

Abstract
Objective
Reduction of oxidized methionines is emerging as a major protein repair pathway. The lack of methionine sulfoxide reductase A (MsrA) exacerbates cardiovascular disease phenotypes driven by increased oxidative stress. However, the role of MsrA on maintaining cellular homeostasis in the absence of excessive oxidative stress is less well understood.
Methods and results
Constitutive genetic deletion of MsrA increased formation of p62-containing protein aggregates, activated autophagy, and decreased a marker of apoptosis in vascular smooth muscle cells (VSMC). The association of Keap1 with p62 was augmented in MsrA-/- VSMC. Keap1 targets the transcription factor Nrf2, which regulates antioxidant genes, for proteasomal degradation. However, in MsrA-/- VSMC, the association of Nrf2 with Keap1 was diminished. Whereas Nrf2 mRNA levels were not decreased in MsrA-/- VSMC, we detected decreased ubiquitination of Nrf2 and a corresponding increase in total Nrf2 protein in the absence of biochemical markers of oxidative stress. Moreover, nuclear-localized Nrf2 was increased under MsrA deficiency, resulting in upregulation of Nrf2-dependent transcriptional activity. Consequently, transcription, protein levels and enzymatic activity of glutamate-cysteine ligase and glutathione reductase were greatly augmented in MsrA-/- VSMC.
Summary
Our findings demonstrate that reversal of methionine oxidation is required for maintenance of cellular homeostasis in the absence of increased oxidative stress. These data provide the first link between autophagy and activation of Nrf2 in the setting of MsrA deletion.
Abbreviations: ARE, antioxidant response element; CaMKII, Ca2+/calmodulin-dependent kinase II; GCLC, glutamate-cysteine ligase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; IκK, inhibitor of nuclear factor kappa-B kinase; Keap-1, Kelch-like ECH-associated protein 1; Msr, Methionine sulfoxide reductase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, Nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; SQSTM1, p62/sequestome; TRAF6, TNF receptor associated factor 6; VSMC, vascular smooth muscle cells
Keywords: Methionine, Methionine sulfoxide reductase, Smooth muscle, Nrf2, Autophagy, Ubiquitination
Graphical abstract
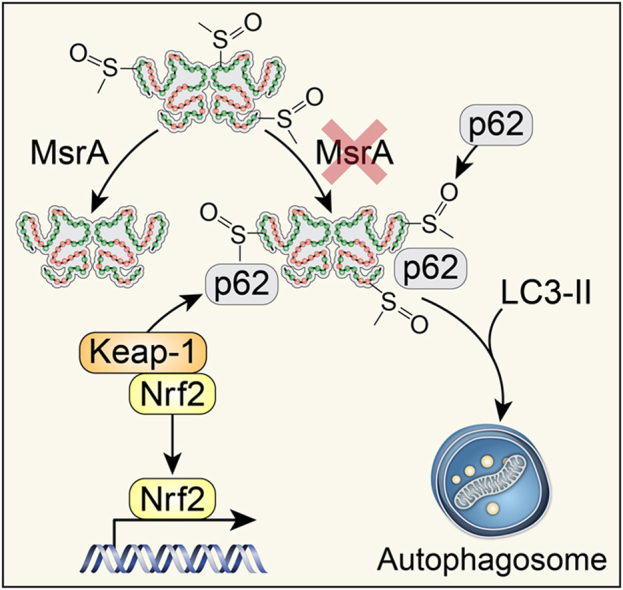
Highlights
-
•
With MsrA deletion, p62-containing intracellular protein aggregates are increased.
-
•
Autophagy is increased in proliferating MsrA-/- smooth muscle cells.
-
•
Increased association of Keap-1 with p62 derepresses Nrf2 in MsrA-/- cells.
-
•
Nrf2 activation in MsrA-/- cells occurs without increased oxidative stress.
1. Introduction
Methionine sulfoxide reductases (Msrs) are antioxidant repair proteins that convert methionine sulfoxide to methionine. In the last decade, oxidation of distinct methionine residues has emerged as a posttranslational modification that alters protein function [1], [2], [3], [4]. Examples include oxidization of distinct methionines in calmodulin [5], calcineurin [6], the Ca2+/calmodulin-dependent kinase II (CaMKII) [3] and voltage-gated potassium channels [7], [8]. On the other hand, oxidation of intracellular methionines on a “global” cellular level has been proposed to serve as a sink for excess reactive oxygen species (ROS) [9]. However, the impact of oxidized methionine residues that do not directly affect protein activity on cellular physiology has been difficult to ascertain.
Many studies to dissect the implication of methionine oxidation have used models of defective methionine sulfoxide repair, in particular, genetic models that are lacking Msrs. The S-enantiomer of methionine sulfoxide is reduced by one enzyme, methionine sulfoxide reductase A (MsrA), whereas several MsrBs reduce R-enantiomers [10]. MsrA deletion has been linked to decreased life span, although initial findings could not be reproduced in later studies and the mechanistic insight remained incomplete [11], [12], [13]. Several studies have described increased cell death in disease states related to oxidative stress and coined the concept that MsrA is a major antioxidant defense protein [14], [15]. Accordingly, the expression of antioxidant proteins via the Nrf2-antioxidant response element (ARE)-signaling pathway is upregulated when MsrA is missing [15], [16], [17]. Moreover, low activity of MsrA has been associated with formation of protein aggregates in Parkinson's and Alzheimer's disease as well as inclusion body myositis [18], [19], [20], [21], but the implications of these protein aggregates on repair and cell death pathways in MsrA deletion models has remained unstudied. In models of neurodegenerative diseases and proteinopathies of the liver, misfolded or unrepaired defective proteins colocalize with p62, also called sequestosome 1 (SQSTM1), a ubiquitin-binding scaffold protein [22], [23]. p62 delivers ubiquitinated proteins to the proteasome for degradation or targets them for autophagy.
Autophagy is a cellular process by which damaged proteins, distinct protein targets, organelles, and bacteria are degraded [24]. Recently, the crosstalk between autophagy and antioxidant response has been dissected: the antioxidant transcription factor Nrf2 is constantly ubiquitinated by the Cullin3–Keap1 ubiquitin E3 ligase complex and rapidly degraded in proteasomes [25]. Upon exposure to electrophilic and oxidative stresses, reactive cysteine residues of Keap1 become modified, leading to a decline in the E3 ligase activity, stabilization of Nrf2 and robust induction of a battery of cytoprotective antioxidant genes [26]. In addition to this canonical pathway, p62/SQSTM1 competitively binds to Keap1. Consequently, sequestering Keap1 via increased binding to p62 derepresses and activates Nrf2 [27], [28].
Thus, increased autophagy and activation of Nrf2-mediated transcriptional activity are mechanistically linked when p62-containing protein aggregates are abundant. Interestingly, in a model of Lamin A/C congenital muscular dystrophy, p62 protein complex formation and ARE transcription occur even in the absence of oxidative stress [29]. These findings support that Nrf2 can be activated by a mechanism that does not require oxidative or electrophilic stress in states where protein degradation is defective or pathologically abundant. These findings prompted us to hypothesize that MsrA deficiency may upregulate the accumulation of p62-containing protein aggregates, augment autophagy and Nrf2-dependent ARE gene transcription under baseline conditions when oxidative stress levels are not elevated. Our previous work focused on MsrA and its effect in the response to mechanical vascular injury, with a particular emphasis on smooth muscle cell proliferation and migration [30]. Here, we investigated whether MsrA deletion affects smooth muscle survival and repair processes at baseline.
