

Abstract
The DNA papillomaviruses infect squamous epithelium and can cause persistent, benign and sometimes malignant hyperproliferative lesions. Effective antiviral drugs to treat human papillomavirus (HPV) infection are lacking and here we investigate the anti-papillomavirus activity of novel epigenetic targeting drugs, BET bromodomain inhibitors.
Bromodomain and Extra-Terminal domain (BET) proteins are host proteins which regulate gene transcription, they bind acetylated lysine residues in histones and non-histone proteins via bromodomains, functioning as scaffold proteins in the formation of transcriptional complexes at gene regulatory regions. The BET protein BRD4 has been shown to be involved in the papillomavirus life cycle, as a co-factor for viral E2 and also mediating viral partitioning in some virus types. We set out to study the activity of small molecule BET bromodomain inhibitors in models of papillomavirus infection. Several BET inhibitors reduced HPV11 E1ˆE4 mRNA expression in vitro and topical therapeutic administration of an exemplar compound I-BET762, abrogated CRPV cutaneous wart growth in rabbits, demonstrating translation of anti-viral effects to efficacy in vivo. Additionally I-BET762 markedly reduced viability of HPV16 infected W12 cells compared to non-infected C33A cells. The molecular mechanism for the cytotoxicity to W12 cells is unknown but may be through blocking viral-dependent cell-survival factors. We conclude that these effects, across multiple papillomavirus types and in vivo, highlight the potential to target BET bromodomains to treat HPV infection.
Keywords: Human Papillomavirus, Epigenetics, Bromodomain Extra-Terminal domain Protein, BRD4, BET inhibitor
1. INTRODUCTION
Papillomaviruses are non-enveloped, small, circular dsDNA viruses which infect squamous epithelium causing benign and sometimes malignant hyperproliferative lesions. There are over 180 human papillomavirus (HPV) types with specific anatomical tropisms for cutaneous or mucosal sites (Howley and Lowy, 2007). HPV infections usually self resolve, but latent virus can be maintained in basal keratinocytes through low level replication of extrachromosomal genomes (episomes) and when reactivated cause new lesions. Ablative strategies or innate immune activators such as Imiquimod and interferon are employed to treat persistent HPV lesions but these treatments can be painful and recurrence is common (Cirelli and Tyring, 1994; Schofer et al., 2006; Stanley, 2012; Stern et al., 2012; Syrjanen, 1998). Additionally some HPV types (e.g. HPV16, HPV18 called ‘high risk’) may integrate into host chromosomes, leading to increased expression of viral oncogenes and cell transformation. HPV is the major cause of cervical cancer, is increasingly associated with head and neck epithelial cancers and whilst vaccination is used to prevent high risk HPV infection therapeutic vaccines are not yet available (Chaturvedi et al., 2011). Overall there remains a clinical need for effective and safe therapeutics to eliminate persistent papillomavirus infections and ideally with cross-strain activity.
Papillomavirus genomes are complexed with host histone proteins forming chromatin like structure and exhibit dynamic histone modifications at viral promoters indicating epigenetic regulation of viral gene expression (del Mar Pena and Laimins, 2001; Favre et al., 1977; Wooldridge and Laimins, 2008). In recent years epigenetic targeting drugs have emerged and in this article we evaluate the anti-papillomavirus activity of one such group of compounds, BET inhibitors.
The Bromodomain and Extra-Terminal domain (BET) proteins are BRD2, BRD3, BRD4 and BRDT. They share a common domain structure, tandem bromodomains which confer binding to acetylated lysines in histones and non-histone proteins and other regions with adapter function; they act as scaffolds to coordinate recruitment of transcriptional complexes to chromatin thereby regulating gene expression (Chiang, 2009; Filippakopoulos and Knapp, 2012). Small molecule ‘BET inhibitors’ were discovered which occupy the BET bromodomains and compete with histone binding (Chung et al., 2011; Ferri et al., 2016; Filippakopoulos et al., 2010). These compounds ameliorated pathology in a range of disease models (e.g. cancer, inflammation and fibrosis), highlighting their therapeutic potential and are now in early clinical development (Prinjha et al., 2012; Shi and Vakoc, 2014; Theodoulou et al., 2016).
BRD4 is intimately involved in the life cycle of papillomaviruses through interaction with the viral E2 protein (reviewed in (McBride and Jang, 2013). The proteins bind via conserved residues in the c-terminus of BRD4 and n-terminus of E2 (Abbate et al., 2006; Baxter et al., 2005; McBride et al., 1991; Muller et al., 2012) and multiple studies show that BRD4 acts as a transcriptional co-factor for E2 directing permissive or repressive activity (Helfer et al., 2014; Ilves et al., 2006; Lee and Chiang, 2009; McPhillips et al., 2006; Schweiger et al., 2007; Senechal et al., 2007; Wu et al., 2006; Yan et al., 2010). BRD4 c-terminus is also involved in genome partitioning of Bovine Papillomavirus (BPV1), HPV16 and HPV31 (Abbate et al., 2006; You, 2010; You et al., 2004; You et al., 2005). Of note, although BRD4 engages E2 proteins of all papillomaviruses, there are differences in binding strength of these interactions suggestive of varied functions, the alphapapillomavirus E2 proteins bind BRD4 weakly whereas E2 proteins from BPV1, HPV1 and CRPV bind more tightly (Jang et al., 2015; McPhillips et al., 2006). Additionally recent work showed a phosphorylated region in BRD4 binds E2 of high risk HPVs mediating viral and host gene expression (Wu et al., 2016). Lastly BET inhibitor JQ1 was shown to block HPV16 and BPV1 early gene expression in cell lines carrying episomal virus demonstrating that BET bromodomain inhibitors exhibit anti-papillomavirus activity (Helfer et al., 2014).
