) FBS (see Materials and Methods). In that medium, CXCL10 showed no antimicrobial activity against the wild-type parent or the ΔftsX or ΔftsE mutant strains of B. subtilis or against wild-type S. pneumoniae D39 at the highest concentrations tested (5.8 and 11.6 μM) (Fig. S7D and E and S8A). The lantibiotic nisin did show antibacterial activity against S. pneumoniae in DMEM–10% (vol/vol) FBS, but its estimated IC50 was ∼20-fold higher than in NPB (Fig. S8A and Table S2). Additionally, CXCL10 did not kill S. pneumoniae when assayed at 3.5 μM in DMEM without serum (Fig. S8B). Taken together, these results demonstrate that CXCL10 binding to FtsX or disruption of FtsEX function is not involved in the mechanism by which CXCL10 kills B. subtilis.
FIG 6.
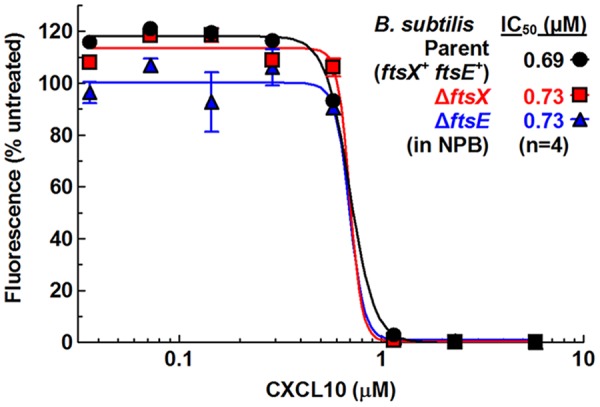
B. subtilis ΔftsX or ΔftsE mutants show no difference in CXCL10 sensitivity from that shown by the B. subtilis ftsX+ ftsE+ parent strain. Sensitivities of ΔftsX (IU12981) and ΔftsE::Tn10 spec (IU12166) mutants to CXCL10 relative to the B. subtilis ftsX+ ftsE+ parent (IU12153) were determined using a fluorescence-based antimicrobial assay in NPB buffer (described in Materials and Methods). Each point represents the mean ± SEM of results of 4 independent experiments. Dose-response curves and IC50s were obtained from duplicate wells from all independent experiments and fitted to a dose-response curve (log of inhibitor versus response-variable slope) using GraphPad Prism. See Table S3 for 95% CI values.
To test whether dysfunction of FtsX imparts CXCL10 resistance in S. pneumoniae, we assayed ftsX mutants of S. pneumoniae D39, where FtsX is an essential protein (57, 61). The large extracellular loop (ECL1) of S. pneumoniae FtsX interacts with the PcsB PG hydrolase (B. E. Rued, unpublished result) (62), whereas the small extracellular loop (ECL2) plays a role in activation of PcsB PG hydrolase activity (57, 61). ftsX(W183L) changes an amino acid at the base of the distal stem of ECL1, leading to suppression of temperature-sensitive mutations in the coiled-coil region of PcsB (61). ftsX(N269T) changes an amino acid in ECL2 required for PcsB activation (61, 82). The ftsX(W183L) and ftsX(N269T) mutants showed reduced growth rates compared to the S. pneumoniae D39 ftsX+ parent, and both mutants formed morphologically defective cells that were enlarged, blocky, and sometimes oddly shaped, indicative of loss of FtsEX::PcsB function (Fig. 7A and B). However, in NPB, the ftsX mutants showed levels of sensitivity to CXCL10 or LL-37 similar to those shown by the ftsX+ parent (Fig. 7C). The ftsX(W183L) and ftsX(N269T) mutants and several other ftsX point mutants also showed sensitivity to N-CXCL10 similar to that shown by the ftsX+ parent in TGS (Table S4). However, unlike the results seen with CXCL10 and LL-37, the ftsX(N269T) mutant was notably resistant to nisin compared to the other two strains in NPB (Fig. 7C). We conclude that misregulation of FtsEX::PcsB does not impart CXCL10 resistance to S. pneumoniae D39.
FIG 7.

S. pneumoniae ftsX mutants show growth and morphology defects indicative of loss of FtsEX function but show no difference in resistance to CXCL10 or LL-37 relative to the ftsX+ parent. (A) Growth in BHI broth of WT S. pneumoniae D39 (IU1690), the ftsX+ Pc-[kan-rpsL+] parent (IU10748), the ftsX (W183L) Pc-[kan-rpsL+] (IU9008) mutant, and the ftsX (N269T) Pc-[kan-rpsL+] (IU9004) mutant. Calculations of three independent growth curves were performed with similar results, and a representative curve is shown (log plot at the top; linear plot below). (B) Montage of representative phase-contrast images of strains grown to mid-log phase in BHI broth. Scale bars = 1 μm. (C) Sensitivity of the parent (IU10748), ftsX (W183L) (IU9008), and ftsX (N269T) (IU9004) strains to CXCL10 (top), LL-37 (middle), and nisin (bottom) was determined using a fluorescence-based antimicrobial assay in NPB buffer (described in Materials and Methods). Each bar represents the mean ± SEM of results of 2 independent experiments, each with duplicate reactions.
DISCUSSION
In this paper, we present a report of a systematic, quantitative study of the inhibition of S. pneumoniae serotype strains and several other pathogenic Streptococcus species, including S. mitis, S. mutans, and S. sanguinis, by the chemokine CXCL10 and two major AMPs. This study was undertaken to fill knowledge gaps and resolve inconsistencies in the literature about the sensitivity of S. pneumoniae and other Streptococcus species to chemokines and AMPs and to test hypotheses about possible resistance mechanisms. One general conclusion is that different incubation media used to assay AMP sensitivity give vastly different results (Fig. 2). In this study, we determined that the low-salt incubation buffer NPB (10 mM sodium phosphate [pH 7.4] with 1% [vol/vol] BHI broth) is compatible with a resazurin (alamarBlue) fluorescence-based assay to determine the chemokine and AMP sensitivity of isogenic S. pneumoniae WT and mutant strains. This fluorescence-based assay was validated by its tight correlation with results from CFU assays.
S. pneumoniae and other Streptococcus species tested for chemokine sensitivity showed high levels of killing (IC50 = 0.03 to 0.06 μM) in commonly used TGS buffer (10 mM Tris-HCl [pH 7.4] and 5 mM glucose), in which S. pneumoniae cells are metabolically static. In comparison, S. pneumoniae cells assayed for killing in NPB were generally less sensitive to the chemokines and AMPs tested and were metabolically active for resazurin reduction, even though the cell numbers, cell masses, and percentages of viable cells remained similar after 2 h of incubation. There is no evidence to suggest that CXCL10 and AMPs would be appreciably less stable or available in NPB than TGS in the time frame of these assays. This alternative explanation is further countered by our results showing that certain S. pneumoniae D39 mutations impart resistance or increased sensitivity to CXCL10 in NPB that is not detected in TGS (Fig. 3) (Table 1). The lower CXCL10, LL-37, and nisin sensitivity of metabolically active compared to metabolically static wild-type S. pneumoniae D39 cells is consistent with induction of metabolic or stress responses that regulate the survival of S. pneumoniae D39 in assays and, conceivably, within the human lower respiratory tract (Fig. 8A).
