

Abstract
Owing to the spread of multidrug resistance (MDR) and extensive drug resistance (XDR), there is a pressing need to identify potential targets for the development of more-effective anti-M. tuberculosis (Mtb) drugs. PafA, as the sole Prokaryotic Ubiquitin-like Protein ligase in the Pup-proteasome System (PPS) of Mtb, is an attractive drug target. Here, we show that the activity of purified Mtb PafA is significantly inhibited upon the association of AEBSF (4-(2-aminoethyl) benzenesulfonyl fluoride) to PafA residue Serine 119 (S119). Mutation of S119 to amino acids that resemble AEBSF has similar inhibitory effects on the activity of purified Mtb PafA. Structural analysis reveals that although S119 is distant from the PafA catalytic site, it is located at a critical position in the groove where PafA binds the C-terminal region of Pup. Phenotypic studies demonstrate that S119 plays critical roles in the function of Mtb PafA when tested in M. smegmatis. Our study suggests that targeting S119 is a promising direction for developing an inhibitor of M. tuberculosis PafA.
Keywords: M. Tuberculosis, Pupylation, PafA, 4-(2-aminoethyl) benzenesulfonyl fluoride
Highlights
-
•
The pupylation activity of purified M. tuberculosis PafA is almost completely inhibited upon the association of AEBSF.
-
•
The AEBSF binding site, Ser 119 plays critical roles in both the pupylation and depupylation activity of purified M. tuberculosis PafA.
-
•
Disruption of purified M. tuberculosis PafA Ser 119 causes a dramatic reduction in Pup binding.
Drug-resistant tuberculosis is a major challenge worldwide, there is an urgent need to identify potential drug targets for developing more effective anti-tubercular drugs. M. tuberculosis ubiquitin-like protein ligase PafA is an attractive drug target, however, effective PafA inhibitors have not yet been identified. Here, we show that interruption of a single amino acid, S119, causes dramatic loss of PafA activity. S119 could thus serve as a promising precise target for developing M. tuberculosis PafA inhibitors.
1. Introduction
Tuberculosis (TB), caused by M. tuberculosis (Mtb), is one of the most common causes of death worldwide (WHO, 2016). At present, this global health problem is exacerbated by the emergence and spread of M. tuberculosis strains that are resistant to antibiotics including multidrug resistant (MDR) and extensively drug resistant (XDR) strains. According to the latest statistics, only 52% of patients with MDR-TB and 28% with XDR-TB can be treated effectively (WHO, 2016). In the past 50 years, only two new drugs, bedaquiline (Goel, 2014) and delamanid (Hoagland et al., 2016), have been successfully developed to address MDR-TB (Zumla et al., 2013, Mdluli et al., 2015). To obtain more effective treatment options for MDR-TB, there is an urgent need to develop new drugs with different mechanisms of action.
Ubiquitin-dependent protein degradation in eukaryotes plays a central role in many cellular functions, such as post-translational quality control, cell proliferation, differentiation and development (Grabbe et al., 2011, Yau and Rape, 2016). Ubiquitin is covalently attached to specific lysine residues of target proteins through a complicated multi-step ligation reaction and eventually delivers doomed proteins for proteasomal degradation (Hershko et al., 2000). Similar to this process in eukaryotic cells, proteins are targeted to the proteasome via a prokaryotic ubiquitin-like protein modifier termed Pup in Mtb (Pearce et al., 2008, Striebel et al., 2009). The inactive form of Pup has a C-terminal glutamine: conversion of this residue to glutamate (PupE) by the enzyme Dop (Striebel et al., 2009) activates Pup for ligation. Activated Pup is then attached to target proteins by PafA, the sole ligase in the Pup-proteasome System (PPS) (Pearce et al., 2006, Pearce et al., 2008, Striebel et al., 2009, Sutter et al., 2010, Guth et al., 2011). Pupylated proteins are then directed into the proteasome via recognition of Pup by Mycobacterium proteasomal ATPase (Mpa) (Sutter et al., 2009, Wang et al., 2009, Striebel et al., 2010, Wang et al., 2010). Analogous to deubiquitination, depupylation also occurs in Mtb and is catalyzed by Dop (Burns et al., 2010, Imkamp et al., 2010b) and PafA (Zhang et al., 2017). Previous studies showed that the Pup-proteasome System (PPS) of Mtb is required for resistance to nitric oxide and is essential for Mtb to cause lethality in mice (Darwin et al., 2003, Darwin et al., 2005, Lamichhane et al., 2006, Gandotra et al., 2007, Samanovic et al., 2015). To our knowledge, the PPS is only present in the Nitrospira and Actinobacteria (Imkamp et al., 2015) and is not present in most other bacteria, including gut microbiota. These unusual properties of the Mtb PPS make it an attractive target for drug development. Previous strategies for inhibiting the Mtb PPS focused on the 20S proteasome (Lin et al., 2009, Cheng and Pieters, 2010, Lin et al., 2010, Clements et al., 2013, Lin et al., 2013, Zheng et al., 2014, Totaro et al., 2017), however, owing to the high degree of mechanistic and structural conservation of mammalian and mycobacterial proteasomes, inherent toxicity is inevitable (Cheng and Pieters, 2010). On the other hand, PafA shares no homology with ubiquitin ligases in eukaryotes (Festa et al., 2007, Burns et al., 2009, Bode and Darwin, 2014), suggesting that there may be no or few side effects for drugs that target PafA. Unfortunately, to date, effective inhibitors of PafA have not been identified.
Here, we show that the serine protease inhibitor, AEBSF (4-(2-aminoethyl) benzenesulfonyl fluoride), is a potent inhibitor of purified Mtb PafA. We further show that this compound binds to PafA via S119. Biochemical analysis demonstrated that substitution of S119 with aromatic amino acid residues, imitating the binding of AEBSF, almost completely abolishes the pupylase and depupylase activity of Mtb PafA. Further structural analysis showed that this inhibition of PafA activity is a consequence of defective Pup binding to PafA even though this residue is far from the PafA catalytic site. Finally, phenotypic studies demonstrated that S119 is critical for the function of Mtb PafA and essential for Msm △PafA survival under nitrogen limitation and in macrophages. Overall, our work shows that S119 is situated within a critical, small-molecule accessible region of PafA whose modification inhibits PafA activity and is thus a highly promising target for the development of inhibitors of M. tuberculosis PafA.
2. Materials and Methods
2.1. Protein Cloning, Expression, and Purification
All genes were cloned from the Mtb reference strain H37Rv. PafA was cloned into pTrc99a as described previously (Striebel et al., 2009). All PafA variants were constructed using a QuikChange® Site-Directed Mutagenesis Kit (Agilent Technologies). C-terminal Flag-His6-tagged Mpa and PanB were cloned into pET28a. Sequences of primers used in this study are given in Table S1. All recombinant proteins were expressed in E. coli BL21 by growing recombinant E. coli BL21 cells in 1 L LB medium to an A600 of 0.6 at 37 °C. Protein expression was induced by the addition of 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) before incubating cells overnight at 16 °C. Proteins were purified on Ni-NTA affinity columns and stored at −80 °C. PupE and N-terminal 5-Carboxyfluorescein-cys-PupE were synthesized by GL Biochem, Shanghai, China.
2.2. Inhibitor Profile of PafA Pupylase Activity
All chemicals or inhibitors were purchased from Sigma-Aldrich. PafA was incubated with different inhibitors at 25 °C for 0.5 h in pupylation buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 20 mM MgCl2 and 10% (v/v) glycerol). Pupylation reactions included PanB-Flag (8 μM), PupE (10 μM) and PafA (0.5 μM) pre-incubated with inhibitors and were incubated at 25 °C for 6 h with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining and western blotting with an anti-Pup monoclonal antibody (Abmart) and an anti-Flag antibody (Sigma-Aldrich Cat# P2983, RRID: AB_439685).
