

Abstract
The accumulation of misfolded α-synuclein (aSyn) and neuron loss define several neurodegenerative disorders including Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). However, the precise relationship between pathology and neurotoxicity and why these processes disproportionately affect certain neuron subpopulations are poorly understood. We show here that Math2-expressing neurons in the hippocampal Cornu ammonis (CA), a region significantly affected by aSyn pathology in advanced PD and DLB, are highly susceptible to pathological seeding with pre-formed fibrils (PFFs), in contrast to dentate gyrus neurons, which are relatively spared. Math2+ neurons also exhibited more rapid and severe cell loss in both in vitro and in vivo models of synucleinopathy. Toxicity resulting from PFF exposure was dependent on endogenous aSyn and could be attenuated by N-acetyl-cysteine through a glutathione-dependent process. Moreover, aSyn expression levels strongly correlate with relative vulnerability among hippocampal neuron subtypes of which Math2+ neurons contained the highest amount. Consistent with this, antisense oligonucleotide (ASO) mediated knockdown of aSyn reduced neuronal pathology in a time-dependent manner. However, significant neuroprotection was observed only with early ASO intervention and a substantial reduction aSyn pathology, indicating toxicity occurs after a critical threshold of pathological burden is exceeded in vulnerable neurons. Together, our findings reveal considerable heterogeneity in endogenous aSyn levels among hippocampal neurons and suggest that this may contribute to the selective vulnerability observed in the context of synucleinopathies.
Keywords: α-synuclein, Lewy pathology, differential vulnerability, primary hippocampal neurons, pre-formed fibrils
Introduction
The deposition of misfolded proteins in select cell populations is a feature shared by many age-related neurodegenerative diseases [38]. Intracellular inclusions containing α-synuclein (aSyn) define a group of neurological conditions collectively known as α-synucleinopathies that includes Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [42]. Underlining their significance in disease, aSyn inclusions in neuronal cell bodies (Lewy bodies) and processes (Lewy neurites) are present in the vast majority of both sporadic and familial PD patients [59], and mutations and multiplication of the aSyn gene (SNCA) cause autosomal dominant PD [reviewed in 72]. Moreover, triplication of wildtype (wt) SNCA results in higher aSyn expression and earlier disease onset compared to PD resulting from gene duplication, indicating that pathogenesis is also closely linked to aSyn expression levels [15, 24, 30, 64].
The clinical progression of PD correlates closely with the appearance of aSyn inclusions throughout the brain in a stereotypical pattern affecting a restricted number of regions [12, 21]. Although the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) is a major contributor to motor symptoms that define PD, populations belonging to glutamatergic, serotonergic, noradrenergic, and cholinergic systems are also frequently impacted [25, 40, 66]. Furthermore, there is considerable heterogeneity in aSyn pathology burden and degeneration between different CNS regions. Indeed, Lewy pathology and cell loss appear to be disconnected in some brain regions in PD. For example, the tuberomammilary nucleus of the hypothalamus develops Lewy bodies without observable neuronal loss [41]. Conversely, neuronal loss with little to no Lewy pathology has been reported in the supraoptic nucleus [4]. Thus, the identities of vulnerable neurons and the relationship between development of pathology and degeneration remain enigmatic [67].
A major obstacle to untangling these questions has been the lack of models that concomitantly recapitulate the accumulation of pathological aSyn inclusions typical of PD brains in both catecholaminergic and non-catecholaminergic neurons that precedes progressive neurodegeneration in clinically-relevant neuronal subpopulations. For example, commonly used neurotoxins induce aSyn-mediated catecholaminergic neuron loss in nuclei such as the SNpc, but Lewy-like inclusions are observed only after chronic exposure [19, 27, 37]. On the other hand, viral-mediated or transgenic aSyn overexpression models recapitulate Lewy-like pathology more reliably but involve ectopic overexpression or introduction of mutations that may potentially distort the distribution of pathology and neuron loss [75]. In both wt rodents and non-human primates, recombinant aSyn pre-formed fibrils (PFFs) seed the conversion of endogenous aSyn into intracellular inclusions that exhibit multiple morphological and biochemical properties that are characteristic of human Lewy pathology and elicit motor deficits [1, 47, 48, 51, 57, 60, 63, 79]. PFF-seeded neuritic and cell body inclusions in cell-based systems are also linked to impaired neuronal activity and trafficking, oxidative stress, and is followed by cell loss [23, 69, 77]. These features therefore allow for the examination of the relationship between aSyn aggregation and neurodegeneration in cells and in vivo.
The fact that such models display both pathological spread and time-dependent toxicity led us to hypothesize that neuronal toxicity is dependent on aSyn aggregation. We provide here not only evidence supporting this hypothesis in non-dopaminergic neurons but also demonstrate that glutamatergic neurons positive for the transcription factor Math2 in the Cornu ammonis (CA), a known hotspot for aSyn pathology in PD with dementia (PDD) and DLB, are particularly vulnerable compared to neurons from the dentate gyrus (DG) [6, 32]. Surprisingly, toxicity resulting from PFF-seeded aSyn pathology does not require the presence of astrocytes or microglia in vitro. Furthermore, vulnerable Math2+ neurons contain higher aSyn protein levels compared to other hippocampal subtypes, and antisense oligonucleotide (ASO)-mediated knock-down of aSyn expression at various time points after PFF treatment revealed that early and marked reduction of aSyn levels reduced pathology most robustly and rescued toxicity. Neuroprotection and pathology reduction both declined with delayed ASO treatment suggesting a cellular threshold for pathology determined by endogenous levels of aSyn within an individual neuron and beyond which toxicity is irreversible.
Materials and Methods
Animals
All animal procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and conformed to the National Institute of Health Guide for Care and Use of Laboratory Animals. Timed-pregnant CD1 mice (purchased from Charles River Laboratories) or Snca−/− mice [2] maintained on a C3H background were used to prepare primary neuronal cultures. Female C57Bl6/C3H mice (purchased from Jackson Laboratories) were used for in vivo PFF-injection studies.
Reagents and Chemicals
Z-VAD-fmk (ZVAD; broad caspase inhibitor) was purchased from Enzo Life Sciences. DPQ (PARP1/ Parthanatos inhibitor) was purchased from Santa Cruz Biotechnology. Necrostatin-1 (Nec-1; RIPK1/ necroptosis inhibitor) and Ferrostatin-1 (Fer-1; Ferroptosis inhibitor) were purchased from Calbiochem. IU1 (USP14 inhibitor), N-acetyl cysteine (NAC; antioxidant), nicotinamide (NAM; NAD supplement), MK-801 (NMDA receptor antagonist), and Cytosine arabinofuranoside (Ara-C) were purchased from Sigma-Aldrich. Antisense oligonucleotides (ASOs) targeting aSyn (5′-TTTAATTACTTCCACCA-3′), which binds within intron 4 of the Snca gene, or control (5′-CCTATAGGACTATCCAGGAA-3′) were generously provided by Tracy Cole and Hien Zhao (Ionis Pharmaceuticals, Carlsbad CA). Complete ASO chemistry information is as follows: aSyn ASO (Tes Teo Aks Ads Tds Ads mCds Tds mCds mCko Aeo mCes Ae) and control ASO (mCes mCeo Teo Aeo Tes Ads Gds Ads mCds Tds Ads Tds mCds Aeo Geo Aes Ae) where capital letters indicate base abbreviation, m=5-methylcytosine, e=2′-O-methoxyethylribose (MOE), k= (S)-2′,4′-constrained 2′-O-ethyl (cEt), d=deoxyribose, s=phosphorothioate, and o=phosphodiester [54, 62].
Recombinant aSyn production, purification, and PFF fibrilization
Wildtype and chimeric aSyn protein production, purification, and fibrilization were conducted as previously described [46]. Briefly, Huwt, Mswt, and HuS87N aSyn constructs were expressed in E. coli BL21 (DE3) RIL cells and purified. PFF assembly was achieved by diluting the purified aSyn to 5 mg/ml (360 μM) in sterile Dulbecco’s PBS (pH 7.0; Mediatech) and then agitated constantly at 1000 rpm at 37°C for 7 days.
Primary hippocampal neuron cultures and fibril transduction
Primary neuronal cultures were prepared from either CD1 or Snca−/− embryos on E16-18, as previously described [78]. Tissue culture plates and coverslips were coated with Poly-D-lysine (Sigma) before addition of cells. Neurons were plated either on glass coverslips (round 12 mm diameter; Carolina Biological Supplies) in 24-well plates (50,000 cells/cm2), directly in 12-well plates for biochemical studies (75,000/cm2), or in 96-well plates (60,000/cm2). Cultures were maintained in Neurobasal medium supplemented with B27 (Invitrogen) unless otherwise indicated. PFF treatment was performed at 7 days in vitro (DIV). aSyn PFFs were diluted in sterile PBS without Ca2+/Mg2+ (Corning) and sonicated with a Biorupter bath sonicator (Diagenode) for 10 cycles (highest power, 30s on, 30s off at 10°C) then diluted in neuronal media before being added to cultures. PFF concentrations are expressed as the total equivalent aSyn monomer content in the preparation. To control for different cell plating densities in different formats, cultures were treated with ~25pg per cell (equivalent to 200 nM in a 96 well plate or 350 nM in a 24 well plate) unless otherwise indicated. At 7 days post transduction (DPT) 0.5 volume of the media in each well was replaced with fresh media. Ara-C (1μM) was added to neuronal cultures (single treatment at 4 DIV) to inhibit astrocyte growth for indicated experiments.
Stereotaxic PFF injection
Injections were performed as previously described with minor modifications [47]. Anesthetized mice received a single unilateral stereotactic injection of sonicated PFFs (5 μg) into the anterior hippocampus (coordinates: -2.5 mm relative to Bregma; 2 mm from midline; 2.4 mm beneath the dura). Animals were monitored regularly after surgery and sacrificed at the indicated time points by overdose with ketamine/xylazine. PFF injected mice were sacrificed at either 45- or 90 days post-injection (DPI), and PBS injected mice were sacrificed at 180 DPI. The brain and spinal cord were removed after transcardial perfusion with heparinized PBS and fixed overnight in 70% ethanol in 150mM NaCl (pH 7.4). Tissues were then embedded in paraffin for sectioning.
