

Abstract
Receptor-interacting protein 3 (Ripk3)-mediated necroptosis contributes to cardiac ischaemia-reperfusion (IR) injury through poorly defined mechanisms. Our results demonstrated that Ripk3 was strongly upregulated in murine hearts subjected to IR injury and cardiomyocytes treated with LPS and H2O2. The higher level of Ripk3 was positively correlated to the infarction area expansion, cardiac dysfunction and augmented cardiomyocytes necroptosis. Function study further illustrated that upregulated Ripk3 evoked the endoplasmic reticulum (ER) stress, which was accompanied with an increase in intracellular Ca2+ level ([Ca2+]c) and xanthine oxidase (XO) expression. Activated XO raised cellular reactive oxygen species (ROS) that mediated the mitochondrial permeability transition pore (mPTP) opening and cardiomyocytes necroptosis. By comparison, genetic ablation of Ripk3 abrogated the ER stress and thus blocked the [Ca2+]c overload-XO-ROS-mPTP pathways, favouring a pro-survival state that ultimately resulted in the inhibition of cardiomyocytes necroptosis in the setting of cardiac IR injury. In summary, the present study helps to elucidate how necroptosis is mediated by ER stress, via the calcium overload /XO/ROS/mPTP opening axis.
Keywords: Necroptosis, Ripk3, XO, ROS, mPTP, ER stress
Graphical abstract
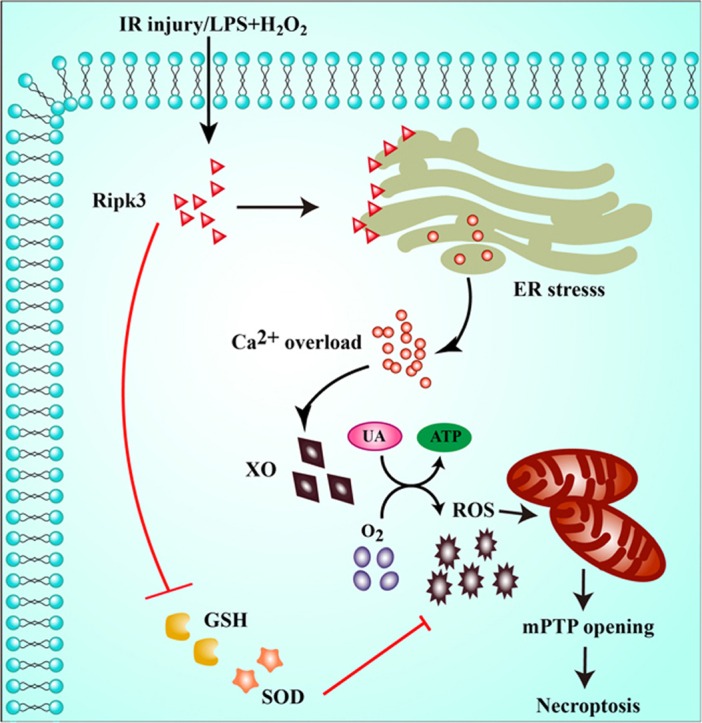
Ripk3 was strongly upregulated in murine hearts subjected to IR injury and cardiomyocytes treated with LPS and H2O2. The upregulated Ripk3 may evoke the ER stress, which was accompanied with intracellular calcium overload and XO expression. Activated XO raised cellular reactive oxygen species (ROS) that mediated the mPTP and cardiomyocytes necroptosis.
Highlights
-
•
ER stress is activated by Ripk3 in cardiac IR injury.
-
•
ER stress induces calcium overload which triggers XO-dependent ROS overproduction.
-
•
ROS outburst promotes mPTP opening that accounts for the necroptosis.
-
•
Inhibiting ER stress favors cardiomyocytes survival and protects cardiac function.
1. Introduction
The percutaneous coronary intervention (PCI) is the primary treatment strategy for acute myocardial infarction (AMI). It works via dredging blocked vessels and restoring cardiac circulation, which significantly reduces cardiovascular mortality in patients with AMI [1], [2]. Although PCI opens the blocked coronary vessels, however, it also causes reperfusion damage to the heart, referred to as ischaemia-reperfusion (IR) injury [3], in approximately 30% of patients with AMI, per our clinical studies [4], [5]. IR injury aggravates the irreversible cardiomyocyte death and infarcted zone expansion, predisposing AMI patients to adverse left-ventricular remodeling and poor clinical prognosis [6]. Therefore, investigations into the molecular basis of IR-mediated cardiomyocytes may open a pathway to new treatment modalities, which are desperately needed for cardiac IR-injury treatment in clinical practice.
Necroptosis is now recognized as an important contributor to necrotic damage in cardiac IR injury. Necroptosis is considered a subtype of necrosis, morphologically indistinguishable from other types but defined by a specific mode of pathway activation [7]. At the molecular level, necroptosis is reported to be regulated by Ripk3 [8]. However, the sequence of events downstream of Ripk3 that ultimately transmits the necroptotic signal to cardiomyocytes is still muddled in the case of reperfusion injury. An association between Ripk3 upregulation and mPTP opening has been illustrated in several studies, which confirms the necessity of mPTP for Ripk3-mediated necroptosis [9], [10]. One consequence of mPTP opening is mitochondrial potential collapse, oxidative phosphorylation attest, and ATP undersupply; following this, the energy shortage induces cardiomyocytes to swell and rupture. These findings indicate that necroptosis is initiated by Ripk3 and executed by mPTP opening. However, the question still needs to be answered how Ripk3 regulates mPTP opening, especially in the cardiac IR-injury setting.
Many previous studies have found that ROS overproduction contributes to mPTP opening via mitochondrial ATP-sensitive potassium channels [11], [12] and voltage-dependent anion channel-1 (VDAC1) oligomerization [13], suggesting that ROS outburst functions as the upstream molecular mechanism for controlling mPTP opening and consequent cellular necroptosis. In addition, recent work by our team showed that IR-mediated ROS generation was primarily due to the upregulation of xanthine oxidase (XO) [14], [15]. Under hypoxia, cellular ATP was gradually decomposed to ADP, AMP, adenosine, inosine, and hypoxanthine, whereas reoxygenation simultaneously promoted the metabolism of hypoxanthine to uric acid by XO and evoked excessive electrons to bind to oxygen and other electron acceptors, ultimately generating excessive ROS [16]. However, it remains unknown whether XO-induced ROS promotes cellular necroptosis via mPTP opening in response to IR injury.