2.3. Pupylation Assays
PanB pupylation assay reactions were carried out in pupylation buffer containing PanB-Flag (8 μM), PupE (10 μM) and PafA (0.5 μM) at 25 °C for 6 h, while Mpa pupylation assay reactions were carried out in pupylation buffer containing Mpa-Flag (6 μM), PupE (10 μM) and PafA (0.5 μM) at 25 °C for 2 h with 5 mM ATP. For pupylation assays using lysate as the substrate, reactions included Msm △PafA lysates (10 μg), PupE (10 μM) and PafA (0.5 μM) and were incubated at 25 °C for 20 min with 5 mM ATP in pupylation buffer. In AEBSF inhibitory assays, reactions were carried out as described above, except that PafA (0.5 μM) was pre-incubated in AEBSF for 30 min at 25 °C. Unbound AEBSF was removed by dialysis using a Spectra/Por(R) Dialysis Membrane (Sangon Biotech, Shanghai, China). Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining and western blotting.
2.4. Identification of the AEBSF Binding Site on PafA by LC-MS/MS
PafA (0.5 μM) was incubated with AEBSF (0.25 mM or 0.5 mM) at 25 °C for 0.5 h in pupylation buffer. After SDS-PAGE and Coomassie staining, PafA bands were cut out and in-gel digested with trypsin. The tryptic peptide digests of the proteins were analyzed using an LC system (Nano Pump, Ultimate 3000, Dionex, Thermofisher) coupled with an ESI-Q-TOF mass spectrometer (MaXis, Impact, Bruker Daltonik, Germany). The peptide sequences were determined by searching MS/MS spectra against the Protein database using the Mascot (version 2.4, Matrix Science) software suite with a precursor ion mass tolerance of 20 ppm and fragment ion mass tolerance of 0.05 Da. Carbamidomethyl (C) was set as the fixed modification, and oxidation (M) was set as the variable modification. For modification of AEBSF, +183.0354 Da was set as a variable modification on serine, and a maximum of two missed cleavage of trypsin was chosen. MS data are available (“ProteomeXchange: PXD007045 and PXD006930”).
2.5. Quantitative Pupylation Assays
Quantitative pupylation assays were performed according to a previously described procedure (Ofer et al., 2013), except that N-terminal 5-Carboxyfluorescein-cys-PupE was used instead of N-terminal 5-iodoacetamidofluorescein-cys-PupE. The fluorescence intensity of each sample was measured with a Synergy 2 microplate reader (Biotek Instruments) at 494 nm (excitation) and 522 nm (emission).
2.6. Determination of PafAMtb Pupylase Activity in M. smegmatis
We cloned PafAMtb and its variants with a C-terminal Flag-tag into the mycobacterial expression vector pMV261 or pSMT3 via BamHI and HindIII restriction sites. These plasmids were then transformed into WT M. smegmatis (Msm) or Msm △PafA. Msm cells were grown at 37 °C in 100 ml 7H9 medium containing 30 μg/ml Kan or 50 μg/ml Hyg with 0.5% glycerol, 0.05% Tween-80 to an OD600 of 2.0. Cells were collected by centrifuging at 5000 rpm for 10 min and were then lysed in pupylation buffer with a high-pressure cracker (Union-Biotech, Shanghai, China). These lysates were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining and western blotting.
2.7. Equilibrium MD Simulations
The initial structure used for equilibrium simulations was the crystal structure of the C. glutamicum PafA/Pup complex (pdb: 4BJR) (Barandun et al., 2013). Alanine and phenylalanine mutants at S126 were generated using VMD (Humphrey et al., 1996). Force-field parameters for covalent attachment of AEBSF to the hydroxyl group of serine were generated using CGenFF (Vanommeslaeghe et al., 2010, Yu et al., 2012) and the initial structure of the modified protein was generated using VMD. All proteins were solvated in TIP3 water in 0.15 M NaCl and minimized and equilibrated using VMD/NAMD and the CHARMM 27 force field (MacKerell et al., 1998, Phillips et al., 2005). The particle mesh Ewald algorithm was employed to treat electrostatic interactions, and van der Waal interactions were treated with a cut-off of 12 Å. Langevin dynamics were employed to maintain a constant temperature of 310 K and a Nose-Hoover Langevin piston was used to maintain a constant pressure of 1 atm. The integration step was set to 2 fs. Extended simulations (35 ns) were performed for each complex. In the movies of the simulations, adjacent frames of the 80 ps/frame simulations were smoothed to facilitate understanding of the conformational changes.
2.8. Lysine Pupylation Assays
Pupylation reactions were carried out in pupylation buffer containing free lysine (80 mM), PupE (10 μM) and PafA variants (0.5 μM each) to which 5 mM ATP was added, and were incubated at 25 °C for 15 min. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining. The reactions in the AEBSF inhibitory assay were carried out as above, except that serially diluted PafA (0.5 μM) that had been pre-incubated in AEBSF in pupylation buffer at 25 °C for 0.5 h was used instead of untreated PafA (0.5 μM). Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining.
2.9. Bio-layer Interferometry of Pup and PafA Interaction Kinetics
The kinetics of Pup and PafA binding was measured using a ForteBio 70 Octet system. PupE was biotinylated using an EZLink Sulfo-NHS-LC-Biotin protein biotinylation kit (Thermo Scientific, Bremen, Germany) according to the manufacturer's instructions. Biotinylated PupE was tethered on the tip surface of a streptavidin-coated sensor, and then exposed to its binding partners in sample dilution (SD) buffer (1 × PBS, pH 7.4 with 0.02% Tween-20 and 0.1% BSA). Binding was measured by the coincident change in the interference pattern. Super strepavidin (SSA) tips (Pall ForteBio, Menlo Park, USA) were prewet in SD buffer, which served as the background buffer for immobilization. The immobilization process involved establishing a stable baseline (60 s), loading of 100 μg/ml biotinylated PupE (300 s), balancing tips with SD buffer (60 s), association with 500 nM PafA variants or PafA pre-incubated with 2 mM AEBSF (dissolved in SD buffer) (300 s) and then eluting non-specific binding with SD buffer (300 s). BLI data were analyzed using ForteBio Data Analysis Software 7. Data were fit to a 1:1 binding model to calculate an association and dissociation rate, and the KD was calculated using the ratio Kdis/Kon.
2.10. Protein Depupylation Assays
In PanB depupylation assays, PanB-Flag-PupE was enriched using an anti-FLAG M2 Affinity gel (Sigma-Aldrich, Saint Louis, USA). PanB-PupE (0.75 μM) was depupylated by PafA variants (1.5 μM each) with 5 mM ADP in phosphate buffer (50 mM Na2PO4 pH 8, 100 mM NaCl, 20 mM MgCl2 and 10% (v/v) glycerol) for 6 h at 25 °C. In the AEBSF inhibitory assay, the reactions were carried out as above, except that PafA (0.5 μM) was pre-incubated with AEBSF in pupylation buffer at 25 °C for 0.5 h. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining and western blotting.
2.11. Msm Survival Under Nitrogen Limitation
Nitrogen limitation assays were performed according to a previously described procedure (Elharar et al., 2014) with slight modification. Briefly, the Msm MC2155 strain was grown at 37 °C in a defined minimal nitrogen medium containing 40 mM K2HPO4, 22 mM KH2PO4, 1.7 mM sodium citrate, 0.4 mM MgSO4, 0.4% glycerol (v/v), and 0.05% Tween-80 (v/v).
2.12. Msm Survival in THP-1 Cell
Human THP-1 cell lines were grown in RPMI 1640 medium (Invitrogen, California, USA) with 10% fetal calf serum (Invitrogen, California, USA). Cultures of THP-1 cells were maintained at final gross of 4 × 106 cells and were treated with 10 ng/ml polymethyl acrylate (PMA) for 12 h to induce a macrophage-like state. After removing the suspension, adherent cells grew for 8 h, and then infected with Msm expressing PafAMtb with different mutations at OD600 = 0.7 for 2 h at a multiplicity of infection (MOI) of 10. The culture plate wells were then washed with incomplete RPMI 1640 medium and the cells were incubated in fresh complete RPMI 1640 medium with 10 ng/ml gentamicin for 12, 24, or 48 h. Cells were harvested for CFU counting and then washed three times with 1× PBS. Cells used for CFU counting were lysed in 7H9 broth containing 0.05% SDS for 10 min. Three sets of tenfold serial dilutions of the lysates were prepared at each time point in 0.05% Tween-80, and portions were plated on 7H10 agar plates. Plates were placed at 37 °C for 3–5 days before counting.