Immunocytochemistry and antibodies
Cultured cells were fixed by replacing media with warm 4% paraformaldehyde (in PBS/4% sucrose) for 15 min at room temperature (RT). Fixed neurons were washed 3 times with PBS and then blocked (3% BSA, 3% FBS in PBS) for 1 hour at RT or overnight at 4°C. Cells were then incubated in primary antibodies diluted in blocking buffer for 4 hours at RT or overnight at 4°C. Primary antibodies used in this study were: p-S129 aSyn (pSyn; CNDR mouse monoclonal IgG2a 81A; 1:2000); NeuN (Millipore A60; 1:2000); Prox1 (Biolegend 925201; 1:500); CTIP2 (Abcam ab18465; 1:500); Math2 (Abcam ab85824; 1:500); GFAP (CNDR rat monoclonal 2.2B10; 1:500); Iba1 (Wako 019-19741; 1:250); aSyn (CNDR mouse monoclonal IgG2a 9027; 1:5k); Neurofilament L (NFL; CNDR rabbit monoclonal; 1:2000); MAP2 (CNDR rabbit monoclonal 17028: 1:2000); GAD 65/67 (Chemicon AB1511; 1:1000); GAD 67 (Millipore MAB5406; 1:500). This was followed by washing with PBS three times and incubating for 1 hour at RT or overnight at 4°C in blocking buffer containing Alexa-Fluor conjugated isotype-specific secondary antibodies (Thermo Fisher). Coverslips were mounted on microscope slides using Fluoromount G containing DAPI (eBioscience) and scanned on a Perkin Elmer Lamina scanner or were captured on a Nikon Ds-Qi1Mc digital camera attached to a Nikon Eclipse Ni microscope. Cells in 96-well plates were washed three times with PBS and incubated 15 min at RT with DAPI (0.4 μg/ml in PBS) and then imaged using an InCell 2200 (GE Healthcare Life Sciences). Quantification of cells grown on coverslips was performed using HALO software (Indica Labs). 96-well plate quantification was done using InCell Toolbox Analyzer software (GE Healthcare Life Sciences).
Protein extraction and immunoblot analysis of neuron lysates
Cells were scraped in SDS lysis buffer (2% SDS in 50mM Tris, 150mM NaCl, pH 7.6) containing phosphatase and protease inhibitor cocktail, sonicated, and centrifuged at 100,000 × g for 30 min at 22°C. The supernatant from this step was resolved by SDS-PAGE (10 or 15 μg total protein per lane), transferred to nitrocellulose membranes, and blocked in 7.5% BSA in TBS for 1 hour at room temperature. For immunoblotting, membranes were incubated overnight at 4°C with constant agitation in blocking buffer containing the following primary antibodies: GAD 65/67 (Chemicon AB1511; 1:1,000); β-III tubulin (Invitrogen 32-2600; 1:5,000); aSyn (CNDR mouse monoclonal IgG2a Syn9027; 1:20,000). Membranes were washed in TBS with 0.05% Tween20 three times for 10 minutes each then incubated in diluted secondary antibodies (Li-Cor Biosciences; 1:20,000 in blocking buffer) for 1 hour at RT on an orbital shaker. Membranes were then washed as before and imaged using a Li-Cor Odyssey infrared imaging system.
Immunofluorescence
Immunofluorescence was performed on 6 μm thick sections as previously described [47]. Primary antibodies used were the same as above. Sections stained for Math2 were blocked (TBS with 3% BSA, 0.3% TritonX-100) and subjected to citrate buffer antigen retrieval. Sections stained for NeuN and pSyn were blocked in TBS containing BSA (3%) and FBS (2%). Primary and secondary antibodies were diluted in blocking buffer. Immunoreactivity was determined by using the appropriate fluorescent secondary antibodies. Images were captured on a Nikon Ds-Qi1Mc digital camera attached to a Nikon Eclipse Ni microscope or scanned using a Perkin Elmer Lamina Scanner. Neurons in hippocampal subregions were quantified from evenly spaced sections (1:40) of NeuN/81a or Math2 stained sections through Bregma -1.6mm – 3.3mm. HALO software was used to determine the number of immunoreactive nuclei or total area occupied.
Image analysis
Coverslips were stained for aSyn and transcription factors (TFs) as indicated above at 8 DIV. Laser intensity was set so that all detectable aSyn remained below the maximum threshold in 3 sample images. Nine images were taken in a 3 × 3 grid pattern from each coverslip to ensure coverage of the whole coverslip. All images were 8-bit images and were captured on a Nikon Ds-Qi1Mc digital camera attached to a Nikon Eclipse Ni microscope. The cell body outlines determined by the TF staining were set using the Analyze Particle function in Fiji (NIH) so that the outlines of the TF staining were saved on the ROI manager. These outlines were then applied to the aSyn channel and the intensity of each neurons of a given subtype was determined using the Analyze Particle function.
Statistical analysis
In vivo cell viability and pSyn data as a function of time and PFF dose was transformed using a Box-Cox transformation to uphold the assumption of a normally distributed outcome for regression. A weighted least squares regression analysis was then performed on the viability and pSyn data using treatment, treatment duration (DPT), and replicate as covariates, with an interaction between treatment and DPT included. This interaction was then used to determine whether each treatment group differed significantly from the untreated group at each time point. The inverse of the variance of each treatment group at each time point was used as the weight in the weighted least squares regression. This analysis was used to account for high variability in the observations so that groups with higher variability were down-weighted in the model. Calculations were performed using the MASS and multcomp packages in R-studio (Version 3.2.4). The significance level was adjusted to correct for multiple hypothesis testing using the Bonferroni approach. Only values from 9-15 DPT are reported for cell viability data. Data from 7 DPT was used as baseline for analyses for viability data and included in the analyses for pathology data to compare pathological burden. Other statistical analyses were performed using GraphPad Prism software (Version 4). Non-parametric (Kruskall-Wallis) tests were used where variance significantly differed between samples.
Results
PFF-induced toxicity is dependent on the levels pathological aSyn
We have previously shown that exposure to PFFs results in the aggregation of endogenously expressed aSyn and subsequent neuronal toxicity in primary neuronal cultures and non-transgenic mice [47, 79]. Since this experimental model reliably results in Lewy-like pathology and neuron death, we first examined whether toxicity is proportional to the total pathology burden in primary hippocampal neurons by inducing de novo pathology across a range of concentrations of PFF seeds. We utilized PFFs assembled from recombinant human wt (Huwt) aSyn or a chimeric mutant (HuS87N), which recruits endogenous aSyn into inclusions more efficiently compared to PFFs containing Huwt and other aSyn sequences when administered to hippocampal cultures or in vivo [46]. If toxicity is proportional to aSyn pathology, HuS87N PFF treatment should result in increased neuron loss compared to Huwt PFF treatment at each concentration, whereas similar neuron loss would indicate that these processes are independent. Immunostaining for pathological aSyn phosphorylated at Ser129 (pSyn), a disease-specific post-translational modification found in PD and in models of seeded synucleinopathy [31, 48], we observed that the amount of pathology increased in a dose- and time-dependent manner in Huwt PFF-treated neurons. However, by 7 DPT, HuS87N PFF treatment at all doses tested induced higher pathological burden than the highest Huwt PFF concentration used, confirming that HuS87N PFFs are more pathogenic (Fig. 1a-c and Fig. S1a).
Figure 1. PFF-induced toxicity is dose-dependent and requires pathological aSyn conversion.
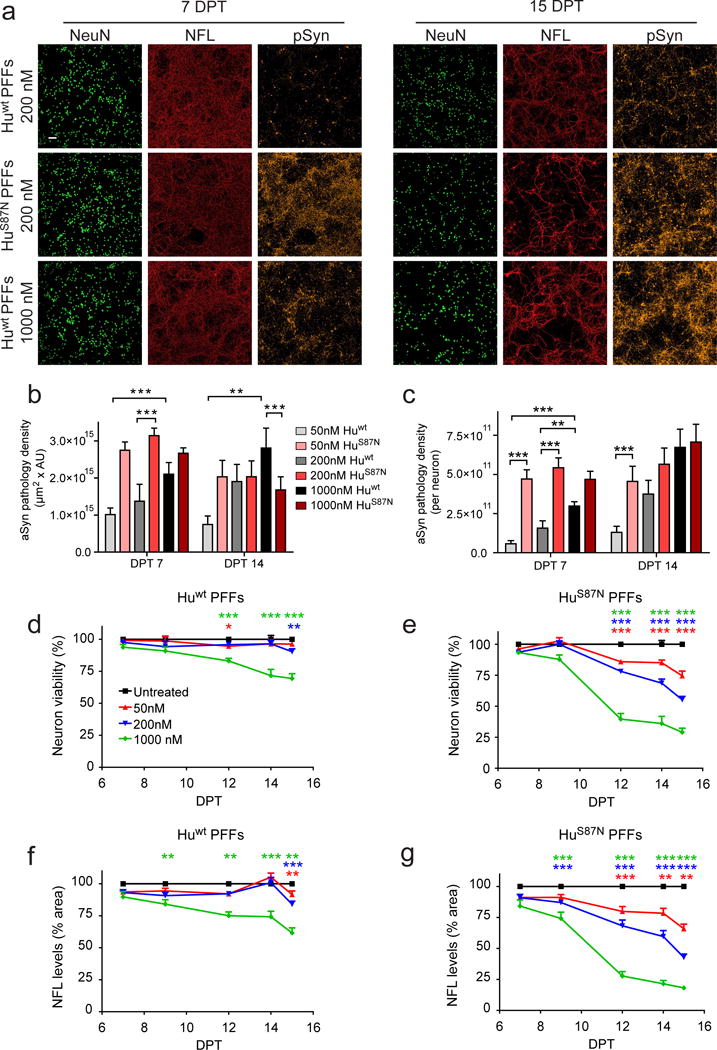
a, Primary hippocampal neurons treated with Huwt or HuS87N PFFs and immunostained for NFL, NeuN, pSyn, and DAPI. b, c, pSyn staining area × intensity and pSyn staining density relative to surviving NeuN+ neurons at 7 and 14 DPT. d, e, Neuronal viability determined by the number of NeuN+ cells remaining per well relative to untreated wells. f, g, Total NFL staining density measured relative to untreated wells. N= 14-18 wells from 3 biological replicates for each group. Weighted least squares regression analysis after Box-Cox transformation. All data are mean ± SEM. P-value significance determined after correcting for multiple hypothesis testing. *p<0.05, **p<0.01, ***p<0.001. Scale bar, 100 μm.
