

Abstract
Obstructive sleep apnea (OSA) is a highly prevalent disease across the lifespan, is characterized by chronic intermittent hypoxia and sleep fragmentation, and has been independently associated with substantial cardiometabolic morbidity. However, the reversibility of end-organ morbidity with treatment is not always apparent, suggesting that both tissue remodeling and epigenetic mechanisms may be operationally involved. Here, we review the cumulative evidence focused around murine models of OSA to illustrate the temporal dependencies of cardiometabolic dysfunction and its reversibility, and more particularly to discuss the critical contributions of tissue macrophages to adipose tissue insulin resistance and vascular atherogenesis. In addition, we describe initial findings potentially implicating epigenetic alterations in both the emergence of the cardiometabolic morbidity of OSA, and in its reversibility with treatment. We anticipate that improved understanding of macrophage biology and epigenetics in the context of intermittent hypoxia and sleep fragmentation will lead to discovery of novel therapeutic targets and improved cardiovascular and metabolic outcomes in OSA.
Keywords: obstructive sleep apnea, sleep fragmentation, intermittent hypoxia, adipose tissue, insulin resistance, macrophage, epigenetics
Obstructive sleep apnea (OSA) is a common disease affecting a high proportion of the population and is strongly associated with obesity. Large epidemiological cohorts have consistently shown the presence of relatively prominent and independent associations between OSA and cardiovascular comorbidities, including systemic hypertension, stroke and ischemic heart disease [1]. In addition, compelling evidence on the putative independent association between OSA and metabolic dysfunction, including insulin resistance (IR), diabetes and dyslipidemia, has also emerged in the last decade [2,3]. Notwithstanding the flurry of epidemiological and intervention studies examining these associations, the mechanisms that putatively mediate the direct of effect of OSA on target cardiovascular and metabolic organs have not been as extensively explored.
To better understand the pathophysiological links between OSA and vascular and metabolic perturbations, animal models of OSA have been developed. The initial studies employed invasive models of tracheally-implanted balloons in big animals, such as dogs and pigs, to reproduce repetitive pharyngeal obstruction mimicking apneas [4,5]. Upper airways obstruction was achieved through implantation of tracheal tubes with balloons whose inflation and deflation was remotely controlled by computers to elicit repetitive occlusions of the endotracheal tube balloon. Alternatively, other investigators used collagen injections into the pharyngeal walls leading to narrowed airway diameter, decreased upper airway volume, along with increased resistance and collapsibility. However, several scientific, economic and ethical issues progressively reduced the popularity of such models. Instead, and considering the large framework of genetically engineered rodent models, the easier access to rats and mice in research laboratories worldwide, and the improved ability to miniaturize instrumentation enabling characterization of sleep and other cardiovascular and metabolic phenotypes in mice, the latter have progressively become the most frequently used species in OSA research. As a consequence, alternative techniques to surgical airway obstruction have been implemented in rodent models and are predicated upon the knowledge of the immediate consequences of apneic events on blood oxygenation and sleep architecture. In human disease, each apnea is associated with a fall in oxygen saturation followed by a quick re-oxygenation upon upper airway opening. Each apnea is also traditionally ended by an arousal from sleep that allows for a more effective control of the upper airway muscles leading to airway opening and recovery of the airflow. Thus, OSA is associated with an intermittent hypoxemia and sleep fragmentation. Implementation of these paradigms in murine experimental settings has provided unique insights into the cardiometabolic consequences of OSA and will be discussed here (Figure 1).
Figure 1. Putative mechanisms of adipose tissue dysfunction and macrophage polarization resulting from intermittent hypoxia and sleep fragmentation as suggested by animal models.
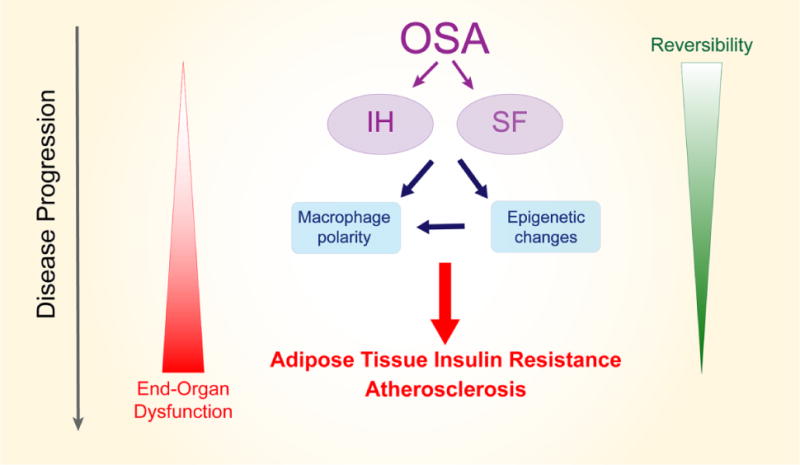
Obstructive sleep apnea is characterized by chronic intermittent hypoxia (IH) and sleep fragmentation (SF) and both perturbations can induce direct changes in macrophage polarity and function toward a pro-inflammatory phenotype, as well as promote the emergence of epigenetic changes which also lead to macrophage polarization and facilitate persistent, and possibly less reversible inflammation. Consequently, vascular wall and adipose tissue infiltration by pro-inflammatory macrophages will result in end-organ structural changes and altered function, as reflected by insulin resistance and atherosclerosis.
Intermittent hypoxia alters adipose tissue insulin sensitivity, macrophage polarity and lipid processing
Prospective epidemiological studies have shown a clear independent association between OSA and IR and diabetes [2,3]. Especially, most of the studies showed that nocturnal desaturations have the most robust relationship with impairments in glucose homeostasis [3,6,7]. Animal models have further confirmed the supposition that intermittent hypoxia is causally involved in IR pathophysiology.
Intermittent hypoxia is the most widely used animal models of OSA. Intermittent hypoxemia and re-oxygenation are achieved by varying nitrogen and oxygen concentration in mice cages, usually via automated computer-controlled gas-exchange systems. Most models employ short cycles (30 seconds to 2 minutes) of hypoxia usually with a nadir environmental oxygen concentration of 5-7% and aim to elicit arterial oxyhemoglobin levels revolving around 75-80%, i.e., corresponding to the levels seen in moderate to severe OSA in humans. Although there is no upper airway occlusion in this model, some respiratory efforts (intermittent tachypnea) occur, corresponding to a fluctuating hyperventilation that follows the intermittent hypoxia cycles. The intermittent hypoxia stimulus is usually applied during the daytime, as rodents preferentially sleep during this period. Some models have adopted a sleep-driven imposition of rapid oxygen environmental reductions, and although these models have provided useful insights into the physiologic adaptations to such closed circuit approaches, their throughput was too low to accommodate the numbers of experimental subjects required for mechanistic studies. Notwithstanding, substantial differences between studies have emerged regarding the duration of daytime exposures (ranging from 4 hours/day to 12 or 24 hours/day), the overall duration of intermittent hypoxia (from days to months) and the frequency of cycling of desaturating events during these periods (i.e., 10-60/h), such that comparisons across published studies can only be qualitative rather than more quantitative in nature. Nevertheless, even short term intermittent hypoxia has been shown to reproduce the major characteristics of the pathophysiological consequences of OSA, including increases in blood pressure, decreased insulin sensitivity and sympathetic activation [8,9]. When applied for more extended periods of time, intermittent hypoxia during the rest period will induce atherosclerosis and insulin resistance, and when combined with high fat diets or when using transgenic mice it may even reproduce the metabolic syndrome phenotype described in severe human OSA patients [8,9].