3. Results
3.1. AEBSF Inhibits Mtb PafA Pupylase Activity
During initial preliminary experiments on the function of PafA, we noted the presence of background proteolytic activity and so examined a range of well-known protease inhibitors to block this activity. Unexpectedly, we discovered that one of these compounds, AEBSF (4-(2-aminoethyl) benzenesulfoninyl fluoride), an irreversible inhibitor of serine proteases, strongly inhibited Mtb PafA pupylation of PanB (Fig. S1), a well-studied substrate of PafA pupylation activity (Pearce et al., 2006, Striebel et al., 2009). To confirm this result, pupylation assays were carried out with serially diluted AEBSF, and we found that the loss of PafA-mediated pupylation of PanB is indeed proportional to the concentration of AEBSF (Fig. 1a). To rule out the possibility that this reduction is due to the binding of AEBSF to PanB, we pre-incubated PafA with AEBSF, then used dialysis to remove unbound AEBSF. As AEBSF inhibits serine proteases by irreversible covalent binding, we reasoned that its inhibition of PafA might be through a similar mechanism and thus that the inhibition would persist even after extensive dialysis. Indeed, we found significant and dose-dependent reduction of pupylation even after dialysis (Fig. S2a). These results clearly demonstrate that the reduced pupylation of PanB was due to covalent attachment of AEBSF to PafA. We also observed a similar reduction in the pupylation of Mpa, another well-studied PafA substrate (Pearce et al., 2006, Delley et al., 2012) in the presence of AEBSF (Fig. 1b), further supporting a PafA-specific effect.
Fig. 1.

AEBSF is an inhibitor of Mtb PafA pupylase activity.
(a) PafA was incubated with serially diluted AEBSF, i.e., 2, 1, 0.5, or 0.25 mM, at 25 °C for 0.5 h in pupylation buffer. Pupylation reactions included PanB-Flag (8 μM), PupE (10 μM) and PafA (0.5 μM; pre-incubated with AEBSF), and were incubated at 25 °C for 6 h with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining and western blotting with an anti-flag antibody. Quantitation of the pupylation level of PanB based on CBB staining is shown in the lower panel. Data are representative of three independent biological replicates (mean and s.e.m. of n = 3 samples), *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
(b) PafA was incubated with serially diluted AEBSF at 25 °C for 0.5 h in pupylation buffer. Pupylation reactions included Mpa-Flag (6 μM), PupE (10 μM) and PafA (0.5 μM; pre-incubated with AEBSF) and were incubated at 25 °C for 2 h with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining and western blotting with an anti-flag antibody.
(c) PafA was incubated with serially diluted AEBSF at 25 °C for 0.5 h in pupylation buffer. Pupylation reactions included Msm △PafA lysates (10 μg), PupE (10 μM) and PafA (0.5 μM; pre-incubated with AEBSF) and were incubated at 25 °C for 20 min with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining to serve as a loading control and western blotting with an anti-Pup or an anti-PafA antibody. The asterisk denotes a cross-reactive band of the rabbit polyclonal anti-pup antibody.
Having confirmed the inhibitory effect of AEBSF on PafA using affinity-purified proteins (PanB and Mpa), we then tested the global effect of AEBSF on pupylation using total proteins (cell lysates) as the substrate. As many proteins in Mtb and M. smegmatis (Msm), a fast-growing non-pathogenic relative of Mtb used as a model organism for Mtb, are highly homologous, and cell lysates of Msm are more convenient to prepare, we performed this experiment using Msm cell lysates. We first constructed a Msm PafA knock-out strain (Msm △PafA) to knockout endogenous protein pupylation in Msm, as Msm PafA exhibits high overall sequence similarity to Mtb PafA (94% identity, 100% similarity) and is also active in pupylation. The total lysate of the Msm △PafA strain was then used to test the inhibitory effect of AEBSF on exogenously added Mtb PafA. As expected, no pupylation was observed in the lysate of Msm △PafA (Fig. 1c). Following incubation with Mtb PafA, we found that Mtb PafA efficiently pupylated many proteins in Msm △PafA cell lysates. Furthermore, PafA activity was inhibited by AEBSF in a dose dependent manner (Fig. 1c). In addition, we prepared purified Msm PafA and found that its pupylase activity could also be inhibited by AEBSF when the Msm △PafA cell lysates were used as the substrate (Fig. S2b). Overall, these findings indicate that AEBSF is an effective inhibitor of the pupylase activity of purified Mtb PafA.
3.2. AEBSF Covalently Binds to S119 of Mtb PafA
To identify the binding site of AEBSF in purified Mtb PafA, we incubated PafA with AEBSF and then performed liquid chromatography–mass spectrometry (LC-MS/MS) analysis. We found that S119 is the predominant residue covalently bound by AEBSF (Fig. 2a).
Fig. 2.

S119 is critical for Mtb PafA pupylase activity.
(a) PafA S119 was identified as an AEBSF binding site by LC-MS/MS analysis. PafA (0.5 μM) was incubated with AEBSF (0.5 mM) for 0.5 h in pupylation buffer at 25 °C, then trypsin digested and subjected to MS/MS analysis. A 183 Da molecular weight increase is predicted on AEBSF-mediated sulfonation.
(b) Pupylation reactions included PanB-Flag (8 μM), PupE (10 μM) and PafA variants (0.5 μM each) and were incubated at 25 °C for 6 h with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining and western blotting with an anti-flag antibody. Quantitation of the pupylation level of PanB was based on CBB staining and is shown in the lower panel. Data are representative of one experiment three independent biological replicates (mean and s.e.m. of n = 3 samples), *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
(c) Pupylation reactions included Mpa-Flag (6 μM), PupE (10 μM) and PafA variants (0.5 μM each) and were incubated at 25 °C for 2 h with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining and western blotting with an anti-flag antibody.
(d) Pupylation reactions included Msm △PafA lysates (10 μg), PupE (10 μM) and PafA variants (0.5 μM each) and were incubated at 25 °C for 20 min with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining to serve as a loading control and western blotting with an anti-Pup of anti-PafA antibody. The asterisk denotes a cross-reactive band of the rabbit polyclonal anti-pup antibody.
(e) Steady-state rates of PanB-Flag pupylation by WT PafA (black), S119A (blue) and S119F (red). Data are representative of three independent biological replicates (mean and s.e.m. of n = 3 samples). Data were fitted to the Michaelis-Menten equation.
We next investigated if S119 is critical for Mtb PafA pupylase activity. As AEBSF has a benzene ring, we speculated that the significant reduction in activity upon AEBSF binding may due to steric hindrance from the relatively large benzene ring. We suspected that mutation of serine to other aromatic amino acids, such as phenylalanine (F), tyrosine (Y), or tryptophan (W), that also have a benzene ring, might mimic the steric hindrance of AEBSF, and so constructed mutants S119F, S119Y and S119W. To determine the absolute requirement of this S119 in the pupylation reaction, we also investigated a mutation of S119 to alanine. Mutants of S115 (that does not interact with AEBSF) were used as controls. Strikingly, the S119F, S119Y and S119 W PafA mutants, similar to the well-known D57N PafA mutant that causes inactivation of PafA (Cerda-Maira et al., 2010), almost entirely lost their pupylase activity using PanB as the substrate (Fig. 2b; Fig. S3). Interestingly, PafA S119A exhibited approximately half of the pupylase activity of that of WT PafA or PafA S115 variants, which all exhibited similar activity as that of WT PafA (Fig. S3). We next examined the kinetic activity of the S119F, S119A and WT PafA using Michaelis-Menten assays and found that WT PafA exhibits a higher Vmax than that of the S119A and S119F mutants (Fig. 2e). It is noteworthy that the Km of WT PafA and S119A were similar, indicating that S119 is not directly involved in PanB binding.