The aim of our study was to determine whether BET inhibitors may offer a new therapeutic antiviral strategy for treating persistent HPV infection and we addressed this using in vitro and in vivo models. We tested several chemically distinct BET inhibitors and chose three papillomaviruses that show contrasting E2-BRD4 binding affinities; alphapapillomaviruses (HPV11 and HPV16) in which E2-BRD4 interactions are weak, and CRPV, a papillomavirus containing an E2 that binds with high affinity to BRD4.
2. MATERIALS and METHODS
2.1 BET inhibitor compounds
Compounds were synthesised and supplied (> 95% purity) by GlaxoSmithKline (GSK) and the structures are shown in S1. They were I-BET762 (GSK525762A, (Nicodeme et al., 2010)), I-BET151 (GSK1210151A, (Dawson et al., 2011)) and GSK2794033A, all reversible competitive ligands of the tandem bromodomains of BRD2, BRD3, BRD4, and BRDT; and Control-768 (GSK525768A), an inactive enantiomer of I-BET762 that does not bind the bromodomains due to steric hindrance.
2.2 Cells and Virus
HPV11 virions were obtained from infected human xenografts grown in athymic mice as previously described (Kreider et al., 1987). A431 cells were purchased from ATCC (A431-CRL1555). The HPV16 cervical carcinoma cell line W12 (#20850) was obtained from GSK central cell bank. C33A cells were purchased from ATCC (C33A-HTB-31).
2.3 HPV11 E1ˆE4 gene expression assay
Anti-viral testing using an in vitro transient infection assay and HPV11 virions was conducted as previously described (Culp and Christensen, 2003; Squiquera et al., 2017). Media and reagents were from Life Technologies unless specified. A431 cells were maintained in Dulbecco’s modified Eagles Medium (DMEM) supplemented with 2mM Glutamax, NaHC03, Hepes, non-essential amino acids, Na Pyruvate (Quality Biologicals Inc.), antibiotics (Penicillin/Streptomycin) and 10% v/v Foetal bovine Serum (FBS, Atlanta Biologicals). Cells were incubated at 37 °C / 5% v/v CO2. 106 A431 cells per well were cultured in DMEM containing 10% v/v FBS in 6-well dishes for 48 hours. All subsequent assay steps were conducted in DMEM containing 2.5% v/v FBS. Cells were infected with HPV11 at MOI 150 particles per cell in 1mL media. 24 hours later 1mL of additional media was added to give a total of 2mL per well. 48 hours post infection fresh media was added containing BET inhibitors or DMSO vehicle (final assay concentration 1% v/v DMSO). 72 hours after compound treatment the cell cultures were harvested, lysed with Trizol reagent (Life Technologies) and RNA prepared.
Quantitative RT-PCR was conducted to measure viral E1ˆE4 and TATA-binding protein (TBP) transcripts. Amplification of both transcripts was performed using a multiplex format in 50μL. Reactions were performed in duplicate using 200ng Total RNA and the Quantitect Probe RT-PCR kit (Qiagen). Amplification efficiencies and relative quantities of viral target cDNA (relative to host reference gene TBP) were determined using REST software™ (Pfaffl, 2001). Primers and probes were as previously described, primers were synthesized at either the Penn State College of Medicine Core facility or Integrated DNA Technologies (IDT) and probes were purchased from IDT (Culp and Christensen, 2003). Relative E1ˆE4 expression and Total RNA yield were expressed as % inhibition of infected untreated cells. Significance values were computed using Student T-Test (unpaired, 2 tailed, unequal variance). Samples were each compound n=3 and DMSO combined from across all the compound assays (n=9).
To note the kinetics of HPV11 virion entry were previously studied for several different cell lines which showed that complete entry occurred 48 hours after addition of virions (Culp and Christensen, 2003). The infection assay was optimised accordingly to test compounds for antiviral testing and we thus add compounds 48 hours after the addition of virions to prevent possible compound effects on binding, uptake and entry (Squiquera et al., 2017). We have used this assay to test more than 100 compounds for antiviral assessment and have observed that toxic compounds reduce both E1ˆE4 transcripts and Total RNA to similar extents at the same concentration in contrast to antiviral compounds which show marked reductions in E1ˆE4 compared with Total RNA (reference data are shown in S2).