In otherwise wild type young adult non-obese mice, exposures to intermittent hypoxia were shown to impair glucose homeostasis, resulting in altered glucose and insulin tolerance tests [10,11] and increased homeostatic model assessment of insulin resistance index (HOMA-IR) [12,13], all of which partially improve with cessation of intermittent hypoxia exposure [14]. The impact of intermittent hypoxia is clearly more pronounced when applied to mice with genetically- or diet-induced obesity [10,12]. In this context, it has become apparent that a central role in these metabolic manifestations is played by adipose tissue (AT), particularly in chronic intermittent hypoxia models. AT is an important and highly adaptive organ playing a myriad of functional roles, but importantly involved in energy storage. AT functions as a key endocrine regulator involved in numerous metabolic functions through the secretion of numerous hormones, cytokines and chemokines referred as adipokines [15]. Emerging evidence suggests that white AT becomes dysfunctional in the context of both obesity and intermittent hypoxia, and that similar pathways may be operational in these 2 conditions, particularly when considering that local fluctuating AT hypoxia occurs in obese individuals [16]. In the context of evolving obesity, AT mass increases at a faster rate than the regional angiogenesis, resulting in areas with low vascular supply, and consequently low oxygenation [17]. In the context of OSA, the intermittent hypoxemia is systemic, and studies have shown that the amplitudes of blood saturation cycling are markedly dampened in AT. Indeed, cycles of 90 seconds of hypoxia (6.3% FIO2) and 90 seconds of normoxia resulted in in-vivo variations of local AT tissue oxygen tensions ranging from 20 mmHg to 40 mmHg [18]. Interestingly, such chronic oscillations in AT oxygen tension do not appear to enable sustained recruitment and transcription of hypoxia-inducible factor 1 signaling, leading to vascular rarefaction in AT. This is in marked contrast with chronic sustained hypoxia, whereby both marked increases in AT vasculogenesis and tissue perfusion are accompanied by increased AT insulin sensitivity despite similar magnitude of tissue hypoxia [18].
AT IR following intermittent hypoxia has been now repeatedly documented ex-vivo and in-vivo. Eight weeks of intermittent hypoxia exposures have been shown to decrease Akt phosphorylation in response to insulin, a surrogate reporter of insulin receptor sensitivity, in both epididymal and mesenteric AT [13]. Similar decreases in Akt phosphorylation were recently documented at the cellular level in 3T3 adipocytes exposed in vitro to intermittent hypoxia [19].
Since AT inflammation has been implicated in the emergence of IR, evidence suggesting the possibility that intermittent hypoxia may induce and propagate local AT inflammation has also been explored. Indeed, in mice subjected to intermittent hypoxia exposures mimicking sleep apnea, evidence for increased inflammation and oxidative stress becomes clearly apparent in AT, and contributes to the metabolic dysfunction. The inflammatory processes in the AT of mice exposed to intermittent hypoxia are illustrated by a wide range of changes in the numbers and types of inflammatory cells present in AT [20]. Through a process involving collagenase digestion, filtration and centrifugation, it has become possible to isolate the stromal-vascular fraction of the AT, and characterize the changes in leucocyte populations using specific antibodies and flow cytometry. These experiments revealed that in mice exposed to intermittent hypoxia, despite a lower body weight and lower AT mass, intermittent hypoxia mimicking OSA was accompanied by global increases in macrophage counts. Notably, increased numbers of pro-inflammatory M1 and of pro-inflammatory metabolic CD36+ macrophages [21] were present in AT of intermittent hypoxia-exposed mice along with reciprocal decreases in anti-inflammatory M2 macrophages [11,13,22]. Interestingly, the percentage of M1 macrophages correlated with the degree of IR, the latter being assessed by insulin tolerance tests [19]. At the cellular level, 3T3 adipocytes exposed in vitro to intermittent hypoxia showed increased TNF-α, IL-8 and IL-6 mRNA expression suggesting a direct impact of intermittent hypoxia on macrophage M1 inflammatory polarization [19]. AT macrophages of mice exposed to intermittent hypoxia also exhibited increased reactive oxygen species production, and substantial changes in electron transport chain function, indicating dysfunctional bioenergetics and mitochondrial autophagy processes [22]. The deleterious implications of oxidative stress and inflammation induced by chronic intermittent hypoxia are also suggested by the protective effects of resveratrol, a putative anti-inflammatory and anti-oxidant natural compound, which elicited significant ameliorations in intermittent hypoxia-induced systemic and AT metabolic dysfunction and in macrophage migration and polarization within visceral white AT [13]. We should also remark that recent evidence has pointed to both intermittent hypoxia and sleep fragmentation inducing significant changes in gut microbiota that are also accompanied by altered permeability of the colonic epithelium, which then translates into systemic inflammation and metabolic dysfunction [23–25]. Preliminary support for these observations in murine models translating to humans has been garnered from a recent study in children with OSA, whereby plasma levels of lipopolysaccharide binding protein, likely reflecting the increased permeability of colonic epithelium, were elevated in these children, correlated with insulin resistance, and improved after treatment [26].
Although we have focused thus far on intermittent hypoxia-induced AT dysfunction, we should also mention that intermittent hypoxia can also alter muscular and liver insulin sensitivity [11]. Sympathetic excitation occurs in OSA, and increases in both tonic and reactive sympathetic outflow are readily detected in intermittent hypoxia-exposed rodents [27]. Increased sympathetic activation potentially induces lipolysis [28] as suggested by increased circulating free fatty acid levels [12]. Circulating fatty acids in turn can accumulate in peripheral muscles and liver leading to decreased “insulin receptor substrate” phosphorylation, and consequent insulin induced glucose uptake [29,30]. Furthermore, pharmacological inhibition of lipolysis by acipimox ameliorates insulin sensitivity in mice subjected to intermittent hypoxia [31]. However, surgical or pharmacological inhibition of sympathetic activity is not sufficient to completely prevent intermittent hypoxia-induced IR, suggesting that complementary mechanisms are involved [32]. intermittent hypoxia induced dyslipidemia can also be related to decreased lipoprotein lipase expression and activity that may be mediated by changes in HIF-1 signaling, particularly via one of HIF-1 downstream regulated genes, namely angiopoietin-like 4 [33,34]. Finally, we should emphasize that intermittent hypoxia decreases adiponectin circulating levels in rodents as well as in in-vitro exposed adipocytes [35,36]. Considering that adiponectin is the most abundant white AT secreted adipokines, and that it possesses insulin-sensitizing, anti-inflammatory and anti-atherogenic properties, the reductions in adiponectin following intermittent hypoxia are likely to be involved in the emergence of IR [37,38]. However, the role of intermittent hypoxia on leptin levels is more controversial. Short term intermittent hypoxia increases leptin levels [39], whereas decreased levels have been shown in chronic intermittent hypoxia models, potentially reflecting the reduction in AT mass associated with long intermittent hypoxia exposures [40]. Leptin is another important metabolic cytokine secreted by AT, with both local and remote functional roles, particularly in satiety signaling in the hypothalamus. Leptin signaling pathway inhibition foster orexigenic behaviors and body mass accrual, but a direct implication of leptin on insulin signaling has also been suggested [41]. How leptin pathways are affected by intermittent hypoxia and their role in intermittent hypoxia-induced IR will require further investigation.
Besides inflammatory cellular changes and functional alterations, structural histological changes are apparent in the visceral AT of mice exposed to intermittent hypoxia. Exposures to 6 weeks of intermittent hypoxia resulted in whitening of epididymal AT, which contrasted with a browning of AT observed in animals exposed to sustained hypoxia [22]. White AT and brown AT have essentially antagonistic functions: white AT stores excess energy as triglycerides, and is involved in metabolic regulation, whereas brown AT is specialized in the dissipation of energy through the production of heat [42]. As mentioned above, differential transcriptional activity temporal trajectories of HIF-1α between intermittent hypoxia and sustained hypoxia were proposed to explain some of the histological and metabolic differences imposed by these two hypoxic exposures [22]. intermittent hypoxia was also associated with a rarefication of AT vascularization and the presence of vascular dysfunction as attested by a decrease in eNOS expression and reduced eNOS phosphorylation at the activation site (serine 1177). Another typical characteristic feature of dysfunctional AT in obesity is the formation of crown-like structures consisting of macrophages surrounding necrotic adipocytes [15]. Similar to obesity, AT in lean animals treated with intermittent hypoxia showed an increased amount of crown-like structures compared to normoxic controls [19].