2. Methods
2.1. Mice
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Iowa (IACUC# 1111234/4101189). Age-matched male and female MsrA-/- mice [31] were supplied by the MMRRC-Mutant Mouse Resource & Research Centers supported by the National Institute of Health and back-crossed for more than 5 generations in C57BL/6J background. MsrA-/- and,wild-type littermates (MsrA+/+, referred to as WT) at 10- to 12-weeks of age were used for cell isolation. Genotyping was done by PCR using MsrA and Neo primers (see Supplemental Table 1).
Nrf2-/- mice (C57BL/6 background, #017009, Jackson Laboratory) were crossbred with MsrA-/- mice to generate double knockout Nrf2-/- x MsrA-/- mice. Genotyping was performed as recommended by Jackson Laboratory.
2.2. Cell culture
Mouse aortic smooth muscle cells were isolated from 2 to 3 10–12-week old male and female mice per isolation by enzymatic dispersion as previously described [30]. Briefly, the adventitia was removed by incubating with elastase (0.74 U/ml) and collagenase (1 mg/ml) in HBSS (#14175–095, Gibco) and peeled away from the medial layers of the aorta. The aorta was cut and the endothelial layer was scraped away. The remaining medial layer was digested in collagenase (1.36 mg/ml) in complete media (described below) for 1 h. Cells were cultured in high glucose (4.5 g/L) DMEM (#11965–092, Gibco) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin, 8 mM HEPES, 1× MEM vitamins, and 1% non-essential amino acids at 37 °C in a humidified 95% air and 5% CO2 incubator. Mouse VSMC at passages 3–10 were used for experiments, with passage number for MsrA+/+ and MsrA-/- VSMCs matched with each experiment.
2.3. Transfection/infection
For adenoviral transduction, VSMC (1 × 106) were plated in a 100 mm dish and grown to 40% confluency. VSMC were transduced with Ad5 LC3-GFP adenovirus (kindly provided by Dr. E. Dale Abel, University of Iowa) at an MOI of 100. Media was changed 8–16 h after initial transduction. VSMC were grown 72 h post transduction prior to imaging. To some samples, bafilomycin A1 (50 nM, #S1413, SellectChem) was added at 48 h after transduction for additional 24 h.
For transfections, VSMC were transfected using Lipofectamine RNAi MAX transfection reagent (#13778030, ThermoFisher Scientific) by following the protocol for Lipofectamine 3000 (#L3000001, ThermoFisher Scientific). The ARE TATA-Inr luciferase reporter plasmid (pARE-Luc) [26], a gift from Dr. Francis Miller (Duke University), contained the 41-bp ARE sequence from the mouse glutathione S-transferase Ya subunit gene cloned behind a minimal TATA-Inr promoter in pGL2-basic (Promega). The Renilla luciferase expression plasmid, pRL-TK, contained the herpes simplex virus thymidine kinase promoter region upstream of the Renilla luciferase cDNA (Promega).
2.4. Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from VSMC per the manufacturer's recommendations (#74104, Qiagen) followed by digestion with DNase I to eliminate genomic DNA contamination. cDNA was transcribed from 75 ng RNA using Superscript III enzyme (#18080051, Invitrogen) and random hexomer primers. Message expression was quantified using Power SYBR Green master mix (#4367218, Applied Biosciences) on an iQ Lightcycler (BioRad) with 1 µL of 10 µM forward and reverse primers. Primer sequences are listed in Supplemental Table 1. All samples were run in triplicate and were normalized to acidic ribosomal phosphoprotein (ARP) mRNA. Results were quantitated using the comparative cycle threshold (ΔΔCt) method [32].
2.5. Cell lysis and nuclear/cytoplasmic fractionation
For total cell lysates, VSMC were washed with ice-cold PBS, then lysed in ice-cold lysis buffer (20 mM Tris/HCl pH 8, 137 mM NaCl, 1% NP-40, 2 mM EDTA) supplemented with protease and phosphatase-inhibitor cocktail, sonicated on ice and centrifuged at 4 °C for 5 min at 16,000×g. Protein concentrations were determined by modified Lowry assay (Bio-Rad DC protein assay).
Nuclear and cytoplasmic fractionation was performed following the manufacturer's protocol for NE-PER (#78835, ThermoFisher Scientific). Briefly, cells were grown on a 100 mm dish to 70% confluency. Cells were trypsinized and pelleted at 500×g for 5 min. Next, 100 µL CER I was added per 10 µL cell pellate. The cells were then vortexed at maximum speed for 15 s and incubated on ice for 10 min. For every 100 µL of CER I, 5.5 µL CER II was added and cells were vortexed and incubated on ice for 1 min. The lysate was centrifuged at 16,000×g for 5 min. The supernatant (cytoplasmic fraction) was transferred to a clean tube and frozen at −80 °C. The pellate was then resuspended in 50 µL of NER for every 100 µL of CER I that was added. The nuclei were washed in PBS and centrifuged at 16,000×g for 5 min, followed by vortexing at maximum speed for 15 s every 10 min for a total of 40 min. The lysate was centrifuged at 16,000×g for 5 min and the supernatant (nuclear fraction) was transferred to a clean tube and stored at −80 °C. Samples were analyzed by immunoblotting as described above.
2.6. Immunoblotting
Protein lysates (20–40 µg) were separated by electrophoresis on a 4–20% SDS polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. After blocking with 5–10% BSA, membranes were incubated with primary antibodies for 1 h at room temperature or overnight at 4 °C. The following primary antibodies were used in this study: anti-dinitrophenol (#S7150, as part of the OxyBlot Protein Oxidation Detection Kit, EMD Millipore, 1:150), anti-SQSTM1/p62 (#7695, Cell Signaling, 1:500), anti-Nrf2 (#D1Z9C, Cell Signaling, 1:1000), anti-Keap1 (#ab150654, Abcam, 1:1000), anti-TOPO II β (#ab125297, Abcam, 1:1000), anti-LC3 (#D50G8, Cell Signaling, 1:1000), anti-ubiquitin (#sc-8017, Santa Cruz, 1:250), anti-GCLC (#PA5–44189, Invitrogen, 1:1000), anti-glutathione reductase (#ab16801, Abcam, 1:1000), anti-Bcl2 (#sc-7382 Santa Cruz, 1:1000), anti-GAPDH (#5174, Cell Signaling, 1:2,000), and anti-actin (#sc-1616, Santa Cruz, 1:500). The blots were then incubated with a horseradish peroxidase-conjugated secondary antibody (1:10,000) and developed using a chemiluminescence detection system (Santa Cruz Biotechnology or LiCor Premium substrate). Band intensity was determined by densitometry with NIH Image J software (v1.48h3) using mean gray value minus background.