2.4 W12 and C33A viability assays
W12 cells were maintained in media (75% v/v DMEM, 25% v/v F12) supplemented with 2.5% v/v FBS, 24 μg/mL adenine, 0.4 μg/mL hydrocortisone, 5 μg/mL bovine insulin, 8.4 ng/mL Cholera toxin and 10 ng/mL epidermal growth factor. They were grown in the presence of murine 3T3 feeder cells treated with mitomycin C. C33A cells were maintained in Eagle’s Minimum Essential Media supplemented with 10% v/v FBS.
For viability assays cells were seeded at 15,000 cells per well in black sided, 96 well, flat bottomed, sterile plates and incubated for 30 minutes at 37°C / 5% v/v CO2 to adhere. Compounds were added to the cells (final assay concentration of vehicle DMSO was 0.3% v/v). Samples were analysed immediately (T0) or incubated at 37°C / 5% v/v CO2 for 2 days. [ATP] were assayed using CellTitre-Glo (Promega) according to the manufacturer’s instructions and read on a PerkinElmer EnVision 2104 Multilabel reader. Data were analysed using Prism 5.0.4 using a non-linear regression curve fit, log (inhibitor) vs. response analysis-variable slope (four parameters).
2.5 CRPV cutaneous wart model
We used the rabbit CRPV cutaneous wart model which has been optimised for topical compound testing. We have many years experience with this model and have optimised the infectivity methods to reduce rabbit-to-rabbit and wart-to-wart variability using a delayed scarification technique (Cladel et al., 2008; Christensen et al., 2000). Antiviral treatments have been standardised for topical treatments using groups of 4 or 5 rabbits and daily application of compounds on left-side papillomas leaving right-side papillomas untreated. These procedures allow dose-response treatments and control for systemic effects of compounds. Some variability in outcome from rabbit to rabbit is still observed, and is likely to reflect the outbred nature of the individual rabbits used in these studies. We have tested over 50 different compounds using the CRPV/rabbit model (Christensen et al., 2000)
Studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and reviewed and approved by the Penn State University, College of Medicine IACUC (protocol #2010-061). Adult male, New Zealand White rabbits were purchased from Robinson Inc. (North Carolina). On day -3 each rabbit was sedated and 4 sites on the back scarified to an area of 1cm2. On day 0, the rabbits were again sedated, and the scarified sites inoculated with viral DNA as previously described (Cladel et al., 2008). Two sites received 5μg/site wild type (wt-) CRPV DNA (L1, R1) and two sites received 5μg/site mE8CRPV DNA (L2, R2). mE8CRPV is a mutant genome in which the ATG of E8 is mutated to prevent E8 protein expression, leading to slow-growing and small papillomas (Hu et al., 2002). L1 and L2 sites were treated with drug or vehicle and R1 and R2 sites remained untreated. Dosing started two weeks after infection, when the infection was still subclinical. Drug solution or vehicle was applied to the surface of each wart (100μL), once daily, for five consecutive days per week for weeks 3, 4 and 5 and four consecutive days per week for weeks 6 and 7. I-BET762 was tested at 3 formulation strengths w/v 0.015%, 0.15% and 1.5% made up in vehicle solution (groups A, B, C respectively). Group D received vehicle solution (60:40 v/v Ethanol:Britton Robinson pH5 buffer). Group E received Cidofovir 0.3% w/v in cremophor during weeks 3 and 4, as a positive control treatment.
The primary end point was wart growth rate, determined weekly by measuring the size of warts across 3 axes (length × width × height). Blood was sampled 2 or 3 hours following the first dose each week for groups B, C, D and assayed for I-BET762 concentrations. Blood was not sampled for group A since the estimated drug exposure was below the limit of quantification. At the end of the study animals were terminated and wart tissue harvested for histopathology. Data were analysed using a repeated measures analysis of variance model.
2.6 Histopathology
CRPV wart biopsy samples were taken at termination and formalin fixed prior to undergoing processing, paraffin wax embedding and sectioning at 5μm. Sections were stained with Harris Haematoxylin and eosin using standard methods. Macroscopic investigations were undertaken at a magnification of 10x. 2 slides were examined per biopsy sample, and histopathology represented on both treated and untreated papillomas scored.
3. RESULTS
3.1 BET bromodomain inhibitors inhibited HPV-11 E1ˆE4 gene expression in vitro
BET inhibitors (I-BET762 and I-BET151) and an inactive control compound (Control-768) were tested in vitro in an HPV11 infection model. The compound structures and activity in displacing BRD4-histone binding in cells are shown in S1. The expression of reference mRNA TBP was unaltered by compound treatments and thus was used for normalisation of relative gene expression. I-BET762 and I-BET151 reduced E1ˆE4 mRNA expression in a concentration dependent manner, with 3μM I-BET-762 and 3μM and 0.3μM I-BET151 showing marked inhibition (≥ 88%) (Figure 1A). Concurrent cell viability was assessed by quantifying total RNA yield at the end of the assay (Figure 1B). Only the top concentration of I-BET151 (3μM) significantly reduced total RNA yield (34% reduction) in the assay (compared with DMSO treated control) indicating there may be some effect of BET bromodomain inhibition on cell viability at this higher concentration. Control-768 was inactive in the HPV-11 assay as expected. In summary I-BET762 and I-BET151 blocked viral gene transcription at concentrations that did not significantly affect cell viability demonstrating anti-HPV11 activity. Preliminary experiments with a third BET inhibitor also showed anti-viral activity in this assay (S3A).