Notably, our early description confirmed that XO activation and upregulation are signalled by cytoplasmic Ca2+ ([Ca2+]c) overload resulting from ER dysfunction. This indicates that endoplasmic reticulum (ER)-Ca2+ balance is the key determinant of ROS levels and mPTP opening rate. Interestingly, other studies have noted the potential association between ER stress and Ripk3. ER-related heat-shock protein 90 (HSP90) is the upstream activator of Ripk3 [17], [18], and the dissociation of HSP90 from its cochaperone CDC37 leads to the inhibition of Ripk3-dependent necroptosis [19]. Moreover, HSP90 activity is required for MLKL oligomerization and membrane translocation and the induction of necroptotic cell death [20]. These studies and our previous study show that Ripk3-mediated necroptosis may be associated with ER stress in cardiac IR injury. Accordingly, the aim of the present study is to explore the role of ER stress in Ripk3-triggered necroptosis, with a focus on calcium overload, oxidative stress, and mPTP opening.
2. Materials and methods
2.1. Animal and cardiac IR injury in vivo
The present study was approved by the PLA General Hospital Institutional Animal Care and Use Committee. Ripk3-/- mice with a C57BL/6 background were generated as previously described [21]. IR surgery was performed on wild type (WT) and Ripk3-/- male mice (12-wk-old, n = 140) in accordance with a previous study [22]. The mice were anaesthetized with isoflurane, and the heart was exposed via a left thoracotomy. Next, a 7–0 silk suture was tied around the left anterior descending coronary artery (LAD) with a slipknot and left for 45 min to induce ischaemia. This was followed by 0–24 h of reperfusion. Pravastatin (5 mg/kg, Sigma-Aldrich, USA) and tunicamycin (2 mg/kg, Sigma-Aldrich, USA) were administered intraperitoneally 1 h before IRI [23]. After the reperfusion period, a blood sample was collected for LDH, troponin T, and CK-MB measurements, as previously described [24]. The infarct size was measured by 2% Evans blue and 1% TTC, following the manufacturer's instructions.
2.2. Electron microscopy and echocardiography
Electron microscopy was conducted as previously described [5]. After treatment, the samples were dehydrated using acetonitrile and graded methanol, embedded in epoxy resin (EMbed-812; Electron Microscopy Sciences, USA) and polymerized at 70 °C overnight. Hitachi H600 Electron Microscope (Hitachi, Japan) was used to capture the images. The samples were imaged using a Hitachi H600 Electron Microscope (Hitachi, Japan). At least 30 cells in at least 5 randomly selected fields were observed. Echocardiography was performed in all mice at 6 h after reperfusion by echocardiogram (14.0 MHz, Sequoia C512; Acuson, Germany).
2.3. Cardiomyocytes isolation, cells culture and IR injury induction in vitro
Cardiomyocytes were isolated from the hearts of Ripk3-/- and WT mice using the enzyme dissociation method as previously described [25]. Cardiomyocytes were cultured in DMEM (high glucose, Gibco) containing 20% foetal bovine serum (FBS, HyClone, USA) at 37 °C with 5% CO2 and 95% air. At 70–80% confluence, the cells were incubated with LPS (10 µg/ml) and H2O2 (0.1 mM) in DMEM to induce IR injury. Tunicamycin (100 nM, Sigma-Aldrich, USA) and pravastatin (10 µM, Sigma-Aldrich, USA), the agonist and antagonist for ER stress, respectively [26], were administered to pretreat cardiomyocytes for 12 h before LPS and H2O2 stimulation. Febuxostat (10 µM, Selleck, USA), the inhibitor of XO, was administered to pretreat cardiomyocytes for 12 h before LPS and H2O2 stimulation.
2.4. Cell shortening and relengthening
The mechanical properties of myocytes were assessed using a SoftEdge Myocam system (IonOptix, Milton, MA). SoftEdge software was used to capture changes in cardiomyocyte length during shortening and re-lengthening. The myocytes were photographed with a MyoCam camera and displayed on a computer monitor. Cell shortening and re-lengthening were assessed using the following indices: peak shortening (PS), the amplitude myocytes shortened on electrical stimulation, which is indicative of peak ventricular contractility; time-to-PS (TPS), the duration of myocyte shortening, which indicates duration of contraction; time-to-90% relengthening (TR90), which represents the duration of cardiomyocyte relaxation (90% was used rather than 100% to avoid a noisy signal at baseline concentration), and maximal velocity of shortening (+ dL/dt) and relengthening (− dL/dt), the maximal slope (derivative) of the shortening and relengthening phases, which indicate the maximal velocities of rise and fall of ventricular pressure.
2.5. Cellular necroptosis detection
Cell suspension was seeded in 6-well cell-culture plates at a density of 106 cells/well and incubated at 37 °C for 24 h. After treatment, the cells were treated with 0.4% trypan blue, a vital stain used to selectively color dead cells blue. Trypan-blue–positive cell images were acquired and analysed using a fluorescence microscope. To discriminate cellular necroptosis, the samples were simultaneously stained with 2 µL of calcein AM working solution and 4 µL of 2-mM ethidium homodimer-1 for 15–20 min at room temperature using the LIVE/DEAD Viability/Cytotoxicity Kit (L3224, Molecular Probes, USA).
Cellular necroptosis was also detected quantitatively using the FITC Annexin V Apoptosis Detection Kit (556547, BD Bioscience). The cells were stained with 5 µL of FITC Annexin V and PI and incubated for 15 min at RT (25 °C) in the dark. The stained cells were subsequently analysed using a BD FACS-Calibur cytometer (BD Bioscience).
2.6. Reactive oxygen species (ROS), malondialdehyde (MDA), superoxide dismutase (SOD) and glutathione (GSH) assays
The ROS was measured with 2′,7′-dichlorofluorescein-diacetate (DCFHDA, Beyotime Institute of Biotechnology, Jiangsu, China) and dihydroethidium (DHE, Invitrogen, San Diego, CA, USA) staining, and observed using a microscope or analysed by flow cytometry [27]. DCFHDA was alternately excited at the wavelengths of 488 nm and 525 nm and DHE was alternately excited at the wavelengths of 300 nm and 535 nm following the manufacturer's instructions. MDA content, SOD activity and GSH concentration were measured using commercial kits (Beyotime Institute of Biotechnology, China) following the manufacturer's instructions.
2.7. Calcium imaging and [Ca2+]c detection
After treatment, cardiomyocytes were loaded with 1 μM Fura-2/AM for 30 min at room temperature. Then, the cells were observed on an inverted microscope (Olympus America, Melville, NY, USA), and Fura-2 fluorescence was alternately excited at 340 and 380 nm wavelengths using a monochromator (TILL Photonics, Polychrome V, Munich, Bavaria, Germany). The intracellular free calcium concentration was calculated according to a previous study [28]. For quantification of the concentration changes of [Ca2+]c, Fura-2 flow cytometry was used.