An important question has arisen in recent years as to whether long-term metabolic reductions in AT insulin sensitivity are completely reversible upon return to normoxic environments, i.e., ideally optimal treatment of OSA in patients. One of the major reasons for such question originates from the fact that the majority of the patients with OSA have likely suffered from this condition for many years, and therefore remodeling of end-organ tissues may have occurred and preclude full and complete functional recovery. The currently available evidence is scarce. However, even though 6-week intermittent hypoxia exposures were accompanied by normalization of IR in AT and also by complete restoration of vascular integrity in mice [43], only partial improvements were present in either metabolic AT dysfunction or in vascular inflammation and atherogenesis after very lengthy intermittent hypoxia exposures (20 weeks) [44], suggesting that some of the processes may progressively become either irreversible or only partially reversible. Since after normoxic recovery, evidence for persistent inflammatory processes as illustrated by pro-inflammatory macrophage polarity was detectable, the possibility that epigenetic changes in macrophages may be occurring and dictating some of the dampening of the recovery processes was raised.
Epigenetics in macrophages
Macrophages were originally described more than a century ago as large phagocytic cells which ingests foreign material [45]. Extensive research throughout the years revealed that macrophages are de facto key components of the innate immune system, and play a key role in maintaining systemic and tissue homeostasis [46]. In particular, monocytes and macrophages are crucial to the regulation of immune responses and inflammation by acting as secretory cells and presenting antigens to helper T cells[45].
The current mononuclear phagocyte theory states that migratory macrophages are differentiated cells which do not proliferate, but are rather re-populated by circulating monocytes derived from myeloid precursors originating in the bone marrow [47]. In addition, a different cell lineage of macrophages, tissue resident macrophages, displays the capacity for cell division and replication, and originates from a different embryonic cell line. In general, monocytes develop to tissue-specific macrophages in response to local microenvironment signals, and are present in virtually all the tissues in the body [48]. Tissue-specific macrophages have a high degree of pleiotropy, with a remarkable adaptability to react to environmental stimuli [49]. However, such plasticity contributes to the development of several pathologies by altering molecular profiles and subsequently activation states of the macrophages, as observed in sepsis, cancer, atherosclerosis, obesity, rheumatoid arthritis and neurodegenerative diseases [50]
The macrophage population within any given tissue or organ is highly variable and presents a continuum of phenotypes representing different activation states [51]. At the two ends of the spectrum, two major phenotypes have been described: the classically activated phenotype or M1, and the alternatively activated phenotype or M2 [52]. While M1 macrophages present a pro-inflammatory phenotype, M2 macrophages have different metabolic functions and promote healing and growth, as seen in wound healing and tissue remodeling [53–55]. M1 macrophages respond to lipopolysaccharide (LPS) and interferon gamma (IFNγ) stimuli, present a pro-inflammatory profile, and express inducible NO synthase (iNOS), interleukin (IL)-1,and TNF-α [53,54]. Conversely, several sub-types of M2 macrophages are triggered by different stimuli: IL-4 and IL-13 lead to M2a phenotype, immune complexes to M2b, and glucocorticoids and transforming growth factor beta (TGF-β) to M2c [53,54]. M2 macrophages express a group of characteristic genes, the “M2 genes”, such as chitinase-like protein (Ym1), found in inflammatory zone 1(Fizz1), arginase-1(Arg)-1, IL-10 and TGF-β. M1 and M2 macrophages are usually characterized by their protein markers (reviewed in [53]). Identified markers for M1 macrophages are CD64, IDO, SCOCS1, CXCL10 in human, and CXLC9, CXCL10, CXCL11 and NOS2 in mouse. For M2 macrophages the most commonly used markers are MRC1, TGM2, CD23 and CCL22 in humans, and Mrc1, Tgm2, Fizz1, Ym1/2 and Arg1 in mouse. Our group has shown that intermittent hypoxia promotes the emergence of insulin resistance and the increased presence of a pro-inflammatory metabolic macrophage phenotype in both visceral white adipose tissue (VWAT) and aorta, as illustrated by increased macrophage counts and increased M1 polarization [18,22,56,57].
The importance of epigenetic mechanisms in the activation and regulation of macrophage function is becoming widely accepted and numerous studies have provided evidence in this direction [58–62]. Epigenetics refers to the heritable changes in phenotype that occur without changes in the DNA sequence [63]. Epigenetic processes regulate early cellular differentiation through interactions between genes and the environment, modulating gene transcription and leading to changes in cellular phenotype [64]. Epigenetic studies focus on three related molecular mechanisms for genome regulation: DNA modifications, histone modifications and non-coding RNAs (ncRNAs). DNA modification is the covalent modification of a nucleotide in the DNA sequence. The most studied DNA modifications are the addition of a methyl or hydroxylmethyl group at the 5′ position of the cytosine nucleotide (C), termed as 5-methylcytosine (5mC) and 5-hydroxylmethylcytosine (5hmC), respectively [65,66]. In the mammalian genome, these additions occur almost exclusively in the context of a cytosine-guanine (CG) dinucleotide and may regulate the expression of the cognate genes [67]. The second epigenetic mechanism in genome regulation is determined by the organization of the histones in the nucleosomes. Histone modifications include methylation, acetylation, phosphorylation, ubiquitination, sumoylation, citrullination, and ADP-ribosylation. Either working as individual marks or in a combinatorial pattern, histone modifications are major regulators of gene expression [68] and can act as activating or repressing marks. The third layer of epigenetic regulation is given by ncRNAs. This category includes several types of RNA molecules which are not coding for proteins, but may have a function on genome regulation, such as micro RNAs (miRNA), small interfering RNAs (siRNA) and long non-coding RNAs (lncRNA) [69,70]. All of these epigenetic changes and potential functional implications have thus far been reported for macrophages in a variety of settings [54]. As will be discussed later in this review, increasing evidence in animal models and clinical studies supports a major role for epigenetic alterations in macrophages in OSA-induced metabolic and cardiovascular alterations.
Epigenetic changes in macrophages in adipose tissues and their contribution to insulin resistance
AT exhibit marked cellular heterogeneity, including adipocytes pre-adipocytes, fibroblasts, endothelial cells and tissue resident and migrated macrophages. In obesity, excessive amounts of visceral white AT lead to autocrine and paracrine dysregulation contributing to metabolic dysfunction and chronic inflammation. As discussed above, peripheral AT insulin resistance results from a combination of altered functions of insulin target cells and the accumulation of macrophages that secrete pro-inflammatory mediators [71].
Among AT macrophages, both phenotypes, M1 and M2, have been found depending on the physiological conditions [72–75]. Moreover, a novel metabolically active M1-like macrophage phenotype has been described in the context of obesity-related AT inflammation and insulin resistance [21], whereby positive CD36 surface labeling conclusively identified this metabolic M1 macrophage phenotype. In lean individuals, AT macrophages present the alternatively activated, M2 phenotype, whereas AT macrophages in obese individuals present the pro-inflammatory, M1 phenotype [72,73,76]. Macrophage phenotype and function highly depend on the microenvironment [49], and polarization is the result of complex molecular mechanisms involving transcriptional regulation and chromatin remodeling. At the molecular level, insulin resistance is promoted by a transition from an alternative M2-like macrophage activation state maintained by STAT6 and PPARs to the classical M1 activation state driven by NF-kappaB, AP1, and other signal-dependent transcription factors that play crucial roles in innate immunity [72–75]. Increasing evidence shows that epigenetics mechanisms play a major role in the AT macrophage polarization. Yang and collaborators showed that elevated saturated fatty acids enhance DNMT3b expression in obesity, leading to DNA methylation at the PPARγ1 promoter, which may contribute to deregulated AT macrophage polarization, inflammation, and insulin resistance [62]. Furthermore, several studies described the importance of chromatin remodeling in the M2-M1 switch, highlighting the action of histone modification enzymes in activating or repressing the expression of genes (reviewed in [58,60]. For example, the Jumonji family of demethylases (in particular Jmjd3) is recruited in LPS-treated macrophages leading to decreased expression of M2-associated genes [58,77]. In contrast, it has been shown that the action of histone deacetylase HDAC3 leads to the M1 phenotype by inhibiting M2 polarization [78,79]. Comprehensive studies (reviewed in [80]) have identified three major effects of intermittent hypoxia exposures in macrophages from VWAT: i) induction of a pro-inflammatory phenotype with polarization of adipose tissue macrophages towards a M1-pro-inflammatory subtype, ii) upregulation and secretion of numerous pro-inflammatory adipokines and cytokines, and iii) downstream impairment of the insulin-signaling pathway, similar to the alterations observed in adipose tissue dysfunction in obesity. Very interestingly, extended recovery under normoxic conditions can only partially reverse these changes [22].