In some experiments, cells were treated with bafilomycin A1 (50 nM, #S1413, SellectChem) at 37 °C prior to lysis.
2.7. Immunoprecipitation
For immunoprecipitation, 200 µg of protein lysate was immunoprecipitated overnight at 4 °C with 2 µg of anti-Keap1 or anti-Nrf2 antibody in the presence of 20 µL Dynabeads (#58095, Invitrogen). Beads with protein bound were washed with PBS. Bound protein was eluded by incubating beads in 15 µL 2× Laemmli Sample Buffer (350001052, Bio-Rad) at 98 °C for 5 min, followed by analysis by immunoblotting as described above. In some experiments, WT cells were treated with the proteasome inhibitor MG132 (25 μM, #474790, Calbiochem) at 37 °C for 2 h.
2.8. Luciferase assay
In WT and MsrA-/- VSMC, luciferase activity was measured using the Promega Luciferase assay system (#E1501, Promega) per the manufacturer's recommendations. Briefly, VSMC plated on a 100 mm plate at 50–70% confluency were washed in HBSS and harvested in 900 µL 1× lysis buffer. Cells were vortexed at high speed for 15 s and centrifuged at 12000 × g for 2 min at 4 °C. Cell lysate (20 µL) was added to 100 µL of luciferase assay reagent, vortexed briefly and placed into the luminometer. Luminescence was measured using Femptomaster FB 12 luminometer (Zylux Corporation) every 10 s for 60 s. The relative light units over the 60-second period were averaged and normalized to total protein levels as determined by DC assay (#5000111, Bio-Rad).
2.9. ROS measurements
2.9.1. CellROX assay
To assess intracellular pro-oxidants such as superoxide, hydrogen peroxide, and hydroxyl radicals, VSMC were plated at 30% confluency on a chamber slide and allowed to attach overnight. Cells were treated with 5 µM CellROX Deep Red reagent (diluted in DMSO) for 30 min at 37 °C (#C10422, ThermoFisher). The stained cells were washed with PBS and fixed in 4% PFA for 15 min. The fixed cells were then imaged on an LSM 510 confocal microscope (Carl Zeiss).
2.9.2. L012 assay
Additionally, we measured the oxidation of L012, a surrogate marker of intracellular superoxide levels. VSMC at 50–75% confluency in a 100 mm dish were trypsinized, pelleted and washed with PBS. Immediately before the assay, cells were resuspended in 500 µL lysis buffer. Lysate (100 µL, approximately 20 µg) was mixed with 100 µL of 1 mM NADPH and 795 µL 1× PBS. Next, 5 µL of 20 mM L012 was added to the mixture and the sample was analyzed using a luminometer as described above.
2.9.3. Dihydroethidium (DHE) fluorescence
Moreover, we assessed the oxidation of DHE, another a surrogate marker of intracellular superoxide. For this purpose, stock solutions of DHE (20 nM) were made using 25 mg DHE (D1168, Invitrogen) dissolved in 3.95 ml DMSO and aliquoted under Argon vapor. DHE working solution was made using 8 µL DHE stock solution in 20 ml of PBS; solution was protected from light. Slides were incubated for 15 min at room temperature in DHE working solution and protected from light. Following incubation, slides were quickly rinsed and cover slipped with PBS. The sections were then imaged on an LSM 510 confocal microscope (Carl Zeiss).
2.9.4. Carbonylation assay (OxyBlot)
Carbonylated proteins were determined using the OxyBlot protein oxidation detection kit (#S7150, EMD Millipore) per the manufacturer's instructions. Briefly, 20 µg of protein lysate was reacted with 10 µL 1× DNPH or derivatization control (negative control) in 5% SDS for 15 min. To stop the reaction, 7.5 µL of neutralization solution was added. The samples were then separated by SDS-PAGE and analyzed by immunoblotting as described above.
2.10. Immunofluorescence
VSMC (1000 cells) were plated on an 8-well chamber slide and allowed to attach overnight. Cells were washed with HBSS and fixed in 4% PFA for 15 min, followed by permeabilization with 0.1% Triton X-100 in cold PBS for 5 min. After blocking cells in PBS containing 5% FBS and 0.3% Triton X-100 for 60 min, cells were incubated with anti-SQSTM1/p62 (1:150) overnight at 4 °C, followed by incubation with Alexa Fluor 488-conjugated secondary antibody (#A-11034, ThermoFisher, 1:500) for 1.5 h at room temperature. Nuclei were stained with TOPRO III (#T3605, Invitrogen, 1:2000). Cells were imaged on an LSM 510 confocal microscope (Carl Zeiss). In some experiments, cells were treated with bafilomycin A1 (#S1413, SellectChem, 50 nM) as indicated at 37 °C prior to fixation.
Paraffin sections were mounted with VectaShield mounting media (VectaShield). Sections were blocked in 5% milk and egg substitution. Sections were probed with Mouse anti-actin (Santa Cruz, #sc-32251, 1:500) for 45 min at RT, and Rabbit anti-SQSTM1/p62 (D10E10) (#7695, Cell Signaling, 1:100) overnight at 4 °C. Sections were incubated with secondary antibodies goat anti-mouse IgG-biotinylated (#BMK-2202, Vector Labs, 1:500) for 10 min, Goat anti-rabbit IgG, F(ab')2 fragment-Alexa Fluor 488 conjugated (#A11070, Life Technologies, 1:500) overnight at 4 °C, and Streptavidin-Alexa Fluor 568 conjugated (#S11226, Life Technologies, 1:500) for 1 h at RT. Nuclei were stained with TOPRO III (#T3605, Invitrogen, 1:2000) for 5 min at RT. Images were acquired with a Zeiss 710 Confocal Microscope at 100× magnification. All images for a given experiment were acquired with the same imaging settings.
2.11. GSH/GSSG measurements
Total GSH and GSSG content were determined on samples prepared in 5% 5-sulfosalicylic acid as described previously [33]. Glutathione disulfide (GSSG) was determined by adding a 1:1 mixture of 2-vinylpyridine and ethanol to the samples and incubating for 2 h before assaying as described previously [34]. In this recycling assay, the rates of the reaction were compared with similarly prepared GSH and GSSG standard curves. Both determinations were normalized to the protein content of the insoluble pellet from the 5-sulfosalicylic acid treatment, dissolved in 0.1 N NaOH with 1% SDS, using the BCA Protein Assay Kit (Thermo Scientific).
2.12. Glutathione reductase (GR) and GST activity assays
GR activity was measured using a protocol that was adapted from a previously described assay [35] The following solutions were mixed, Reagent A: 100 mM potassium phosphate buffer / 3.4 mM EDTA (PB/EDTA) pH 7.6, Reagent B: 30 mM oxidized glutathione substrate solution (GSSG), Reagent C: 0.8 mM NADPH (β-NADPH), Reagent D: 1% bovine serum albumin (BSA). The absorbance was measure (A340 nm) until constant (approximately 5 min). Samples were added to cuvet and A340 nm was measured for 3 min. The ΔA340 nm /minute were obtained using the maximum linear rate for blank and sample. Activity was calculated as follows.