2.8. Real-time PCR
RT-PCR (Q-PCR) was performed according to a standard protocol. The following primers were used for polymerase chain reaction: XO, forward, AAGTAGAGGGGGCATTTGTCCAG, reverse, TGATGGCAAAGAAGATAGAGGAAGC; GADPH, forward, TGGAGTCTACTGGCGTCTT, reserve, TGTCATATTTCTCGTGGTTCA. The mRNA levels were determined by qRT-PCR in triplicate for each of the independently prepared RNAs, and GADPH was used as the endogenous control [29].
2.9. Western blot analysis
First, mouse heart tissues and cardiomyocytes after treatment were homogenized and sonicated in a lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 0.1% sodium dodecyl sulfate, and a protease inhibitor cocktail [30]. Subsequently, samples were centrifuged for approximately 10 min at 4 °C to obtain the supernatant. Next, the protein concentrations were detected via the BCA Protein Quantification Kit. Equivalent protein amounts (generally 50 or 100 μg) were separated by SDS-PAGE and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane [31]. After the blocking step, the membranes were incubated with the following primary antibodies at 4 °C overnight: anti-mouse polyclonal antibodies: PAGM5 (1:1000, Abcam, ab126534), GRP78 (1:1000, Abcam, ab21685), GADPH (1:1000, Abcam, ab8245); anti-mouse monoclonal antibodies: p-MLKL (1:1000, Abcam, ab196436), XO (1:1000, Cell Signaling Technology, 5865), MLKL (1:1000, Cell Signaling Technology, 37,705), Ripk3 (1:1000, Cell Signaling Technology, 95,702), CHOP (1:1000, Cell Signaling Technology, 2895), PERK(1:1000, Cell Signaling Technology, 3192). Secondary antibodies including Anti-mouse IgG (1:1000, 14,709) and Anti-rabbit IgG (1:1000, 14,708) were purchased form Cell Signaling Technology.
The antigen-antibody complexes were detected by an enhanced chemiluminescence (ECL) substrate kit (Thermo, USA). The bands were scanned and quantified by ImageJ (Bio-Rad, USA) analysis software.
2.10. Immunofluorescence staining
After treatments, the samples were washed three times with PBS and were fixed with 4% paraformaldehyde for 30 min at room temperature [32]. Subsequently, the samples were incubated with the primary antibody at 4 °C overnight. Next, PBS was used to wash the samples three times, and subsequently the samples were stained with fluorescent second antibody at 37 °C for 30 min. DAPI was used for nuclear staining. The pictures were acquired using a fluorescence microscope (Olympus Corporation, Tokyo, Japan) with standard excitation filters. The following primary antibodies were used for immunofluorescence staining: Ripk3 (1:1000, Cell Signaling Technology, 95,702), GRP78 (1:1000, Abcam, ab21685), and Cardiac Troponin T (1:1000, Abcam, ab50576). Anti-mouse IgG (1:500, 4408, green), anti-mouse IgG (1:500, 4409, red), anti-rabbit IgG (1:500, 4412, green) and anti-rabbit IgG (1:500, 4413, red) were purchased from Cell Signaling Technology. DAPI (Sigma-Aldrich, USA) and ER-Tracker™ Green (Molecular Probes, USA) were used to detect the nuclei and ER, respectively.
2.11. Mitochondrial membrane potential (ΔΨm) measurement, mitochondrial permeability transition pore (mPTP) opening evaluation
The ΔΨm was visualized using the JC-1 Kit (Beyotime, China). The red fluorescence was alternately excited at the wavelengths of 585 nm and 590 nm and the green fluorescence was alternately excited at the wavelengths of 514 nm and 529 nm following the manufacturer's instructions. The mPTP opening was assessed as the rapid dissipation of tetramethylrhodamine ethyl ester (TMRE) fluorescence. TMRE fluorescence was alternately excited at the wavelengths of 550 nm and 575 nm. The arbitrary mPTP opening time was determined as the time when TMRE fluorescence intensity decreased by half between initial and residual fluorescence intensity according to a previous study [28].
2.12. Lactate dehydrogenase (LDH) release, MTT and Cytokines measurement
LDH release was measured using an LDH release kit (Beyotime, Beijing, China). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure cell viability as previously described [33]. Cells were plated onto 96-well plates (104 cells/well). To examine the amount of IL-6, TNF-a and IL-10 in the heart tissues, measurements were carried out using commercially available ELISA kits (Sen-Xiong Company, Shanghai, China) [34].
2.13. RNA interference
XO were knocked down by transient transfection of small interfering RNA (siRNA). These siRNAs and their non-targeting sequences (negative controls) were purchased from GenePharma Co., Ltd. (Shanghai, China). The expression levels of proteins in transfected cells were determined by Western blot analysis [35].
2.14. Statistical analysis
All results were expressed as the means ± standard error of mean (SEM). We performed statistical analysis using one- or two-way ANOVA and Student-Newman-Keuls post hoc tests or t-test. A value of P ≤ 0.05 was considered to be statistically significant.
3. Results
3.1. Ripk3-mediated necroptosis contributed to cardiac IR injury
First, western blot was used to analyse the alterations of Ripk3 before and after IR injury. Ripk3 expression was primarily upregulated at reperfusion stage (Fig. 1A–B) in comparison to the sham group. We thus asked whether elevated Ripk3 played a causal role in cardiac IR injury, so the Ripk3-deficient mice (Ripk3-/-) were used. Meanwhile, given that Ripk3 expression was highest after 6 h of reperfusion, 45 min of ischaemia and 6 h of reperfusion were used in the following experiments. Through analysis of the infarction area, we confirmed that Ripk3-deleted mice demonstrated a significant reduction in infarction zone over the WT group (Fig. 1C–D). Loss of Ripk3 also reduced the secretion of pro-inflammatory TNF-a, IL-6, and increased the secretion of anti-inflammatory IL-10 in the infract heart (Supplemental Fig. 1A–C). Ripk3 genetic ablation also attenuated the cardiac necroptosis caused by IR injury, as evidenced by reduced phosphoglycerate mutase 5 (PGAM5) and p-MLKL (Fig. 1E–F). These data show that Ripk3 is activated by IR injury and contributed to the cardiac necroptosis.
Fig. 1.

Ripk3-mediated necroptosis contributed to cardiac IR injury (n = 6/group). (A–B) Western blot analysis was performed to detect the expression of Ripk3 after 45-min ischaemia and 0–24-h reperfusion. (C–D) Representative images of heart sections with TTC and Evans Blue staining of the infarcted area. Bar graph indicates the infarct size. (E–F) Quantitative analysis of the relative expression of Ripk3, p-MLKL, T-MLKL and PGAM5. (G–H) Loss of Ripk3 reduced the number of trypan blue-positive cells in cardiomyocytes. (I–J) The effects of Ripk3 depletion on the necroptosis of cardiomyocytes by flow cytometry with Annexin V/PI staining. Necroptosis group: the percentage of PI+ cells. *P < 0.05 vs the sham group (in vivo) or WT group (in vitro), #P < 0.05 vs the WT+IR injury (in vivo) or WT+LPS+H2O2 group (in vitro).