Epigenetic changes in macrophages and atherosclerosis
Vascular tissue macrophages play a major role in atherogenesis. During the initiation and progression of atherosclerosis, macrophages show intrinsic activation, changes in polarity and phenotype, production of cytokines, and signaling to other cells in the vessel wall [81–85]. M1-like macrophages accumulate as the atheroma plaque grows, and are associated with adverse disease outcome [81–85]. CD36-mediated activation of aortic macrophages was found in pro-atherogenic disease models [44]. Different epigenetic mechanisms have been shown to determine the phenotype and activity profiles of macrophages, and more specifically the pro- or anti-atherogenic profile of macrophages in vascular areas prone to atherosclerosis [79,86,87]. In the context of intermittent hypoxia, Castro-Grattoni and collaborators showed that the atherogenic process is potentially reversible with return to normoxia following short intermittent hypoxia exposures[43]. However as shown by Cortese et al, such reversibility is markedly hampered following prolonged intermittent hypoxia exposures [44], suggesting that epigenetic alterations may have occurred in intermittent hypoxia-exposed macrophages. In the context of sleep fragmentation, Carreras et al described marked elastic fiber disruption and fiber disorganization in sleep fragmentation-exposed mice, along with increases in the numbers of foam cells and macrophages within the aorta wall. Senescence markers further showed evidence for reduced TERT and cyclin A, and increased p16INK4a expression, with higher IL-6 plasma levels in sleep fragmentation-exposed mice [88].
As discussed below, shared epigenetic mechanisms may be operational in both vascular and adipose intermittent hypoxia-exposed tissues in the context of macrophages.
The peroxisome proliferator-activated receptor-gamma (PPARγ) pathway. A shared mechanism of epigenetic-mediated macrophage activation in adipose and vascular tissues
The peroxisome proliferator-activated receptor-gamma (PPARγ) pathway. PPARγ pathway represents one of the major molecular and biochemical pathways underlying the prevention of M1 macrophage phenotype polarization and preservation of M2 phenotype in visceral white AT [49]. Indeed, PPARγ-deficient macrophages are resistant to M2 polarization and promote insulin resistance [89,90]. PPARγ regulates the expression of downstream genes by binding to PPAR-response elements (PPAREs) located in the promoter and enhancer of these genes [91]. For example, it directly controls the expression of numerous adipocyte-specific genes, such as adiponectin (Adipoq), fatty acid binding protein 4 (Fabp4, also known as adipocyte protein 2 or aP2), resistin (Retn), and glucose transporter 4 (Glut4). In macrophages, PPARγ is a master regulator of lipid metabolism, and it has long been known to inhibit pro-inflammatory gene expression through several mechanisms, including the trans-repression of NF-kappaB [92,93]. In AT macrophages, PPARγ is constitutively expressed, but can be also induced by IL-4 and IL-13 [94]. A crosstalk between PPARγ and the IL-4/STAT6 axis, which might coordinate the control of the M2-like phenotype, has been also evoked [95]. An alternative pathway for crosstalk involves IL-4-mediated stimulation of PPARγ activation through the synthesis of putative endogenous PPARγ ligands [94]. Pharmacological PPARγ agonists (i.e. thiazolidinediones, TZD) are used to treat type 2 diabetic patients leading to an improved glycemic control and enhanced insulin sensitivity [96]. These drugs also improve the lipid profile of patients at risk of developing atherosclerosis and reduce circulating levels of inflammatory markers [96].
PPARγ is activated in a ligand-dependent manner, switching the binding partner from a co-repressor complex to a co-activator complex on PPAREs. Both PPARγ complexes contain molecules with known epigenetic activities which act as chromatin remodeling factors, such as the histone acetyltransferase p300/CBP[97], histone deacetylases [98], the methyltransferase CARM1 [99] and a SWI/SNF complex [100]. Moreover, it has been shown that the PPARγ complex has the ability to direct local DNA de-methylation at PPAREs [101]. On PPAREs, the PPARγ complex is poly (ADP-ribosyl)ated (PARylation) and Tet proteins present in the complex catalyze the conversion from a repressive 5-methylcytosine (5-mC) DNA modification to an active 5-hydroxylmethylcytosine (5-hmC), inducing local DNA demethylation and gene expression [101].
Evidence for epigenetic modifications in OSA patients and in intermittent hypoxia-exposed mice
As discussed above, the cellular and molecular mechanisms involved in OSA-induced metabolic and cardiovascular derangements are not fully understood. However, the insofar cumulative evidence supports a major role for macrophages and inflammation in the development of OSA morbidity. In this context, recent work from our laboratory has now described activation of pro-atherogenic pathways involving a complex interplay of histone modifications in functionally-relevant biological pathways, such as inflammation and oxidative stress in aorta macrophages [44]. One of the major pathways regulated by long-term intermittent hypoxia and exhibiting extensive epigenetic alterations was the PPARγ pathway, thereby providing initial support for conceptual framework that epigenetic modifications occur in intermittent hypoxia, and may not only contribute to end-organ morbidity, but may also dictate the potential reversibility of such alterations upon cessation of intermittent hypoxia.
To further elucidate these issues, we studied DNA methylation profiles in blood monocytes of obese patients with severe OSA before and 6 weeks after initiation of treatment with positive airway pressure [102]. We identified over 1,800 differentially methylated regions. Analysis of biochemical pathways and gene networks demonstrated that differentially methylated regions were associated with immune responses, and particularly with mechanisms governing gene regulation by PPARγ [102]. Of note, we have also detected specific gene methylation changes in genes involved in immune regulation and vascular function in children with OSA [103,104].
Other groups have also highlighted the importance of the PPARγ pathway in in OSA-induced metabolic and cardiovascular disorders. Using a microarray-based transcriptome profiling, Gharib and colleagues identified a number of gene sets that were up-regulated in the adipose tissue of OSA patients, including those in the pro-inflammatory NF-κB pathway and the proteolytic ubiquitin/proteasome pathways, whereas genes in the PPAR pathway were down-regulated in OSA patients [105]. Sharma and collaborators studied alveolar macrophages in obese subjects with and without OSA [106], and showed that PPARγ expression and functional activity were significantly reduced in obese subjects with OSA compared to those without OSA. More recently, Zhang et al have shown that PPARγ gene is downregulated in mice with High Fat Diet (HFD)-induced obesity, resulting in oxidative stress and inflammation in response to chronic intermittent hypoxia contributing to endothelial dysfunction. Endothelial-dependent vasodilation was impaired in HFD-obese mice exposed to chronic intermittent hypoxia, compared with Low Fat Diet (LFD)-fed mice and to HFD-mice not exposed to intermittent hypoxia, but endothelial function was restored by concomitant pioglitazone treatment, a known PPAR-γ agonist[107].
Thus, epigenetic alterations are detectable in experimental models of OSA, as well as in actual patients suffering from this condition, further reinforcing the need for continued exploration of this avenue to identify their contributions to disease morbidity and response to therapy.