GST activity was measured as previously described [36]: The following was mixed in a cuvette, 900 µL 100 mM Phosphate buffer with 1 mM Diethylenetriamine penta-acetic acid (DETAPAC) (Sigma #D-6518), 40 µL 25 mM 1-Chloro-2,4-dinitrobenzene (CDNB) (Sigma #C-6396, 27 µL GSH 93.7 μM (Sigma #G-4251)), and 33 µL sample. The A340 nm was measured for 2.5 min.
2.13. Apoptosis assays
2.13.1. TUNEL staining
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining was performed with the InSitu Cell Death Detection Kit, TMR Red Kit as recommended by the manufacturer (12156792910, Roche). Nuclei were counterstained with TOPRO III (#T3605, Invitrogen). Cells were imaged on an LSM 510 confocal microscope (Carl Zeiss). TUNEL positive cells were counted using NIH Image J software and calculated as the percentage of TUNEL-positive cells relative to the total number of cells.
2.13.2. Annexin V
VSMC were trypsinized and incubated with 100 µL MUSE Annexin V Dead Cell Kit (#MCH100105, Millipore) for 20 min at room temperature in the dark. Cells were then run on the MUSE flow cytometer.
2.14. Statistical analysis
Data are shown as mean ± SE. Statistical significance of multiple treatments was determined using GraphPad Prism Software Version 7 by Student's t-test or ANOVA. A probability value ≤ 0.05 was considered significant.
3. Results
3.1. p62-positive inclusion bodies are increased with MsrA deletion
To extend findings from other models of protein aggregation disease [22], [23], we first determined whether VSMC with constitutive MsrA deletion contain increased levels of p62-positive protein aggregates. First, total protein levels of p62 were significantly increased in cultured MsrA-/- VSMC compared to cells isolated from WT littermates (Fig. 1A, B). Similarly, p62 protein levels were elevated in lysates from in carotid arteries (Fig. 1C, D). As predicted, immunofluorescent staining of cultured VSMC and carotid artery sections revealed an increase in the p62-positive aggregates (Fig. 1E-G). Addition of bafilomycin, an autophagy inhibitor that prevents degradation of protein aggregates, led to an increase in p62-positive inclusion bodies in both WT and MsrA-/- VSMC, suggesting that formation of p62 aggregates is a dynamic process (Fig. 1E). To assess whether the increase in p62 protein is due to increased transcription, we performed quantitative real time PCR and detected no significant difference in p62 mRNA levels (Fig. 1H).
Fig. 1.

p62 is elevated in MsrA-deficient VSMC and arteries. (A, B) Representative immunoblot (A) and summary data (B) for p62 in whole cell lysates of VSMC isolated from MsrA-/- and WT mice. Data were normalized to GAPDH loading control and expressed relative to p62 levels in WT VSMC (n = 9 biological replicates). (C, D) Representative immunoblot (C) and summary data (D) for p62 in whole cell lysates of carotid arteries isolated from MsrA-/- and WT mice (n = 10 biological replicates). E) Representative immunofluorescent images of p62 (green) and nuclei (TOPRO, blue) in VSMC from MsrA-/- and WT mice with or without treatment with bafilomycin a1 (Baf) for 24 h. Scale bars 20 µm. (F) Quantification of p62 aggregates from (E). Arbitrary aggregate score was calculated as the mean GFP fluorescence intensity per cell in at least 5 images per biological replicate (1–5 cells/image; n = 5 biological replicates). (G) Representative immunofluorescent images of p62 (green), smooth muscle actin (red) and nuclei (TOPRO, blue) in carotid artery sections from MsrA-/- and WT mice. 100 × , scale bar 10 µm. NC denotes negative control without primary antibody, p62 inset with p62 (green) only. Arrows denote p62 aggregates. (H) mRNA expression of p62 in VSMC from MsrA-/- and WT mice by qRT-PCR; data were normalized to ARP and expressed relative to p62 in WT VSMC (n = 5 biological replicates). * p < 0.05 versus WT by two-tailed t-test. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Autophagy is upregulated in MsrA-/- VSMC
Since high levels of p62 are often associated with impaired autophagic flux, we next tested the autophagy activity in MsrA-/- VSMC. At baseline, we detected an increase in LC3-II, the active form of LC3 that is necessary for formation of the autophagosome, in MsrA-/- VSMC compared to WT (Fig. 2A, B). To determine if the autophagic flux is preserved, cells were incubated with bafilomycin, an inhibitor of the late phase of autophagy that prevents maturation of autophagic vacuoles by inhibiting the fusion between autophagosomes and lysosomes. Upon treatment of WT VSMC with bafilomycin, we detected a strong increase in active LC3-II (Fig. 2A, B). In MsrA-/- cells, LC3-II was also increased after administration of bafilomycin compared to baseline, suggesting that autophagic flux is preserved. Moreover, protein levels of p62 were elevated in MsrA-/- VSMC at baseline and 24 h after bafilomycin treatment (Fig. 2C, D).
Fig. 2.

Autophagy is increased in MsrA-/- VSMC. (A) Immunoblot of autophagy marker LC3 in MsrA-/- and WT VSMC with or without bafilomycin a1 (Baf) for indicated times. Upper band represents full-length LC3-I and lower band active, cleaved LC3-II. Positive control for autophagy: treatment with serum-free media (SF) for 2 h. (B) Quantification of LC3-II in MsrA-/- and WT VSMC relative to LC3-II in WT VSMC at 0 h; data were normalized to GAPDH (n = 4–5 biological replicates). * p < 0.05 versus WT by 2-way ANOVA. (C) Immunoblots for p62 and GAPDH in in MsrA-/- and WT VSMC with or without bafilomycin a1 (Baf) for 24 h. (D) Data in (C) were normalized to GAPDH and expressed relative to p62 in WT VSMC at 0 h. n = 8 biological replicates * p < 0.05 versus WT at untreated and # p < 0.05 vs MsrA-/- untreated by 1-way ANOVA. (E) MsrA-/- and WT VSMC were transduced with an adenovirus expressing LC3-GFP for 72 h. To some samples, bafilomycin was added for 24 h. Nuclei: blue (TOPRO). Scale bars 20 µm. (F) GFP puncta were quantified per cell, adjusted for cell area and expressed per 1000 µm2. n = 5 biological replicates. * p < 0.05 versus WT untreated # p < 0.05 vs MsrA-/- untreated by 1-way ANOVA.
To substantiate that autophagy is indeed active in MsrA-/- conditions, we transduced WT and MsrA-/- VSMC with a GFP-LC3 adenovirus and assessed the formation of punctate LC3, which indicates autophagosomal localization [37]. As shown in Fig. 2E, F, there was a 7-fold increase in fluorescent puncta in VSMC with MsrA deletion, and this was strongly augmented by bafilomycin treatment in MsrA-/- VSMC. These data indicate that autophagy is induced in MsrA-/- even in the absence of an additional stimulus that activates protein degradation or turnover.