The regulatory role of Ripk3 in cardiac IR injury is further evidenced by cellular experiments isolating cardiomyocytes from WT (called WT-cells) and Ripk3-/- mice (called Ripk3-/--cells). Notably, we used LPS and H2O2 co-treatment to mimic animal reperfusion injury because inadequate oxygen supply is not the primary factor in IR injury and, more importantly, is not enough to induce Ripk3 upregulation and cellular necroptosis in vitro (unpublished data). Accordingly, to determine the role of Ripk3 in vitro, we applied LPS and H2O2 co-treatment. As shown in Supplemental Fig. 1K–L, LPS and H2O2 increased the cardiomyocyte necroptosis index, as shown by upregulated Ripk3, PGAM5, and p-MLKL, and this effect was nullified in the Ripk3-/--cells. Furthermore, the decrease in cellular necroptosis was closely associated with an increase in cellular survival via MTT assay and LDH release (Supplemental Fig. 1D–E). Moreover, to observe the cellular death directly, we used trypan blue staining, and fewer dead cells appeared under LPS and H2O2 co-treatment in Ripk3-/--cells than in WT-cells (Fig. 1G–H). Subsequently, Annexin V/PI flow-cytometry analysis was also used to precisely quantify the cellular necroptosis. As shown in Fig. 1I–J, LPS and H2O2 co-treatment elevated the ratio of PI+ cardiomyocytes, and this effect was reduced in Ripk3-/--cells. Altogether, these data indicate that increased Ripk3 expression aggravates the cardiomyocytes’ reperfusion death, and this is partially mediated by activating cellular necroptosis.
3.2. Genetic ablation of Ripk3 sustained cardiac function after IR injury
Apart from the cellular necroptosis, we also observed cardiac function under IR injury with Ripk3 deletion. Through echocardiography analysis, we demonstrated that IR injury reduced the left-ventricular ejection fraction (LVEF) and left-ventricular fractional shortening (LVFS). By contrast, the left-ventricular diastolic dimension (LVDd) increased in response to IR injury. Interestingly, Ripk3 deletion largely reversed the cardiac function parameters, as evidenced by increased LVEF and LVFS and decreased LVDd (Fig. 2D–F). The cardiac dysfunction may be a result of extensive myocardial injury, given the higher levels of LDH, troponin T, and CK-MB that appeared in the reperfused hearts, whereas Ripk3 deficiency was positively associated with less LDH, troponin T, and CK-MB contents after IR injury (Fig. 2A–C). Furthermore, the functional change would be derived from the structural alteration, so electron microscopy was used to observe the ultra-structural destruction of myocardium after IR injury. As shown in Fig. 2M, myocardial breakdown and Z-line disappearance could be identified in the WT group but not in the Ripk3-/- group.
Fig. 2.

Genetic ablation of Ripk3 sustained cardiac function after IR injury (n = 6/group). (A–C) Content of cardiac damage markers, including LDH, Troponin T, and CK-MB. (D–F) Cardiac function is evaluated through echocardiography. Quantitative analysis of the data derived from echocardiography. (G) Resting cell length. (H) Peak shortening (normalized to cell length). (I) Maximal velocity of shortening (+ dL/dt). (J) Maximal velocity of relengthening (− dL/dt). (K) Time-to-peak shortening (TPS). (L) Time-to-90% relengthening (TR90). (M) TEM was used to observe the ultrastructural changes after IR injury in vivo. *P < 0.05 vs the sham group; #P < 0.05 vs IR+WT group.
Solid evidence was also obtained from the analysis of cardiomyocyte contraction properties via acute isolation of cardiomyocytes from the WT and Ripk3-/- mice, following our previous study [36]. Neither IR injury nor Ripk3 knockdown obviously affected the resting cell length in cardiomyocytes (Fig. 2G). However, cardiomyocytes from WT mice exposed to IR injury displayed significantly depressed PS, ± dL/dt, TR90, and TPS (Fig. 2H–I). By contrast, the ablation of Ripk3 did not affect the mechanical parameters tested; it apparently reduced or reversed IR-induced cardiomyocyte dysfunction (Fig. 2H–I). Collectively, these data confirm that Ripk3 deletion maintains cardiac function during IR injury.
3.3. Ripk3 initiated necroptosis via ROS-mediated mPTP opening
To figure out the mechanism underlying the Ripk3-mediated necroptosis, the mitochondrial mPTP opening rate was evaluated, as previous studies have identified mPTP opening as the upstream trigger of cellular necroptosis [9], [37], [38]. By analyzing the mPTP opening time, we demonstrated that LPS and H2O2 co-treatment increased the time of mPTP opening, which was reduced by Ripk3 deficiency (Fig. 3A–B). A similar result was observed in mitochondrial potential, which would be dissipated upon mPTP opening (Fig. 3C–D). CsA, the inhibitor of mPTP opening, was used as the negative control group. Notably, application of CsA could further maintain cellular viability under LPS and H2O2 stress (Fig. 3E), similar to the results of Ripk3 ablation, suggesting that mPTP closing was helpful for necroptosis inhibition.
Fig. 3.

Ripk3 initiated necroptosis via ROS-mediated mPTP opening (n = 6/group). (A–B) Arbitrary mPTP opening time was determined as the time when the TMRE fluorescence intensity decreased by half between the initial and residual fluorescence intensity. (C–D) The change in membrane potential (∆Ψm) by JC-1 staining. (E) The cellular viability was evaluated by MTT assay. (F–G) The ROS production was detected by flow cytometric analysis of ROS-DCFHDA in cardiomyocytes. (H) The change of arbitrary mPTP opening time. (I) Cell death was determined by calcein AM/EthD-1 flow cytometry. The x-axis shows calcein AM staining, and the y-axis show EthD-1 staining. *P < 0.05 vs WT (in vitro), #P < 0.05 vs the WT+LPS+H2O2 group (in vitro).