Sleep fragmentation and adipose tissue insulin resistance, macrophages and lipid processing
Notwithstanding the metabolic perturbations described heretofore in the context of intermittent hypoxia, intermittent hypoxia alone cannot explain all of the vascular and metabolic consequences of OSA [108]. As indicated, apneic episodes are usually accompanied by bursts of increased brain activity (arousals) leading to repetitive conscious or unconscious awakenings, resulting in sleep discontinuity, i.e., sleep fragmentation. Epidemiological studies have shown that the number of arousals per hour of sleep is associated with fasting insulin levels and IR, even after adjustments for the major confounders [109,110]. Experimental studies in humans have further indicated that mechanical or auditory disruption of sleep decreases insulin sensitivity [111,112].
Murine models have been developed with the aim to reproduce the repetitive arousals from sleep that occur at the end of apneas and hypopneas in OSA. Generation of fragmented sleep is usually achieved by intermittent tactile stimulation using a near-silent motorized horizontal bar sweeping just above the cage floor from one side to the other at a frequency adapted to reproduce the arousal index of moderate to severe OSA (about 30 arousals per hour of sleep) [113,114]. Mice exposed to sleep fragmentation manifest increased food intake that leads over time to accelerated weight accrual even when mice are fed normal chow [115]. However, even before increased AT mass occurs in the context of sleep fragmentation, evidence of IR is detected in mice, as attested by altered glucose tolerance tests and also by reduced glycemic responses during insulin tolerance tests, along with increased HOMA-IR index in fasting animals [23,116–118].
IR was confirmed at the AT level by the presence of reduced p-Akt/Akt ratios in responses to exogenous insulin in mice exposed to sleep fragmentation [23,119]. Of note, the increases in AT mass in the context of sleep fragmentation can be explained by increased bone marrow recruitment, proliferation and differentiation of adipocyte progenitors in the AT [118]. Similar to intermittent hypoxia, sleep fragmentation is also associated with a pro-inflammatory AT cellular profile with increases in M1pro-inflammatory and CD36+ dysmetabolic macrophages, and a reduction in anti-inflammatory M2 macrophages and T-regulatory lymphocytes [117,119]. The pro-inflammatory macrophage profile was further confirmed by their increased ex-vivo ability to release TNF-α. In addition, prolonged sleep fragmentation was also accompanied by remodeling of the vasculature, increased deposition of macrophages in the vascular wall, and increased expression of senescence markers, attesting to incipient atherogenesis [120].
Interestingly, chronic sleep fragmentation also induced reversible gut microbiota changes characterized by the preferential growth of highly fermentative members of Lachnospiraceae and Ruminococcaceae and a decrease of Lactobacillaceae families [23]. Those modifications in the gut microbiome ecosystem may be a critical initiator of the sleep fragmentation-induced systemic and AT inflammation, and consequently altered insulin sensitivity, most likely via enhanced colonic epithelium barrier disruption and increased permeability to LPS ad other bacterial moieties. Of note, the major sleep fragmentation-induced inflammatory alterations within the AT were prevented by administration of resveratrol, a putative sirtuin 1 agonist and insulin sensitizing agent [121]. AT metabolic dysfunction was also restored by administration of TUG891, a potent and selective free fatty acid 4 receptor agonist. It is now well established that FFA4 mediates important functional aspects of both adipocyte and macrophage function, involving stimulation of glucose uptake and adipogenesis, as well as mitigation of inflammatory processes. Notably, FFA4 plasma levels were shown to be reduced in pediatric OSA and in obese children without OSA, and to strongly correlate with the degree of systemic insulin resistance in pediatric patients [122].
Sleep fragmentation was also shown to impact leptin signaling pathways. In this context, despite increases in plasma leptin levels [119], sleep fragmentation was associated with obesogenic behaviors and weight gain. Those findings are likely explained by emergence of leptin resistance induced by sleep fragmentation at the hypothalamic level [123]. Indeed, sleep fragmentation was shown to activate endoplasmic reticulum stress in the hypothalamus ultimately leading to increased protein tyrosine phosphatase 1B (PTP-1B) activity, a negative regulator of leptin signaling pathway in the hypothalamus [124]. Recent work in transgenic mice and using PTP-1B inhibitors have further confirmed the important role of PTP-1B in fostering both central leptin resistance and macrophage polarity changes in sleep fragmentation-exposed mice [125].
Figure 2.
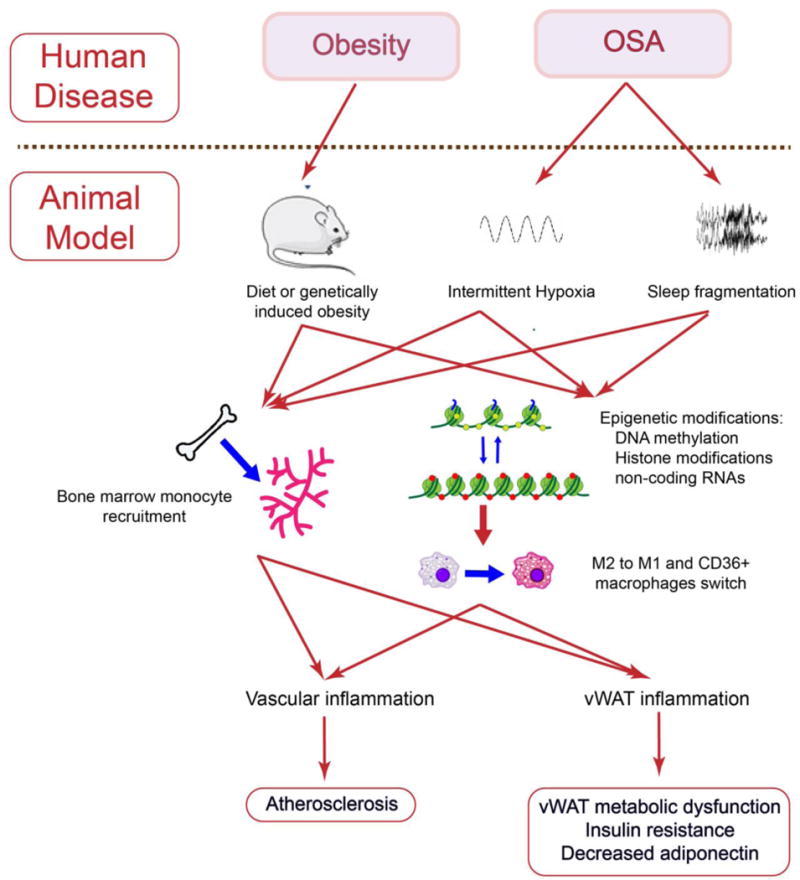
Summary of the pathophysiological processes linking obesity and sleep apnea to vascular atherogenesis and vWAT metabolic dysfunction.
Summary and Translational Implications.
Experimental murine models mimicking characteristic components of OSA lead to the emergence of IR and atherogenesis, and such morbid consequences appear to involve the recruitment and polarity changes of macrophages to a pro-inflammatory phenotype, as well as epigenetic processes (Figure 2). The putative role of such epigenetic changes in dictating the potential for reversibility of the cardiometabolic morbid consequences of OSA is also described, and hopefully will prompt future research efforts in these directions. Based on the current evidence, it is possible that with increased duration of disease, OSA will be accompanied by a progressively reduced reversibility of cardiometabolic morbid phenotypes, particularly those involving atherosclerosis and end-organ insulin resistance. If such assumptions are indeed correct, a complete redesign of randomized controlled therapeutic trials in OSA will be necessary and either take into account antecedent disease duration (a difficult and nearly impossible challenge) or revise down targeted effect sizes and anticipated beneficial outcomes of treatments such as continuous positive pressure therapy or intra-oral appliances, and consequently adjust cohort size needs for adequate power estimates.
Acknowledgments
Financial support: DG is supported by the Herbert T. Abelson Chair in Pediatrics.
Abbreviations
- AT
adipose tissue
- LPS
lipopolysaccharide
- OSA
obstructive sleep apnea
- PPARγ
peroxisome proliferator-activated receptor-gamma
- PTP-1B
protein tyrosine phosphatase 1B
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ Contributions: WT and RC and DG were involved in the redaction of the manuscript. All authors have reviewed and approved the final version of the manuscript.