FIG 8.

Summary of S. pneumoniae responses to chemokines and antimicrobial peptides. (A) Metabolically active S. pneumoniae cells are able to mount a stress response to chemokines or AMPs, whereas metabolically inactive cells are not. This results in up to 100-fold-greater sensitivity to chemokines and AMPs. (B) Three possible mechanisms for resistance of S. pneumoniae ΔamiA-F mutants to killing by CXCL10 or nisin. For simplicity, only CXCL10 is shown. (i) Facilitated-insertion model, where the AmiA-F transporter initially binds to CXCL10 and initiates its insertion into the cell membrane. (ii) Direct-uptake model, where AmiA-F directly transports CXCL10 into the cytoplasm, where CXCL10 is toxic to S. pneumoniae cells. (iii) Indirect-effect model, where the normal pleiotropic function of AmiA-F leads to a physiological state that makes cells susceptible to killing by CXCL10. For models 1 to 3, greatly reduced binding and/or uptake of truncated N-CXCL10 could account for the reduced killing of wild-type S. pneumoniae D39 and the marginal resistance of ΔOPT mutants observed. dlt operon-mediated d-alanylation of teichoic acids imparts resistance of S. pneumoniae D39 to CXCL10 and AMPs through charge repulsion or increased density of the cell wall, such that Δdlt operon mutants have increased sensitivity to CXCL10 and AMP killing. In addition, serotype 4 S. pneumoniae TIGR4 is intrinsically >10-fold more sensitive to CXCL10 killing than serotype 2 S. pneumoniae D39 in metabolically active cells. See the text for additional details.
Previous studies demonstrated that resistance to AMPs and chemokines is imparted to bacteria through a variety of mechanisms, including degradation by external proteases, repulsion through cell surface charge modifications, and transport by Opp-like ABC transporters for cytoplasmic degradation (39, 40, 41, 49). Additionally, interactions with specific bacterial proteins, such as FtsEX and lipid II, may increase the antimicrobial activity of some AMPs and chemokines against certain bacterial species (28, 53, 54). On the basis of that previous work, we tested candidate mutations in S. pneumoniae D39 for resistance or sensitivity to CXCL10, LL-37, and nisin. Mutants lacking external serine proteases (ΔhtrA or ΔprtA) did not demonstrate changes in sensitivity to CXCL10, LL-37, or nisin relative to S. pneumoniae D39 (data not shown). For a control, we performed tests and confirmed that Δdlt operon mutants, which are unable to add positively charged d-alanine residues to teichoic acids and thus result in an overall reduction in cell surface charge, increase the relative levels of sensitivity of D39 S. pneumoniae to CXCL10, LL-37, and nisin (Fig. 5A; see also Fig. S5A and C in the supplemental material). However, a cardiolipin synthase (Δcls) mutant, which lacks the doubly negatively charged phospholipid cardiolipin in its cell membrane, did not show changes in CXCL10, LL-37, or nisin sensitivity relative to S. pneumoniae D39 (IC50 of the Δcls mutant for CXCL10; see Fig. S3C, top).
We were unable to generalize the CXCL10 resistance of ftsX and ftsE mutants reported in B. anthracis (53, 54) to B. subtilis or S. pneumoniae. As in B. anthracis (53), ftsX and ftsE are not essential in B. subtilis (56, 83); however, unlike in B. anthracis, ΔftsX or ΔftsE mutants of B. subtilis showed the same sensitivity to CXCL10 as their parent strain in NPB or TGS (Fig. 6; see also Fig. S7). Unlike in the two Bacillus species, ftsX and ftsE are essential in S. pneumoniae (57, 61). Yet ftsX point mutations that compromise FtsX function in cell division and/or PG remodeling showed the same sensitivity to CXCL10 as the wild-type parent S. pneumoniae D39 strain (Fig. 7C). The lack of killing of wild-type and mutant B. subtilis and S. pneumoniae in DMEM–10% (vol/vol) FBS, coupled with the lack of antimicrobial assays of B. anthracis in either NPB or TGS, makes direct comparisons between our results and the previous work in B. anthracis difficult. However, a transposon screen performed in E. coli for analysis of CXCL10 resistance did not yield ftsE or ftsX mutants, and transcriptomic analysis of S. pneumoniae challenged with LL-37, nisin, or bacitracin did not show changes in expression of ftsE or ftsX (52, 84). We conclude that FtsX targeting by CXCL10 does not occur in all bacterial species and may, in fact, be confined to B. anthracis. One interesting observation from these assays was that the S. pneumoniae D39 ftsX(N269T) mutant, which alters ECL2 in FtsX, was more resistant to nisin than the parent ftsX+ strain. This result suggests a possible link between certain FtsX functions and the availability of external lipid II or the susceptibility of the membrane to nisin.
Mutants lacking the general AmiA-F oligopeptide permease (ΔamiA-F and ΔOPT) showed increased resistance to CXCL10 and nisin relative to the S. pneumoniae D39 parent (Fig. 3; see also Fig. S5C). In contrast, mutants of oligopeptide substrate-binding proteins (ΔaliA and ΔaliB) or of another putative peptide uptake system (Δ[spd_1167-spd_1170]) did not show altered sensitivity to CXCL10, LL-37, or nisin compared to S. pneumoniae D39 (Fig. S4B). There are precedents for peptide transport systems binding to cationic peptides, transporting oligopeptides similar in length to AMPs and chemokines, imparting resistance to AMPs, and having pleiotropic effects on physiology (70, 75, 85, 86). E. coli OppA, which is a homologue of the S. pneumoniae AmiA substrate-binding protein, preferentially binds small, positively charged peptides (86). The Opp oligopeptide permease system in Lactococcus lactis, which is a homologue of S. pneumoniae AmiA-F, binds and transports peptides up to 35 amino acids long, with little selectivity for amino acid side chain composition (75). An analog of hBD-3 interacts directly with SapA, the substrate-binding protein of the Sap uptake system in nontypeable H. influenzae (70), and LL-37 and hBD-3 are imported by the Sap system for degradation in the cytoplasm (49). Mutations in S. pneumoniae Opp (AmiA-F) cause numerous pleiotropic effects, including reduced amino acid and oligopeptide uptake, altered cell adherence to human epithelial cells, decreased competence, reduced quorum sensing and colonization, and reduced virulence in animal models of infection (76, 87–93). Moreover, expression of amiA-F is upregulated when S. pneumoniae cells bind to human lung epithelial cells (92). Since cytoplasmic amino acid pools activate global regulatory proteins to change gene expression, uptake of oligopeptides by AmiA-F has been proposed to play a major role in how S. pneumoniae cells sense and respond to their nutrient environment (85). Last, a previous study showed that the AmiA-F transporter has pleiotropic effects on phospholipid composition (94). Treatment of wild-type S. pneumoniae with the drug aminopterin leads to an increased amount of negatively charged cardiolipin in the cell membrane, whereas the phospholipid composition remains unchanged in amiA mutants after aminopterin treatment (94). The increased CXCL10 resistance of ΔamiA-F mutants may be due at least in part to these pleiotropic effects, as discussed below.