ROS has been reported as the primary factor activating mPTP opening and has been implicated in the process of Ripk3-induced cellular necroptosis in cardiac IR injury [39], [40], [41]. Therefore, we asked whether mPTP opening was derived from the ROS outburst. To address this question, we first observed the ROS alterations under IR injury with Ripk3 ablation. Through DHE staining, we demonstrated that ROS content was elevated after IR injury and was reduced to normal levels by Ripk3 deletion (Supplemental Fig. 1F–G). In addition, IR injury produced more MDA and consumed a large amount of anti-oxidant factors, such as GSH/SOD, which was reversed by Ripk3 deletion (Supplemental Fig. 1H–J). To provide more direct evidence, we measured ROS production by DCFHDA flow-cytometry analysis in vitro (Fig. 3F–G). ROS content was elevated after LPS/H2O2 treatment and was reduced to normal levels by Ripk3 deletion. Together, these results suggest that ROS production is regulated by Ripk3. We subsequently investigated whether ROS eruption played a causal role in mPTP opening. NAC, an ROS scavenger, was used to neutralize excessive ROS under LPS/H2O2 treatment. After the fatal ROS in cardiomyocytes was cleared, the mPTP opening time was strongly reduced, similar to the results of Ripk3 deletion (Fig. 3H). Furthermore, Ripk3 deletion and NAC also promoted cell survival under LPS/H2O2 stress, indicated by calcein AM/EthD-1 flow-cytometry analysis (Fig. 3I). This verified our hypothesis that ROS was regulated by Ripk3 and contributed to the mPTP opening that mediated cellular necroptosis under IR injury.
3.4. Ripk3 induced ROS overproduction via ER -calcium-XO signaling pathways
Our previous study demonstrated that cardiac IR injury-related ROS outburst was primarily derived from ER-calcium-XO signaling [42]. IR injury induced the release of ER-calcium into the cytoplasm, which activated calcium-dependent XO expression, leading to ROS overproduction. On this basis, we wondered whether this signal pathway was implicated in Ripk3-mediated ROS eruption. First, we verified that calcium balance was manipulated by Ripk3, because loss of Ripk3 limited cellular calcium overload under LPS/H2O2 stress (Fig. 4A). Subsequently, we found that the inhibitory effect of Ripk3 on calcium overload further attenuated expression of XO, a calcium-dependent protein, via western blots (Fig. 4B) and qPCR assay (Fig. 4C). Last, to determine whether the higher XO expression was associated with ROS outburst, we used siRNA and Febuxostat (a XO inhibitor) against XO. Inhibiting XO with siRNA and Febuxostat was able to repress the excessive ROS generation, as in the results for Ripk3 ablation (Fig. 4D). Collectively, these data show that IR-mediated ROS overproduction is governed by Ripk3 via ER-calcium-XO pathway.
Fig. 4.

Ripk3 induced ROS overproduction via ER -calcium-XO signaling pathways. (n = 6/group). (A) The Ca2+ levels under LPS and H2O2 were observed by fluo-2. (B) The change of XO expression by western blot. (C) The change of XO mRNA expression by PCR. (D) ROS production was observed by DHE staining. *P < 0.05 vs WT group, #P < 0.05 vs the WT+LPS+H2O2 group.
3.5. Ripk3 contributed to calcium overload via activating ER stress
What remains unclear is how Ripk3 evokes ER-calcium liberation into the cytoplasm. The ER lumen is the main storage of intracellular Ca2+, and ER dysfunction promotes calcium output from the ER into the cytoplasm, termed “calcium overload”. To answer this question, we first observed the ER stress markers. In vivo, IR injury contributed to overexpression of GRP78, CHOP, and PERK in comparison with the control group, as indicated by immunofluorescence (Fig. 5A–B) and western blot (Fig. 5C–F). Interestingly, this phenomenon was reversed by Ripk3 deletion. Similar results were observed in vitro (Fig. 5G–H). These data suggest that Ripk3 upregulation is closely associated with ER stress.
Fig. 5.

Ripk3 contributed to calcium overload via activating ER stress (n = 6/group). (A–B) Loss of Ripk3 reversed the upregulated GRP78 in response to IR injury as indicated by immunofluorescence. (C–F) The changes of ER stress markers, including PERK, CHOP and GRP78, in vivo were measured by Western blotting. (G–H) The changes of ER stress markers, including PERK, CHOP and GRP78, in vitro were measured by Western blotting. (I) The change in Ca2+ concentration was measured by flow cytometry. *P < 0.05 vs the sham group (in vivo) or WT group (in vitro), #P < 0.05 vs the WT+IR injury (in vivo) or WT+LPS+H2O2 group (in vitro).
Subsequently, to explore whether ER stress contributed to Ripk3-induced calcium overload, we used tunicamycin (TM), an activator of ER stress, in Ripk3-deleted cells to recall ER stress. In contrast, we applied pravastatin (Pr), an inhibitor of ER stress, in WT-cells. Through immunofluorescence assay of CHOP, we confirmed that ER stress (CHOP expression) was triggered by LPS and H2O2 co-treatment or TM and was inhibited by Ripk3 deletion and Pr (Supplemental Fig. 1N). Moreover, ER-stress activation was coupled to calcium overload via flow cytometry in WT-cells, the effect of which was cancelled by Pr, similar to the results of Ripk3 deletion (Fig. 5I). By contrast, reactivation of ER stress by TM could re-elevate the cytoplasmic calcium in Ripk3-/- cells. Together, these results suggest to us that Ripk3 causes calcium overload via ER stress.
Next, we asked how Ripk3 induces ER stress. Ripk3 has been reported to have a high affinity with cellular membrane [43], [44]. Thus, we used immunofluorescence to test whether Ripk3 could translocate to ER under LPS/H2O2 stress. The results, shown in Supplemental Fig. 1M, demonstrated that more Ripk3 migrated onto the ER under LPS/H2O2 in co-treatment in WT-cells, and this effect was reversed by Ripk3 deletion. This experiment showed the mechanism by which Ripk3 may translocate onto the ER and mediate ER stress.
3.6. Inhibition of ER stress improved cardiac function following IR injury
Finally, to examine whether ER stress is required for Ripk3-mediated cardiac function in vivo, an animal IR-injury model was evaluated. Fig. 6A–D showed that the inhibition of ER stress, or Ripk3 depletion, can reduce cardiac damage and dysfunction after IR injury. However, the reactivation of ER stress abolished the cardiac protection provided by Ripk3 depletion. To obtain further evidence for the role of ER stress in Ripk3-induced cardiac dysfunction, we observed the cardiomyocyte properties again and observed that the inhibition of ER stress apparently reduced or abrogated LPS and H2O2-induced TPS and TR90 changes (Fig. 6E–F). To explore the potential mechanism of action underlying the impaired contractile dysfunction in cardiomyocytes under the inhibition of ER stress, we evaluated intracellular Ca2+ handling. Functionally, Fura-2/AM was used to observe changes in intracellular Ca2+ transients in spontaneously beating cardiomyocytes (Fig. 6G–I). The intracellular resting calcium level increased following treatment with LPS and H2O2 but decreased to a normal level upon the loss of Ripk3 or treatment with pravastatin. The activation of ER stress in Ripk3-/- mice induced a rebound of resting calcium. By contrast, the intracellular Ca2+ transient amplitude was reduced after LPS and H2O2 treatment but increased after the deletion of Ripk3. Interestingly, the activation of ER stress in Ripk3-/- mice could alleviate the intracellular Ca2+ transient amplitude. These data suggest that ER stress is responsible for Ripk3-induced Ca2+ dis-homeostasis.