Disclosure Statement: The authors declare no conflicts of interest in relation to this work.
References
- 1.Javaheri S, Barbe F, Campos-Rodriguez F, Dempsey JA, Khayat R, Javaheri S, et al. Sleep Apnea: Types, Mechanisms, and Clinical Cardiovascular Consequences. J Am Coll Cardiol. 2017;69:841–58. doi: 10.1016/j.jacc.2016.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kendzerska T, Gershon AS, Hawker G, Tomlinson G, Leung RS. Obstructive sleep apnea and incident diabetes. A historical cohort study. Am J Respir Crit Care Med. 2014;190:218–25. doi: 10.1164/rccm.201312-2209OC. [DOI] [PubMed] [Google Scholar]
- 3.Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol. 2004;160:521–30. doi: 10.1093/aje/kwh261. [DOI] [PubMed] [Google Scholar]
- 4.Dematteis M, Godin-Ribuot D, Arnaud C, Ribuot C, Stanke-Labesque F, Pépin J-L, et al. Cardiovascular consequences of sleep-disordered breathing: contribution of animal models to understanding the human disease. ILAR J. 2009;50:262–81. doi: 10.1093/ilar.50.3.262. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Carreras A, Gozal D. Encycl Sleep Kushida C. Academic Press; 2013. Phylogeny and Animal Models. [Google Scholar]
- 6.Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep. 2006;29:777–83. doi: 10.1093/sleep/29.6.777. [DOI] [PubMed] [Google Scholar]
- 7.Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med. 2009;179:228–34. doi: 10.1164/rccm.200804-608OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jun J, Polotsky VY. Metabolic consequences of sleep-disordered breathing. ILAR J. 2009;50:289–306. doi: 10.1093/ilar.50.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanagy NL. Vascular effects of intermittent hypoxia. ILAR J. 2009;50:282–8. doi: 10.1093/ilar.50.3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, et al. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol. 2003;552:253–64. doi: 10.1113/jphysiol.2003.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carreras A, Kayali F, Zhang J, Hirotsu C, Wang Y, Gozal D. Metabolic effects of intermittent hypoxia in mice: steady versus high-frequency applied hypoxia daily during the rest period. Am J Physiol Regul Integr Comp Physiol. 2012;303:R700–709. doi: 10.1152/ajpregu.00258.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obes Silver Spring Md. 2011;19:2167–74. doi: 10.1038/oby.2011.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carreras A, Zhang SXL, Almendros I, Wang Y, Peris E, Qiao Z, et al. Resveratrol attenuates intermittent hypoxia-induced macrophage migration to visceral white adipose tissue and insulin resistance in male mice. Endocrinology. 2015;156:437–43. doi: 10.1210/en.2014-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polak J, Shimoda LA, Drager LF, Undem C, McHugh H, Polotsky VY, et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep. 2013;36:1483–1490. 1490A–1490B. doi: 10.5665/sleep.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diab Rep. 2011;11:203–10. doi: 10.1007/s11892-011-0183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasarica M, Rood J, Ravussin E, Schwarz J-M, Smith SR, Redman LM. Reduced oxygenation in human obese adipose tissue is associated with impaired insulin suppression of lipolysis. J Clin Endocrinol Metab. 2010;95:4052–5. doi: 10.1210/jc.2009-2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gozal D, Gileles-Hillel A, Cortese R, Li Y, Almendros I, Qiao Z, et al. Visceral White Adipose Tissue after Chronic Intermittent and Sustained Hypoxia in Mice. Am J Respir Cell Mol Biol. 2017;56:477–87. doi: 10.1165/rcmb.2016-0243OC. [DOI] [PubMed] [Google Scholar]
- 19.Murphy AM, Thomas A, Crinion SJ, Kent BD, Tambuwala MM, Fabre A, et al. Intermittent hypoxia in obstructive sleep apnoea mediates insulin resistance through adipose tissue inflammation. Eur Respir J. 2017;49 doi: 10.1183/13993003.01731-2016. [DOI] [PubMed] [Google Scholar]
- 20.Poulain L, Thomas A, Rieusset J, Casteilla L, Levy P, Arnaud C, et al. Visceral white fat remodelling contributes to intermittent hypoxia-induced atherogenesis. Eur Respir J. 2014;43:513–22. doi: 10.1183/09031936.00019913. [DOI] [PubMed] [Google Scholar]
- 21.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–25. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gileles-Hillel A, Almendros I, Khalyfa A, Nigdelioglu R, Qiao Z, Hamanaka RB, et al. Prolonged Exposures to Intermittent Hypoxia Promote Visceral White Adipose Tissue Inflammation in a Murine Model of Severe Sleep Apnea: Effect of Normoxic Recovery. Sleep. 2017;40 doi: 10.1093/sleep/zsw074. [DOI] [PubMed] [Google Scholar]
- 23.Poroyko VA, Carreras A, Khalyfa A, Khalyfa AA, Leone V, Peris E, et al. Chronic Sleep Disruption Alters Gut Microbiota, Induces Systemic and Adipose Tissue Inflammation and Insulin Resistance in Mice. Sci Rep. 2016;6:35405. doi: 10.1038/srep35405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreno-Indias I, Torres M, Sanchez-Alcoholado L, Cardona F, Almendros I, Gozal D, et al. Normoxic Recovery Mimicking Treatment of Sleep Apnea Does Not Reverse Intermittent Hypoxia-Induced Bacterial Dysbiosis and Low-Grade Endotoxemia in Mice. Sleep. 2016;39:1891–7. doi: 10.5665/sleep.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno-Indias I, Torres M, Montserrat JM, Sanchez-Alcoholado L, Cardona F, Tinahones FJ, et al. Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur Respir J. 2015;45:1055–65. doi: 10.1183/09031936.00184314. [DOI] [PubMed] [Google Scholar]
- 26.Kheirandish-Gozal L, Peris E, Wang Y, Tamae Kakazu M, Khalyfa A, Carreras A, et al. Lipopolysaccharide-binding protein plasma levels in children: effects of obstructive sleep apnea and obesity. J Clin Endocrinol Metab. 2014;99:656–63. doi: 10.1210/jc.2013-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao G, Metreveli N, Li R, Taylor A, Fletcher EC. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol Bethesda Md. 1985;1997(83):95–101. doi: 10.1152/jappl.1997.83.1.95. [DOI] [PubMed] [Google Scholar]
- 28.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res. 2009;50:3–21. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 2010;1801:209–14. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Cooke AA, Connaughton RM, Lyons CL, McMorrow AM, Roche HM. Fatty acids and chronic low grade inflammation associated with obesity and the metabolic syndrome. Eur J Pharmacol. 2016;785:207–14. doi: 10.1016/j.ejphar.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 31.Weiszenstein M, Shimoda LA, Koc M, Seda O, Polak J. Inhibition of Lipolysis Ameliorates Diabetic Phenotype in a Mouse Model of Obstructive Sleep Apnea. Am J Respir Cell Mol Biol. 2016;55:299–307. doi: 10.1165/rcmb.2015-0315OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin M-K, Han W, Bevans-Fonti S, Jun JC, Punjabi NM, Polotsky VY. The effect of adrenal medullectomy on metabolic responses to chronic intermittent hypoxia. Respir Physiol Neurobiol. 2014;203:60–7. doi: 10.1016/j.resp.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drager LF, Yao Q, Hernandez KL, Shin M-K, Bevans-Fonti S, Gay J, et al. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am J Respir Crit Care Med. 2013;188:240–8. doi: 10.1164/rccm.201209-1688OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drager LF, Li J, Shin M-K, Reinke C, Aggarwal NR, Jun JC, et al. Intermittent hypoxia inhibits clearance of triglyceride-rich lipoproteins and inactivates adipose lipoprotein lipase in a mouse model of sleep apnoea. Eur Heart J. 2012;33:783–90. doi: 10.1093/eurheartj/ehr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He Q, Yang Q, Zhou Q, Zhu H, Niu W, Feng J, et al. Effects of varying degrees of intermittent hypoxia on proinflammatory cytokines and adipokines in rats and 3T3-L1 adipocytes. PloS One. 2014;9:e86326. doi: 10.1371/journal.pone.0086326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiori CZ, Martinez D, Baronio D, da Rosa DP, Kretzmann NA, Forgiarini LF, et al. Downregulation of uncoupling protein-1 mRNA expression and hypoadiponectinemia in a mouse model of sleep apnea. Sleep Breath Schlaf Atm. 2014;18:541–8. doi: 10.1007/s11325-013-0916-2. [DOI] [PubMed] [Google Scholar]