The mechanism explaining how the absence of the AmiA-F transporter imparts CXCL10 and nisin resistance to S. pneumoniae D39 remains to be determined. On the basis of aspects of mechanisms proposed for killing by other AMPs (22), three general models for resistance can be postulated (Fig. 8B). In the first, facilitated-insertion model, the AmiA-F transporter initially binds to CXCL10 or nisin and initiates its insertion into the cell membrane, where complexes of CXCL10 or nisin are formed that disrupt the membrane. This model is a variation of a current mechanism for nisin killing where lipid II serves as an initiator of membrane insertion (26, 27). In the second, direct-uptake model, AmiA-F transports CXCL10 or nisin into the cytoplasm, where CXCL10 or nisin is toxic to S. pneumoniae cells. In the third, indirect-effect model, the normal pleiotropic function of AmiA-F leads to a physiological state that makes cells susceptible to killing by CXCL10 or nisin. For each model, reduced binding and/or uptake of truncated N-CXCL10 could account for the observed reduced killing of wild-type S. pneumoniae D39 by N-CXCL10 and the marginal resistance of the ΔOPT mutant to the fragment. A corollary of this interpretation is that the C-terminal α-helix of CXCL10 is required for strong binding of intact chemokine CXCL10 to AmiA-F.
MATERIALS AND METHODS
Strains of S. pneumoniae and other Streptococcus species and growth conditions.
The strains used are described in Table S1 in the supplemental material. S. pneumoniae strains were derived from the following: IU1690, an encapsulated serotype 2 D39 strain (95); IU1945 (D39 Δcps); IU1781 (D39 rpsL1); or IU11966, an encapsulated serotype 4 TIGR4 strain (96). S. mitis, S. sanguinis, and S. mutans strains were acquired from the ATCC. Strains with antibiotic resistance cassettes were constructed as described previously (97–99) using overlapping fusion PCR to synthesize linear DNA amplicons. Error-prone amplicons were generated as described in reference 61. Amplicons were transformed into S. pneumoniae D39 cells induced with competence-stimulating peptide 1 (CSP-1) as described previously (99) or with CSP-2 (Anaspec; catalog no. AS-63877) for TIGR4 cells. Transformation of TIGR4 strains was carried out similarly to the method used for the D39 strains, except for the following: 100 ng of DNA was transformed into 200 μl of recipient cells at an OD620 of ∼0.05 in 900 μl of transformation mix containing 90% (vol/vol) BHI broth, 10% (vol/vol) heat-inactivated horse serum, 0.18% (wt/vol) glucose, and 500 ng CSP-2. Strains with markerless alleles were constructed using intermediates containing the Janus cassette (Pc-[kan-rpsL+]) (100). Oligonucleotide primers used to synthesize amplicons are described in Table S1. Streptococcus species were grown on Trypticase soy agar II plates containing 5% (vol/vol) defibrinated sheep blood (TSAII-BA; catalog no. 221239/221261, Becton-Dickinson BBL) at 37°C in 5% CO2. Antibiotic selection on plates used concentrations as described previously (98, 99). PCR and DNA sequencing of relevant chromosomal regions were completed to confirm the constructed strains.
Streptococcus species were grown planktonically in Becton-Dickinson Bacto brain heart infusion (BHI) broth (catalog no. 237500) at 37°C in 5% CO2 without shaking. Growth was measured by monitoring the OD620 as described previously (99). In all experiments, Streptococcus cells were taken from glycerol stocks stored at −80°C, serially diluted in BHI broth, and incubated 12 to 16 h at 37°C in 5% CO2 under static conditions (overnight cultures).
Chemokines, antimicrobial peptides, and media used.
During stock preparation and antimicrobial assays, all chemokine and antimicrobial peptide (AMP) stocks were prepared with and stored in protein LoBind tubes (catalog no. 022431081, Eppendorf), and all mixing of chemokines and AMPs was done with gentle pipetting. CXCL10 (recombinant human IP-10; catalog no. 300-12, Peprotech) was diluted to 57.8 μM (500 μg/ml) in H2O with 0.015% (wt/vol) human serum albumin (HSA; catalog no. A3782-500MG, Sigma), divided into aliquots, and stored at −80°C until use. CXCL9 (recombinant human MIG; catalog no. 300-26, Peprotech) and CXCL14 (recombinant human BRAK; catalog no. 300-50, Peprotech) were diluted to 85.5 and 106.4 μM, respectively (1 mg/ml), in H2O with 0.03% (wt/vol) HSA, divided into aliquots, and stored at −80°C until use. The N-terminal (aa 1 to 54) and C-terminal (aa 55 to 77) domains of CXCL10 were synthesized (Fisher), diluted to 92.5 μM in H2O with 0.006% (wt/vol) HSA, divided into aliquots, and stored at −80°C until use. LL-37 was synthesized and generously provided by Cheng Kao (IUB) as described previously (101), diluted to 100 μM in H2O with 0.015% (wt/vol) HSA, divided into aliquots, and stored at −80°C until use. Nisin (catalog no. N5764-1G, Sigma) was diluted to 298 μM (1 mg/ml) in H2O with 0.015% (wt/vol) HSA fresh for each use. The media or buffers used for assays were TGS (10 mM Tris-HCl, 5 mM glucose, pH 7.4), NPB (10 mM sodium phosphate [pH 7.4], 1% [vol/vol] BHI broth), and DMEM ± 10% (vol/vol) FBS (high-glucose DMEM, catalog no. 11-965-092, Fisher; FBS, catalog no. SH30910, HyClone; PBS, catalog no. BP2944-100, Fisher).
Resazurin-based fluorescence assay to measure bacterial reductive metabolism.
Overnight cultures of Streptococcus strains were diluted to an OD620 of ∼0.003 in 6 ml fresh BHI broth and grown at 37°C in 5% CO2 without shaking until an OD620 of ∼0.1 to 0.13 was reached. Cells (3 ml divided into two 1.5-ml tubes) were centrifuged for 5 min at 21.1K × g at room temperature. To evaluate the fluorescence signals obtained in each buffer in the absence of compound addition, S. pneumoniae D39 was resuspended by vortex mixing in 2 ml of one of the following solutions: BHI broth, NPB, DMEM–10% (vol/vol) FBS, TGS, or PBS. Cells were centrifuged again, and the pellet was resuspended in 3 ml fresh buffer. Cells were diluted to an OD620 of ∼0.05 in 4 ml of buffer (1 × 107 CFU/ml [estimated]). Cells (150 μl) were further diluted to 1 ml (6 × 104 CFU/40 μl [estimated]). All reactions were performed in duplicate in a black, flat-bottom 96-well plate with a nonbinding surface (catalog no. 3993, Corning). Cells (40 μl) were added to wells containing 10 μl buffer with 0.003% (wt/vol) HSA. At time zero, 5 μl resazurin dye (alamarBlue; catalog no. BUF012A, Bio-Rad) was added and reaction volumes were gently mixed by pipetting. For each buffer condition, blank wells containing 40 μl buffer, 10 μl buffer with 0.003% (wt/vol) HSA, and 5 μl dye were also prepared. Reaction mixtures were incubated at 37°C in 5% CO2 without shaking, and fluorescence was monitored periodically for 4 h on a Biotek Synergy H1 plate reader. For each buffer condition, raw fluorescence values were subtracted from values obtained from blank wells to give subtracted blank values. Subtracted blank values were then averaged and normalized to the mean value determined for cells in BHI broth at 4 h, which was defined as 100%. Additionally, time plots of fluorescence signal were calculated to display the average subtracted blank values (expressed in arbitrary fluorescence units [AFU]) as a function of time. The plate reader specifications were as follows: detection, fluorescence; read type, endpoint; excitation wavelength, 530 nm; emission wavelength, 590 nm; optics position, top; gain, 50; read height, 7.00 mm.