3.3. The association of p62 and Keap1 is increased with MsrA deletion
To establish the effect of MsrA on Nrf2 activation via the alternative pathway whereby p62 competes with Nrf2 for Keap1 binding [27], we first immunoprecipitated Keap1 and probed for p62. In MsrA-/- VSMC, we detected an increased association between the p62 and Keap1 (Fig. 3A, B). These results predict that Nrf2 is released from its inhibition by Keap1 when MsrA is deleted. To test this hypothesis, we immunoprecipitated Nrf2 and detected reduced association of Keap1 with Nrf2 under MsrA-/- conditions (Fig. 3C, D). Thus, we anticipated increased nuclear localization of Nrf2 and antioxidant response elements (ARE)-dependent transcriptional activity in MsrA-/- VSMC. More Nrf2 protein was detected in the nucleus of MsrA-/- VSMC as compared to WT by immunoblot (Fig. 3E, F). Additionally, Nrf2 protein was increased in cytosolic fractions (Fig. 3E, G). Lastly, we detected significantly increased Nrf2 transcriptional activity using an ARE-dependent luciferase reporter (Fig. 3H).
Fig. 3.

MsrA deletion abrogates the interaction of Nrf2 with Keap1, resulting in Nrf2 nuclear localization and transcriptional activity. (A, B) Representative immunoblots (A) and quantitation (B) of the interaction of Keap1 with p62 in MsrA-/- and WT VSMC. Immunoprecipitation for Keap1, immunoblots for p62 and Keap1. n = 6 biological replicates. (C, D) Representative immunoblots (C) and quantitation (D) of the interaction of Keap1 with Nrf2. Immunoprecipitation for Nrf2, immunoblots for Keap1 and Nrf2. n = 4 biological replicates. (E-G) Representative immunoblot (E) and summary data for Nrf2 localization in nuclear (F) and cytoplasmic fractions (G) of MsrA-/- and WT VSMC. TOPO IIβ: nuclear marker; GAPDH: cytoplasmic marker. n = 7 biological replicates. (H) Nrf2-dependent transcriptional activity as determined by quantification of ARE-dependent luciferase activity in MsrA-/- and WT VSMC expressing ARE-luciferase reporter. n = 6 biological replicates. * p < 0.05 versus WT by two-tailed t-test.
3.4. Increased Nrf2 protein levels in MsrA-/- VSMC are mediated by decreased ubiquitination
To provide mechanistic insight into the increase in Nrf2 protein levels, we performed confirmatory immunoblots in whole cell lysates and determined the mRNA expression levels in WT and MsrA-/- VSMC. Whereas the total Nrf2 protein levels were consistently increased under MsrA deficiency (Fig. 4A, B), no difference in Nrf2 mRNA levels was seen in passage-matched samples (Fig. 4C). These data suggest that the strong increase in Nrf2 protein is due to changes in protein turnover. Nrf2 is targeted for degradation after Keap1-mediated ubiquitination. Since the association of Nrf2 with Keap1 is reduced with MsrA deletion, we tested the ubiquitination of Nrf2 and detected decreased ubiquitination in MsrA-/- compared to WT VSMC (Fig. 4D, E).
Fig. 4.

Elevated Nrf2 protein expression under MsrA deficiency is due to protein stabilization rather than increased transcription. (A, B) Representative immunoblot (A) and quantification (B) for Nrf2 protein levels in whole cell lysates from MsrA-/- and WT VSMC; n = 3 biological replicates. (C) Nrf2 mRNA levels in VSMC by qRT-PCR; n = 5 biological replicates. (D) Representative immunoprecipitation of Nrf2 followed by immunoblot for ubiquitin in MsrA-/- and WT VSMC. IgG: IP with IgG, WT + MC132: IP with anti-Nrf2 in WT VSMC incubated with MG132, WCL: whole cell lysate of WT VSMC as controls. (E) Quantification of (D); n = 7 biological replicates. (F) p62 mRNA levels by qRT-PCR in aortic samples WT, MsrA-/- and MsrA-/- x Nrf2-/- mice; n = 7, 9 biological replicates. (G) Representative Immunoblots for Nrf2 and GAPDH in aortic samples from WT, MsrA-/- and MsrA-/- x Nrf2-/- mice. (H) Quantification of (G) n = 7 biological replicates. (E) * p < 0.05 versus WT by two-tailed t-test, H) p < 0.05 versus WT by 1-way ANOVA.
p62 has been proposed as a transcriptional target of Nrf2 [38]. Since our data demonstrate that MsrA deficiency results in increased p62 expression and Nrf2-dependent transcription, we examined the impact of dual MsrA and Nrf2 knockout on p62 expression. First, mRNA levels of p62 were similar in MsrA-/- aorta with or without Nrf2 deletion (Fig. 4F). These data suggest that the increase in p62 under MsrA deficiency is not explained by elevated Nrf2-dependent transcription. Accordingly, p62 protein levels were not significantly different in MsrA-/- x Nrf2-/- aortic samples as compared to MsrA-/- samples (Fig. 4G).
3.5. Glutathione synthesis is increased in MsrA-/- VSMC
Nrf2 drives the ARE-dependent transcription of many antioxidant genes [39], [40], [41]. An increase in ARE-dependent gene transcription has been described with acute MsrA knockdown [17]. While previous data reported on increases in heme oxidase 1, a detailed understanding of the MsrA-driven ARE response is missing. Here, we concentrated on glutathione synthesis and recovery since it represents a major cellular antioxidant pathway that affects the cellular redox state. Transcription of glutamate-cysteine ligase (GCLC), the first enzyme in the glutathione synthesis pathway, and glutathione reductase (GR) is dependent on Nrf2. Accordingly, the mRNA and protein levels of GCLC and GR were significantly increased in MsrA-/- VSMC as compared to WT (Fig. 5A-E). Moreover, the activity of GR was increased (Fig. 5F). As a result, the intracellular concentration of reduced glutathione (GSH) was increased in MsrA-/- VSMC (Fig. 5G), whereas the concentrations of oxidized glutathione (GSSG) were not different between genotypes (Fig. 5H). The resultant ratio of GSH/GSSG was higher in MsrA-/- VSMC (Fig. 5I). Lastly, the activity of glutathione transferase was increased (Fig. 5J). Collectively, these data illustrate that in MsrA-/- VSMC, the glutathione-driven antioxidant response is elevated.
Fig. 5.

Expression and activity of Nrf2-regulated genes are elevated under MsrA deletion. (A, B) GCLC (A) and GR (B) mRNA levels in MsrA-/- and WT VSMC by qRT-PCR, normalized to ARP; n = 3 biological replicates. (C-E) Representative immunoblot (C) and summary data for GCLC (D) and GR (E) protein levels in whole cell lysates from MsrA-/- and WT VSMC. Data were normalized to GAPDH; n = 6–7 biological replicates. (F) GR activity, normalized to total protein concentration; n = 4 biological replicates. (G, H) Levels of reduced glutathione (GSH, G) and oxidized glutathione (GSSG, H), normalized to total protein; n = 4 biological replicates. (I) Ratio of reduced/oxidized glutathione. (J) Glutathione transferase (GST) activity, normalized to total protein; n = 7 biological replicates. * p < 0.05 versus WT by two-tailed t-test.