We also found that PafA S119F was completely inactive in Mpa pupylation (Fig. 2c). To further confirm the effect of the S119F and S119A mutants, Msm △PafA total lysates were also used as substrates (mutant D57N being used as a control). Results indicated that the S119A mutant exhibited lower pupylase activity than WT PafA while the S119F mutant exhibited almost no pupylase activity in this assay (Fig. 2d).
Thus, taken together, these results indicate that AEBSF covalently attaches to S119, and that changes in the chemical nature of this residue, whether by AEBSF binding or by mutation to aromatic acids, can almost completely abolish the pupylase activity of purified Mtb PafA.
3.3. S119 is Critical for Mtb PafA Pupylase Activity in Bacterial Cells
To investigate whether PafA S119 contributes to Mtb PafA pupylase activity in bacterial cells, we performed experiments in model organism Msm which also has the Pup-proteasome system (Knipfer and Shrader, 1997, Imkamp et al., 2010a). Previous studies have shown that PafA is the only enzyme in mycobacteria that catalyzes protein pupylation (Pearce et al., 2008, Striebel et al., 2009). Consistent with this, we did not detect any bands in the Msm △PafA strain using an anti-pup antibody (Fig. 3). We transformed Msm △PafA with plasmids encoding different Flag-tagged PafAMtb variants (Table S2) under the control of a mycobacterial hsp60 promoter. PafAMtb expression in Msm △PafA almost completely restored the levels of pupylation to that of WT Msm. We found that Msm △PafA/pMV·PafAMtb S119F exhibited a very low level of pupylation while Msm △PafA/pMV·PafAMtb S119A exhibited similar pupylation level to that of the Msm △PafA/pMV·PafAMtb strain. These data are consistent with our observations on PafAMtb pupylase activity in the above biochemical experiments. Thus, we conclude that S119 contributes to Mtb PafA pupylase activity in bacterial cells.
Fig. 3.
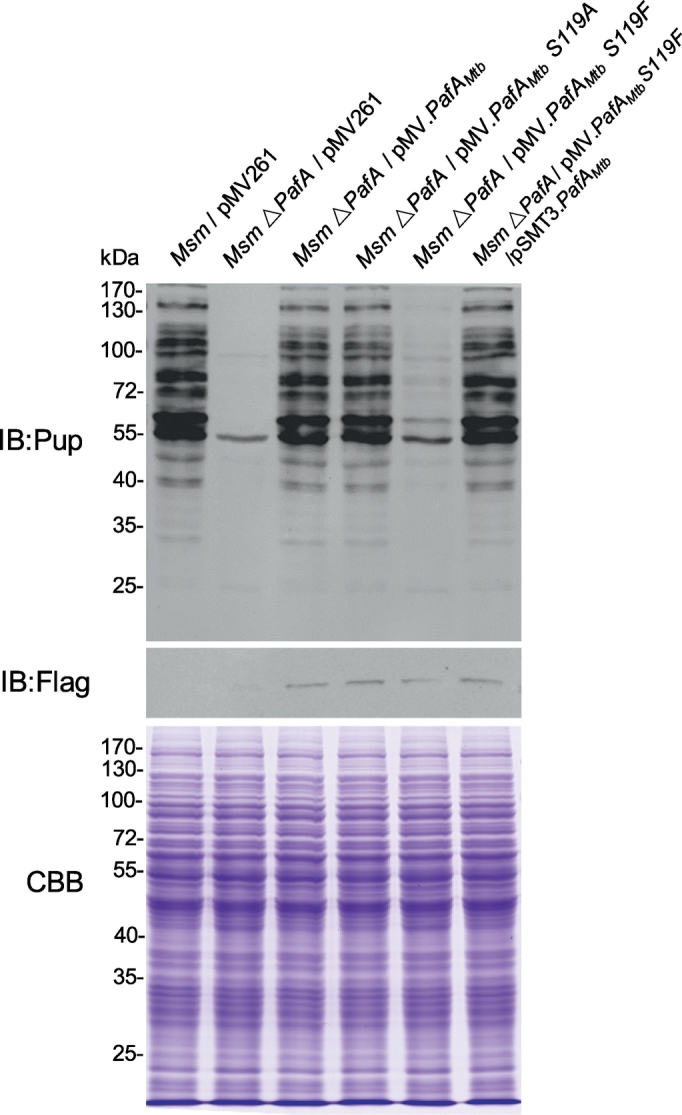
S119 is critical for Mtb PafA pupylase activity in bacterial cells.
WT Msm or Msm △PafA strains were transformed with plasmids expressing Flag-tagged Mtb PafA variants. Samples were collected at an OD600 of ~2, then analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-Pup or anti-Flag antibody.
3.4. Structural Basis for the Involvement of S119 on Mtb PafA Activity
To determine the mechanistic role of S119 in pupylation, we aligned the Mtb PafA amino acid sequence with PafA from representative actinomycetes and C. glutamicum (Cglu). We found that the sequences surrounding S119, including S119 itself, are highly conserved (Fig. 4a; Fig. S4a). Since there is no available crystal structure of PafAMtb, we examined the known structure of PafA from Cglu that exhibits high overall sequence similarity to PafAMtb (53% identity, 92% similarity).
Fig. 4.

Structural basis for the involvement of S119 in Mtb PafA activity.
(a) Alignment of residues 111–130 of Mtb PafA to that of PafA from C. glutamicum. Sequences were compiled from the National Center for Biotechnology Information server and aligned by means of ClustalW.
(b) Crystal structure of the C. glutamicum PafA/Pup complex. Pup (orange) wraps around PafA (cyan) via two small α-helices and a short C-terminal tail. Binding of Pup to substrate proteins occurs via its C-terminus. Also shown in stick representation are the S126 pocket residues and the S126-loop (purple) that define an extended interface of the Pup-binding groove of PafA.
(c) Close-up view of the S126-pocket of the WT, S126F and S126-AEBSF initial structures. Simply changing the S126 side chain to phenylalanine or attaching AEBSF results in significant steric overlap with other pocket resides. Thus, these modifications must disrupt the organization of this pocket. Phenylalanine is colored white and the carbon, oxygen, sulfur, nitrogen, and hydrogen atoms in the AEBSF-linked residue are colored cyan, red, yellow, blue, and white, respectively.
(d) Snap-shot of the equilibrium MD simulations of the WT and S126F PafACglu/Pup complexes, with the PafA proteins overlapped. The interaction of PafA with Pup is significantly weakened in S126F as a result of the disruption of the S126-pocket compared with the WT. This weakened interaction occurs at both the Pup C-terminal tail, which directly contacts the S126-loop, and its α-helices, which are distant from the S126-loop.
(e) BLI data for the binding of PupE to PafA variants or PafA pre-incubated with 2 mM AEBSF and their interaction kinetics. PupE was immobilized on streptavidin-coated biosensors and exposed to binding partners in buffer. Binding was measured by coincident changes in the interference pattern. The KD (nM), Kon (1/Ms) and Kdis (1/s) are shown in the table to the right.
(f) Pupylation reactions included free lysine (80 mM), PupE (10 μM) and PafA variants (0.5 μM each) and were incubated at 25 °C for 15 min with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining.
(g) As in Fig. 4f, except that PafA (0.5 μM) pre-incubated with AEBSF at 25 °C for 0.5 h was used instead of PafA variants.