Neuronal viability did not differ between PFF-treated and control cells at 7 and 9 DPT, but neurons loss was apparent by 12 DPT with the degree of toxicity being proportional to the concentration of PFFs added (Fig. 1d, e). Neurons treated with Huwt PFFs showed markedly less toxicity compared to HuS87N treated neurons with only the 1000 nM condition displaying any significant toxicity by 12 DPT (Fig. 1d). Mouse wt (Mswt) aSyn PFFs also reduced neuron survival in a dose dependent manner, but toxicity was less than that of HuS87N PFFs at each dose (Fig. S1b, c), in agreement with Mswt PFFs inducing an intermediate level pathology between that of Huwt and HuS87N PFFs, and further supporting that neuron toxicity is dependent on and proportional to aSyn pathology [46]. Both Huwt and HuS87N PFFs failed to induce aSyn inclusions or neuron loss in Snca−/− neurons indicating toxicity requires the pathological conversion of endogenous aSyn regardless of the type of PFFs used (Fig. S2a) [79]. Monomeric HuS87N aSyn, which lacks seeding activity, was also unable to induce either pathology or toxicity (Fig. S2b). Staining density of neurofilament light chain (NFL), a structural protein expressed mainly in axons, closely mirrored the dose dependent reduction in NeuN+ cell counts under each treatment condition (Fig. 1f, g), confirming that the observed NeuN loss in PFF-treated cultures was due to neuron death rather than downregulation of NeuN.
aSyn pathology-induced toxicity affects specific neuronal subtypes
The largest decrease in neuronal viability occurred between 9-12 DPT and did not appreciably decline afterwards even at the highest PFF doses, suggesting that a significant subset of neurons continued to persist despite prolonged exposure to PFFs. This observation prompted us to determine if differences in vulnerability between neuronal subtypes present in this culture system were responsible for this effect. Glutamatergic neurons derived from CA regions express the transcription factor Math2 while DG neurons contain Prox1. Another transcription factor, CTIP2, is found in both CA1 and DG neurons [5, 7, 65]. Immunostaining showed that ~60% of NeuN+ neurons co-express Math2 indicating CA origin; ~10% were co-positive for Prox1 (Fig. S3a). Thus, the majority of neurons in this culture system are glutamatergic with GABAergic neurons and other subtypes representing ~30% of neurons.
All three major glutamatergic subpopulations declined significantly by 15 DPT after HuS87N PFF treatment (Fig. 2a). However, compared to Math2+ and CTIP2+ neurons (in which viability declined by 54% and 51%, respectively), the Prox1+ population declined by 32% relative to PBS controls (Fig. 2b), indicating that these neurons are relatively resistant to PFF-induced toxicity. To confirm this, we treated primary cultures with 5-fold higher concentration of HuS87N PFFs, which reduced Math2+ neuron viability by >85% while approximately half of Prox1+ neurons still remained (Fig. 2c-e). Thus, Prox1+ neurons are more resistant to PFF-induced toxicity than Math2+ neurons, although they are susceptible in the presence of a sufficiently high pathological burden. Furthermore, we also found that the proportion of neurons that contained cell body inclusions was significantly higher among Math2+ and CTIP2+ neurons compared with Prox1+ neurons (Fig. 2f, g), consistent with the notion that neuronal subpopulations that are most susceptible to developing pathology are also more vulnerable to cell death.
Figure 2. Math2+ neurons are vulnerable to aSyn pathology and toxicity relative to Prox1+ neurons.
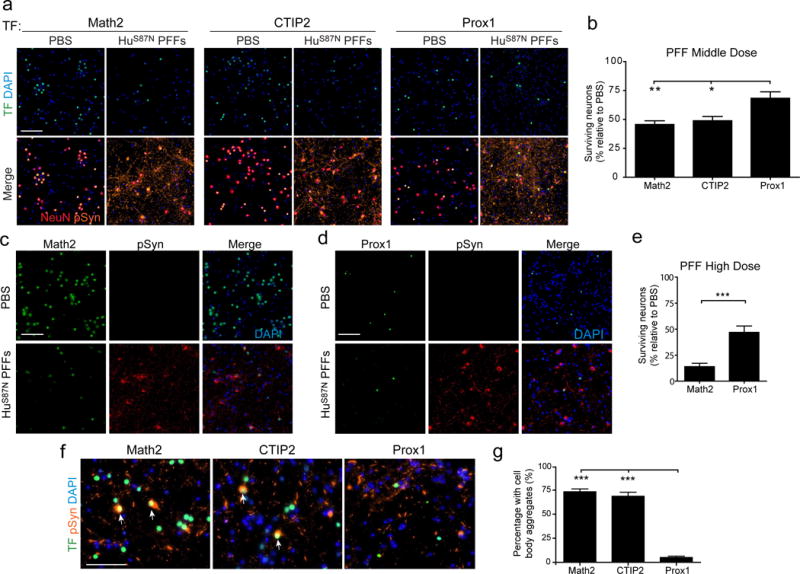
a, Neurons treated with 350 nM HuS87N PFFs were immunostained at 15 DPT for NeuN, pSyn, DAPI, and indicated transcription factors (TF). b, Survival of neuronal subpopulations in PFF-treated cultures relative to PBS treatment. N= 12-16 wells from 3-4 biological replicates per group. One-way ANOVA with Tukey’s multiple comparison test. c, d, Neurons treated with 1.7 μM HuS87N PFFs and co-labeled for Math2 or Prox1 and pSyn at DPT 15. e, Survival of neuronal subpopulations in 1.7 μM HuS87N PFF-treated cultures relative to PBS treatment. N= 9 wells from 3 biological replicates. Unpaired two-tailed t-test with Welch’s correction. f, TF, pSyn, and DAPI staining at 4 DPT. g, Percentage of cell body aggregates found in neuronal subpopulations. Arrows point to cells that contain both cell body aggregates and are positive for the TF. N= 15-21 wells from 3 biological replicates per group. Kruskall-Wallis Test with Dunn’s Multiple Comparison Test. All Data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. Scale bar, 100μm.
It has been previously reported that glutamatergic neurons develop increased aSyn pathology compared to GABAergic neurons when treated with PFFs due to their higher levels of aSyn expression [68]. We did not detect cell body inclusions in GABAergic neurons indicating that they are resistant to pathology formation similar to Prox1+ neurons (Fig. S3b). We did find that GABAergic neurons do contain aSyn in this system suggesting that they may be capable of exhibiting PFF-induced toxicity (Fig. S3c). We therefore measured GABAergic neuron viability by immunostaining for GAD 65/67, an enzyme in the GABA synthesis pathway. At 15 DPT, there was no statistical difference in GAD 65/67 staining (Fig. S3d, e). This contrasted with a ~25% reduction in overall neuronal viability measured by NeuN counts during this same time period after HuS87N PFF treatment (Fig. S3f). Normalized GAD 65/67 protein levels actually increased by ~50% at 14 DPT (Fig. S3g, h), indicating that GABAergic neurons make up a larger percentage of the surviving neuronal subpopulations and thus are resistant to PFF-induced toxicity.
Math2 neurons are susceptible to PFF-induced pathology and toxicity in vivo and in vitro
To further validate these findings, we next examined if hippocampal neurons in vivo also display differential vulnerabilities to PFF-induced toxicity. Although less pathogenic than HuS87N PFFs in vitro (Fig. S1c), we have previously observed that Mswt PFFs efficiently seed inclusion formation and result in SNpc neuron loss in vivo [46]. Mice given a single unilateral injection of Mswt PFFs into the anterior hippocampal hilum displayed pathology that was localized predominantly to the CA3 region of the ipsilateral hippocampus with CA1-2 regions showing moderate pSyn staining by 45 DPI (Fig. 3a). Pathology was also present in the corresponding contralateral structures but to a lesser extent. In PBS-injected control mice, no pathology or NeuN-positive cell loss was detectable between the ipsilateral and contralateral hippocampal regions. In contrast, NeuN loss was observed as early as 45 DPI in the ipsilateral CA3 region of PFF-injected mice and in the contralateral CA3 later at 90 DPI (Fig. 3b, arrows), consistent with our in vitro data showing that CA-derived neurons are lost in a time- and pathology-dependent manner. Immunoreactivity for pSyn declined significantly between 45 DPI and 90 DPI on the ipsilateral side. Interestingly, pSyn was also increased at 45 DPI as well on the contralateral side and trended toward a decrease by 90 DPI. (Fig. 3c). The decrease in NeuN+ cells corresponded with the decline in pathology similar to what was observed in the primary cultures that occurred after cell loss (Fig. 3d). Inclusions containing pSyn were also detectable in regions synaptically connected to the hippocampal injection site and appeared on the ipsilateral side including the lateral entorhinal cortex, but regions that are known to be permissive to pathology but unconnected to the injection site such as the SNpc were spared (Fig. 3e, f).
Figure 3. aSyn PFFs induce toxicity in CA3 neurons in vivo.