Fig. 6.

Inhibition of ER stress improved cardiac function followed IR injury (n = 6/group). (A, B) Cardiac function was evaluated through echocardiography. Quantitative analysis of the data derived from echocardiography. Tunicamycin (TM), an activator of ER stress was used to recall ER stress. Pravastatin (Pr), an inhibitor of ER stress, was used to inhibit ER stress. (C–E) The contractile properties of cardiomyocytes, including TPS and TR90, in WT and Ripk3-/- mice in the context of IR injury. (F) Representative tracings of F340/F380 fluorescence ratio. (G) [Ca2+]i transient baseline. (H) Amplitude of [Ca2+]i transient. Data are presented as the means ± SEM (n = 6, for each group). *P < 0.05 vs the WT+IR injury (in vivo) or control group (in vitro); #P < 0.05 vs the Ripk3-/-+IR injury (in vivo) or LPS+H2O2 group (in vitro); &P < 0.05 vs the Ripk3-/-+LPS+H2O2 group (in vitro).
4. Discussion
Necroptosis contributes to approximately 50% of cellular death in the context of cardiac IR injury. By comparison, apoptosis contributes to ~ 30% of IR-injury–induced cellular death [8]. It is well-known that Ripk3 plays the critical role in the activating process of necroptosis. In the current study, through loss- and gain-of-function assays of Ripk3, we illustrate the comprehensive role of Ripk3 in cardiac IR injury: (1) Ripk3 was increased in the setting of cardiac IR injury and was positively involved in cardiac damage. (2) Functional assay demonstrated that loss of Ripk3 dramatically improves cardiac function via strongly reducing cardiomyocytes necroptosis. (3) Mechanically, Ripk3 may evoke ER stress via direct interaction with ER. (4) As a result of ER stress, [Ca2+]c was greatly elevated and subsequently promoted XO activation and ROS outburst. (5) The increase in cellular ROS contributed to the opening of mPTP. (6) Through the regulation of ER stress, [Ca2+]c overload, XO, and mPTP pathways, Ripk3 initiated necroptosis under IR-injury stimulation. To our knowledge, this is the first study to explain the mechanism by which Ripk3 induces necroptosis via ER stress, calcium overload, XO-mediated oxidative stress, and mPTP opening. These findings advance our understanding of the molecular signaling underlying Ripk3-mediated necroptosis in response to cardiac IR injury.
Mechanistically, the formation of “necrosome” by Ripk3 induces cellular necroptosis during IR injury [45], [46], [47]. Recent studies have confirmed that myocardial necroptosis is distinct from that of other cell types during IR injury in being dependent on the activation of Ripk3 [8] and the mPTP opening [9], [10]. Nevertheless, the fundamental mechanisms underlying Ripk3-induced mPTP opening are poorly understood. This study fills this gap and confirms that Ripk3 activates mPTP opening via the ER stress/calcium overload/XO/ROS pathway.
mPTP opening has been identified as an important therapeutic target for limiting myocardial infarct size, inhibiting cardiac IR injury, and alleviating hypertension [48], [49], [50], [51]. mPTP opening is activated by mitochondrial calcium overload, ROS, mitochondrial ROS (mROS), and the constituent components of the channel itself [52]. Recently, cyclophilin D (CypD) has been regarded as a critical regulator of the mPTP and as promoting mPTP opening by activating mROS and inducing mitochondrial dysfunction [50], [51]. Several studies have confirmed that inhibiting CypD with cyclosporine A reduces myocardial infarct size but does not have a detrimental effect on left-ventricular (LV) remodeling in patients with AMI [53], [54], [55], [56]. The present study confirms that mPTP opening is caused by ROS, which is consistent with previous studies.
The production of ROS is very complicated. It is now appreciated that there are various sources of ROS, including mitochondria [57], [58], NAD(P)H oxidase [57], [59], [60], XO [61], [62], and autoxidation of diverse substances [63]. XO, a calcium concentration-dependent superoxide-producing enzyme, is localized to the ER and generates ROS by promoting the metabolism of hypoxanthine to uric acid. The present study confirms that XO-induced ROS plays a critical role in Ripk3-mediated necroptosis in cardiac IR injury. Whether other ROS-producing enzymes also contribute to necroptosis in cardiac IR injury is unclear. Further experimental evidence is needed.
We have also determined that ER stress is the mediator of cytoplasmic calcium overload. Previous studies on ER stress focused mainly on the unfolded protein response (UPR) for ER stress-mediated cellular apoptosis [64], [65]. However, few were found defining the relationship between ER stress and necroptosis. Our present research identifies ER stress as the key regulator of Ripk3-mediated necroptosis. It is also well-known that ER stress contributes to cellular apoptosis in response to cardiac IR injury. We believe that ER stress, as a common downstream signal, participates in the regulation of both apoptosis and necroptosis in response to cellular stress. This conclusion suggests that ER stress is an important therapeutic target for attenuating IR-mediated myocardial damage by interrupting apoptosis and necroptosis simultaneously.
The significance of this article is that we discovered the relationship between ER stress, ROS overproduction, and mPTP opening in necroptosis. Previous studies focused primarily on protein interaction (Ripk3 and Ripk1) in necroptosis [66]. We show that necroptosis requires cellular organelle interaction (ER and mitochondria). Accordingly, this study extends our understanding of the molecular mechanisms underlying necroptosis.
In summary, the present study helps to elucidate how necroptosis is mediated by ER stress, which transmits the death signal to mitochondria via the Ca2+/XO/ROS axis. This study links traditionally independent injury factors, including calcium overload and oxidative stress, with the ER and mitochondria during IR injury and suggests that Ripk3 is the upstream signal for these factors, thereby updating these traditional concepts.
Acknowledgements
This study was financially supported by grants from the National Natural Science Foundation of China (Nos. 81770237). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
Disclosure statement
The authors declare no potential disclosure statement.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.02.019.
Contributor Information
Hao Zhou, Email: zhouhao301@outlook.com.
Yundai Chen, Email: cyundai@vip.163.com.
Appendix A. Supplementary material
Supplemental Fig. 1.

(A–C) Cardiac levels of IL-10, TNF-a and IL-6 by ELISA. (D) LDH release. (E) Cell viability was measured by MTT. (F–G) ROS content was measured by DHE staining. (H–J) The changes of GSH, SOD and MDA were detected by ELISA in cardiac tissue after IR injury. (K–L) The expression of Ripk3, PGAM5, p-MLKL and T-MLKL were measured by western blot. (M) More Ripk3 migrated on the ER under LPS/H2O2 in co-treatment in WT-cell, which was reversed by Ripk3 deletion. (N) The reactivation of ER stress under Ripk3 knockout re-upregulated the expression of CHOP by immunofluorescence in response to LPS and H2O2. *P < 0.05 vs WT group, #P < 0.05 vs the WT+LPS+H2O2 group. n = 6/group.