3.6. Reactive oxygen species production is not increased with constitutive MsrA deletion
MsrA deletion impairs the response to pro-oxidant stress. In studies of NIH3T3 mouse fibroblasts and rat VSMC, acute MsrA knockdown increased ROS production and Nrf2 activity [17]. To establish how constitutive MsrA deletion affects oxidative status, we determined acute changes in intracellular oxidation with CellRox and L-012 in cultured VSMC. However, no increase in oxidation of these probes occurred (Fig. 6A-C). We then measured DHE oxidation in carotid arteries of WT and MsrA-/- mice. The intensity of DHE staining was comparable in the two genotypes (Fig. 6D, E). Lastly, we determined the level of carbonylated proteins in MsrA-/- VSMC and tissue samples from MsrA-/- mice. Again, no difference in protein carbonylation was detected in VSMC or arterial samples (Fig. 6F-H). However, in accordance to published data, protein carbonylation was increased in kidney lysates from MsrA-/- mice as compared to WT (Fig. 6G, H). These data demonstrate that constitutive deletion of MsrA does not appear to lead to increases in steady-state levels of ROS, although ARE-dependent gene transcription is induced.
Fig. 6.

MsrA deletion does not increase oxidative stress in vascular smooth muscle cells or aorta. (A, B) Representative images (A) and quantification (B) of Cell ROX fluorescence (red) in MsrA-/- and WT VSMC. Nuclei: blue (TOPRO). Data were quantified as the fold-change relative to WT; n = 4 biological replicates. Scale bars 20 µm. (C) Superoxide levels in MsrA-/- and WT VSMC by L-012. Data were normalized to total protein; n = 4 biological replicates* p < 0.05 versus WT by two-tailed t-test. (D, E) Representative images (D) and summary data (E) of DHE staining (red) in aortic sections from MsrA-/- and WT mice; n = 11 biological replicates. Scale bars 50 µm. (F, G) Representative immunoblots (F) and summary data (G) for oxidized proteins in the indicated tissues from MsrA-/- and WT mice. Oxidized proteins were normalized to GAPDH (loading control); n = 4 biological replicates. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.7. Apoptosis is diminished in MsrA-/- VSMC
Autophagy and apoptosis both control the turnover of organelles and proteins within cells. Whereas low levels of cellular stress are believed to elicit autophagy, increased stress conditions induce apoptosis. Since autophagy generally blocks the induction of apoptosis, we postulated that apoptosis would be decreased in MsrA-/- VSMC since we detected increased autophagy in Fig. 2. Accordingly, Annexin-V immunofluorescence and TUNEL labeling were decreased in MsrA-/- VSMC (Fig. 7A-D). Moreover, we detected higher protein levels of the anti-apoptotic protein BCl-2 (Fig. 7E, F). However, the addition of 100 μM H2O2 for 60 min readily induced abundant apoptosis in MsrA-/- but not WT VSMC (Fig. 7G), substantiating previous findings that MsrA deficiency increases the sensitivity to stress-induced apoptosis [42].
Fig. 7.

Apoptosis is decreased at baseline in MsrA-deficient vascular smooth muscle cells. (A, B) Representative dot plot (A) and summary data (B) of Annexin V by flow cytometry in MsrA-/- and WT VSMC. Data were quantified as the number of Annexin V positive cells relative to the total number of cells; n = 7–9 biological replicates. (C, D) Representative images (C) and summary data (D) TUNEL staining (red) in MsrA-/- and WT VSMC. Nuclei: blue (TOPRO); n = 5 biological replicates. Scale bars 50 µm. (E, F) Representative immunoblot (E) and summary data (F) for Bcl-2 expression in whole cell lysates from MsrA-/- and WT VSMC. Data were normalized to GAPDH; n = 6–7 biological replicates. (G) Oxidative stress-induced apoptosis by Annexin V staining (green) in MsrA-/- and WT VSMC treated with the indicated concentrations of H2O2 for 60 min (H) Summary data for G. n = 7–12 replicates. Nuclei: red (TOPRO). (B, D, F) * p < 0.05 versus WT by two-tailed t-test. (H) # p < 0.05 versus WT by multiple t-tests, one per row. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
Here, we demonstrate abundant p62-containing aggregates in cultured, dedifferentiated VSMC in vitro and in arterial MsrA-/- VSMC in vivo. Concomitantly, autophagic activity in VSMC was increased. The association of p62 and Keap1 was higher in MsrA-/- versus WT VSMC. As a result, the protein levels and transcriptional activity of Nrf2 were enhanced, resulting in enhanced transcription of ARE-mediated detoxifying genes. MsrA deletion did not appear to affect hydroxyl radical and superoxide anion production. The upregulation of p62 was not altered when Nrf2 was deleted in MsrA-/- samples, suggesting that p62 complex formation under these conditions is independent of the antioxidant response. These data demonstrate that MsrA deletion in the absence of any additional stress stimuli affects cellular homeostasis via accumulation of protein aggregates, increased autophagy and antioxidant response. In the absence of increased oxidative stress, it is unlikely that Keap1 is degraded as a result of cysteine oxidation, the canonical step by which Keap1 releases Nrf2 repression and degradation. We propose that the abundant p62 competes for Keap1. Our findings thereby link two observations that have been made in studies of MsrA biology; impaired protein degradation with formation of intracellular protein inclusion bodies and the induction of Nrf2-dependent transcription.
Repair of oxidized methionine by Msrs has been a target of increased interest in the last decade. As the reduction of methionine sulfoxide is an evolutionarily well-conserved mechanism, Msrs and their orthologs have been investigated in a wide range of organisms spanning from yeast [43] and bacteria [44], to drosophila [11], C. elegans [45] and mice [31]. However, progress towards dissecting Msr function has been hampered by the absence of straightforward systematic methods to identify Msr protein substrates [46], [47], [48]. Therefore, Msrs have been investigated in mammalian models of diseases that are driven by high oxidative stress, such as asthma, neurodegenerative diseases and various forms of cardiovascular disease [18], [30], [49], [50], [51], [52], [53]. Methionine oxidation also occurs with environmental stress, for example diesel exhaust particles. A recent study demonstrated that diesel exhaust particles activate MsrA-driven protein repair and activate autophagy and proteasomal protein degradation [54]. Our study contributes to the understanding of Msr biology under baseline conditions [55] when oxidative stress is not excessive by dissecting how the loss of reduction of methionine sulfoxide affects protein repair mechanisms and intersects with antioxidant signaling. As such, we posit that our observed effects of MsrA deficiency on autophagy and antioxidant signaling represent baseline shifts in cellular homeostasis, which when superimposed with excess oxidative stress, are responsible for the various pathologies that have been linked with Msr deficiency. In fact, several prior studies detected effects of MsrA deficiency at baseline without further characterizing the mechanism [12], [52], [56]. We believe that our findings may also be helpful in interpreting the various genome-wide association studies that have identified Msrs as targets in a range of diseases from obesity and cardiovascular disease to deafness and schizophrenia [57], [58], [59], [60].