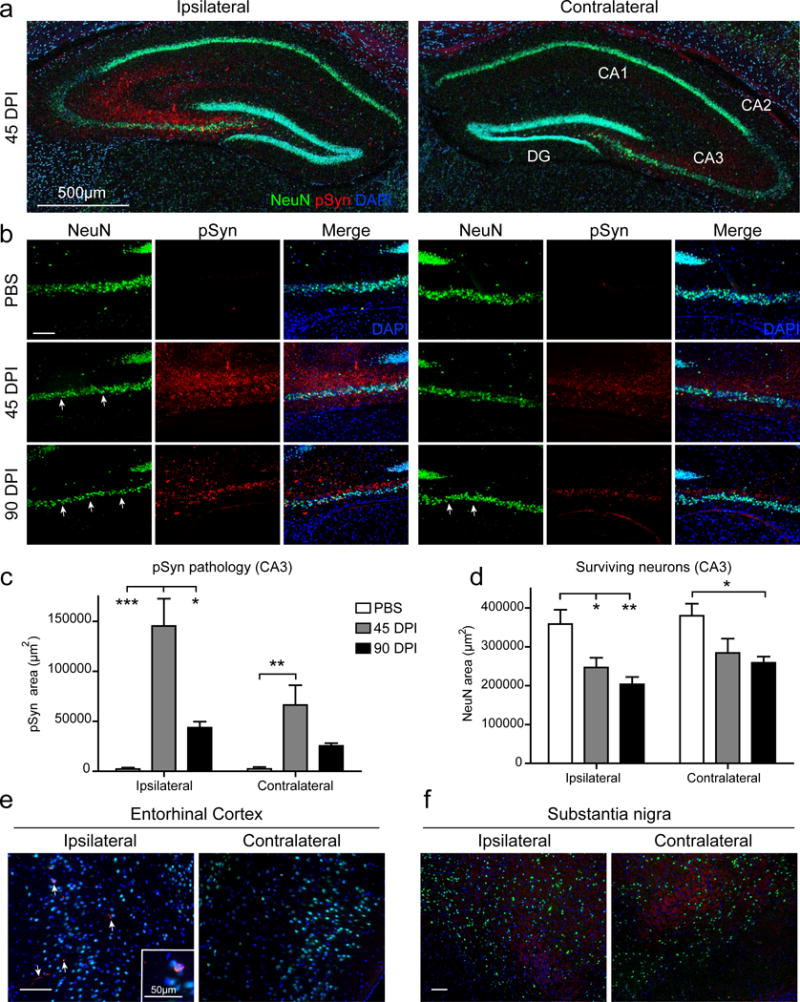
a, pSyn and NeuN staining of ipsilateral and contralateral hippocampus. b, High magnification image of ipsilateral (left) and contralateral (right) CA3 regions. Arrows indicate areas of NeuN+ cell loss. c, Area of pSyn pathology in both ipsilateral and contralateral CA3. d, NeuN+ area of both ipsilateral and contralateral CA3. PBS N=4 mice, 45 dpi N=4 mice, 90 dpi N=4 mice. NeuN and pSyn staining in lateral entorhinal cortex (e) and substantia nigra (f). Arrows indicate pathological pSyn aggregates. All data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, one-Way ANOVA with Tukey’s multiple comparison test for each side. Scale bar, 100μm unless otherwise noted.
Given the increased susceptibility of Math2+ neurons to aSyn pathology in vitro, we also quantified their number in wt mice after hippocampal PFF-injections. As expected, Math2 was predominantly expressed in CA neurons in PBS-injected controls (Fig. 4a). However, Math2 staining in PFF-injected mice was markedly reduced in the ipsilateral CA3 region at 45 dpi and remained lower at 90 dpi (Fig. 4b, c), confirming their vulnerability. Quantification revealed a decrease in the number of contralateral Math2+ neurons although this did not reach statistical significance (Fig 4c).
Figure 4. Math2 neurons are susceptible to PFF induced toxicity and Math2 loss precedes neurodegeneration.
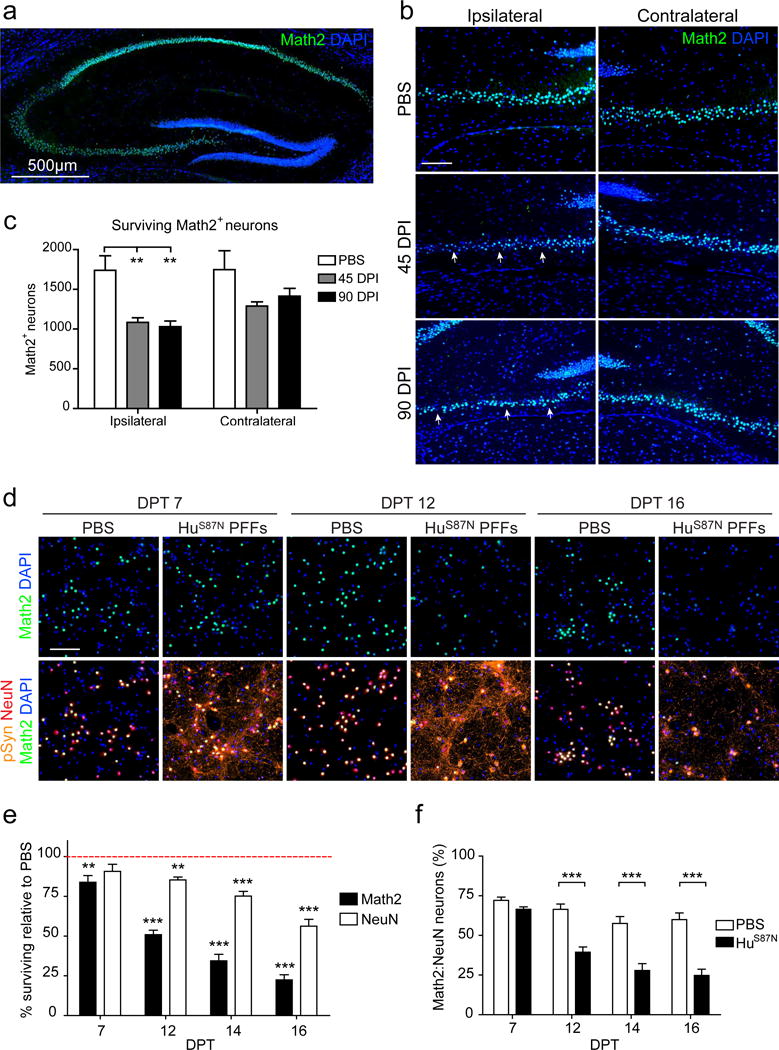
a, Math2 and DAPI staining of PBS-injected hippocampus. b, Math2 and DAPI staining of CA3 regions after PBS or PFF treatment. Arrowheads point to areas with reduced Math2 staining. c, Math2+ cell counts in ipsilateral and contralateral CA3. PBS N=4 mice; 45 dpi N=4 mice; 90 dpi N=4 mice. One-Way ANOVA with Tukey’s multiple comparison test for each side. d, WT hippocampal neurons treated with 350 nM HuS87N PFF immunostained for Math2, NeuN, pSyn, and DAPI at indicated time points. e, Percentage of surviving Math2+ and NeuN+ neurons in (d) relative to PBS (Normalized as 100% at each time point; red dashed line). f, Ratio of Math2 to NeuN expressing cells in d. N=10-12 wells from 3 biological replicates. Two-way ANOVA with Bonferroni post-tests. All data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. Scale bar, 100 μm unless noted otherwise.
Math2 has been reported to play a role in maintaining redox and mitochondrial homeostasis [9, 73, 74]. Given that oxidative stress has been implicated as a mechanism for aSyn toxicity in this model [23, 69], we therefore asked whether Math2 might be perturbed following the development of aSyn pathology. We detected a reduction in Math2 shortly prior to NeuN loss and neurodegeneration in PFF-treated hippocampal cultures (Fig. 4d). In fact, by 12 DPT Math2 levels were 50% lower in HuS87N PFF treated cells and continued to decrease with time (Fig. 4e). By contrast, NeuN immunoreactivity decreased only slightly by (15% compared to PBS controls) at 12 DPT and was reduced by 46% at 16 DPT. Furthermore, the ratio of Math2+ neurons to the total NeuN+ neuronal population decreased over time from ~65% at 7 DPT, similar to PBS-treated controls, to 32% at 16 DPT (Fig. 4f). Thus, Math2 loss precedes neurodegeneration.
Prox1+ neurons express low levels of aSyn and are resistant relative to Math2+ subpopulations
Given their relative resistance to PFF-induced toxicity in vitro, we asked if Prox1+ DG neurons are spared while neighboring CA3 neurons degenerate after PFF-injection. We observed neuritic aSyn pathology in the DG after injection with PFFs but not PBS, and this pathology was predominantly restricted to the ipsilateral hemisphere (Fig. 5a). Again, pathology was increased at 45 DPI which trended toward a decrease by 90 DPI (Fig. 5b). NeuN staining area was decreased by 90 DPI in the DG showing that neurons in the DG are susceptible to pathology induced toxicity similar to what we found in vitro (Fig 5c). Interestingly, this loss was not evident until 90 DPI whereas NeuN loss was observed as early as 45 DPI in the CA3 again suggesting that DG neurons degenerate more slowly and are thus more resistant to pathology compared to CA3 Math2+ neurons.
Figure 5. Neuronal susceptibility to PFF-induced toxicity correlates with aSyn expression levels.