Previous studies reported that the binding site for the C-terminal region of Pup is close to the N-terminal end of PafACglu β6 (Ozcelik et al., 2012, Barandun et al., 2013), which is close to the location of S126 (the homologous residue of PafAMtb S119). More specifically, the PafA-binding region of Pup consists of two small α-helices and a short C-terminal tail (Fig. 4b). Pup wraps around PafACglu, with Serine 126 located within a loop that defines one side of a groove on PafACglu that interacts with the Pup C-terminal tail (Fig. 4b). Interestingly, the S126 side-chain does not face into this groove but is instead buried within a pocket in the interior of PafACglu, where it makes close contact with Asn119, Val120, Arg128, and His188 (which, with S126, we will refer to as the “S126-pocket residues”) (Fig. 4c).
To determine how modification of S126 might perturb PafACglu pupylation, we examined its effects on the binding of Pup to PafACglu by performing all-atom molecular dynamics (MD) simulations. To this end, we studied equilibrium simulations of the structures of WT PafACglu, S126F and S126A mutants, and PafACglu to which AEBSF is covalently attached at S126 (S126-AEBSF), using the aforementioned Pup-PafACglu atomic model as the starting structure for the simulations. Strikingly, even before the simulations began, simply changing the S126 side-chain to that of phenylalanine or attaching AEBSF to the S126 side-chain hydroxyl group results in a significant steric overlap with S126-pocket residues, particularly Arg185 (Fig. 4c). Thus, this observation suggests that these modifications might disrupt the close-packed organization of the S126-pocket, and with it, the Pup-binding groove in PafACglu, which might then lead to weakened binding of Pup.
Indeed, extended simulations of these structures showed that, while the S126-pocket in WT PafACglu is highly stable, it collapses and is significantly more unstable in S126F and S126-AEBSF, S126F exhibiting similar significant fluctuation as in S126-AESBF, and to a lesser extent, in S126A (Fig. S4b, Movie S1). As a result, both the C-terminal region and the helices of Pup exhibit significant fluctuations in the S126F and S126-AEBSF structures, with smaller changes in S126A, while the PafACglu/Pup complex is quite stable (Fig. S4b, Movie S2). Thus, these results suggest that modifications to S126 perturb pupylation as a result of destabilization of this Pup-binding groove in PafA, thereby weakening the binding strength of Pup to PafA.
To test this suggestion directly, the strength of the interactions between Mtb PafA and Pup was examined using Bio-Layer Interferometry (BLI). We found that PafA S119A exhibits a slightly lower affinity for Pup than WT PafA, and that PafA S119F or PafA co-incubated with 2 mM AEBSF exhibited equilibrium dissociation constants (KD) that are twice as high as that of WT PafA (Fig. 4e). Therefore, these results support the notion that the inhibitory effect of AEBSF is due to defective Pup binding.
To obtain a better understanding of defective Pup binding in Mtb PafA S119 mutants and to exclude possible effects on protein-substrate binding, we examined the ability of Mtb PafA to pupylate free lysine under different conditions. Clearly, the docking of free lysine in the PafA active site does not require most of the residues involved in the interaction of PafA with protein targets (Regev et al., 2016). We found that PafA S119A shows a weakened pupylase activity on free lysine compared to that of the WT enzyme, while lysine pupylation by PafA S119F, S119Y and S119 W was undetectable (Fig. 4f). Pre-incubation of PafA with increasing concentrations of AEBSF resulted in a gradual loss of pupylation on free lysine (Fig. 4g).
Taken together, these results suggest that S119 plays a critical role in the Pup C-teminal binding pocket in Mtb PafA and that it can function as an effective inhibitor binding site.
3.5. S119 is Critical for Mtb PafA Depupylase Activity
Recently, Zhang et al. found that Mtb PafA also exhibits depupylase activity on pupylated proteins (Zhang et al., 2017). This depupylation activity is also expected to require a Pup-binding pocket to bind the pupylated protein. Thus, we speculated that S119 may also be critical for Mtb PafA depupylase activity.
To differentiate depupylation from pupylation, we performed pupylation and depupylation reactions in two different buffer systems (Zhang et al., 2017). The biggest difference between the two reactions is the concentration of PO43− (see Materials and Methods). We examined the depupylase activity of purified Mtb PafA variants for pupylated PanB. Consistent with this earlier work, we also found that PafA exhibits depupylase activity on pupylated PanB (Fig. 5a). Of note, PafA S119F exhibits almost no depupylase activity while the depupylase activity of PafA S119A is significantly reduced. Next, we examined the depupylase activity of PafA pre-incubated with AEBSF on pupylated PanB. As with pupylation, we observed that AEBSF inhibited depupylation in a dose-dependent manner (Fig. 5b). Thus, S119 also contributes to the depupylase activity of purified Mtb PafA.
Fig. 5.

S119 is critical for Mtb PafA depupylase activity.
(a) PanB-PupE (0.75 μM) were depupylated by PafA variants (1.5 μM each) with 5 mM ADP in phosphate buffer for 6 h at 25 °C. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-flag antibody. Quantitation of PanB-Flag based on CBB staining is shown in the lower panel. Data are representative of three independent biological replicates (mean and s.e.m. of n = 3 samples), *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
(b) As in Fig. 5a, except that the PafA (1.5 μM) pre-incubated with AEBSF at 25 °C for 0.5 h was used instead of PafA variants.
3.6. S119 is Critical for Function of Mtb PafA that Essential for Msm △PafA Survival Under Nitrogen Limitation and in Macrophages
To determine whether the defective activity of Mtb PafA S119 variants could cause phenotypic changes, Msm △PafA strains with different PafAMtb variants (Table S2) were tested under nitrogen limitation and also in macrophages. As shown above, the level of pupylation in the Msm △PafA strains is due to the introduced pupylase activity of the PafAMtb variants (Fig. 3). In addition, it is also known that the PPS plays an important role under nitrogen limitation (Elharar et al., 2014, Fascellaro et al., 2016). Thus, we expected the growth of these strains to be proportional to their pupylase activity under nitrogen limitation. Results demonstrated that this is indeed the case (Fig. 6a). Further, when we complemented WT PafAMtb in the PafAMtb S119F strain, bacterial colony-forming unit (CFU) numbers recovered to a level similar to that of the WT PafAMtb strain. These findings indicate that S119 is critical for the function of Mtb PafA, which itself plays an essential role in Msm △PafA survival under nitrogen limitation.
Fig. 6.

S119 is critical for the function of Mtb PafA that is essential for Msm △PafA survival under nitrogen limitation and in macrophages.
(a) Exponentially growing cultures (OD600 = 0.5) of Msm expressing PafAMtb with different mutations were centrifuged and resuspended in media lacking nitrogen sources. Following resuspension, aliquots were collected at the indicated time points for determination of live cell concentration (n = 3).
(b) Survival of Msm expressing PafAMtb with different mutations in primary human monocyte-derived macrophages infected for 0–48 h. Following macrophage disruption, aliquots were collected at the indicated time points for determination of live Msm concentration (n = 3). *P < 0.05 and **P < 0.01 (two-tailed unpaired t-test).
Considering that PPS is required for Mtb resistance to nitric oxide and is essential for Mtb survival in mice, we reasoned that the PPS pathway of Msm may also be required for Msm survival in macrophage cells (Darwin et al., 2003, Lamichhane et al., 2006, Gandotra et al., 2007, Samanovic et al., 2015). To test this, THP-1 macrophage cells were infected with Msm △PafA strains bearing different PafAMtb variants (Fig. 6b, Table S2). The survival of Msm strains in these cells was monitored at three time points, namely 12 h, 24 h, and 48 h. Markedly lower survival rates were observed in the Msm △PafA/pMV261 and Msm △PafA/pMV·PafAMtb S119F strains, but not in the Msm △PafA/pMV·PafAMtb and Msm △PafA/pMV·PafAMtb S119A strains, compared to the WT Msm strain at all the three time points. In addition, when PafAMtb was re-introduced into the Msm △PafA and Msm △PafA/pMV·PafAMtb S119F strains, these strains recovered their survival ability to a level similar to that of WT Msm.