CFU survival assay.
S. pneumoniae D39 cells were cultured and prepared as described above to obtain a culture at an OD620 of ∼0.05 in 4 ml of the appropriate buffer. In assays performed with TGS, 20 to 45 μl of cells was further diluted to a final volume of 900 μl (0.9 × 104 to 2 × 104 CFU/40 μl [estimated]; approximately 2 to 4 × 105 CFU/ml) (31, 33). In assays performed with NPB, 150 μl of cells was further diluted to 1 ml (6 × 104 CFU/40 μl [estimated]; approximately 1.2 × 106 CFU/ml) (similarly to the method described in reference 35). For the assays whose results are presented in Fig. 1B, Fig. 2, and Table S2 comparing different buffer conditions, 150 μl of cells was further diluted to 1 ml (6 × 104 CFU/40 μl [estimated]) under all conditions. Assays performed with S. pneumoniae in DMEM (see Fig. S8B in the supplemental material) used cells at an OD620 of ∼0.05 (4.5 × 105 CFU/40 μl [estimated]) (similarly to the method described in reference 54).
In antimicrobial assays, chemokines and AMPs were contained in 1.5-ml protein LoBind tubes and all mixing of chemokines and AMPs was done with gentle pipetting. Reactions were performed in duplicate in either 1.5-ml protein LoBind tubes or a black, flat-bottom 96-well plate with a nonbinding surface. Cells (40 μl) were added with gentle mixing to reaction mixtures containing 10 μl chemokine or AMP stock at 5× the final concentration in assay buffer with 0.003% (wt/vol) HSA. For untreated samples, 40 μl of cells was added with gentle mixing to 10 μl of assay buffer with 0.003% (wt/vol) HSA. After incubation at 37°C in 5% CO2 without shaking for 2 h, cells were serially diluted 10-fold in PBS. Assays performed with S. pneumoniae in DMEM (Fig. S8) used 5.8 h of incubation (similarly to the method described in reference 54). Cells were plated by mixing the appropriate dilution with 3 ml molten soft agar incubated at 50°C (the soft agar consisted of 0.7% [wt/vol] Bacto agar and 0.8% [wt/vol] Difco nutrient broth; catalog no. 234000, Becton-Dickinson) and pouring the mixture onto a TSAII-BA plate. CFU counts were performed after 24 h of incubation at 37°C in 5% CO2. For each strain, survival is expressed relative to that of the untreated control, which was defined as 100% survival. An additional untreated control was plated without incubation to assess the effect of the incubation on cell survival.
Fluorescence-based antimicrobial assay.
Overnight cultures of Streptococcus strains were grown in BHI broth, centrifuged to remove BHI broth, and diluted to an OD620 of ∼0.05 (1 × 107 CFU/ml [estimated]) in the appropriate buffer as described for the CFU survival assay. In the assays performed with NPB, 150 μl cells was further diluted to 1 ml (6 × 104 CFU/40 μl [estimated]; approximately 1.2 × 106 CFU/ml, as used in a previous fluorescence-based antimicrobial assay performed with S. pneumoniae [35]). Assays performed with S. pneumoniae in DMEM ± 10% (vol/vol) FBS (Fig. S8) used cells at an OD620 of ∼0.05 (4.5 × 105 CFU/40 μl [estimated]) (similarly to the method described in reference 54).
Reaction mixtures were prepared in a black 96-well plate and incubated as described for the CFU survival assay. Duplicate blank wells containing 40 μl buffer and 10 μl buffer with 0.003% (wt/vol) HSA were also prepared. After the 2 h of incubation, 5 μl of resazurin dye was added to each reaction mixture. Cells were incubated for an additional 2.5 h, and fluorescence was monitored approximately every 30 min as described above. Assays performed with S. pneumoniae in DMEM without FBS (Fig. S8B), resazurin dye was added after 3.8 h of incubation, and then fluorescence was read after an additional 2 h of incubation, similarly to the method described in reference 54. Raw fluorescence values were subtracted from values obtained from blank wells to give subtracted blank values. Subtracted blank values were then averaged and normalized to the mean value determined for an untreated control for each strain, which was defined as 100%. Time plots of fluorescence signal were calculated to display the average subtracted blank values (in arbitrary fluorescence units [AFU]) as a function of time.
B. subtilis strains, growth assays, and antimicrobial assays.
B. subtilis strains were derived from the ancestral B. subtilis NCIB3610 strain (102). B. subtilis strains were grown planktonically in lysogeny broth (LB) containing 10 g/liter Bacto tryptone (catalog no. 211705, Becton-Dickinson), 5 g/liter Bacto yeast extract (catalog no. 212750, Becton-Dickinson), and 5 g/liter NaCl (catalog no. SX0420-3, EMD) at 37°C with shaking, with 100 μg/ml spectinomycin or 5 μg/ml kanamycin, if needed. B. subtilis strains were grown on plates with LB containing 15 g/liter Bacto agar (catalog no. 214010, Becton-Dickinson) at 37°C. In all experiments, B. subtilis strains were taken from glycerol stocks stored at −80°C, inoculated into 5 ml of LB with appropriate antibiotics, and incubated for 12 to 16 h at 37°C with shaking (overnight cultures).
For antimicrobial assays of B. subtilis strains, overnight cultures were diluted to an OD620 of ∼0.01 in 5 ml fresh LB with appropriate antibiotics and grown at 37°C without CO2 and with shaking until an OD620 of ∼0.5 was reached. Cells were washed and resuspended in 3 ml of the appropriate assay buffer (TGS, NPB, or DMEM–10% [vol/vol] FBS) as described for the Streptococcus species. The OD620 was determined, and cells were diluted to an OD620 of ∼0.2 (2 × 107 CFU/ml for B. subtilis [estimated]) in 4 ml.
In both CFU survival and fluorescence-based antimicrobial assays performed using NPB, 200 μl cells was further diluted to 900 μl (1.8 × 105 CFU/40 μl [estimated]). In CFU survival assays and fluorescence-based antimicrobial assays performed using DMEM–10% (vol/vol) FBS, 37.5 μl cells was further diluted to 1 ml (3 × 104 CFU/40 μl [estimated]) (similarly to experiments performed previously with B. anthracis [19, 53]). For assays performed using both NPB and DMEM–10% (vol/vol) FBS, reaction mixtures were set up in a 96-well plate as described above for the Streptococcus species. After 2 h (for assays performed with NPB) or 3.4 h (for assays performed using DMEM–10% [vol/vol] FBS) of incubation in 37°C in 5% CO2 without shaking, 5 μl resazurin was added to each well. Reaction mixtures were incubated at 37°C in 5% CO2 without shaking, and fluorescence was monitored periodically for either 4 h (NPB assays) or 1.8 h (DMEM–10% [vol/vol] FBS assays) as described above. After the final fluorescence reading, cells were serially diluted 10× in PBS and spread on LB agar plates. Duplicate untreated samples were plated before incubation as well. CFU counts were performed after 24 h of incubation at 37°C without CO2. CFU and fluorescence data were analyzed as described above.