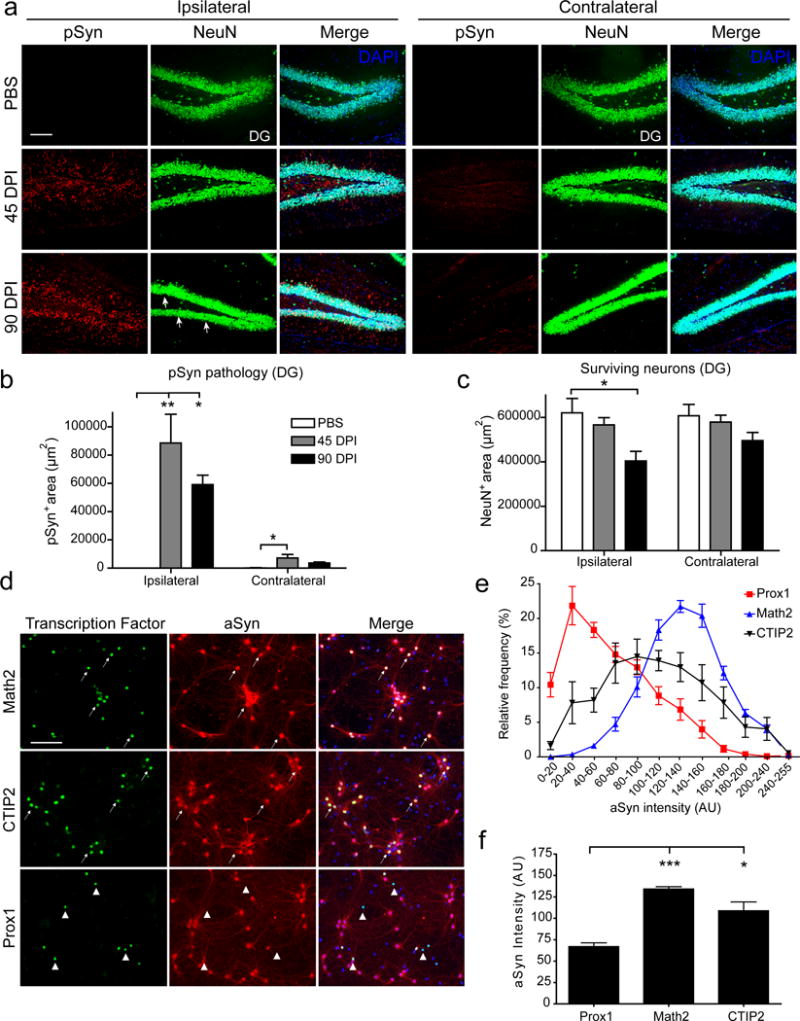
a, DG staining for NeuN and pSyn in ipsilateral and contralateral hippocampus of PFF- or PBS-injected mice. b, Quantification of pSyn pathology area. c, Quantification of NeuN-immunoreactive area in ipsilateral and contralateral DG. One-way ANOVA with Tukey’s multiple comparison test for each side. PBS N = 4 mice; 45 dpi N = 4 mice, 90 dpi N = 4 mice. d, Immunocytochemistry of 8 DIV WT hippocampal neurons for aSyn, and either Math2, Prox1, or CTIP2. Arrows and arrowheads point to the same cells expressing aSyn or not detected, respectively. e, Histogram of binned relative aSyn-immunoreactivity of each neuronal subpopulation (AU: arbitrary units). f, Comparison of aSyn intensity of Math2, Prox1, and CTIP2 populations. N = 8 coverslips per group from 3 experiments. Kruskall-Wallis Test with Dunn’s Multiple Comparison Test. Data are mean ± SEM. *p<0.05, **p<0.01. Scale bar, 100 μm.
Because pathology and toxicity are dependent on aSyn expression (Fig. S2a) we asked whether vulnerable neurons express higher aSyn protein levels compared to resistant ones, thereby facilitating more rapid pathological conversion and increasing pathology burden. To determine relative expression levels, cultures were co-stained for aSyn and the TFs that specify hippocampal glutamatergic neurons at 7-8 DIV, corresponding to the time at which PFFs were added in our toxicity studies and when neuronal aSyn is mainly localized to the soma [81], allowing for accurate assignment to individual neurons. Neurons that stained most intensely for aSyn were overwhelmingly Math2+, while some Prox1+ cell bodies expressed lower or undetectable levels of aSyn (Fig. 5d, e). Staining intensities of CTIP2+ neurons were generally between those of Math2+ and Prox1+ populations (Fig. 5e). Comparison of the means of the 3 groups also showed that Math2+ neurons expressed the highest levels of aSyn (mean = 134 AU) followed by CTIP2 (mean = 109 AU) with Prox1+ populations having the lowest relative aSyn expression (mean = 67 AU) (Fig. 5f). Thus, the most resistant subpopulation in this model expresses lower levels of aSyn compared to vulnerable Math2 and CTIP2 populations, suggesting relative aSyn expression is a significant driver of vulnerability.
PFF-induced toxicity is dependent on aSyn expression levels, does not require astrocytes or microglia in vitro, and is mediated by oxidative stress
Given that our data show that neuronal toxicity is dependent on pSyn pathology and that neuronal susceptibility is strongly correlated with aSyn expression levels, we examined whether decreasing aSyn expression could reduce pathology formation and rescue cell viability. We used ASOs directed against aSyn mRNA to decrease aSyn expression over an extended length of time. The ability of the ASO to reduce aSyn expression was first confirmed by treating primary neuronal cultures at 8 DIV (equivalent to time of PFF addition) and determining aSyn protein levels at 21 DIV (13 DPT). aSyn ASO treatment significantly decreased aSyn levels in a dose-dependent manner at concentrations ranging from 0.1 - 5.0 μM leading to 57 – 73% reductions in normal expression levels after a single administration (Fig. 6a).
Figure 6. Reduction aSyn expression with ASOs preserves neuronal viability.
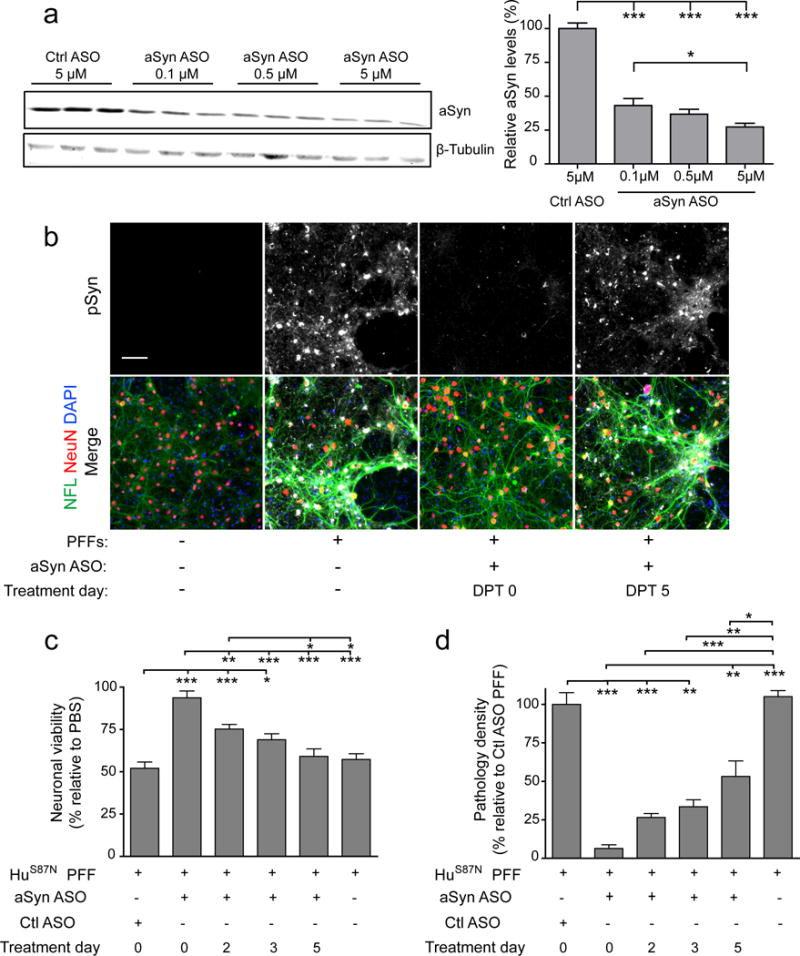
a, Immunoblot showing aSyn expression in neurons after treatment with aSyn or control ASOs. One-way ANOVA with Tukey’s Multiple Comparison Test, N = 9 wells from 3 biological replicates per group. b, Immunostaining for pSyn, NeuN and NFL in neurons treated with 200 nM HuS87N PFFs and 0.5 μM ASO. c, Neuronal viability measured by NeuN counts relative to PBS with same treatment and (d) normalized pSyn density relative to Control (Ctl) ASO treated with PFFs. N = 14-15 wells from 3 biological replicates. Data are mean ± SEM with one-way ANOVA with Tukey’s multiple comparison test (c) or Kruskall-Wallis Test with Dunn’s Multiple Comparison Test (d). *p<0.05, **p<0.01, ***p<0.001. Scale bar, 100μm.
We proceeded to apply ASOs to PFF-treated neuron cultures at various time points after PFF treatment between 0 DPT (i.e., concurrent with PFFs) to 5 DPT (by which time pSyn pathology can be detected) and examined the neurons at 14-15 DPT in order to test if ASO treatment timing affected pathology formation and ultimately viability. Pathology was reduced in a time-dependent manner after ASO treatment with simultaneous application of aSyn ASO and PFFs showing the greatest effect (Fig. 6b). Consistent with a direct relationship between aSyn pathology and neurotoxicity, simultaneous co-treatment with ASO and PFF reduced pathology by ~95% compared to untreated controls and almost completely preserved neuronal viability while the protective effect of ASOs was reduced over time whereby no rescue was evident when treatment was started at 5 DPT. Surprisingly, even when ASO treatment was started at 2 DPT and pathology levels decreased by ~75%, we observed only a mild rescue of viability. Similarly, ASO treatment at DPT 5 reduced pathology by 47% but had no protective effect (Fig. 6c, d).
Because neurodegeneration in this model is driven by the aggregation of endogenous aSyn which in turn correlates with aSyn expression levels in neuronal subtypes, we also examined if toxicity is dependent on non-neuronal cell types. The primary hippocampal cultures used here contain a small proportion of non-neuronal cells (mostly astrocytes), raising the possibility that glial cells mediate the neuron loss observed after PFF treatment. In addition, aSyn PFFs have been shown to bind, enter, and activate microglia via a variety of receptors (including CD11b, TLR-2 and -4 receptors) resulting in the release of neurotoxic factors [22, 26, 39, 80]. Although they do not express appreciable levels of aSyn, glial cells themselves might also be lost due to PFF exposure. To mitigate these scenarios, we first determined whether astrocyte or microglia activation occurs either following PFF treatment or at the onset of neurodegeneration by monitoring GFAP and Iba1 levels. In all treatment conditions, GFAP signal increased steadily over the duration of the experiment (Fig. 7a), although GFAP staining was comparable between PBS- and PFF-treated cultures at the time-points examined (2, 9, and 14 DPT), even in the presence of decreased neuronal viability after HuS87N PFF treatment (Fig. 7b, c). Thus astrocytes in these cultures proliferate and/or activate over the course of incubation but do so independently of PFFs and neuronal toxicity.