We noted that under “Nitrogen limitation” conditions, the bacteria grew ~ 2 logs more after 3 days, while in THP-1 cells, most bacterial strains died by 48 h. We also noted that Msm grew much slower under the “Nitrogen limitation” condition than under normal conditions, and our results were consistent with others (Elharar et al., 2014, Fascellaro et al., 2016). We used a “Nitrogen limitation” condition to partially mimic the stress environment that Mtb faces in macrophage cells. However, actual pressures inside macrophages are much more complicated, and may arise from reactive nitrogen intermediates, reactive oxygen intermediates, nutrient restriction and other factors. The difference in Msm survival and growth between the “Nitrogen limitation” condition and macrophage cells may be due to the different stresses that the bacteria are facing.
Thus, we conclude that S119 is critical for the function of Mtb PafA which is itself essential for Msm △PafA survival in macrophages.
4. Discussion
In this study, we identified Mtb PafA S119 as an effective inhibitor binding site using the serine protease inhibitor, AEBSF. We found that purified PafA variants of S119 mutated to aromatic amino acids, mimicking AEBSF-binding, present no or only very low catalytic activity in pupylation and depupylation reactions. Furthermore, Mtb PafA S119 variants with defective enzymatic activity could not compensate for the significant growth defects of Msm △PafA under nitrogen limitation and in macrophages.
Guided by the PafAcglu crystal structure (Ozcelik et al., 2012, Barandun et al., 2013), we found that Serine 126 of PafAcglu, the homologous residue of PafAMtb S119, is a highly conserved residue that is located close to the groove that binds the Pup C-terminal. MD simulations of PafA mutants or with AEBSF covalently attached to PafA clearly revealed disruption of this binding groove and a weakening of the interaction with both the C-terminal tail and α- helices of Pup. These predictions were verified using BLI, which showed that Pup-binding of PafA S119 variants or of PafA co-incubated with AEBSF was significantly reduced compared to that of WT PafA. These results are also consistent with the clearly defective pupylation of lysine, whose binding does not involve substantial contact with PafA (Regev et al., 2016), by PafA S119 variants or PafA co-incubated with AEBSF, as compared to that of WT PafA. Thus, although we cannot completely rule out that the reduced pupylation may result from a catalytic defect, our data strongly support the proposal that the inhibitory effect of the interference of the PafA S119-pocket, a non-active site location, is due to a reduced binding of Pup. There are a number of advantages for non-active site-based inhibitor design. One main advantage is avoidance of direct binding competition with substrates of high affinity which often result in off-target effects (Nussinov and Tsai, 2013). In this respect, we suggest that attempts to interfere with the PafA S119-pocket should be an attractive approach for acquiring a highly efficient inhibitor for PafA.
Recently, Darwin et al. found that PafAMtb possesses depupylase activity in vitro in addition to its pupylase activity (Zhang et al., 2017). Here, we propose that the Pup binding pocket participates in bifunctional catalytic activities. Considering our findings and the structure base of PafA S119, we speculate that S119 is also critical for the depupylase activity of Mtb PafA. Indeed, PafA S119F almost completely lost depupylase activity and PafA S119A lost the majority of its depupylase activity. As the crystal structure of PafAMtb is not yet available, our findings may pave the way to reveal the depupylase mechanism of PafAMtb.
MS analysis showed that AEBSF reacts with the hydroxy group of the Mtb PafA S119 residue to form a sulfonyl derivative. However, as AEBSF is a general serine protease inhibitor and may bind to many proteins, it can not be treated as an anti-Mtb lead compound and tested in vivo directly. Potential strategies could be applied to enhance the selectivity and sensitivity of AEBSF for PafA, for example, by modifying the reactive side chain but retaining the sulfonyl fluoride of AEBSF. There are many successful cases of other inhibitor/enzyme complexes that have followed this strategy, for example, the enhancement of the specificity of the inhibitor for an intracellular lipid binding protein (Chen et al., 2016). Alternatively, since PafA does not have a canonical serine protease active site, it should be possible to screen or rationally design molecules that would interact with S119 but not conventional serine proteases. Such site-specific inhibitor design has been proven to be a successful strategy (Ostrem et al., 2013, Ostrem and Shokat, 2016, Dawidowski et al., 2017), including in the development of mutant-specific RAS inhibitors (Stephen et al., 2014, Ostrem and Shokat, 2016, Janes et al., 2018). In this case, to discriminate the inhibition of both the WT and the mutant KRAS, Shokat et al. developed a series of site-based inhibitors that covalently attach to the mutant cysteine residue of KARS (Ostrem et al., 2013). The cysteine acting as a nucleophile to form a covalent bond with the inhibitors is reminiscent of the interaction of AEBSF with serine observed here in PafA.
Most of the assays performed in this study were at the biochemical or bacteria cell levels. To strengthen the clinical significance of the major finding that S119 is critical for Mtb PafA, it will be important in future to carry out some in vivo assays, especially with the Mtb PafA mutants, using pathogenic Mtb strains. Since PafA (especially PafA S119) is highly conserved between Mtb and Msm, it is reasonable to argue that what we observed using Msm will be similar in Mtb. The role of PafA in Mtb virulence has already been demonstrated in vitro and in vivo (Darwin et al., 2003, Festa et al., 2007, Samanovic et al., 2015). When Mtb PafA activity is significantly disrupted by the interruption targeting S119 location, it is anticipated that the virulence of Mtb will be reduced dramatically in vivo. In addition, in an on-going study based on the findings of this study, we have already identified several compounds using a computer assisted drug screening strategy that targets the S119 location, which show ~20 μM efficacy on BCG and H37Ra (data not shown). To further strengthen the clinical potential of our findings, the best way is to screen compounds targeting PafA S119 and validate the compounds that we have already identified in vivo using a pathogenic Mtb strain, or even MDR/XDR strains. However, performing such studies are not currently feasible and out of the scope of the present study.
Taken together, our results demonstrate that S119 could serve as a promising precise target for developing inhibitors of M. tuberculosis PafA. Both pupylase and depupylase activities of purified PafA can be significantly inhibited upon modification of S119, and these modifications profoundly affect the growth of the bacteria under conditions important for its survival. Our results provide clear direction for developing inhibitors of Mtb PafA, thus holding promise for developing new compounds for combating Mtb.
The following are the supplementary data related to this article.
A zoomed-in view of the changes within the S126-pocket during simulations of the WT (left), S126F (middle), and S126-AEBSF (right) proteins. The pocket residues are in stick representation and are colored as in Fig. 4c. The C-terminal tail for the three complexes is colored orange, red, and pink, respectively.
A comparison of the WT and S126F structures, showing the weakened binding of Pup with S126F PafACglu. The PafA/Pup complexes are colored cyan/orange and yellow/red in the WT and S126F structures, respectively.
Inhibitor profile of Mtb PafA pupylase activity.
PanB-Flag (8 μM) was pupylated by PafA (0.5 μM) and PupE (10 μM) at 25 °C for 6 h in pupylation buffer followed by incubation of PafA with the indicated inhibitors at 25 °C for 0.5 h. Samples were collected and analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-Pup or an anti-Flag antibody.
AEBSF is an inhibitor of PafA pupylase activity.
(a) PafA was incubated with serially diluted AEBSF, i.e., 2, 0.5 (mM) at 25 °C for 0.5 h in pupylation buffer followed by dialysis to remove any unbound inhibitor. Pupylation reactions included PanB-Flag (8 μM), PupE (10 μM) and PafA (0.5 μM; pre-incubated with AEBSF) and were incubated at 25 °C for 6 h in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-flag antibody.
(b) Pupylation reactions included Msm △PafA lysates (10 μg), PupE (10 μM) and PafAMsm (0.5 μM each) (0.5 μM) and were incubated at 25 °C for 20 min with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining to serve as a loading control and western blotting with an anti-Pup or anti-PafA antibody.
S119 is critical for Mtb PafA pupylase activity.
As in Fig. 1a, except that PafA variants (0.5 μM each) were used instead of the PafA pre-incubated with AEBSF.
Structural basis for the involvement of S119 in Mtb PafA activity.