Figure 1. BET inhibitors show anti-viral activity against HPV-11 in vitro.
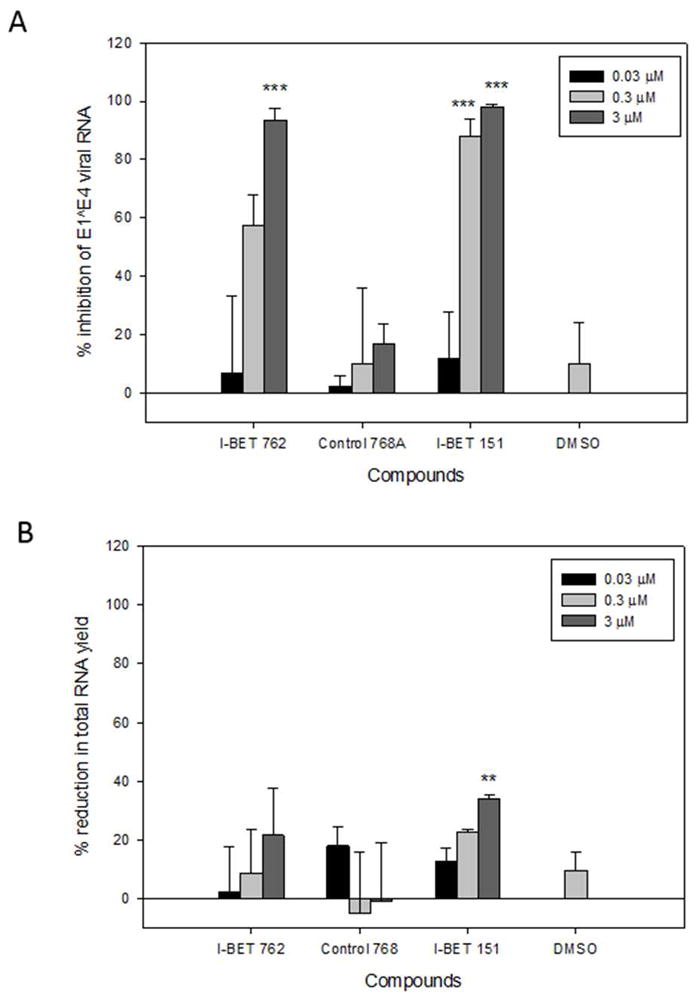
A. Relative HPV-11 E1ˆE4 mRNA and B. Total cellular RNA yields.
Assays performed following 72 hours treatment with drugs or DMSO vehicle.
The plots show mean percent change ± SEM. Percent change was calculated relative to cultures infected with virus alone. Compound Treated samples were each n=3 and DMSO was n=9 (composite from all the experiments).
Significance relative to DMSO control was determined by Student T-test. ** = p<0.01, *** = p<0.001
These compounds were next tested in a CRPV E1ˆE4 gene expression assay, preliminary results indicated that they also blocked CRPV E1ˆE4 gene expression without cytoxicity which encouraged us to employ the CRPV rabbit wart model for a subsequent in vivo investigation (S3B and S44). Of note, we found the 1% v/v DMSO vehicle control showed variable, weak effects on relative E1ˆE4 expression in the HPV11 and CRPV assays, albeit differentiated from the strong effects of the BET inhibitors.
3.2 Therapeutic topical dosing of I-BET762 reduced wart growth in the CRPV cutaneous wart model
To evaluate the potential of BET inhibitors to treat papillomavirus warts, we tested the effect of topical delivery of I-BET762 in the CRPV cutaneous wart model in rabbits, which has been successfully used to evaluate topical anti-viral drug treatments and vaccines (Christensen, 2005; Christensen et al., 2001; Christensen et al., 2000; Kreider et al., 1992; Wolfgang et al., 2009).
Dosing was by a therapeutic schedule as illustrated in Figure 2A. Figure 2B shows the mean data for treated wt-CRPV warts (L1). The positive control Cidofovir (0.3%) blocked wart growth in all rabbits as expected. 1.5% I-BET762 significantly reduced wart growth from week 5 through to 7. I-BET762 at 0.15% and 0.015% were no different from vehicle control. There was an observed small reduction in mean wart size with 0.015% I-BET762 at week 6, but given this was a single time effect we conclude this was an anomaly. No skin irritancy was observed with I-BET762 treatments. Figure 2C shows untreated (R1) wt-CRPV warts for each group, these grew progressively and were similar across rabbits, confirming that none of the drug treatments had a systemic effect which impacted wart growth. Since I-BET762-treated warts (1.5%) grew more slowly than the contralateral untreated warts this confirmed a local effect of drug treatment (Figure 2D).
Figure 2. Topical administration of I-BET762 reduced wt-CRPV wart growth in rabbits.