Figure 7. Astrocytes and microglia are unaffected by PFF induced pathology and toxicity.
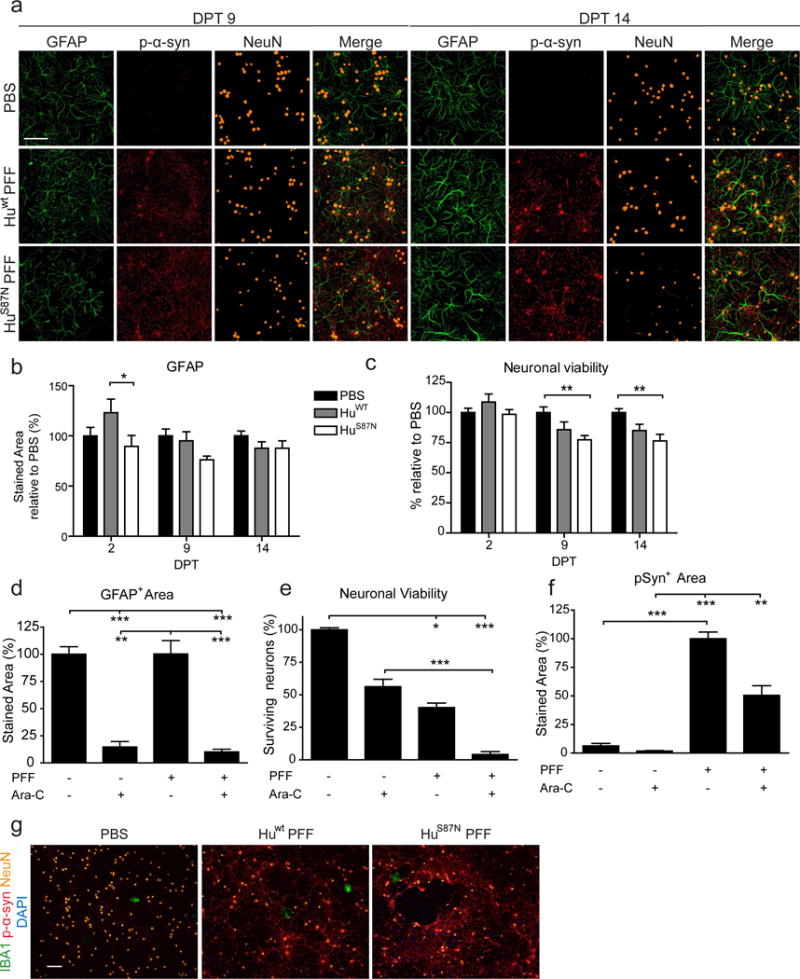
a, Immunostaining of WT primary hippocampal neurons treated with 350nM HuS87N PFFs for GFAP, pSyn, and NeuN. b, Average GFAP+ staining area normalized to PBS for each time point. c, Neuronal viability was determined by the number of NeuN+ cells relative to the PBS for each time point. N = 11-12 wells from 3 biological replicates per group. Two-way ANOVA with Bonferroni post-tests. d, e, f, Quantification of GFAP, neuron viability (NeuN), and pSyn pathology at DPT 15. N = 9-13 wells from 3 biological replicates per group. Kruskall-Wallis Test with Dunn’s Multiple Comparison Test. All data are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. g, Immunostaining of 16 DPT primary hippocampal neurons for Iba1, pSyn, and NeuN. Scale bars, 100 μm.
To further determine if astrocytes are necessary for toxicity, a subset of cultures were pre-treated with Ara-C to minimize astrocyte proliferation. As expected, cultures containing Ara-C showed a significant decrease in GFAP expression (Fig. 7d). Treatment with Ara-C alone was also accompanied by a moderate reduction in neuronal viability, suggesting that astrocytes normally contribute to neuron health in this system or that Ara-C is intrinsically and mildly toxic to neurons. Nonetheless, PFF-induced toxicity was still prominent in Ara-C treated neurons (Fig. 7e). In line with the decreased neuronal viability, pSyn immunoreactivity was also reduced after PFF administration in Ara-C treated cells (Fig. 7f). Microglia are rare in these cultures, as indicated by Iba1 stain (<0.001% of the cells; Fig. 7g), suggesting that toxicity in vitro is independent of microglia activation.
We then addressed the mechanism that links aSyn aggregation toxicity, by treating neurons with a panel of pharmacological agents targeting pathways implicated in aSyn models and in PD [82]. Compounds were administered daily starting at 9-10 DPT, when aSyn pathology had already been established and viability was assessed at 14-15 DPT. We were unable to detect rescue with inhibitors against known execution pathways for cell-death including PARP, caspases, RIPK1, NMDA excitotoxicity, wallerian degeneration, ferroptosis, and deubiquitinase activity (Fig. S4). Among the compounds tested, only treatment with 100 μM N-acetyl cysteine (NAC), an antioxidant, significantly increased viability relative to PFF-exposed neurons treated with vehicle. Other antioxidants such as Trolox and mitotempo did not rescue toxicity at any concentration tested suggesting that NAC may be affecting viability in a more specific manner. We hypothesized that NAC may be preventing toxicity through replenishing glutathione levels in the cells. To test this, we co-treated neurons with 100 μM NAC and the glutathione depleting agent BSO and found that co-treatment prevented NAC mediated rescue (Fig. S4a, b). Overall these data suggest that PFF induced toxicity is at least partially mediated by glutathione depletion that develops to a critical point after pathology has exceeded the cells ability to cope with the aggregates.
Discussion
Despite its central role in the pathogenesis of several major neurological disorders, our understanding of how aSyn aggregation intersects with neurodegeneration remains limited. Resolving why certain neurons are preferentially susceptible to this process is critical towards elucidating the pathogenesis and progression of synucleinopathies. The lack of disease models that reliably develop intracellular aSyn inclusions and cell death in selectively vulnerable populations has contributed to this knowledge gap. In this study, we utilized a model of seeded α-synucleinopathy that recapitulates these two key processes to address the relationship between aSyn misfolding and cellular toxicity. While others have previously shown that dopaminergic SNpc neurons are susceptible to toxicity in fibril-based models as is observed in PD [47, 51, 57, 60], this finding has not previously been examined in neocortical and hippocampal regions, which are particularly affected in DLB and PDD [10, 13, 83]. This approach, which leverages the natural heterogeneity that exists in the CNS, has yielded new insights into how Lewy-like pathology may be linked to toxicity and a working hypothesis of differential vulnerability between neuron subtypes.
Our data strongly support the notion that toxicity is closely linked to total aSyn pathology burden in neurons. We further demonstrate that the kinetics of pathology formation are largely determined by: 1) the availability of substrate (i.e., intraneuronal aSyn levels) and 2) the pathogenicity of the seeding agent (e.g., conformational strain). Using both wt and human-mouse chimeric aSyn PFFs with distinct pathogenicities, we observed that the degree of neuron loss is proportional to the initial seeding efficiency and the resultant pathological load. Interestingly, although chimeric (HuS87N) PFFs clearly increased pathology formation by as early as 7 DPT, toxicity was not evident until 12 DPT in all treatment conditions, a considerable period after the induction of inclusion pathology and in stark contrast to the rapid cell loss typically observed following excitotoxicity or axonal damage in cultured neurons [55, 58]. Consistent with its milder pathogenicity, Huwt PFFs at the highest dose tested induced toxicity comparable to that seen with the lowest concentration of HuS87N PFFs, while Mswt PFFs showed an intermediate level of potency, further supporting that aSyn pathology correlates directly with toxicity in hippocampal neurons.
Given that lower concentrations of Huwt PFFs also induce aSyn inclusions, albeit at a slower rate, we speculate that degeneration would ensue with extended incubation periods that are beyond the period these neurons could be maintained under the conditions used in this study. These data are also consistent with our previous finding that HuS87N PFFs accelerate pathology and toxicity in the SNpc after injection into the dorsal striatum, while others have also reported time-dependent toxicity in vivo, illustrating that toxicity eventually follows the accumulation of aSyn inclusions in multiple CNS regions with known susceptibility in PD and DLB [46, 50, 51, 53]. Taken together with the existing body of evidence from post-mortem studies supporting the clinical correlation of PD symptoms with aSyn pathology spread, our data indicate that aSyn aggregation directly contributes to neurotoxicity and is a marker of eventual neurodegeneration [12, 21].
Even after prolonged exposure to PFFs, degeneration does not occur uniformly among neurons from a single neuroanatomical region such as the hippocampus. Indeed, aSyn-mediated neurotoxicity appears to level off by 12-14 DPT after which, a considerable proportion of neurons continue to survive. Given that primary neuronal cultures are comprised of multiple neuronal subtypes as well as non-neuronal cells, this would suggest differential vulnerabilities between subpopulations. Previous work suggests that GABAergic neurons develop pathology less readily after PFF treatment compared to their glutamatergic counterparts [68]. We have further established that glutamatergic subpopulations differ widely in their vulnerability to PFF-induced toxicity, a property that correlates closely with the rate at which they develop aSyn pathology. In particular, Math2 expressing neurons from the CA region are highly vulnerable to PFF-induced toxicity compared to their DG-derived Prox1+ neighbors in both in vitro and in vivo settings. Similarly, the majority of pathology that forms in vivo following hippocampal PFF-injection occurs in CA2 and CA3 regions, where Math2+ neurons reside. This coincides with the prominent aSyn pathology found in these regions in DLB and PDD brains whereas DG neurons are relatively spared [6, 32]. More recent work also shows that pathology and loss of CA1 neurons, another Math2+ subset which we identified as vulnerable from our in vitro data, correlates strongly with cognitive decline in PDD [3].