For CFU survival assays performed using TGS, cells were diluted to 1.8 × 105 CFU/40 μl and reaction mixtures were set up in 1.5-ml protein LoBind tubes, as described above. Reaction mixtures were incubated for 2 h at 37°C without CO2 and with shaking (220 rpm; tubes had lids closed), and then cells were serially diluted 10× in PBS and spread on LB agar plates. Duplicate untreated samples were plated before incubation as well. CFU counts were performed after 24 h of incubation at 37°C without CO2 and analyzed as described above.
Quellung assay.
The presence or absence of capsule was determined for selected strains during the antimicrobial assay. The S. pneumoniae D39 strains assayed for the presence of capsule included the following: S. pneumoniae D39 (IU1690), D39 rpsL1 (IU1781), D39 ΔdltA (IU12470), and D39 rpsL1 ΔamiA-F (IU13780). The S. pneumoniae D39 strains assayed for absence of capsule included the following: D39 Δcps (IU1945) and D39 rpsL1 cps2E (ΔA) (IU3309). TIGR4 (IU11966) and TIGR4 Δcps (IU12001) strains were assayed for the presence and absence of type 4 capsule, respectively. When overnight cultures diluted in BHI broth reached an OD620 of ∼0.1, 1 μl of culture was mixed with 1 μl of type 2 (for D39 strains) or type 4 (for TIGR4 strains) antisera (Statens Serum Institut) and was viewed with phase-contrast microscopy after 5 min (see below). Swollen cells with visible capsule indicated a positive result (capsule present). Strains D39 (IU1690) and D39 Δcps (IU1945) were used as positive and negative controls, respectively.
Phase-contrast microscopy.
Cell morphology was examined using phase-contrast images as described in reference 103. Briefly, 500 μl of cells growing in BHI broth at an OD620 of ∼0.1 to 0.2 was centrifuged at 21,100 × g for 3 min at room temperature. A 450-μl volume of the supernatant was removed, the pellet was resuspended in the remaining 50 μl by vortex mixing, and 1.5 μl was placed onto a slide with a glass coverslip.
Live-dead staining of S. pneumoniae D39 cells in different assay buffers.
Live-dead staining was performed as previously described (61), using a LIVE/DEAD BacLight bacterial viability kit (catalog no. L7007; Molecular Probes). Briefly, cultures of S. pneumoniae D39 were grown in BHI broth, washed, and resuspended in 3 ml BHI broth, NPB, or TGS as described above for the resazurin-based fluorescence assay. Cells were diluted to an OD620 of ∼0.05 in the appropriate buffer, and then a 2-ml volume was centrifuged for 4 min at 21,100 × g at room temperature. Supernatant was removed, and cell pellets were resuspended in 20 μl buffer. A 1.5-μl volume of a 1:1 (vol/vol) mixture of Syto-9 and propidium iodide was added, and cells were incubated in the dark at room temperature for 10 min. Cells were visualized with fluorescence microscopy as described previously (61). Averages of 1,600 cells were counted for each buffer condition from 2 biologically independent replicates.
Statistical analysis.
Dose-response curves were fitted to data using the log (inhibitor) versus response-variable slope function in GraphPad Prism, with no constraints. IC50s and 95% confidence intervals of the IC50 were calculated using this function.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lok-To (Chris) Sham and members of the Winkler laboratory for critical comments and discussion, Cheng Kao (IUB) for the generous gift of LL-37 and for thoughtful discussions, and Andrew Burrage, Katherine Hummels, and Daniel Kearns (IUB) for constructing isogenic Bacillus subtilis strains and for helpful discussions.
This work was supported by NIH grant R01GM114315 to M.E.W. and by predoctoral Quantitative and Chemical Biology (QCB) NIH institutional training grant T32 GM109825 (to B.E.R.).
K.E.B., H.-C.T.T., and M.E.W. were responsible for the conception or design of the study; K.E.B., B.E.R., H.-C.T.T., and M.E.W. performed acquisition, analysis, or interpretation of data; K.E.B., H.-C.T.T., and M.E.W. wrote the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00745-17.
REFERENCES
- 1.Luster AD. 1998. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med 338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 2.Kufareva I, Salanga CL, Handel TM. 2015. Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol Cell Biol 93:372–383. doi: 10.1038/icb.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller MC, Mayo KH. 2017. Chemokines from a structural perspective. Int J Mol Sci 18:E2088. doi: 10.3390/ijms18102088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen LT, Vogel HJ. 2012. Structural perspectives on antimicrobial chemokines. Front Immunol 3:384. doi: 10.3389/fimmu.2012.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf M, Moser B. 2012. Antimicrobial activities of chemokines: not just a side-effect? Front Immunol 3:213. doi: 10.3389/fimmu.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaminathan GJ, Holloway DE, Colvin RA, Campanella GK, Papageorgiou AC, Luster AD, Acharya KR. 2003. Crystal structures of oligomeric forms of the IP-10/CXCL10 chemokine. Structure 11:521–532. doi: 10.1016/S0969-2126(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 7.Proudfoot AE, Uguccioni M. 2016. Modulation of chemokine responses: synergy and cooperativity. Front Immunol 7:183. doi: 10.3389/fimmu.2016.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colvin RA, Campanella GS, Manice LA, Luster AD. 2006. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol 26:5838–5849. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. 1996. Chemokine receptor specific for IP-10 and MIG: structure, function, and expression in activated T-lymphocytes. J Exp Med 184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eliasson M, Olin AI, Malmstrom JA, Morgelin M, Bodelsson M, Collin M, Egesten A. 2012. Characterization of released polypeptides during an interferon-gamma-dependent antibacterial response in airway epithelial cells. J Interferon Cytokine Res 32:524–533. doi: 10.1089/jir.2012.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eliasson M, Morgelin M, Farber JM, Egesten A, Albiger B. 2010. Streptococcus pneumoniae induces expression of the antibacterial CXC chemokine MIG/CXCL9 via MyD88-dependent signaling in a murine model of airway infection. Microbes Infect 12:565–573. doi: 10.1016/j.micinf.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Gomez JC, Dang H, Kanke M, Hagan RS, Mock JR, Kelada SNP, Sethupathy P, Doerschuk CM. 2017. Predicted effects of observed changes in the mRNA and microRNA transcriptome of lung neutrophils during S. pneumoniae pneumonia in mice. Sci Rep 7:11258. doi: 10.1038/s41598-017-11638-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann J, Machado D, Terrier O, Pouzol S, Messaoudi M, Basualdo W, Espinola EE, Guillen RM, Rosa-Calatrava M, Picot V, Benet T, Endtz H, Russomando G, Paranhos-Baccala G. 2016. Viral and bacterial co-infection in severe pneumonia triggers innate immune responses and specifically enhances IP-10: a translational study. Sci Rep 6:38532. doi: 10.1038/srep38532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skovbjerg S, Norden R, Martner A, Samuelsson E, Hynsjo L, Wold AE. 2017. Intact pneumococci trigger transcription of interferon-related genes in human monocytes, while fragmented, autolyzed bacteria subvert this response. Infect Immun 85:e00960-16. doi: 10.1128/IAI.00960-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang D. 2003. Many chemokines including CCL20/MIP-3 display antimicrobial activity. J Leukoc Biol 74:448–455. doi: 10.1189/jlb.0103024. [DOI] [PubMed] [Google Scholar]