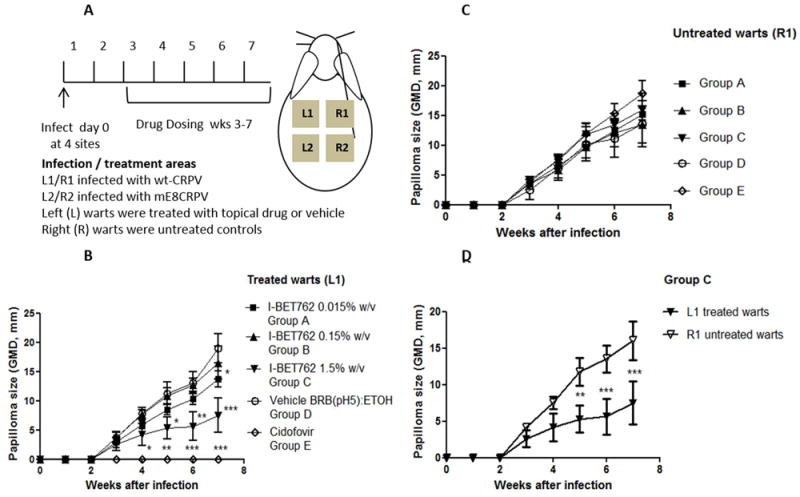
Papillomas were induced at each site with 5μg of wt-CRPV or mE8-mutantCRPV plasmid and treated at the start of week 3 with topical treatments at the site of infection. Treatments were as labelled. Plots show Geometric Mean Diameters +/- SEM for wt-CRPV warts, measured weekly (Groups A-C n=5, Group D n=4, Group E n=3). Significance was determined by repeated measures of analyses of variance. * = p <0.05 ** = p<0.01 *** = p<0.001.
A. Illustration of protocol
B. Wart growth for treated warts (L1), significance shown is for difference relative to vehicle group
C. Wart growth for untreated warts (R1)
D. Comparison of L1 (treated) and R1 (untreated) wart growth for group C animals, significance shown is relative to each other
We further examined the wt-CRPV warts by histopathology (Table 1 and S5). Smaller wart size was consistent with reduced pathological features. Interestingly immune cell infiltrate was still present in the skin of I-BET762 treated warts and it would be valuable to characterise anti-viral immunity in future studies. As expected for biological systems, there was variability in the response of rabbits to treatment with 1.5% I-BET762; one rabbit had no visible wart growth, two showed markedly reduced wart growth, two rabbits had low/no response to drug treatments (Figure 3 and S6).
Table 1.
Histopathology of wt-CRPV papillomas for individual animals
| A | Animal Number | Increased Cell Infiltrate | Acanthosis | Koilocytes | Stratum Corneum Thickening | Parakeratosis |
|---|---|---|---|---|---|---|
| 3422 | X | X | X | X | X | |
| 3423 | X | X | X | X | X | |
| 3424 | X | X | X | X | X | |
| 3425 | X | X | X | X | X | |
| B | Animal Number | Increased Cell Infiltrate | Acanthosis | Koilocytes | Stratum Corneum Thickening | Parakeratosis |
|
| ||||||
| 3412 | X | |||||
| 3413 | X | X | X | X | ||
| 3414 | X | X | X | X | X | |
| 3415 | X | |||||
| 3416 | X | X | X | X | X | |
Presence of histopathology features in wt-CRPV papillomas from animals treated with A. vehicle (group D) and B. 1.5% w/v I-BET762 (group C). Examples of pathology are illustrated in S5.
Figure 3. Visual appearance of warts for individual animals treated with 1.5% I-BET762.

Schematic of wart positions on rabbit back. Papillomas were wt-CRPV (L1, R1) or me8-CRPV (L2, R2). L1 and L2 were treated and R1 and R2 were untreated controls.
Photographs of warts for Group C animals, taken at end of the study 7 weeks post infection.
In contrast to wt-CRPV, mE8CRPV warts did not grow sufficiently well in this experiment to enable drug effects to be assessed (S6).
We measured blood concentrations of I-BET762 to assess delivery of drug (S7). Systemic I-BET762 concentrations were detectable on each sampling occasion over the duration of the study and were higher in group C than B consistent with the dose strength. As I-BET762 was observed in the blood it was inferred that the target cells in the skin had been exposed to the drug although skin concentrations were not directly measured. Unexpectedly I-BET762 was also detected in some blood samples from animals dosed with vehicle alone. After consideration of all possible operational and analytical factors, the definitive reason for this observation remains unknown, however it is not considered to affect the overall conclusion for efficacy of I-BET762.
In summary, these results confirm therapeutic efficacy of topical I-BET762 and to our knowledge this is the first demonstration of a BET inhibitor abrogating papillomavirus infection in vivo.