(a) Alignment of residues 111–130 of M. tuberculosis PafA to that of PafA from representative Actinomycetes. Sequences were compiled from the National Center for Biotechnology Information server and aligned by means of ClustalW.
(b) Changes in the structure of localized regions of the PafACglu/Pup complex observed during MD simulations. The root-mean-squared-deviation (RMSD) of each region (labeled at the top of each panel) throughout the trajectories is shown in each panel. As shown in the top left panel, the modifications significantly change the structure and stability of the S126-pocket residues (as evident from the fluctuations). The top right panel shows the significantly decreased stability of the Pup structure in the modified PafA proteins, while the Pup structure in the WT complex is very stable. The bottom two panels show that the weakened interaction of Pup with PafA in the modified proteins occurs in both the Pup C-terminal tail and the Pup helices.
(c) The entire gel of Fig. 4f is shown.
(d) The entire gel of Fig. 4g is shown.
Supplementary material
Funding Sources
This study was supported in part by the National Key Research and Development Program of China Grant 2016YFA0500600, National Natural Science Foundation of China Grants 31670831, 31370813, 31370750 and 31670722, Shanghai Jiao Tong University Grant 16X120030015.
Conflicts of Interest
The authors declare no conflicts of interest.
Author Contributions
S.C.T. conceived the idea. H.W.J. performed enzyme activity assays and functional analysis. X.D.W., H.N.Z. and T.W. performed protein purification. J.B.W. and D.M.C. performed Equilibrium MD simulations. C.X.L., F.L.W., X.H., Z.W.X., H.C., S.J.G. and Y.L. prepared the Figures with the help of L.J.B, J.Y.D., J.X., J.F.P. and X.E.Z. H.W.J. and S.C.T. wrote the manuscript.
Acknowledgements
We thank Prof. Chuanyou Li of Beijing Chest Hospital for providing the Msm △PafA strain, Dr. Joy Fleming for editing, and Dr. Jing-li Hou of the Instrumental Analysis Center of Shanghai Jiao Tong University for her kind help with Ultra Performance LC & Q-TOF MS (UPLC-MS) analysis.
References
- Barandun J., Delley C.L., Ban N., Weber-Ban E. Crystal structure of the complex between prokaryotic ubiquitin-like protein and its ligase PafA. J. Am. Chem. Soc. 2013;135:6794–6797. doi: 10.1021/ja4024012. [DOI] [PubMed] [Google Scholar]
- Bode N.J., Darwin K.H. The pup-proteasome system of mycobacteria. Microbiol. Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0008-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns K.E., Liu W.T., Boshoff H.I.M., Dorrestein P.C., Barry C.E. Proteasomal protein degradation in mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J. Biol. Chem. 2009;284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns K.E., Cerda-Maira F.A., Wang T., Li H., Bishai W.R., Darwin K.H. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol. Cell. 2010;39:821–827. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda-Maira F.A., Pearce M.J., Fuortes M., Bishai W.R., Hubbard S.R., Darwin K.H. Molecular analysis of the prokaryotic ubiquitin-like protein (pup) conjugation pathway in Mycobacterium tuberculosis. Mol. Microbiol. 2010;77:1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.T., Dong J.J., Plate L., Mortenson D.E., Brighty G.J., Li S.H., Liu Y., Galmozzi A., Lee P.S., Hulce J.J., Cravatt B.F., Saez E., Powers E.T., Wilson I.A., Sharpless K.B., Kelly J.W. Arylfluorosulfates inactivate intracellular lipid binding protein(s) through chemoselective SuFEx reaction with a binding site Tyr residue. J. Am. Chem. Soc. 2016;138:7353–7364. doi: 10.1021/jacs.6b02960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Pieters J. Novel proteasome inhibitors as potential drugs to combat tuberculosis. J. Mol. Cell Biol. 2010;2:173–175. doi: 10.1093/jmcb/mjp053. [DOI] [PubMed] [Google Scholar]
- Clements G.V., Yepikhin A.S., Bashoff H.I., Dowd C.S. Mycobacterium tuberculosis proteasome inhibitors. Abstr. Pap. Am. Chem. Soc. 2013:245. [Google Scholar]
- Darwin K.H., Ehrt S., Gutierrez-Ramos J.C., Weich N., Nathan C.F. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- Darwin K.H., Lin G., Chen Z.Q., Li H.L., Nathan C.F. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 2005;55:561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- Dawidowski M., Emmanouilidis L., Kalel V.C., Tripsianes K., Schorpp K., Hadian K., Kaiser M., Maser P., Kolonko M., Tanghe S., Rodriguez A., Schliebs W., Erdmann R., Sattler M., Popowicz G.M. Inhibitors of PEX14 disrupt protein import into glycosomes and kill Trypanosoma parasites. Science. 2017;355:1416. doi: 10.1126/science.aal1807. [DOI] [PubMed] [Google Scholar]
- Delley C.L., Striebel F., Heydenreich F.M., Ozcelik D., Weber-Ban E. Activity of the mycobacterial proteasomal ATPase Mpa is reversibly regulated by pupylation. J. Biol. Chem. 2012;287:7907–7914. doi: 10.1074/jbc.M111.331124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elharar Y., Roth Z., Hermelin I., Moon A., Peretz G., Shenkerman Y., Vishkautzan M., Khalaila I., Gur E. Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J. 2014;33:1802–1814. doi: 10.15252/embj.201387076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fascellaro G., Petrera A., Lai Z.W., Nanni P., Grossmann J., Burger S., Biniossek M.L., Gomez-Auli A., Schilling O., Imkamp F. Comprehensive proteomic analysis of nitrogen-starved Mycobacterium smegmatis Deltapup reveals the impact of Pupylation on nitrogen stress response. J. Proteome Res. 2016;15:2812–2825. doi: 10.1021/acs.jproteome.6b00378. [DOI] [PubMed] [Google Scholar]
- Festa R.A., Pearce M.J., Darwin K.H. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J. Bacteriol. 2007;189:3044–3050. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandotra S., Schnappinger D., Monteleone M., Hillen W., Ehrt S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 2007;13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel D. Bedaquiline: a novel drug to combat multiple drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 2014;5:76–78. doi: 10.4103/0976-500X.124435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabbe C., Husnjak K., Dikic I. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth E., Thommen M., Weber-Ban E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J. Biol. Chem. 2011;286:4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A., Varshavsky A. The ubiquitin system. Nat. Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- Hoagland D.T., Liu J., Lee R.B., Lee R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 2016;102:55–72. doi: 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(33–8):27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Imkamp F., Rosenberger T., Striebel F., Keller P.M., Amstutz B., Sander P., Weber-Ban E. Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol. Microbiol. 2010;75:744–754. doi: 10.1111/j.1365-2958.2009.07013.x. [DOI] [PubMed] [Google Scholar]
- Imkamp F., Striebel F., Sutter M., Ozcelik D., Zimmermann N., Sander P., Weber-Ban E. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 2010;11:791–797. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imkamp F., Ziemski M., Weber-Ban E. Pupylation-dependent and -independent proteasomal degradation in mycobacteria. Biomol Concepts. 2015;6:285–301. doi: 10.1515/bmc-2015-0017. [DOI] [PubMed] [Google Scholar]
- Janes M.R., Zhang J., Li L.S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., Feng J., Chen J.H., Li S., Li S., Long Y.O., Thach C., Liu Y., Zarieh A., Ely T., Kucharski J.M., Kessler L.V., Wu T., Yu K., Wang Y., Yao Y., Deng X., Zarrinkar P.P., Brehmer D., Dhanak D., Lorenzi M.V., Hu-Lowe D., Patricelli M.P., Ren P., Liu Y. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–589. doi: 10.1016/j.cell.2018.01.006. e17. [DOI] [PubMed] [Google Scholar]
- Knipfer N., Shrader T.E. Inactivation of the 20S proteasome in Mycobacterium smegmatis. Mol. Microbiol. 1997;25:375–383. doi: 10.1046/j.1365-2958.1997.4721837.x. [DOI] [PubMed] [Google Scholar]
- Lamichhane G., Raghunand T.R., Morrison N.E., Woolwine S.C., Tyagi S., Kandavelou K., Bishai W.R. Deletion of a Mycobacterium tuberculosis proteasomal ATPase homologue gene produces a slow-growing strain that persists in host tissues. J. Infect. Dis. 2006;194:1233–1240. doi: 10.1086/508288. [DOI] [PubMed] [Google Scholar]