References
- 1.Hu S.Y., Zhang Y., Zhu P.J., Zhou H., Chen Y.D. Liraglutide directly protects cardiomyocytes against reperfusion injury possibly via modulation of intracellular calcium homeostasis. J. Geriatr. Cardiol. 2017;14(1):57–66. doi: 10.11909/j.issn.1671-5411.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou H., Hu S., Jin Q., Shi C., Zhang Y., Zhu P., Ma Q., Tian F., Chen Y. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J. Am. Heart Assoc. 2017;6(3) doi: 10.1161/JAHA.116.005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kloner R.A., Ganote C.E., Jennings R.B. The "no-reflow" phenomenon after temporary coronary occlusion in the dog. J. Clin. Invest. 1974;54(6):1496–1508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weir R.A., Murphy C.A., Petrie C.J., Martin T.N., Balmain S., Clements S., Steedman T., Wagner G.S., Dargie H.J., Mcmurray J.J. Microvascular obstruction remains a portent of adverse remodeling in optimally treated patients with left ventricular systolic dysfunction after acute myocardial infarction. Circ. Cardiovasc. Imaging. 2010;3(4):360. doi: 10.1161/CIRCIMAGING.109.897439. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H., Zhu P., Guo J., Hu N., Wang S., Li D., Hu S., Ren J., Cao F., Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2017;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degterev A., Zhou W., Maki J.L., Yuan J. Assays for necroptosis and activity of RIP kinases. Methods Enzymol. 2014;545:1–33. doi: 10.1016/B978-0-12-801430-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F., Liu Y., Zheng W., Shang H., Zhang J. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016;22(2) doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 9.Baines C.P., Kaiser R.A., Purcell N.H., Blair N.S., Osinska H., Hambleton M.A., Brunskill E.W., Sayen M.R., Gottlieb R.A., Dorn G.W., Robbins J., Molkentin J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 11.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta. 2010;1797(6–7):897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 12.Zorov D.B., Filburn C.R., Klotz L.O., Zweier J.L., Sollott S.J. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H., Zhang Y., Hu S., Shi C., Zhu P., Ma Q., Jin Q., Cao F., Tian F., Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017 doi: 10.1111/jpi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Zhou H., Wu W., Shi C., Hu S., Yin T., Ma Q., Han T., Zhang Y., Tian F., Chen Y. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med, 2016;95:278–292. doi: 10.1016/j.freeradbiomed.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y., Liao H., Zhong S., Gao F., Chen Y., Huang Z., Lu S., Sun T., Wang B., Li W., Xu H., Zheng F., Shi G. Effect of N-n-butyl haloperidol iodide on ROS/JNK/Egr-1 signaling in H9c2 cells after hypoxia/reoxygenation. Sci. Rep. 2015;5:11809. doi: 10.1038/srep11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry C.E., Hare J.M. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J. Physiol. 2004;555(Pt 3):589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobsen A.V., Silke J. The importance of being chaperoned: HSP90 and necroptosis. Cell Chem. Biol. 2016;23(2):205–207. doi: 10.1016/j.chembiol.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Park S.Y., Shim J.H., Chae J.I., Cho Y.S. Heat shock protein 90 inhibitor regulates necroptotic cell death via down-regulation of receptor interacting proteins. Pharmazie. 2015;70(3):193–198. [PubMed] [Google Scholar]
- 19.Li D., Li C., Li L., Chen S., Wang L., Li Q., Wang X., Lei X., Shen Z. Natural product Kongensin A is a non-canonical HSP90 inhibitor that blocks RIP3-dependent necroptosis. Cell Chem. Biol. 2016;23(2):257–266. doi: 10.1016/j.chembiol.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 20.Jacobsen A.V., Lowes K.N., Tanzer M.C., Lucet I.S., Hildebrand J.M., Petrie E.J., Van Delft M.F., Liu Z., Conos S.A., Zhang J.G., Huang D.C., Silke J. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;e2051 doi: 10.1038/cddis.2015.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 22.Han D., Huang W., Li X., Gao L., Su T., Li X., Ma S., Liu T., Li C., Chen J., Gao E., Cao F. Melatonin facilitates adipose-derived mesenchymal stem cells to repair the murine infarcted heart via the SIRT1 signaling pathway. J. Pineal Res. 2016;60(2):178–192. doi: 10.1111/jpi.12299. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X., Yuan Y., Jiang L., Zhang J., Gao J., Shen Z., Zheng Y., Deng T., Yan H., Li W., Hou W.W., Lu J., Shen Y., Dai H., Hu W.W., Zhang Z., Chen Z. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy. 2014;10(10):1801–1813. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H., Li D., Zhu P., Hu S., Hu N., Ma S., Zhang Y., Han T., Ren J., Cao F., Chen Y. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J. Pineal Res. 2017;63(4) doi: 10.1111/jpi.12438. [DOI] [PubMed] [Google Scholar]
- 25.Zhou H., Yang J., Xin T., Zhang T., Hu S., Zhou S., Chen G., Chen Y. Exendin-4 enhances the migration of adipose-derived stem cells to neonatal rat ventricular cardiomyocyte-derived conditioned medium via the phosphoinositide 3-kinase/Akt-stromal cell-derived factor-1alpha/CXC chemokine receptor 4 pathway. Mol. Med. Rep. 2015;11(6):4063–4072. doi: 10.3892/mmr.2015.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao M., Lu L., Lei S., Chai H., Wu S., Tang X., Bao Q., Chen L., Wu W., Liu X. Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxid. Med. Cell. Longev. 2016;2016:1451676. doi: 10.1155/2016/1451676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel M., Yarlagadda V., Adedoyin O., Saini V., Assimos D.G., Holmes R.P., Mitchell T. Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol. 2017;15:207–215. doi: 10.1016/j.redox.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H., Zhu P., Guo J., Hu N., Wang S., Li D., Hu S., Ren J., Cao F., Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. J. Pineal Res. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H., Du W., Li Y., Shi C., Hu N., Ma S., Wang W., Ren J. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018;64(1) doi: 10.1111/jpi.12450. [DOI] [PubMed] [Google Scholar]