Since PFF-induced toxicity is intrinsically coupled to aSyn expression, high intracellular levels of aSyn should render neurons more permissive to seeding by increasing the rate of pathological conversion [47, 79]. Indeed, relative aSyn expression levels of the three major glutamatergic subpopulations in our primary cultures correlate with their vulnerability to PFF treatment with the most vulnerable Math2+ neurons containing the highest levels and resistant Prox1+ neurons having the least. CTIP2+ neurons, which generally express levels between that of the two former subpopulations, display an intermediate degree of vulnerability. The differences in expression levels we observed by immunostaining is further supported by RNA-seq data showing that aSyn mRNA levels are highest in CA3, relative to DG and CA1 and -2 regions where Prox1+ and CTIP2+ neurons are found (Fig. S5a, b and ref. [14]). Consistent with this pattern, CA3 neurons also exhibited the highest pathology burden and neuron loss compared to the DG and other CA regions in PFF-injected mice. Thus, heterogeneity in aSyn protein levels provides a likely explanation for the selective vulnerability of CA neurons in both our model and disease, although we cannot exclude additional subpopulation-specific properties, such as susceptibility to high intracellular calcium levels and metabolic demands, as contributors to vulnerability [56].
Microgliosis and astrocytosis are commonly observed in PD brains suggesting a role for inflammation in synucleinopathies [28, 36, 52]. Moreover, these non-neuronal cells are readily activated by misfolded aSyn internalized via CD11b and TLR-2/4 receptors [22, 26, 39, 80], and thus may be potential mediators of aSyn toxicity. Our data show that although GFAP staining increases over time in culture, astrocytes are not required in PFF-mediated neurotoxicity. The minute fraction of microglia present also suggests a limited role for them in vitro although studies using PFF-injection in rats suggest a role for microglia in vivo [33, 70]. Additional investigation will be required to address their role, especially in the hippocampus.
At the cellular level, Math2 loss precedes overt neurodegeneration since other neuronal markers (e.g., NeuN) and structural proteins such as NFL decrease later indicating that overt degeneration is occurring and not solely phenotype switching. Math2 is a master transcription factor responsible for a number of processes, including mitochondrial homeostasis and regulation of apoptosis, raising the possibility that transcriptional dysregulation represents an early step of the degeneration cascade [9, 74]. Impairment of axonal and organelle transport by the accumulation of misfolded aSyn may also play a concurrent role in toxicity [16, 77]. Interestingly, transcriptomic analysis of post-mortem brains from Alzheimer’s disease patients, in which aSyn co-pathology is common [71], indicate a reduction in Math2 in the hippocampus relative to healthy controls [35, 61], and genetic analysis has linked certain Math2 polymorphisms to late onset AD [8, 29, 45]. Collectively, these data suggest that Math2 identifies a subset of hippocampal neurons that are vulnerable in multiple neurodegenerative settings, although it remains unclear whether aSyn toxicity is directly related to Math2 expression and function.
For example, Math2 is reported to increase cellular tolerance for oxidative stress by inducing scavenging enzymes such as Gpx1 and SOD2 [73]. Consistent with this and previous work showing elevated oxidative stress in dopaminergic neurons containing aSyn inclusions [23, 69], only an antioxidant that replenishes glutathione levels (NAC) effectively reduced toxicity. However, Math2 is detected in only a few neuronal subpopulations outside of the hippocampus, including in the isocortex, cortical subplate, and a small subset of striatal neurons [44]. Moreover, Math2 is not present in other vulnerable populations in PD such as SNpc neurons, which further decouples Math2 expression from intrinsic vulnerability.
Surprisingly, inhibitors of several established cell death pathway’s executors (e.g., caspases and PARP1) did not alter PFF-induced toxicity, suggesting that cell loss occurs outside of caspase-dependent apoptosis and parthanatos, which have been reported in mice administered with the neurotoxin MPTP or in post mortem PD patient samples [11, 17, 34, 43]. The lack of protection provided by NMDA receptor antagonist and NAD supplement also argues against a major role for excitotoxicity and Wallerian degeneration, respectively, in this aSyn seeding model. This diverges somewhat from previous reports implicating caspase 3, -8 and -9 activation in PFF-triggered toxicity both in cell-lines, primary hippocampal cultures and organotypic slices [20, 49]. However, we note that neuron death in our experimental system requires several-fold lower concentrations of aSyn seeds compared to those used in other studies, although we cannot exclude activation of different mechanisms of toxicity following acute vs. chronic aSyn exposure and/or aggregation. While unlikely, the cell death pathways triggered by the same stressor may also differ between neuron subpopulations.
A number of soluble oligomeric entities ranging from misfolded monomers to protofibrils have been reported to be associated with cellular or neuronal toxicity, and it has also been suggested that exogenous fibrillar aSyn can seed misfolding along the plasma membrane leading to its disruption [18, 49, 76]. Although amyloid forms of aSyn represent the predominant species applied to neurons and in vivo, our data cannot discern if they are solely responsible for toxicity or if additional or transient aSyn species such as oligomers also accumulate over time and contribute to cellular demise. Nonetheless, the observation that aSyn knock out provides neurons with immunity to extended exposure to high concentrations of Mswt and HuS87N aSyn PFFs indicates that intracellular Lewy-like aSyn pathology is necessary for toxicity [47, 79]. Determining whether a similar sequence of events is responsible for neurodegeneration in synucleinopathy patients, where multiple strains likely exist, will be crucial.
Finally, these findings prompted us to test whether PFF-induced toxicity can be attenuated by knocking down aSyn expression with a targeted ASO that significantly suppressed aSyn expression over several days after a single administration. Co-treatment with aSyn ASO dramatically reduces PFF-induced Lewy-like pathology and subsequent toxicity, further supporting that toxicity is directly downstream of aSyn pathology. Intriguingly, delayed ASO treatment after PFF-seeding can still significantly reduce pathology, yet is largely ineffective in restoring neuron viability. This requirement for early knock down suggests that neurons have an intrinsic but finite tolerance for aSyn pathology (or a pathological intermediate species) but toxicity is irreversible after this threshold is exceeded.
In summary, this work provides new insight into determinants that predict neuronal subtype vulnerability to aSyn pathology-related toxicity by leveraging the ability to modulate pathology in permissive neuronal populations and CNS regions. Our findings support a model (summarized in Fig. 8) where increased levels of aSyn expression enhance the vulnerability of different neuronal subtypes to forming Lewy pathology and consequent toxicity. Subpopulations that express lower aSyn levels are likely protected as this pathological process is delayed. Similarly, early intervention in decreasing aSyn prevents toxicity by suppressing pathology burden below the critical threshold for toxicity, whereas delayed intervention only partially inhibits pathology but has a limited effect in preventing toxicity. Elucidation of the cellular processes involved in aSyn pathological aggregation will help optimize the therapeutic window for strategies targeting aSyn.
Figure 8. Proposed model of relation between aSyn pathology formation and neurotoxicity.
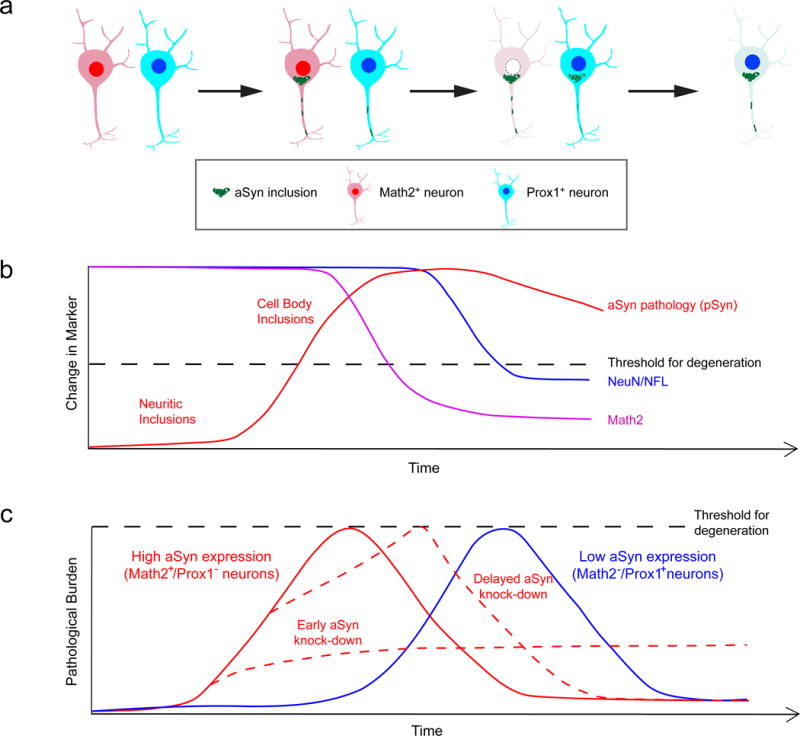
a, Upon exposure to misfolded species neurons develop aSyn pathology in a time-dependent manner after a lag period, starting as neuritic inclusions that coalesce into larger inclusions in the cell body. Pathology formation occurs more rapidly in vulnerable neurons (Math2+ cells in the hippocampus), and leads to more rapid cell death. Formation and accumulation of aSyn pathology in resistant Prox1+ neurons is comparably slower, partially due to lower aSyn expression levels. b, In susceptible Math2+ neurons, degenerative mechanisms are triggered when the critical threshold of aSyn aggregation is reached. In the case of Math2+ neurons, cell death is preceded by the loss of this key transcription factor. Math2+ neurons reach this threshold faster than neighboring Prox1+ neurons due to their higher endogenous aSyn protein levels, which facilitates faster propagation of pathological aSyn. c, Early knockdown of aSyn reduces neurodegeneration in vulnerable neurons by preventing aSyn pathology from reaching critical levels, but knockdown at later stages is not sufficient for rescuing neurons despite reducing overall pathology burden.
Supplementary Material
Acknowledgments
We thank Drs. Xiaolu Yang, Michael Henderson, Dustin Covell, Chao Peng, Kurt Brunden, and John Trojanowski for helpful discussions and insights. This research was funded in part by the NIH grants NS088322, NS053488, T32-AG000255, and a pilot grant from University of Pennsylvania Institute for Translational Medicine and Therapeutics. T.C. was an employee of Ionis Pharmaceuticals at the time when data were generated and interpreted.