- 16.Yung SC, Murphy PM. 2012. Antimicrobial chemokines. Front Immunol 3:276. doi: 10.3389/fimmu.2012.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T. 2002. Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol 169:6985–6991. doi: 10.4049/jimmunol.169.12.6985. [DOI] [PubMed] [Google Scholar]
- 18.Laube DM, Yim S, Ryan LK, Kisich KO, Diamond G. 2006. Antimicrobial peptides in the airway. Curr Top Microbiol Immunol 306:153–182. [DOI] [PubMed] [Google Scholar]
- 19.Crawford MA, Zhu Y, Green CS, Burdick MD, Sanz P, Alem F, O'Brien AD, Mehrad B, Strieter RM, Hughes MA. 2009. Antimicrobial effects of interferon-inducible CXC chemokines against Bacillus anthracis spores and bacilli. Infect Immun 77:1664–1678. doi: 10.1128/IAI.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egesten A, Olin AI, Linge HM, Yadav M, Morgelin M, Karlsson A, Collin M. 2009. SpeB of Streptococcus pyogenes differentially modulates antibacterial and receptor activating properties of human chemokines. PLoS One 4:e4769. doi: 10.1371/journal.pone.0004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 22.Segev-Zarko L, Mangoni ML, Shai Y. 2017. Antimicrobial peptides: multiple mechanisms against a variety of targets, p 119–134. In Wang G. (ed), Antimicrobial peptides: discovery, design and novel therapeutic strategies, 2nd ed CABI, Wallingford, Oxfordshire, United Kingdom. [Google Scholar]
- 23.Barns KJ, Weisshaar JC. 2013. Real-time attack of LL-37 on single Bacillus subtilis cells. Biochim Biophys Acta 1828:1511–1520. doi: 10.1016/j.bbamem.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42:2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung MC, Dean SN, van Hoek ML. 2015. Acyl carrier protein is a bacterial cytoplasmic target of cationic antimicrobial peptide LL-37. Biochem J 470:243–253. doi: 10.1042/BJ20150432. [DOI] [PubMed] [Google Scholar]
- 26.Islam MR, Nagao J, Zendo T, Sonomoto K. 2012. Antimicrobial mechanism of lantibiotics. Biochem Soc Trans 40:1528–1533. doi: 10.1042/BST20120190. [DOI] [PubMed] [Google Scholar]
- 27.Wiedemann I, Benz R, Sahl HG. 2004. Lipid II-mediated pore formation by the peptide antibiotic nisin: a black lipid membrane study. J Bacteriol 186:3259–3261. doi: 10.1128/JB.186.10.3259-3261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, de Kruijff B, Breukink E. 2006. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 313:1636–1637. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- 29.Aprianto R, Slager J, Holsappel S, Veening JW. 2016. Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal transcriptomes during early infection. Genome Biol 17:198. doi: 10.1186/s13059-016-1054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. 1997. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88:553–560. doi: 10.1016/S0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 31.Linge HM, Sastalla I, Nitsche-Schmitz DP, Egesten A, Frick IM. 2007. Protein FOG is a moderate inducer of MIG/CXCL9, and group G streptococci are more tolerant than group A streptococci to this chemokine's antibacterial effect. Microbiology 153:3800–3808. doi: 10.1099/mic.0.2007/009647-0. [DOI] [PubMed] [Google Scholar]
- 32.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. 1996. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85:229–236. doi: 10.1016/S0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 33.Dai C, Basilico P, Cremona TP, Collins P, Moser B, Benarafa C, Wolf M. 2015. CXCL14 displays antimicrobial activity against respiratory tract bacteria and contributes to clearance of Streptococcus pneumoniae pulmonary infection. J Immunol 194:5980–5989. doi: 10.4049/jimmunol.1402634. [DOI] [PubMed] [Google Scholar]
- 34.Ishiguro K, Washio J, Sasaki K, Takahashi N. 2015. Real-time monitoring of the metabolic activity of periodontopathic bacteria. J Microbiol Methods 115:22–26. doi: 10.1016/j.mimet.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 35.Nagaoka I, Kuwahara-Arai K, Tamura H, Hiramatsu K, Hirata M. 2005. Augmentation of the bactericidal activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by amino acid substitutions. Inflamm Res 54:66–73. doi: 10.1007/s00011-004-1323-8. [DOI] [PubMed] [Google Scholar]
- 36.Rampersad SN. 2012. Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors 12:12347–12360. doi: 10.3390/s120912347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiloh MU, Ruan J, Nathan C. 1997. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect Immun 65:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yajko DM, Madej JJ, Lancaster MV, Sanders CA, Cawthon VL, Gee B, Babst A, Hadley WK. 1995. Colorimetric method for determining MICs of antimicrobial agents for Mycobacterium tuberculosis. J Clin Microbiol 33:2324–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole JN, Nizet V. 2016. Bacterial evasion of host antimicrobial peptide defenses. Microbiol Spectr 4:(1). doi: 10.1128/microbiolspec.VMBF-0006-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaRock CN, Nizet V. 2015. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim Biophys Acta 1848:3047–3054. doi: 10.1016/j.bbamem.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovács M, Halfmann A, Fedtke I, Heintz M, Peschel A, Vollmer W, Hakenbeck R, Brückner R. 2006. A functional dlt operon, encoding proteins required for incorporation of d-alanine in teichoic acids in gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae. J Bacteriol 188:5797–5805. doi: 10.1128/JB.00336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer NE, Hasper HE, van den Bogaard PT, Morath S, de Kruijff B, Hartung T, Smid EJ, Breukink E, Kok J, Kuipers OP. 2008. Increased d-alanylation of lipoteichoic acid and a thickened septum are main determinants in the nisin resistance mechanism of Lactococcus lactis. Microbiology 154:1755–1762. doi: 10.1099/mic.0.2007/015412-0. [DOI] [PubMed] [Google Scholar]
- 43.Lysenko ES, Gould J, Bals R, Wilson JM, Weiser JN. 2000. Bacterial phosphorylcholine decreases susceptibility to the antimicrobial peptide LL-37/hCAP18 expressed in the upper respiratory tract. Infect Immun 68:1664–1671. doi: 10.1128/IAI.68.3.1664-1671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, Trieu-Cuot P, Shai Y. 2012. d-Alanylation of lipoteichoic acids confers resistance to cationic peptides in group B Streptococcus by increasing the cell wall density. PLoS Pathog 8:e1002891. doi: 10.1371/journal.ppat.1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beiter K, Wartha F, Hurwitz R, Normark S, Zychlinsky A, Henriques-Normark B. 2008. The capsule sensitizes Streptococcus pneumoniae to alpha-defensins human neutrophil proteins 1 to 3. Infect Immun 76:3710–3716. doi: 10.1128/IAI.01748-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rukke HV, Engen SA, Schenck K, Petersen FC. 2016. Capsule expression in Streptococcus mitis modulates interaction with oral keratinocytes and alters susceptibility to human antimicrobial peptides. Mol Oral Microbiol 31:302–313. doi: 10.1111/omi.12123. [DOI] [PubMed] [Google Scholar]