Modulation of autophagy has emerged as a target for cardiovascular therapy [61]. In models of balloon angioplasty, autophagy is believed to contribute to VSMC dedifferentiation immediately after injury, which would stimulate the formation of neoatherosclerosis [62]. However, defective autophagy also accelerates senescence and promotes ligation-induced neointima formation [63]. A similarly contradictory picture has emerged in atherosclerosis, where autophagy is protective and contributes to plaque stability by degrading damaged intracellular materials in VSMC, [64] similar to our findings in the absence of a disease model. On the other hand, excessive stimulation of autophagy results in autophagic death of VSMC during atherogenesis [65]. Therefore, autophagy formation has different implications under different conditions. Whereas the methods used to alter autophagy in various target cells may be a novel pathway for the therapy of vascular diseases, the findings of this study cannot readily be translated to a particular disease phenotype or treatment strategy.
We and others have detected increased NFκB activity in MsrA-/- mice [66], [67]; however, the mechanism by which NFκB is activated is not entirely understood. One explanation could be decreased IκK degradation by impaired Keap1-dependent ubiquitination [68]. Potential insight also comes from studies reporting that p62 association with TRAF6 enhances the activation of IκK by TRAF6 [69], [70], [71], [72]. Our findings of increased p62 protein levels with MsrA deletion in this study point towards a potential mechanism, in which TRAF6 binding to p62 and subsequent IκK activation may be enhanced. Such experimental evidence for this potential pathway would create a new paradigm in which p62, through competitive binding to transcriptional regulators like Keap1 and TRAF6, mediates transcriptional pathways under MsrA deficiency.
5. Summary
Here, we demonstrate that lack of MsrA promotes formation of p62-containing protein aggregates and autophagy. The enhanced association of Keap-1 with the p62 derepresses Nrf2 by reducing its degradation. As a consequence, Nrf2 protein levels, its nuclear localization and ARE-dependent gene transcription are increased in the absence of enhanced oxidative stress. These findings demonstrate that reduction of intracellular methionine sulfoxide is necessary for cellular homeostasis and survival.
Acknowledgements
We thank Dr. Kristina W. Thiel for assistance in preparation of the manuscript, Dr. Chantal Allamargot, the Central Microscopy Research Facility, University of Iowa for assistance with immunofluorescence detection of p62.
Acknowledgments
Funding
This work was supported by Merit Review Grants (Biomedical Laboratory Research and Development Service) I01 BX000163 (to IMG), by NIH R01 HL 108932 (to IMG), R01 CA182804 and P30 CA086862 (to DRS), by T32 HL007121 (to PRK and SMP) and American Heart Association 14POST19860006 (to PRK). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.04.001.
Appendix A. Supplementary material
Supplementary material
References
- 1.Robison A. Oxidation of calmodulin alters activation and regulation of CaMKII. Biochem. Biophys. Res. Commun. 2007;356(1):97–101. doi: 10.1016/j.bbrc.2007.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu S.X., Stevens J.W., Lentz S.R. Regulation of thrombosis and vascular function by protein methionine oxidation. Blood. 2015;125(25):3851–3859. doi: 10.1182/blood-2015-01-544676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erickson J.R. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciorba M.A. Modulation of potassium channel function by methionine oxidation and reduction. Proc. Natl. Acad. Sci. USA. 1997;94(18):9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones E.M., Squier T.C., Sacksteder C.A. An altered mode of calcium coordination in methionine-oxidized calmodulin. Biophys. J. 2008;95(11):5268–5280. doi: 10.1529/biophysj.108.139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carruthers N.J., Stemmer P.M. Methionine oxidation in the calmodulin-binding domain of calcineurin disrupts calmodulin binding and calcineurin activation. Biochemistry. 2008;47(10):3085–3095. doi: 10.1021/bi702044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahoo N., Hoshi T., Heinemann S.H. Oxidative modulation of voltage-gated potassium channels. Antioxid. Redox Signal. 2014;21(6):933–952. doi: 10.1089/ars.2013.5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su Z. Functional consequences of methionine oxidation of hERG potassium channels. Biochem. Pharmacol. 2007;74(5):702–711. doi: 10.1016/j.bcp.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine R.L. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. 1996;93(26):15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boschi-Muller S., Branlant G. Methionine sulfoxide reductase: chemistry, substrate binding, recycling process and oxidase activity. Bioorg. Chem. 2014;57:222–230. doi: 10.1016/j.bioorg.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Ruan H. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA. 2002;99(5):2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moskovitz J. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. USA. 2001;98(23):12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmon A.B. Lack of methionine sulfoxide reductase A in mice increases sensitivity to oxidative stress but does not diminish life span. FASEB J. 2009;23(10):3601–3608. doi: 10.1096/fj.08-127415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J.I. Protective role of methionine sulfoxide reductase A against ischemia/reperfusion injury in mouse kidney and its involvement in the regulation of trans-sulfuration pathway. Antioxid. Redox Signal. 2013;18(17):2241–2250. doi: 10.1089/ars.2012.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh M.P., Kim K.Y., Kim H.Y. Methionine sulfoxide reductase A deficiency exacerbates acute liver injury induced by acetaminophen. Biochem. Biophys. Res. Commun. 2017;484(1):189–194. doi: 10.1016/j.bbrc.2017.01.025. [DOI] [PubMed] [Google Scholar]
- 16.Singh M.P. Methionine sulfoxide reductase A protects hepatocytes against acetaminophen-induced toxicity via regulation of thioredoxin reductase 1 expression. Biochem. Biophys. Res. Commun. 2017;487(3):695–701. doi: 10.1016/j.bbrc.2017.04.119. [DOI] [PubMed] [Google Scholar]
- 17.Kim J.Y. Methionine sulfoxide reductase A attenuates heme oxygenase-1 induction through inhibition of Nrf2 activation. Arch. Biochem. Biophys. 2012;528(2):134–140. doi: 10.1016/j.abb.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Liu F. Methionine sulfoxide reductase A protects dopaminergic cells from Parkinson's disease-related insults. Free Radic. Biol. Med. 2008;45(3):242–255. doi: 10.1016/j.freeradbiomed.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabbita S.P. Decrease in peptide methionine sulfoxide reductase in Alzheimer's disease brain. J. Neurochem. 1999;73(4):1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 20.Moskovitz J. Methionine sulfoxide reductase A affects beta-amyloid solubility and mitochondrial function in a mouse model of Alzheimer's disease. Am. J. Physiol. Endocrinol. Metab. 2016;310(6):E388–E393. doi: 10.1152/ajpendo.00453.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minniti A.N. The protein oxidation repair enzyme methionine sulfoxide reductase a modulates Abeta aggregation and toxicity in vivo. Antioxid. Redox Signal. 2015;22(1):. 48–62. doi: 10.1089/ars.2013.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H. From autophagy to mitophagy: the roles of P62 in neurodegenerative diseases. J. Bioenerg. Biomembr. 2017;49(5):413–422. doi: 10.1007/s10863-017-9727-7. [DOI] [PubMed] [Google Scholar]