- Lin G., Li D.Y., De Carvalho L.P.S., Deng H.T., Tao H., Vogt G., Wu K.Y., Schneider J., Chidawanyika T., Warren J.D., Li H.L., Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–663. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G., Li D., Chidawanyika T., Nathan C., Li H. Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch. Biochem. Biophys. 2010;501:214–220. doi: 10.1016/j.abb.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G., Chidawanyika T., Tsu C., Warrier T., Vaubourgeix J., Blackburn C., Gigstad K., Sintchak M., Dick L., Nathan C. N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J. Am. Chem. Soc. 2013;135:9968–9971. doi: 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., Joseph-Mccarthy D., Kuchnir L., Kuczera K., Lau F.T., Mattos C., Michnick S., Ngo T., Nguyen D.T., Prodhom B., Reiher W.E., Roux B., Schlenkrich M., Smith J.C., Stote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mdluli K., Kaneko T., Upton A. The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harb. Perspect. Med. 2015;5 doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Tsai C.J. Allostery in disease and in drug discovery. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Ofer N., Forer N., Korman M., Vishkautzan M., Khalaila I., Gur E. Allosteric transitions direct protein tagging by PafA, the prokaryotic ubiquitin-like protein (pup) ligase. J. Biol. Chem. 2013;288:11287–11293. doi: 10.1074/jbc.M112.435842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J.M., Shokat K.M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016;15:771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik D., Barandun J., Schmitz N., Sutter M., Guth E., Damberger F.F., Allain F.H., Ban N., Weber-Ban E. Structures of pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat. Commun. 2012;3:1014. doi: 10.1038/ncomms2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce M.J., Arora P., Festa R.A., Butler-Wu S.M., Gokhale R.S., Darwin K.H. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25:5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce M.J., Mintseris J., Ferreyra J., Gygi S.P., Darwin K.H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev O., Korman M., Hecht N., Roth Z., Forer N., Zarivach R., Gur E. An extended loop of the pup ligase, PafA, mediates interaction with protein targets. J. Mol. Biol. 2016;428:4143–4153. doi: 10.1016/j.jmb.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Samanovic M.I., Tu S., Novak O., Iyer L.M., Mcallister F.E., Aravind L., Gygi S.P., Hubbard S.R., Strnad M., Darwin K.H. Proteasomal control of cytokinin synthesis protects Mycobacterium tuberculosis against nitric oxide. Mol. Cell. 2015;57:984–994. doi: 10.1016/j.molcel.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen A.G., Esposito D., Bagni R.K., Mccormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Striebel F., Imkamp F., Sutter M., Steiner M., Mamedov A., Weber-Ban E. Bacterial ubiquitin-like modifier pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 2009;16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- Striebel F., Hunkeler M., Summer H., Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter M., Striebel F., Damberger F.F., Allain F.H., Weber-Ban E. A distinct structural region of the prokaryotic ubiquitin-like protein (pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett. 2009;583:3151–3157. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Sutter M., Damberger F.F., Imkamp F., Allain F.H., Weber-Ban E. Prokaryotic ubiquitin-like protein (pup) is coupled to substrates via the side chain of its C-terminal glutamate. J. Am. Chem. Soc. 2010;132:5610–5612. doi: 10.1021/ja910546x. [DOI] [PubMed] [Google Scholar]
- Totaro K.A., Barthelme D., Simpson P.T., Jiang X., Lin G., Nathan C.F., Sauer R.T., Sello J.K. Rational design of selective and bioactive inhibitors of the Mycobacterium tuberculosis proteasome. ACS Infect. Dis. 2017;3:176–181. doi: 10.1021/acsinfecdis.6b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Li H., Lin G., Tang C.Y., Li D.Y., Nathan C., Darwin K.H., Li H.L. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure. 2009;17:1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Darwin K.H., Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat. Struct. Mol. Biol. 2010;17:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . 2016. Global Tuberculosis Report 2016. [Google Scholar]
- Yau R., Rape M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016;18:579–586. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- Yu W., He X., Vanommeslaeghe K., Mackerell A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Burns-Huang K.E., Janssen G.V., Li H., Ovaa H., Hedstrom L., Darwin K.H. Mycobacterium tuberculosis proteasome accessory factor a (PafA) can transfer prokaryotic ubiquitin-like protein (pup) between substrates. MBio. 2017:8. doi: 10.1128/mBio.00122-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Jiang X., Gao F., Song J., Sun J., Wang L., Sun X., Lu Z., Zhang H. Identification of plant-derived natural products as potential inhibitors of the Mycobacterium tuberculosis proteasome. BMC Complement. Altern. Med. 2014;14:400. doi: 10.1186/1472-6882-14-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Nahid P., Cole S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A zoomed-in view of the changes within the S126-pocket during simulations of the WT (left), S126F (middle), and S126-AEBSF (right) proteins. The pocket residues are in stick representation and are colored as in Fig. 4c. The C-terminal tail for the three complexes is colored orange, red, and pink, respectively.
A comparison of the WT and S126F structures, showing the weakened binding of Pup with S126F PafACglu. The PafA/Pup complexes are colored cyan/orange and yellow/red in the WT and S126F structures, respectively.
Inhibitor profile of Mtb PafA pupylase activity.
PanB-Flag (8 μM) was pupylated by PafA (0.5 μM) and PupE (10 μM) at 25 °C for 6 h in pupylation buffer followed by incubation of PafA with the indicated inhibitors at 25 °C for 0.5 h. Samples were collected and analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-Pup or an anti-Flag antibody.
AEBSF is an inhibitor of PafA pupylase activity.
(a) PafA was incubated with serially diluted AEBSF, i.e., 2, 0.5 (mM) at 25 °C for 0.5 h in pupylation buffer followed by dialysis to remove any unbound inhibitor. Pupylation reactions included PanB-Flag (8 μM), PupE (10 μM) and PafA (0.5 μM; pre-incubated with AEBSF) and were incubated at 25 °C for 6 h in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or western blotting with an anti-flag antibody.
(b) Pupylation reactions included Msm △PafA lysates (10 μg), PupE (10 μM) and PafAMsm (0.5 μM each) (0.5 μM) and were incubated at 25 °C for 20 min with 5 mM ATP in pupylation buffer. Samples were analyzed by SDS-PAGE, followed by CBB staining to serve as a loading control and western blotting with an anti-Pup or anti-PafA antibody.
S119 is critical for Mtb PafA pupylase activity.
As in Fig. 1a, except that PafA variants (0.5 μM each) were used instead of the PafA pre-incubated with AEBSF.
Structural basis for the involvement of S119 in Mtb PafA activity.
(a) Alignment of residues 111–130 of M. tuberculosis PafA to that of PafA from representative Actinomycetes. Sequences were compiled from the National Center for Biotechnology Information server and aligned by means of ClustalW.
(b) Changes in the structure of localized regions of the PafACglu/Pup complex observed during MD simulations. The root-mean-squared-deviation (RMSD) of each region (labeled at the top of each panel) throughout the trajectories is shown in each panel. As shown in the top left panel, the modifications significantly change the structure and stability of the S126-pocket residues (as evident from the fluctuations). The top right panel shows the significantly decreased stability of the Pup structure in the modified PafA proteins, while the Pup structure in the WT complex is very stable. The bottom two panels show that the weakened interaction of Pup with PafA in the modified proteins occurs in both the Pup C-terminal tail and the Pup helices.
(c) The entire gel of Fig. 4f is shown.
(d) The entire gel of Fig. 4g is shown.
Supplementary material