- 37.Ohashi K, Ouchi N, Matsuzawa Y. Anti-inflammatory and anti-atherogenic properties of adiponectin. Biochimie. 2012;94:2137–42. doi: 10.1016/j.biochi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Gasbarrino K, Gorgui J, Nauche B, Côté R, Daskalopoulou SS. Circulating adiponectin and carotid intima-media thickness: A systematic review and meta-analysis. Metabolism. 2016;65:968–86. doi: 10.1016/j.metabol.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Reinke C, Bevans-Fonti S, Drager LF, Shin M-K, Polotsky VY. Effects of different acute hypoxic regimens on tissue oxygen profiles and metabolic outcomes. J Appl Physiol Bethesda Md. 1985;2011(111):881–90. doi: 10.1152/japplphysiol.00492.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carreras A, Kayali F, Zhang J, Hirotsu C, Wang Y, Gozal D. Metabolic effects of intermittent hypoxia in mice: steady versus high-frequency applied hypoxia daily during the rest period. Am J Physiol Regul Integr Comp Physiol. 2012;303:R700–709. doi: 10.1152/ajpregu.00258.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison CD. Leptin signaling in brain: A link between nutrition and cognition? Biochim Biophys Acta. 2009;1792:401–8. doi: 10.1016/j.bbadis.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saely CH, Geiger K, Drexel H. Brown versus white adipose tissue: a mini-review. Gerontology. 2012;58:15–23. doi: 10.1159/000321319. [DOI] [PubMed] [Google Scholar]
- 43.Castro-Grattoni AL, Alvarez-Buvé R, Torres M, Farré R, Montserrat JM, Dalmases M, et al. Intermittent Hypoxia-Induced Cardiovascular Remodeling Is Reversed by Normoxia in a Mouse Model of Sleep Apnea. Chest. 2016;149:1400–8. doi: 10.1016/j.chest.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Cortese R, Gileles-Hillel A, Khalyfa A, Almendros I, Akbarpour M, Khalyfa AA, et al. Aorta macrophage inflammatory and epigenetic changes in a murine model of obstructive sleep apnea: Potential role of CD36. Sci Rep. 2017;7:43648. doi: 10.1038/srep43648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stefater JA, Ren S, Lang RA, Duffield JS. Metchnikoff’s policemen: macrophages in development, homeostasis and regeneration. Trends Mol Med. 2011;17:743–52. doi: 10.1016/j.molmed.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15:731–44. doi: 10.1038/nri3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128:415–35. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 50.Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. 2016;17:18–25. doi: 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- 51.Mantovani A, Locati M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: lessons and open questions. Arterioscler Thromb Vasc Biol. 2013;33:1478–83. doi: 10.1161/ATVBAHA.113.300168. [DOI] [PubMed] [Google Scholar]
- 52.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou D, Yang K, Chen L, Zhang W, Xu Z, Zuo J, et al. Promising landscape for regulating macrophage polarization: epigenetic viewpoint. Oncotarget. 2017 doi: 10.18632/oncotarget.17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gileles-Hillel A, Almendros I, Khalyfa A, Zhang SX, Wang Y, Gozal D. Early intermittent hypoxia induces proatherogenic changes in aortic wall macrophages in a murine model of obstructive sleep apnea. Am J Respir Crit Care Med. 2014;190:958–61. doi: 10.1164/rccm.201406-1149LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khalyfa A, Qiao Z, Gileles-Hillel A, Khalyfa AA, Akbarpour M, Popko B, et al. Activation of the Integrated Stress Response and Metabolic Dysfunction in a Murine Model of Sleep Apnea. Am J Respir Cell Mol Biol. 2017;57:477–86. doi: 10.1165/rcmb.2017-0057OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahmed M, de Winther MPJ, Van den Bossche J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology. 2016 doi: 10.1016/j.imbio.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 59.Gosselin D, Glass CK. Epigenomics of macrophages. Immunol Rev. 2014;262:96–112. doi: 10.1111/imr.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MCS, van der Velden S, et al. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455:396–402. doi: 10.1016/j.bbrc.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 61.Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PloS One. 2013;8:e78045. doi: 10.1371/journal.pone.0078045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol Baltim Md. 2014;28:565–74. doi: 10.1210/me.2013-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Ct, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293:1103–5. doi: 10.1126/science.293.5532.1103. [DOI] [PubMed] [Google Scholar]
- 64.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 65.Schübeler D. Function and information content of DNA methylation. Nature. 2015;517:321–6. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 66.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghirlando R, Giles K, Gowher H, Xiao T, Xu Z, Yao H, et al. Chromatin domains, insulators, and the regulation of gene expression. Biochim Biophys Acta. 2012;1819:644–51. doi: 10.1016/j.bbagrm.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 69.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–29. doi: 10.1093/hmg/ddl046. Spec No 1. [DOI] [PubMed] [Google Scholar]
- 70.Ritchie W, Rasko JEJ, Flamant S. MicroRNA target prediction and validation. Adv Exp Med Biol. 2013;774:39–53. doi: 10.1007/978-94-007-5590-1_3. [DOI] [PubMed] [Google Scholar]
- 71.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 72.Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–82. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen MTA, Favelyukis S, Nguyen A-K, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–92. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 75.Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2010;30:186–92. doi: 10.1161/ATVBAHA.109.198044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–46. doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ishii M, Wen H, Corsa CAS, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–54. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25:2480–8. doi: 10.1101/gad.175950.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6:1124–32. doi: 10.15252/emmm.201404170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ryan S. Adipose tissue inflammation by intermittent hypoxia: mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J Physiol. 2017;595:2423–30. doi: 10.1113/JP273312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeller I, Srivastava S. Macrophage functions in atherosclerosis. Circ Res. 2014;115:e83–85. doi: 10.1161/CIRCRESAHA.114.305641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peled M, Fisher EA. Dynamic Aspects of Macrophage Polarization during Atherosclerosis Progression and Regression. Front Immunol. 2014;5:579. doi: 10.3389/fimmu.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–16. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. 2015;35:1306–16. doi: 10.1161/ATVBAHA.114.304650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014;34:1731–8. doi: 10.1161/ATVBAHA.114.303887. [DOI] [PubMed] [Google Scholar]
- 87.Neele AE, Van den Bossche J, Hoeksema MA, de Winther MPJ. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol. 2015;763:79–89. doi: 10.1016/j.ejphar.2015.03.101. [DOI] [PubMed] [Google Scholar]
- 88.Carreras A, Zhang SX, Peris E, Qiao Z, Gileles-Hillel A, Li RC, et al. Chronic sleep fragmentation induces endothelial dysfunction and structural vascular changes in mice. Sleep. 2014;37:1817–24. doi: 10.5665/sleep.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 91.Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12:722–34. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 93.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–82. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 95.Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giannini S, Serio M, Galli A. Pleiotropic effects of thiazolidinediones: taking a look beyond antidiabetic activity. J Endocrinol Invest. 2004;27:982–91. doi: 10.1007/BF03347546. [DOI] [PubMed] [Google Scholar]