- 23.Manley S., Williams J.A., Ding W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013;238(5):525–538. doi: 10.1177/1535370213489446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Z., Klionsky D.J. An overview of the molecular mechanism of autophagy. In: Levine B., Yoshimori T., Deretic V., editors. Vol. 335. Springer; Berlin, Heidelberg: 2009. pp. 1–32. (Autophagy in Infection and Immunity. Current Topics in Microbiology and Immunology). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kansanen E. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang D.D., Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 28.Taguchi K. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA. 2012;109(34):13561–13566. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dialynas G. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015;11(5):e1005231. doi: 10.1371/journal.pgen.1005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klutho P.J. Deletion of methionine sulfoxide reductase A does not affect atherothrombosis but promotes neointimal hyperplasia and extracellular signal-regulated kinase 1/2 signaling. Arterioscler. Thromb. Vasc. Biol. 2015;35(12):2594–2604. doi: 10.1161/ATVBAHA.115.305857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moskovitz J. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. USA. 2001;98(23):12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 33.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27(3):502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 34.Griffith O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980;106(1):207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 35.Ray L.E., Prescott J.M. Isolation and some characteristics of glutathione reductase from rabbit erythrocytes (38548) Proc. Soc. Exp. Biol. Med. 1975;148(2):402–409. doi: 10.3181/00379727-148-38548. [DOI] [PubMed] [Google Scholar]
- 36.Spitz D.R., Malcolm R.R., Roberts R.J. Cytotoxicity and metabolism of 4-hydroxy-2-nonenal and 2-nonenal in H2O2-resistant cell lines. Do aldehydic by-products of lipid peroxidation contribute to oxidative stress? Biochem. J. 1990;267(2):453–459. doi: 10.1042/bj2670453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D.J. Klionsky, et al., Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 12(1): p. 1-222. [DOI] [PMC free article] [PubMed]
- 38.Jain A. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285(29):22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paul N. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD (P) H: quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem. J. 2003;374(2):337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thimmulappa R.K. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62(18):5196–5203. [PubMed] [Google Scholar]
- 41.Jaiswal A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004;36(10):1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 42.Sreekumar P.G. Protection from oxidative stress by methionine sulfoxide reductases in RPE cells. Biochem. Biophys. Res. Commun. 2005;334(1):245–253. doi: 10.1016/j.bbrc.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 43.Moskovitz J. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc. Natl. Acad. Sci. USA. 1997;94(18):9585–9589. doi: 10.1073/pnas.94.18.9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh V.K., Moskovitz J. Multiple methionine sulfoxide reductase genes in Staphylococcus aureus: expression of activity and roles in tolerance of oxidative stress. Microbiology. 2003;149(Pt 10):2739–2747. doi: 10.1099/mic.0.26442-0. [DOI] [PubMed] [Google Scholar]
- 45.Minniti A.N. Methionine sulfoxide reductase A expression is regulated by the DAF-16/FOXO pathway in Caenorhabditis elegans. Aging Cell. 2009;8(6):690–705. doi: 10.1111/j.1474-9726.2009.00521.x. [DOI] [PubMed] [Google Scholar]
- 46.Oien D.B. Detection of oxidized methionine in selected proteins, cellular extracts and blood serums by novel anti-methionine sulfoxide antibodies. Arch. Biochem. Biophys. 2009;485(1):35–40. doi: 10.1016/j.abb.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wehr N.B., Levine R.L. Wanted and wanting: antibody against methionine sulfoxide. Free Radic. Biol. Med. 2012;53(6):1222–1225. doi: 10.1016/j.freeradbiomed.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghesquiere B. Redox proteomics of protein-bound methionine oxidation. Mol. Cell Proteom. 2011;10(5) doi: 10.1074/mcp.M110.006866. (M110 006866) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erickson J.R. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He B.J. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011;17(12):1610–1618. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanders P.N. CaMKII is essential for the proasthmatic effects of oxidation. Sci. Transl. Med. 2013;5(195):195ra97. doi: 10.1126/scitranslmed.3006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wassef R. Methionine sulfoxide reductase A and a dietary supplement S-methyl-L-cysteine prevent Parkinson's-like symptoms. J. Neurosci. 2007;27(47):12808–12816. doi: 10.1523/JNEUROSCI.0322-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanski J. Role of glycine-33 and methionine-35 in Alzheimer's amyloid beta-peptide 1-42-associated oxidative stress and neurotoxicity. Biochim. Biophys. Acta. 2002;1586(2):190–198. doi: 10.1016/s0925-4439(01)00097-7. [DOI] [PubMed] [Google Scholar]
- 54.Lai C.H. Protein oxidation and degradation caused by particulate matter. Sci. Rep. 2016;6:33727. doi: 10.1038/srep33727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ugarte N. Proteome alteration in oxidative stress-sensitive methionine sulfoxide reductase-silenced HEK293 cells. Free Radic. Biol. Med. 2013;65:1023–1036. doi: 10.1016/j.freeradbiomed.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Ogawa F. The repair enzyme peptide methionine-S-sulfoxide reductase is expressed in human epidermis and upregulated by UVA radiation. J. Investig. Dermatol. 2006;126(5):1128–1134. doi: 10.1038/sj.jid.5700116. [DOI] [PubMed] [Google Scholar]
- 57.Ma X. A genome-wide association study for quantitative traits in schizophrenia in China. Genes Brain Behav. 2011;10(7):734–739. doi: 10.1111/j.1601-183X.2011.00712.x. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Bermudez M. Association of the methionine sulfoxide reductase A rs10903323 gene polymorphism with cardiovascular disease in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2012;41(5):350–353. doi: 10.3109/03009742.2012.677063. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y. MSRA polymorphism is associated with the risk of rheumatoid arthritis in a Chinese population. Scand. J. Rheumatol. 2013;42(2):91–96. doi: 10.3109/03009742.2012.730626. [DOI] [PubMed] [Google Scholar]
- 60.Ahmed Z.M. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am. J. Hum. Genet. 2011;88(1):19–29. doi: 10.1016/j.ajhg.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gottlieb R.A. Untangling autophagy measurements: all fluxed up. Circ. Res. 2015;116(3):504–514. doi: 10.1161/CIRCRESAHA.116.303787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salabei J.K. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem. J. 2013;451(3):375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grootaert M.O. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. 2015;11(11):2014–2032. doi: 10.1080/15548627.2015.1096485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schrijvers D.M., De Meyer G.R., Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2011;31(12):2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- 65.Levine B., Yuan J. Autophagy in cell death: an innocent convict? J. Clin. Investig. 2005;115(10):2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan H. Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-kappaB signaling pathways through a catalytic antioxidant function. Antioxid. Redox Signal. 2015;22(10):832–847. doi: 10.1089/ars.2014.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu S.X. Protein methionine oxidation augments reperfusion injury in acute ischemic stroke. JCI Insight. 2016;1(7) doi: 10.1172/jci.insight.86460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tian H. Keap1: one stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett. 2012;325(1):26–34. doi: 10.1016/j.canlet.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 69.Zhang P. Activation of IKK by thymosin alpha1 requires the TRAF6 signalling pathway. EMBO Rep. 2005;6(6):531–537. doi: 10.1038/sj.embor.7400433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wooten M.W. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005;280(42):35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- 71.Sanz L. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19(7):1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zotti T. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol. Immunol. 2014;58(1):27–31. doi: 10.1016/j.molimm.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material