- 30.Yoo J.M., Lee B.D., Sok D.E., Ma J.Y., Kim M.R. Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 2017;11:592–599. doi: 10.1016/j.redox.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He D., Zhao M., Wu C., Zhang W., Niu C., Yu B., Jin J., Ji L., Willard B., Mathew A.V., Chen Y.E., Pennathur S., Yin H., He Y., Pan B., Zheng L. Apolipoprotein A-1 mimetic peptide 4F promotes endothelial repairing and compromises reendothelialization impaired by oxidized HDL through SR-B1. Redox Biol. 2017;15:228–242. doi: 10.1016/j.redox.2017.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griesser E., Vemula V., Raulien N., Wagner U., Reeg S., Grune T., Fedorova M. Cross-talk between lipid and protein carbonylation in a dynamic cardiomyocyte model of mild nitroxidative stress. Redox Biol. 2017;11:438–455. doi: 10.1016/j.redox.2016.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Amine R., Germini D., Zakharova V.V., Tsfasman T., Sheval E.V., Louzada R.a.N., Dupuy C., Bilhou-Nabera C., Hamade A., Najjar F., Oksenhendler E., Lipinski M., Chernyak B.V., Vassetzky Y.S. HIV-1 tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2017;15:97–108. doi: 10.1016/j.redox.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D.J., Fu H., Tong J., Li Y.H., Qu L.F., Wang P., Shen F.M. Cholinergic anti-inflammatory pathway inhibits neointimal hyperplasia by suppressing inflammation and oxidative stress. Redox Biol. 2017;15:22–33. doi: 10.1016/j.redox.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulo L.L., Cruz J.C., Zhuge Z., Carvalho-Galvao A., Brandao M.C.R., Diniz T.F., Haworth S.M., Athayde-Filho P.F., Lemos V.S., Lundberg J.O., Montenegro M.F., Braga V.A., Carlstrom M. The novel organic mononitrate NDHP attenuates hypertension and endothelial dysfunction in hypertensive rats. Redox Biol. 2017;15:182–191. doi: 10.1016/j.redox.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Q., Li R., Hu N., Xin T., Zhu P., Hu S., Ma S., Zhu H., Ren J., Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakayama H., Chen X., Baines C.P., Klevitsky R., Zhang X., Zhang H., Jaleel N., Chua B.H., Hewett T.E., Robbins J., Houser S.R., Molkentin J.D. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest. 2007;117(9):2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 39.Kim J.S., Jin Y., Lemasters J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006;290(5):H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 40.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 41.He S., Liang Y., Shao F., Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA. 2011;108(50):20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Zhou H., Wu W., Shi C., Hu S., Yin T., Ma Q., Han T., Zhang Y., Tian F. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med. 2016;95:278–292. doi: 10.1016/j.freeradbiomed.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 43.Fan H., Tang H.B., Kang J., Shan L., Song H., Zhu K., Wang J., Ju G., Wang Y.Z. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience. 2015;311:362–373. doi: 10.1016/j.neuroscience.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 44.Chen W., Zhou Z., Li L., Zhong C.Q., Zheng X., Wu X., Zhang Y., Ma H., Huang D., Li W. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 2013;288(23):16247. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oerlemans M.I., Koudstaal S., Chamuleau S.A., De Kleijn D.P., Doevendans P.A., Sluijter J.P. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int. J. Cardiol. 2013;165(3):410–422. doi: 10.1016/j.ijcard.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 46.Smith C.C., Davidson S.M., Lim S.Y., Simpkin J.C., Hothersall J.S., Yellon D.M. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007;21(4):227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 47.Lalaoui N., Lindqvist L.M., Sandow J.J., Ekert P.G. The molecular relationships between apoptosis, autophagy and necroptosis. Semin. Cell Dev. Biol. 2015;39:63–69. doi: 10.1016/j.semcdb.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Lim S.Y., Hausenloy D.J., Arjun S., Price A.N., Davidson S.M., Lythgoe M.F., Yellon D.M. Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J. Cell. Mol. Med. 2011;15(11):2443–2451. doi: 10.1111/j.1582-4934.2010.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroller-Schon S., Steven S., Kossmann S., Scholz A., Daub S., Oelze M., Xia N., Hausding M., Mikhed Y., Zinssius E., Mader M., Stamm P., Treiber N., Scharffetter-Kochanek K., Li H., Schulz E., Wenzel P., Munzel T., Daiber A. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014;20(2):247–266. doi: 10.1089/ars.2012.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itani H.A., Dikalova A.E., Mcmaster W.G., Nazarewicz R.R., Bikineyeva A.T., Harrison D.G., Dikalov S.I. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67(6):1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hausenloy D.J., Lim S.Y., Ong S.G., Davidson S.M., Yellon D.M. Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc. Res. 2010;88(1):67–74. doi: 10.1093/cvr/cvq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halestrap A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 53.Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., Elbelghiti R., Cung T.T., Bonnefoy E., Angoulvant D., Macia C., Raczka F., Sportouch C., Gahide G., Finet G., Andre-Fouet X., Revel D., Kirkorian G., Monassier J.P., Derumeaux G., Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008;359(5):473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 54.Mewton N., Croisille P., Gahide G., Rioufol G., Bonnefoy E., Sanchez I., Cung T.T., Sportouch C., Angoulvant D., Finet G., Andre-Fouet X., Derumeaux G., Piot C., Vernhet H., Revel D., Ovize M. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J. Am. Coll. Cardiol. 2010;55(12):1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 55.Ivanes F., Rioufol G., Piot C., Ovize M. Postconditioning in acute myocardial infarction patients. Antioxid. Redox Signal. 2011;14(5):811. doi: 10.1089/ars.2010.3354. [DOI] [PubMed] [Google Scholar]
- 56.Cung T.T., Morel O., Cayla G., Rioufol G., Garciadorado D., Angoulvant D., Bonnefoycudraz E., Guérin P., Elbaz M., Delarche N. Cyclosporine before PCI in patients with acute myocardial infarction. New Engl. J. Med. 2015;373(11):1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 57.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daiber A., Di Lisa F., Oelze M., Kroller-Schon S., Steven S., Schulz E., Munzel T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017;174(12):1670–1689. doi: 10.1111/bph.13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rathore R., Zheng Y.M., Niu C.F., Liu Q.H., Korde A., Ho Y.S., Wang Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008;45(9):1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou L., Aon M.A., Almas T., Cortassa S., Winslow R.L., O'rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010;6(1):e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zinkevich N.S., Gutterman D.D. ROS-induced ROS release in vascular biology: redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011;301(3):H647–H653. doi: 10.1152/ajpheart.01271.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schulz E., Wenzel P., Munzel T., Daiber A. Mitochondrial redox signaling: interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014;20(2):308–324. doi: 10.1089/ars.2012.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 65.Hetz C., Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017;13(8):477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez D.A., Weinlich R., Brown S., Guy C., Fitzgerald P., Dillon C.P., Oberst A., Quarato G., Low J., Cripps J.G., Chen T., Green D.R. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23(1):76–88. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]