3.3 I-BET762 reduced viability of HPV-16 infected W12 cells in vitro
Episome maintenance is central to persistent papillomavirus infection and we were interested to evaluate whether BET inhibitors may impact this process. We set out to explore this using W12 cells (clone #20850), derived from a low grade cervical intra-epithelial neoplasia (CIN1) and which contains predominantly episomal HPV16 (Gray et al., 2010; Stanley et al., 1989). However, treatment of W12 cells with I-BET762 lead to loss in cell viability within two days making it unfeasible to study changes in viral load during propagation of the cells (data not shown). W12 cells are immortalized and it has been suggested that they may depend upon HPV16 gene products for survival (Stanley et al., 1989). We therefore thought the rapid reduction in viability with BET inhibitor treatment could be due to modulation of viral dependent processes e.g. viral gene expression or deregulated host-gene expression. To consolidate this hypothesis we evaluated the specificity for the effects on cell viability, comparing effects on W12 versus an HPV-negative cervical epithelial cell line C33A. Cell viability was measured by ATP concentration.
I-BET762 markedly reduced viability of W12 cells in a concentration dependent manner, with IC50 =700 nM and 63% reduction at 30μM (Figure 4A) and slightly reduced the viability of C33A cells 28% at 30μM. Control-768 had no impact on viability of W12 or C33A cells, indicating the effects of I-BET762 were mediated by inhibition of the bromodomains. Furthermore, I–BET-762 treatment resulted in W12 cell depletion over two days as the cell viability at the end of the assay was reduced below that at the start (Figure 4B), in contrast I-BET762 treated C33A cells continued to proliferate, albeit at a slower rate. Control-768 compound had no effect on the growth of W12 or C33A cells as expected. In summary, these experiments show that cervical epithelial cells lines have differential sensitivity to I-BET762 treatment with W12 cells rapidly dying in the presence of the drug in contrast to the C33A cells which exhibited delayed growth, and we conclude that BET proteins are involved in W12 cell survival.
Figure 4. I-BET762 selectively reduced viability of HPV-16 infected cells.
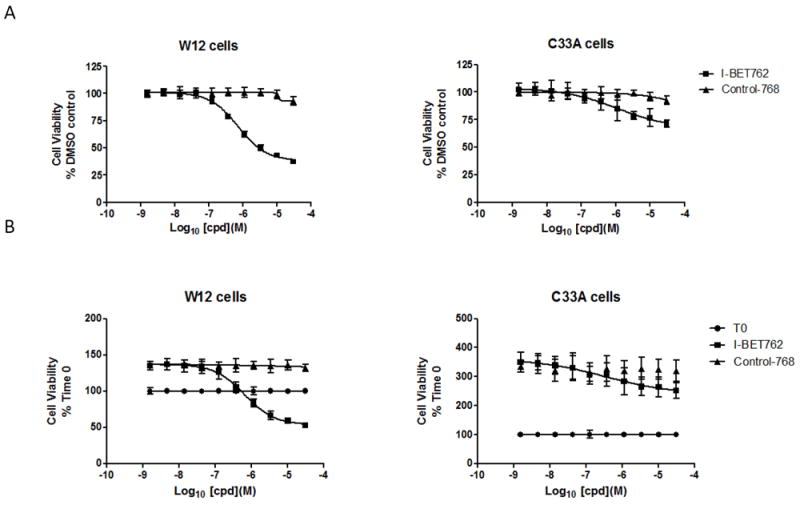
W12 and C33A epithelial cells were treated with BET inhibitors for 2 days or harvested at time 0 as labelled.
A. Viability assayed by [ATP] on day 2 post compound treatment and normalised to DMSO control. Mean data from n=4 technical replicates ± SD. Similar results were obtained in a replicate experiment.
B. Viability assayed by [ATP] on day 2 post compound treatment and normalised to viability on day 0. Mean data from n=4 technical replicates ± SD.
4. DISCUSSION AND CONCLUSIONS
We aimed to establish whether BET inhibitors may offer a new therapeutic anti-viral strategy for treating persistent HPVinfection and addressed this using in vitro and in vivo models.
Several BET inhibitors inhibited HPV11 E1ˆE4 gene expression. This effect is consistent with published work using HPV16 and BPV1 replication models, which showed early papillomavirus gene transcription was impaired by treatment with BET bromodomain inhibitor JQ1 (Helfer et al., 2014). The anti-viral activity we observed with an additional papillomavirus type point to conserved mechanisms. Although we did not monitor viral DNA replication or other viral transcripts, the transient nature of the assay (2 days infection to permit viral entry followed by 3 days of drug treatment) could suggest that the anti-viral effect is post-entry and my potentially target both the initial but limited unlicensed viral DNA replication as well as early viral mRNA expression that occurs when virus particles infect basal keratinocytes (McBride and Jang, 2013). Interestingly it has been shown that a BET inhibitor blocked replication of EBV both by blocking early viral gene expression and lytic genome replication (Keck et al., 2017).