Abbreviations
- aSyn
Alpha-synuclein
- pSyn
p-Ser129 alpha-synuclein
- ASO
Antisense oligonucleotide
- CA
Cornu Ammonis
- DG
Dentate gyrus
- DPI
Days post-injection
- DPT
Days post-transduction
- PD
Parkinson’s disease
- DLB
Dementia with Lewy bodies
- PDD
Parkinson’s disease with dementia
- PFF
Pre-formed fibril
- LB/LN
Lewy body/Lewy neurite
- Ms
Mouse
- Hu
Human
- SNpc
Substantia nigra pars compacta
- wt
wildtype
Footnotes
Author contributions
Conceptualization, EL and KCL; Methodology, EL, AC, BZ, SCD, DMR, and KCL; Investigation, EL, CC, SX, and KCL; Writing – original draft, EL; Writing – review and editing, EL, VMYL and KCL; Funding Acquisition, KCL, VMYL; Resources, KCL; Supervision, KCL.
The remaining authors have no additional financial interests.
References
- 1.Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, et al. alpha-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol Dis. 2017;105:84–98. doi: 10.1016/j.nbd.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 3.Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH. Hippocampal alpha-Synuclein in Dementia with Lewy Bodies Contributes to Memory Impairment and Is Consistent with Spread of Pathology. J Neurosci. 2017;37:1675–1684. doi: 10.1523/JNEUROSCI.3047-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ansorge O, Daniel SE, Pearce RK. Neuronal loss and plasticity in the supraoptic nucleus in Parkinson’s disease. Neurology. 1997;49:610–613. doi: 10.1212/wnl.49.2.610. [DOI] [PubMed] [Google Scholar]
- 5.Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong RA, Kotzbauer PT, Perlmutter JS, Campbell MC, Hurth KM, Schmidt RE, Cairns NJ. A quantitative study of alpha-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm (Vienna) 2014;121:171–181. doi: 10.1007/s00702-013-1084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bagri A, Gurney T, He X, Zou YR, Littman DR, Tessier-Lavigne M, Pleasure SJ. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–4260. doi: 10.1242/dev.129.18.4249. [DOI] [PubMed] [Google Scholar]
- 8.Barral S, Cheng R, Reitz C, Vardarajan B, Lee J, Kunkle B, Beecham G, Cantwell LS, Pericak-Vance MA, Farrer LA, et al. Linkage analyses in Caribbean Hispanic families identify novel loci associated with familial late-onset Alzheimer’s disease. Alzheimer’s Dement. 2015;11:1397–1406. doi: 10.1016/j.jalz.2015.07.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baxter KK, Uittenbogaard M, Chiaramello A. The neurogenic basic helix-loop-helix transcription factor NeuroD6 enhances mitochondrial biogenesis and bioenergetics to confer tolerance of neuronal PC12-NeuroD6 cells to the mitochondrial stressor rotenone. Exp Cell Res. 2012;318:2200–2214. doi: 10.1016/j.yexcr.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, Hefti F, Harper SJ. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. doi: 20026227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 13.Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA. Parkinson’s disease is associated with hippocampal atrophy. Mov Disord. 2003;18:784–790. doi: 10.1002/mds.10444. [DOI] [PubMed] [Google Scholar]
- 14.Cembrowski MS, Wang L, Sugino K, Shields BC, Spruston N. Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. eLife. 2016;5:e14997. doi: 10.7554/eLife.14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 16.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cosi C, Colpaert F, Koek W, Degryse A, Marien M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996;729:264–269. [PubMed] [Google Scholar]
- 18.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R, Burnett M, Parisi JE, Klos KJ, Ahlskog JE. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S1–5. doi: 10.1016/S1353-8020(09)70769-2. [DOI] [PubMed] [Google Scholar]
- 22.Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2014;2:90. doi: 10.1186/s40478-014-0090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 25.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 26.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forno LS, DeLanney LE, Irwin I, Di Monte D, Langston JW. Astrocytes and Parkinson’s disease. Prog Brain Res. 1992;94:429–436. doi: 10.1016/s0079-6123(08)61770-7. [DOI] [PubMed] [Google Scholar]
- 29.Fowler KD, Funt JM, Artyomov MN, Zeskind B, Kolitz SE, Towfic F. Leveraging existing data sets to generate new insights into Alzheimer’s disease biology in specific patient subsets. Sci Rep. 2015;5:14324. doi: 10.1038/srep14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- 31.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 32.Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain. 2014;137:2493–2508. doi: 10.1093/brain/awu193. [DOI] [PubMed] [Google Scholar]
- 33.Harms AS, Delic V, Thome AD, Bryant N, Liu Z, Chandra S, Jurkuvenaite A, West AB. alpha-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol Commun. 2017;5:85. doi: 10.1186/s40478-017-0494-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, Iwaki T, Ohara T, Sasaki T, LaFerla FM, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex. 2014;24:2476–2488. doi: 10.1093/cercor/bht101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iannaccone S, Cerami C, Alessio M, Garibotto V, Panzacchi A, Olivieri S, Gelsomino G, Moresco RM, Perani D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Related Disord. 2013;19:47–52. doi: 10.1016/j.parkreldis.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Jenner P. Functional models of Parkinson’s disease: a valuable tool in the development of novel therapies. Ann Neurol. 2008;64(Suppl 2):S16–29. doi: 10.1002/ana.21489. [DOI] [PubMed] [Google Scholar]
- 38.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kremer HP, Bots GT. Lewy bodies in the lateral hypothalamus: do they imply neuronal loss? Mov Disord. 1993;8:315–320. doi: 10.1002/mds.870080310. [DOI] [PubMed] [Google Scholar]
- 42.Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 43.Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D, Jiang H, Kang SU, Lee BD, Kang HC, et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013;16:1392–1400. doi: 10.1038/nn.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Long J, He T, Belshaw R, Scott J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci Rep. 2015;5:12393. doi: 10.1038/srep12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, Lee VM. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep. 2016;16:3373–3387. doi: 10.1016/j.celrep.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahul-Mellier AL, Vercruysse F, Maco B, Ait-Bouziad N, De Roo M, Muller D, Lashuel HA. Fibril growth and seeding capacity play key roles in alpha-synuclein-mediated apoptotic cell death. Cell Death Differ. 2015;22:2107–2122. doi: 10.1038/cdd.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun. 2014;2:88. doi: 10.1186/s40478-014-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 53.Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–1260. doi: 10.1016/j.celrep.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ostergaard ME, Southwell AL, Kordasiewicz H, Watt AT, Skotte NH, Doty CN, Vaid K, Villanueva EB, Swayze EE, Bennett CF, et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic acids Res. 2013;41:9634–9650. doi: 10.1093/nar/gkt725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr Biol. 2015;25:2349–2360. doi: 10.1016/j.cub.2015.07.050. [DOI] [PubMed] [Google Scholar]
- 57.Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–199. doi: 10.1016/j.nbd.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson C, Neal JH, Cotman CW. Development of N-methyl-D-aspartate excitotoxicity in cultured hippocampal neurons. Brain Res Dev Brain Res. 1989;48:187–195. doi: 10.1016/0165-3806(89)90075-8. [DOI] [PubMed] [Google Scholar]
- 59.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord. 2012;27:831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 61.Satoh J, Yamamoto Y, Asahina N, Kitano S, Kino Y. RNA-Seq data mining: downregulation of NeuroD6 serves as a possible biomarker for alzheimer’s disease brains. Dis Markers. 2014;2014:123165. doi: 10.1155/2014/123165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seth PP, Yu J, Allerson CR, Berdeja A, Swayze EE. Synthesis and biophysical characterization of R-6′-Me-alpha-L-LNA modified oligonucleotides. Bioorganic Med Chem Lett. 2011;21:1122–1125. doi: 10.1016/j.bmcl.2010.12.119. [DOI] [PubMed] [Google Scholar]
- 63.Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M. Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun. 2017;5:12. doi: 10.1186/s40478-017-0413-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 65.Sugiyama T, Osumi N, Katsuyama Y. A novel cell migratory zone in the developing hippocampal formation. J Comp Neurol. 2014;522:3520–3538. doi: 10.1002/cne.23621. [DOI] [PubMed] [Google Scholar]
- 66.Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov Disord. 2013;28:41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18:101–113. doi: 10.1038/nrn.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M. Differential expression of alpha-synuclein in hippocampal neurons. PloS one. 2014;9:e89327. doi: 10.1371/journal.pone.0089327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tapias V, Hu X, Luk KC, Sanders LH, Lee VM, Greenamyre JT. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol Life Sci. 2017;74:2851–2874. doi: 10.1007/s00018-017-2541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC, Lee VMY, Trojanowski JQ, Bjorklund A. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci. 2017;114:E8284–E8293. doi: 10.1073/pnas.1710442114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, et al. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131:393–409. doi: 10.1007/s00401-015-1526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- 73.Uittenbogaard M, Baxter KK, Chiaramello A. The neurogenic basic helix-loop-helix transcription factor NeuroD6 confers tolerance to oxidative stress by triggering an antioxidant response and sustaining the mitochondrial biomass. ASN neuro. 2010;2:e00034. doi: 10.1042/AN20100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uittenbogaard M, Chiaramello A. The basic helix-loop-helix transcription factor Nex-1/Math-2 promotes neuronal survival of PC12 cells by modulating the dynamic expression of anti-apoptotic and cell cycle regulators. J Neurochem. 2005;92:585–596. doi: 10.1111/j.1471-4159.2004.02886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Visanji NP, Brotchie JM, Kalia LV, Koprich JB, Tandon A, Watts JC, Lang AE. alpha-Synuclein-Based Animal Models of Parkinson’s Disease: Challenges and Opportunities in a New Era. Trends Neurosci. 2016;39:750–762. doi: 10.1016/j.tins.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 76.Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- 77.Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Molec Biol Cell. 2014;25:4010–4023. doi: 10.1091/mbc.E14-02-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, Guo M, Wilson B, Hong JS, Zhang J. alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci. 2015;112:E1926–1935. doi: 10.1073/pnas.1417883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 82.Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang W, Yu S. Synucleinopathies: common features and hippocampal manifestations. Cell Molec Life Sci. 2017;74:1485–1501. doi: 10.1007/s00018-016-2411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.