- 47.Kietzman CC, Gao G, Mann B, Myers L, Tuomanen EI. 2016. Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat Commun 7:10859. doi: 10.1038/ncomms10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Llobet E, Tomas JM, Bengoechea JA. 2008. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 154:3877–3886. doi: 10.1099/mic.0.2008/022301-0. [DOI] [PubMed] [Google Scholar]
- 49.Shelton CL, Raffel FK, Beatty WL, Johnson SM, Mason KM. 2011. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog 7:e1002360. doi: 10.1371/journal.ppat.1002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gryllos I, Tran-Winkler HJ, Cheng MF, Chung H, Bolcome R III, Lu W, Lehrer RI, Wessels MR. 2008. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc Natl Acad Sci U S A 105:16755–16760. doi: 10.1073/pnas.0803815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. 2007. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci U S A 104:9469–9474. doi: 10.1073/pnas.0702159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Majchrzykiewicz JA, Kuipers OP, Bijlsma JJ. 2010. Generic and specific adaptive responses of Streptococcus pneumoniae to challenge with three distinct antimicrobial peptides, bacitracin, LL-37, and nisin. Antimicrob Agents Chemother 54:440–451. doi: 10.1128/AAC.00769-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crawford MA, Lowe DE, Fisher DJ, Stibitz S, Plaut RD, Beaber JW, Zemansky J, Mehrad B, Glomski IJ, Strieter RM, Hughes MA. 2011. Identification of the bacterial protein FtsX as a unique target of chemokine-mediated antimicrobial activity against Bacillus anthracis. Proc Natl Acad Sci U S A 108:17159–17164. doi: 10.1073/pnas.1108495108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Margulieux KR, Fox JW, Nakamoto RK, Hughes MA. 2016. CXCL10 acts as a bifunctional antimicrobial molecule against Bacillus anthracis. mBio 7:e00334-16. doi: 10.1128/mBio.00334-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mavrici D, Marakalala MJ, Holton JM, Prigozhin DM, Gee CL, Zhang YJ, Rubin EJ, Alber T. 2014. Mycobacterium tuberculosis FtsX extracellular domain activates the peptidoglycan hydrolase, RipC. Proc Natl Acad Sci U S A 111:8037–8042. doi: 10.1073/pnas.1321812111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meisner J, Montero Llopis P, Sham LT, Garner E, Bernhardt TG, Rudner DZ. 2013. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol Microbiol 89:1069–1083. doi: 10.1111/mmi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sham LT, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Natl Acad Sci U S A 108:E1061–E1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang DC, Peters NT, Parzych KR, Uehara T, Markovski M, Bernhardt TG. 2011. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc Natl Acad Sci U S A 108:E1052–E1060. doi: 10.1073/pnas.1107780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bajaj R, Bruce KE, Davidson AL, Rued BE, Stauffacher CV, Winkler ME. 2016. Biochemical characterization of essential cell division proteins FtsX and FtsE that mediate peptidoglycan hydrolysis by PcsB in Streptococcus pneumoniae. Microbiologyopen 5:738–752. doi: 10.1002/mbo3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Du S, Lutkenhaus J. 2017. Assembly and activation of the Escherichia coli divisome. Mol Microbiol 105:177–187. doi: 10.1111/mmi.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sham LT, Jensen KR, Bruce KE, Winkler ME. 2013. Involvement of FtsE ATPase and FtsX extracellular loops 1 and 2 in FtsEX-PcsB complex function in cell division of Streptococcus pneumoniae D39. mBio 4:e00431-13. doi: 10.1128/mBio.00431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bartual SG, Straume D, Stamsas GA, Munoz IG, Alfonso C, Martinez-Ripoll M, Havarstein LS, Hermoso JA. 2014. Structural basis of PcsB-mediated cell separation in Streptococcus pneumoniae. Nat Commun 5:3842. doi: 10.1038/ncomms4842. [DOI] [PubMed] [Google Scholar]
- 63.Margulieux KR, Liebov BK, Tirumala V, Singh A, Bushweller JH, Nakamoto RK, Hughes MA. 2017. Bacillus anthracis peptidoglycan integrity is disrupted by the chemokine CXCL10 through the FtsE/X complex. Front Microbiol 8:740. doi: 10.3389/fmicb.2017.00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryan JD, Shelver DW. 2009. Streptococcus agalactiae CspA is a serine protease that inactivates chemokines. J Bacteriol 191:1847–1854. doi: 10.1128/JB.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holdren GO, Rosenthal DJ, Yang J, Bates AM, Fischer CL, Zhang Y, Brogden NK, Brogden KA. 2014. Antimicrobial activity of chemokine CXCL10 for dermal and oral microorganisms. Antibiotics 3:527–539. doi: 10.3390/antibiotics3040527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marshall A, Celentano A, Cirillo N, Mignogna MD, McCullough M, Porter S. 2016. Antimicrobial activity and regulation of CXCL9 and CXCL10 in oral keratinocytes. Eur J Oral Sci 124:433–439. doi: 10.1111/eos.12293. [DOI] [PubMed] [Google Scholar]
- 67.Gela A, Kasetty G, Jovic S, Ekoff M, Nilsson G, Morgelin M, Kjellstrom S, Pease JE, Schmidtchen A, Egesten A. 2015. Eotaxin-3 (CCL26) exerts innate host defense activities that are modulated by mast cell proteases. Allergy 70:161–170. doi: 10.1111/all.12542. [DOI] [PubMed] [Google Scholar]
- 68.Lee HY, Andalibi A, Webster P, Moon SK, Teufert K, Kang SH, Li JD, Nagura M, Ganz T, Lim DJ. 2004. Antimicrobial activity of innate immune molecules against Streptococcus pneumoniae, Moraxella catarrhalis and nontypeable Haemophilus influenzae. BMC Infect Dis 4:12. doi: 10.1186/1471-2334-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM. 1998. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 102:874–880. doi: 10.1172/JCI2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mason KM, Bruggeman ME, Munson RS, Bakaletz LO. 2006. The non-typeable Haemophilus influenzae Sap transporter provides a mechanism of antimicrobial peptide resistance and SapD-dependent potassium acquisition. Mol Microbiol 62:1357–1372. doi: 10.1111/j.1365-2958.2006.05460.x. [DOI] [PubMed] [Google Scholar]
- 71.Maisetta G, Batoni G, Esin S, Raco G, Bottai D, Favilli F, Florio W, Campa M. 2005. Susceptibility of Streptococcus mutans and Actinobacillus actinomycetemcomitans to bactericidal activity of human beta-defensin 3 in biological fluids. Antimicrob Agents Chemother 49:1245–1248. doi: 10.1128/AAC.49.3.1245-1248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Velarde JJ, Ashbaugh M, Wessels MR. 2014. The human antimicrobial peptide LL-37 binds directly to CsrS, a sensor histidine kinase of group A Streptococcus, to activate expression of virulence factors. J Biol Chem 289:36315–36324. doi: 10.1074/jbc.M114.605394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mason KM, Munson RS Jr, Bakaletz LO. 2005. A mutation in the sap operon attenuates survival of nontypeable Haemophilus influenzae in a chinchilla model of otitis media. Infect Immun 73:599–608. doi: 10.1128/IAI.73.1.599-608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alloing G, de Philip P, Claverys JP. 1994. Three highly homologous membrane-bound lipoproteins participate in oligopeptide transport by the Ami system of the gram-positive Streptococcus pneumoniae. J Mol Biol 241:44–58. doi: 10.1006/jmbi.1994.1472. [DOI] [PubMed] [Google Scholar]