One model for BRD4 regulation of viral gene expression is via recruitment of host factors such as positive transcriptional elongation factor protein b (P-TEFb) to the viral genome. BRD4 promotes transcription of host genes via pTEFb recruitment, also shown for HPV18 oncogenes and interestingly BRD4 and pTEFb are present on HPV16 and BPV1 episomes (Helfer et al., 2014; Jang et al., 2005; Yan et al., 2010; Yang et al., 2005). Additionally E2 and P-TEFb compete for the same binding location on BRD4, and E2 repressive functions can depend on BRD4 (McBride and Jang, 2013; Wu et al., 2006; Yan et al., 2010). Thus the control of viral gene expression is multifaceted. Given that expression from the papillomaviral promoters is contemporaneous with keratinocyte differentiation, extending these compound studies to assays of late viral gene expression and vegetative viral DNA replication using in vitro organotypic raft cultures, would be insightful (reviewed in (Andrei et al., 2010).
The activity observed with several compounds in the CRPV E1ˆE4 in vitro assay encouraged us to evaluate effects of topical delivery of a BET inhibitor in the CRPV cutaneous wart model in rabbits. We show for the first time in vivo efficacy of a BET inhibitor on papillomavirus infection. 1.5% I-BET762 profoundly impaired wart growth in 3/5 animals, the lack of effect in the contralateral untreated wart confirmed these effects were via the topical delivery route and not through systemic drug levels. Variable responses of warts to treatments in the CRPV rabbit model are not unusual in our experience, most likely due to the outbred nature of laboratory rabbits (Christensen et al., 2000). Since we used a liquid formulation there may also be opportunity to improve efficacy through optimising PK-PD using alternative formulations such as gels or creams, designed specifically for this molecule and disposition in the skin. From a translational perspective, it would be useful to evaluate whether BET inhibitors have the potential to clear virus from the basal epithelial cells and consequently reduce recurrence rates, which can be explored in the CRPV model (Christensen et al., 2001). Moreover, these results in vivo complement research demonstrating a critical role for BRD4 in CRPV tumourigenesis; in which mutation of residues R37 and I73 in CRPV E2, essential for BRD4 binding, and expression of a shRNA targeting BRD4, were shown to reduce CRPV tumour formation in vivo (Delcuratolo et al., 2016; Jeckel et al., 2002).
Additional investigations using the W12 cell line carrying HPV16 episomes, showed these cells were rapidly killed by I-BET 762 treatment in contrast to a non infected cervical epithelial cell line. The mechanism for the cytotoxicity is unknown, but it is plausible that BET inhibition blocked expression of viral and or deregulated host genes in W12 cells, affecting overall cell survival; HPV16 early genes were sensitive to BET inhibition within 15 hours of treatment in the W12 clone (20863) used by Helfer et al 2014, also E7 expressed in W12 cells may promote expression and/or activity of cellular oncogenes such as c-Myc which is a known target gene for BET inhibitors in some cancers (Dawson et al., 2011; Doorbar et al., 1990; Hwang et al., 2002; Wang et al., 2007). ChIP-Seq has been conducted in E2 transfected C33A cells, demonstrating that cellular BRD4 and BPV1 E2 co-localise to transcriptionally active regions in host chromatin and similar analyses in the W12 cells, if technically feasible (with endogenous BRD4/E2), may elucidate novel viral-host interactions which contribute to stable HPV16 infection and latency (Jang et al., 2009).
The role BET proteins play in the life cycle of viruses has been explored for other DNA viruses and retroviruses and BET inhibitors show contrasting effects depending on the virus, for example BET inhibitors reactivated HIV in models of latency and blocked EBV early gene expression and lytic genome replication (Banerjee et al., 2012; Keck et al., 2017; Palermo et al., 2011; Weidner-Glunde et al., 2010; Zhu et al., 2012). Since BRD4/PTEFb dependent activation of HPV oncogenes has been described the effect of BET inhibitors on reducing HPV oncogene expression could also be worthwhile investigating (Yan et al., 2010).
Oral BET inhibitors are in early clinical development in oncology which should provide insights into the safety and tolerability in humans over the next few years. Still preclinical experiments have shown that a BET inhibitor disrupted spermatogenesis, likely through targeting BRDT (Matzuk et al., 2012) and therefore for treatment of benign HPV infection topical formulations which limit systemic exposure and/or BET bromodomain inhibitors with improved selectivity for BRD4 may be desired.
In conclusion, we show BET inhibitors reduced HPV11 viral E1ˆE4 gene expression in vitro and a topical formulation of I-BET762 abrogated CRPV wart growth in vivo. Additionally I-BET762 exhibited a marked cytotoxicity to HPV16 infected W12 cells, suggesting a hypothesis for BET-regulation of HPV-dependent survival factors. Collectively our results highlight the potential to target BET bromodomains to treat HPV infection. We also describe pharmacological tools to investigate the role of BET proteins and associated epigenetic processes in papillomaviral replication in-situ. Further research in this area could give insights to differentiation from current treatments, for example whether BET inhibitors can clear virus from basal cells and reduce recurrent infection.
Supplementary Material
HIGHLIGHTS.
We show anti-papillomavirus effects of BET bromodomain inhibitors, a new class of epigenetic drug.
Several BET inhibitors reduced HVP11 E1ˆE4 transcription in a transient infection model in vitro.
Topical delivery of BET inhibitor abrogated CRPV wart growth in rabbits, demonstrating translation to efficacy in vivo.
BET inhibitor treatment reduced viability of HPV16 infected W12 cells.