- 97.Gelman L, Zhou G, Fajas L, Raspé E, Fruchart JC, Auwerx J. p300 interacts with the N-and C-terminal part of PPARgamma2 in a ligand-independent and-dependent manner, respectively. J Biol Chem. 1999;274:7681–8. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- 98.Zuo Y, Qiang L, Farmer SR. Activation of CCAAT/enhancer-binding protein (C/EBP) alpha expression by C/EBP beta during adipogenesis requires a peroxisome proliferator-activated receptor-gamma-associated repression of HDAC1 at the C/ebp alpha gene promoter. J Biol Chem. 2006;281:7960–7. doi: 10.1074/jbc.M510682200. [DOI] [PubMed] [Google Scholar]
- 99.Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM, et al. CARM1 promotes adipocyte differentiation by coactivating PPARgamma. EMBO Rep. 2008;9:193–8. doi: 10.1038/sj.embor.7401151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Debril M-B, Gelman L, Fayard E, Annicotte J-S, Rocchi S, Auwerx J. Transcription factors and nuclear receptors interact with the SWI/SNF complex through the BAF60c subunit. J Biol Chem. 2004;279:16677–86. doi: 10.1074/jbc.M312288200. [DOI] [PubMed] [Google Scholar]
- 101.Fujiki K, Shinoda A, Kano F, Sato R, Shirahige K, Murata M. PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat Commun. 2013;4:2262. doi: 10.1038/ncomms3262. [DOI] [PubMed] [Google Scholar]
- 102.Cortese R, Zhang C, Bao R, Andrade J, Khalyfa A, Mokhlesi B, et al. DNA Methylation Profiling of Blood Monocytes in Patients With Obesity Hypoventilation Syndrome: Effect of Positive Airway Pressure Treatment. Chest. 2016;150:91–101. doi: 10.1016/j.chest.2016.02.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim J, Bhattacharjee R, Khalyfa A, Kheirandish-Gozal L, Capdevila OS, Wang Y, et al. DNA methylation in inflammatory genes among children with obstructive sleep apnea. Am J Respir Crit Care Med. 2012;185:330–8. doi: 10.1164/rccm.201106-1026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kheirandish-Gozal L, Khalyfa A, Gozal D, Bhattacharjee R, Wang Y. Endothelial dysfunction in children with obstructive sleep apnea is associated with epigenetic changes in the eNOS gene. Chest. 2013;143:971–7. doi: 10.1378/chest.12-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gharib SA, Hayes AL, Rosen MJ, Patel SR. A pathway-based analysis on the effects of obstructive sleep apnea in modulating visceral fat transcriptome. Sleep. 2013;36:23–30. doi: 10.5665/sleep.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sharma S, Malur A, Marshall I, Huizar I, Barna BP, Pories W, et al. Alveolar macrophage activation in obese patients with obstructive sleep apnea. Surgery. 2012;151:107–12. doi: 10.1016/j.surg.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Y, Zhang C, Li H, Hou J. Down-regulation of vascular PPAR-γ contributes to endothelial dysfunction in high-fat diet-induced obese mice exposed to chronic intermittent hypoxia. Biochem Biophys Res Commun. 2017;492:243–8. doi: 10.1016/j.bbrc.2017.08.058. [DOI] [PubMed] [Google Scholar]
- 108.Gottlieb DJ, Punjabi NM, Mehra R, Patel SR, Quan SF, Babineau DC, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med. 2014;370:2276–85. doi: 10.1056/NEJMoa1306766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lesser DJ, Bhatia R, Tran WH, Oliveira F, Ortega R, Keens TG, et al. Sleep fragmentation and intermittent hypoxemia are associated with decreased insulin sensitivity in obese adolescent Latino males. Pediatr Res. 2012;72:293–8. doi: 10.1038/pr.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pogach MS, Punjabi NM, Thomas N, Thomas RJ. Electrocardiogram-based sleep spectrogram measures of sleep stability and glucose disposal in sleep disordered breathing. Sleep. 2012;35:139–48. doi: 10.5665/sleep.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stamatakis KA, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest. 2010;137:95–101. doi: 10.1378/chest.09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gonnissen HKJ, Hursel R, Rutters F, Martens EAP, Westerterp-Plantenga MS. Effects of sleep fragmentation on appetite and related hormone concentrations over 24 h in healthy men. Br J Nutr. 2013;109:748–56. doi: 10.1017/S0007114512001894. [DOI] [PubMed] [Google Scholar]
- 113.Nair D, Zhang SXL, Ramesh V, Hakim F, Kaushal N, Wang Y, et al. Sleep fragmentation induces cognitive deficits via nicotinamide adenine dinucleotide phosphate oxidase-dependent pathways in mouse. Am J Respir Crit Care Med. 2011;184:1305–12. doi: 10.1164/rccm.201107-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ramesh V, Nair D, Zhang SXL, Hakim F, Kaushal N, Kayali F, et al. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-α pathway. J Neuroinflammation. 2012;9:91. doi: 10.1186/1742-2094-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Y, Carreras A, Lee S, Hakim F, Zhang SX, Nair D, et al. Chronic sleep fragmentation promotes obesity in young adult mice. Obes Silver Spring Md. 2014;22:758–62. doi: 10.1002/oby.20616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang SXL, Khalyfa A, Wang Y, Carreras A, Hakim F, Neel BA, et al. Sleep fragmentation promotes NADPH oxidase 2-mediated adipose tissue inflammation leading to insulin resistance in mice. Int J Obes. 2005;2014(38):619–24. doi: 10.1038/ijo.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gozal D, Qiao Z, Almendros I, Zheng J, Khalyfa A, Shimpukade B, et al. Treatment with TUG891, a free fatty acid receptor 4 agonist, restores adipose tissue metabolic dysfunction following chronic sleep fragmentation in mice. Int J Obes. 2005;2016(40):1143–9. doi: 10.1038/ijo.2016.37. [DOI] [PubMed] [Google Scholar]
- 118.Khalyfa A, Wang Y, Zhang SX, Qiao Z, Abdelkarim A, Gozal D. Sleep fragmentation in mice induces nicotinamide adenine dinucleotide phosphate oxidase 2-dependent mobilization, proliferation, and differentiation of adipocyte progenitors in visceral white adipose tissue. Sleep. 2014;37:999–1009. doi: 10.5665/sleep.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carreras A, Zhang SX, Peris E, Qiao Z, Wang Y, Almendros I, et al. Effect of resveratrol on visceral white adipose tissue inflammation and insulin sensitivity in a mouse model of sleep apnea. Int J Obes. 2005;2015(39):418–23. doi: 10.1038/ijo.2014.181. [DOI] [PubMed] [Google Scholar]
- 120.Carreras A, Zhang SX, Peris E, Qiao Z, Gileles-Hillel A, Li RC, et al. Chronic sleep fragmentation induces endothelial dysfunction and structural vascular changes in mice. Sleep. 2014;37:1817–24. doi: 10.5665/sleep.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hausenblas HA, Schoulda JA, Smoliga JM. Resveratrol treatment as an adjunct to pharmacological management in type 2 diabetes mellitus-systematic review and meta-analysis. Mol Nutr Food Res. 2015;59:147–59. doi: 10.1002/mnfr.201400173. [DOI] [PubMed] [Google Scholar]
- 122.Gozal D, Kheirandish-Gozal L, Carreras A, Khalyfa A, Peris E. Obstructive sleep apnea and obesity are associated with reduced GPR 120 plasma levels in children. Sleep. 2014;37:935–41. doi: 10.5665/sleep.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hakim F, Wang Y, Carreras A, Hirotsu C, Zhang J, Peris E, et al. Chronic sleep fragmentation during the sleep period induces hypothalamic endoplasmic reticulum stress and PTP1b-mediated leptin resistance in male mice. Sleep. 2015;38:31–40. doi: 10.5665/sleep.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–24. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 125.Gozal D, Khalyfa A, Qiao Z, Akbarpour M, Maccari R, Ottanà R. Protein-Tyrosine Phosphatase-1B Mediates Sleep Fragmentation-Induced Insulin Resistance and Visceral Adipose Tissue Inflammation in Mice. Sleep. 2017 doi: 10.1093/sleep/zsx111. [DOI] [PubMed] [Google Scholar]