- 75.Doeven MK, Abele R, Tampe R, Poolman B. 2004. The binding specificity of OppA determines the selectivity of the oligopeptide ATP-binding cassette transporter. J Biol Chem 279:32301–32307. doi: 10.1074/jbc.M404343200. [DOI] [PubMed] [Google Scholar]
- 76.Hoover SE, Perez AJ, Tsui HC, Sinha D, Smiley DL, DiMarchi RD, Winkler ME, Lazazzera BA. 2015. A new quorum-sensing system (TprA/PhrA) for Streptococcus pneumoniae D39 that regulates a lantibiotic biosynthesis gene cluster. Mol Microbiol 97:229–243. doi: 10.1111/mmi.13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kerr AR, Adrian PV, Estevao S, de Groot R, Alloing G, Claverys JP, Mitchell TJ, Hermans PW. 2004. The Ami-AliA/AliB permease of Streptococcus pneumoniae is involved in nasopharyngeal colonization but not in invasive disease. Infect Immun 72:3902–3906. doi: 10.1128/IAI.72.7.3902-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V. 2005. d-Alanylation of teichoic acids promotes group A Streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 187:6719–6725. doi: 10.1128/JB.187.19.6719-6725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, Konradsen HB, Nahm MH. 2015. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev 28:871–899. doi: 10.1128/CMR.00024-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tettelin H, Hollingshead SK. 2004. Comparative genomics of Streptococcus pneumoniae: intrastrain diversity and genome plasticity, p 15–29. In Tuomanen TJM, Morrison DA, Spratt BG (ed), The pneumococcus. ASM Press, Washington, DC. [Google Scholar]
- 81.Zafar MA, Hamaguchi S, Zangari T, Cammer M, Weiser JN. 2017. Capsule type and amount affect shedding and transmission of Streptococcus pneumoniae. mBio 8:4e00989-17. doi: 10.1128/mBio.00989-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sham LT, Tsui HC, Land AD, Barendt SM, Winkler ME. 2012. Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opin Microbiol 15:194–203. doi: 10.1016/j.mib.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garti-Levi S, Hazan R, Kain J, Fujita M, Ben-Yehuda S. 2008. The FtsEX ABC transporter directs cellular differentiation in Bacillus subtilis. Mol Microbiol 69:1018–1028. doi: 10.1111/j.1365-2958.2008.06340.x. [DOI] [PubMed] [Google Scholar]
- 84.Schutte KM, Fisher DJ, Burdick MD, Mehrad B, Mathers AJ, Mann BJ, Nakamoto RK, Huges MA. 2016. Escherichia coli pyruvate dehydrogenase complex is an important component of CXCL10-mediated antimicrobial activity. Infect Immun 84:320–328. doi: 10.1128/IAI.00552-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Claverys JP, Grossiord B, Alloing G. 2000. Is the Ami-AliA/B oligopeptide permease of Streptococcus pneumoniae involved in sensing environmental conditions? Res Microbiol 151:457–463. doi: 10.1016/S0923-2508(00)00169-8. [DOI] [PubMed] [Google Scholar]
- 86.Klepsch MM, Kovermann M, Low C, Balbach J, Permentier HP, Fusetti F, de Gier JW, Slotboom DJ, Berntsson RP. 2011. Escherichia coli peptide binding protein OppA has a preference for positively charged peptides. J Mol Biol 414:75–85. doi: 10.1016/j.jmb.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 87.Alloing G, Granadel C, Morrison DA, Claverys JP. 1996. Competence pheromone, oligopeptide permease, and induction of competence in Streptococcus pneumoniae. Mol Microbiol 21:471–478. doi: 10.1111/j.1365-2958.1996.tb02556.x. [DOI] [PubMed] [Google Scholar]
- 88.Chen H, Ma Y, Yang J, O'Brien CJ, Lee SL, Mazurkiewicz JE, Haataja S, Yan JH, Gao GF, Zhang JR. 2007. Genetic requirement for pneumococcal ear infection. PLoS One 3:e2950. doi: 10.1371/journal.pone.0002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cundell DR, Pearce BJ, Sandros J, Naughton AM, Masure HR. 1995. Peptide permeases from Streptococcus pneumoniae affect adherence to eucaryotic cells. Infect Immun 63:2493–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hava DL, Camilli A. 2002. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol 45:1389–1406. doi: 10.1046/j.1365-2958.2002.03106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, Tuomanen EI. 2004. Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun 72:5582–5596. doi: 10.1128/IAI.72.10.5582-5596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song XM, Connor W, Jalal S, Hokamp K, Potter AA. 2008. Microarray analysis of Streptococcus pneumoniae gene expression changes to human lung epithelial cells. Can J Microbiol 54:189–200. doi: 10.1139/W07-133. [DOI] [PubMed] [Google Scholar]
- 93.Trombe MC, Laneelle G, Sicard AM. 1984. Characterization of a Streptococcus pneumoniae mutant with altered electric transmembrane potential. J Bacteriol 158:1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trombe MC, Laneelle MA, Laneelle G. 1979. Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim Biophys Acta 574:290–300. doi: 10.1016/0005-2760(79)90010-9. [DOI] [PubMed] [Google Scholar]
- 95.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 97.Ramos-Montañez S, Tsui HC, Wayne KJ, Morris JL, Peters LE, Zhang F, Kazmierczak KM, Sham LT, Winkler ME. 2008. Polymorphism and regulation of the spxB (pyruvate oxidase) virulence factor gene by a CBS-HotDog domain protein (SpxR) in serotype 2 Streptococcus pneumoniae. Mol Microbiol 67:729–746. doi: 10.1111/j.1365-2958.2007.06082.x. [DOI] [PubMed] [Google Scholar]
- 98.Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. 2010. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol 192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. 2016. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol Microbiol 100:1039–1065. doi: 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sung CK, Li H, Claverys JP, Morrison DA. 2001. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl Environ Microbiol 67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kao C, Lin X, Yi G, Zhang Y, Rowe-Magnus DA, Bush K. 2016. Cathelicidin antimicrobial peptides with reduced activation of Toll-like receptor signaling have potent bactericidal activity against colistin-resistant bacteria. mBio 7:e01418-16. doi: 10.1128/mBio.01418-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rajkovic A, Hummels KR, Witzky A, Erickson S, Gafken PR, Whitelegge JP, Faull KF, Kearns DB, Ibba M. 2016. Translation control of swarming proficiency in Bacillus subtilis by 5-amino-pentanolylated elongation factor P. J Biol Chem 291:10976–10985. doi: 10.1074/jbc.M115.712091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol 191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.