Efficacy across multiple PV types and in vivo highlight potential to target BET bromodomains to treat HPV infection.
Acknowledgments
We are grateful to Heather Greenstone and Christopher Tseng for administrating the project at the National Institutes of Health. We thank Neil Garton for arranging the GSK Material Transfer Agreement and Steven Fox for statistical support.
FUNDING
This work was funded in part by the National Institute of Allergy and Infectious Diseases Antiviral testing program, contracts HHSN2722011000005C and HHSN272201000020I (NDC).
Footnotes
CONTRIBUTIONS OF AUTHORS
MAM, CAC, RCF, KL, RKP, ST, JW, NDC: Conceived and /or designed research.
KKB, SAB, CAC, MXC, RCF, ILH, IDH, UK, MR, JS: Performed experiments and / or delivered reagents.
MAM, KKB, SAB, CAC, MXC, RCF, ILH, IDH, UK, MR, ST, NDC: Analysed data and interpreted results of experiments.
MAM, NDC: Drafted manuscript.
All authors approved final version.
DECLARATION OF INTERESTS
MAM, MXC, RCF, ILH, UK, RKP, MR, JS, ST, JW are employees of GSK.
KKB, SAB, CAC, IDH, KL, NDC have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell. 2006;24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Andrei G, Duraffour S, Van den Oord J, Snoeck R. Epithelial raft cultures for investigations of virus growth, pathogenesis and efficacy of antiviral agents. Antiviral Res. 2010;85:431–449. doi: 10.1016/j.antiviral.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol. 2012;92:1147–1154. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter MK, McPhillips MG, Ozato K, McBride AA. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J Virol. 2005;79:4806–4818. doi: 10.1128/JVI.79.8.4806-4818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, Liu L, Lynch CF, Wentzensen N, Jordan RC, Altekruse S, Anderson WF, Rosenberg PS, Gillison ML. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. 2009;1:98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ND. Cottontail Rabbit Papillomavirus (CRPV) Model System to Test Antiviral and Immunotherapeutic Strategies. Antiviral Chemistry and Chemotherapy. 2005;16:355–362. doi: 10.1177/095632020501600602. [DOI] [PubMed] [Google Scholar]
- Christensen ND, Han R, Cladel NM, Pickel MD. Combination treatment with intralesional cidofovir and viral-DNA vaccination cures large cottontail rabbit papillomavirus-induced papillomas and reduces recurrences. Antimicrob Agents Chemother. 2001;45:1201–1209. doi: 10.1128/AAC.45.4.1201-1209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ND, Pickel MD, Budgeon LR, Kreider JW. In vivo anti-papillomavirus activity of nucleoside analogues including cidofovir on CRPV-induced rabbit papillomas. Antiviral Research. 2000;48:131–142. doi: 10.1016/s0166-3542(00)00124-8. [DOI] [PubMed] [Google Scholar]
- Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- Cirelli R, Tyring SK. Interferons in human papillomavirus infections. Antiviral Research. 1994;24:191–204. doi: 10.1016/0166-3542(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Cladel NM, Hu J, Balogh K, Mejia A, Christensen ND. Wounding prior to challenge substantially improves infectivity of cottontail rabbit papillomavirus and allows for standardization of infection. J Virol Methods. 2008;148:34–39. doi: 10.1016/j.jviromet.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culp TD, Christensen ND. Quantitative RT-PCR assay for HPV infection in cultured cells. Journal of Virological Methods. 2003;111:135–144. doi: 10.1016/s0166-0934(03)00170-8. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Pena LM, Laimins LA. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J Virol. 2001;75:10005–10013. doi: 10.1128/JVI.75.20.10005-10013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcuratolo M, Fertey J, Schneider M, Schuetz J, Leiprecht N, Hudjetz B, Brodbeck S, Corall S, Dreer M, Schwab RM, Grimm M, Wu SY, Stubenrauch F, Chiang CM, Iftner T. Papillomavirus-Associated Tumor Formation Critically Depends on c-Fos Expression Induced by Viral Protein E2 and Bromodomain Protein Brd4. PLoS Pathog. 2016;12:e1005366. doi: 10.1371/journal.ppat.1005366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology. 1990;178:254–262. doi: 10.1016/0042-6822(90)90401-c. [DOI] [PubMed] [Google Scholar]
- Favre M, Breitburd F, Croissant O, Orth G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. Journal of Virology. 1977;21:1205–1209. doi: 10.1128/jvi.21.3.1205-1209.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri E, Petosa C, McKenna CE. Bromodomains: Structure, function and pharmacology of inhibition. Biochem Pharmacol. 2016;106:1–18. doi: 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E, Pett MR, Ward D, Winder DM, Stanley MA, Roberts I, Scarpini CG, Coleman N. In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res. 2010;70:4081–4091. doi: 10.1158/0008-5472.CAN-09-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfer CM, Yan J, You J. The cellular bromodomain protein Brd4 has multiple functions in E2-mediated papillomavirus transcription activation. Viruses. 2014;6:3228–3249. doi: 10.3390/v6083228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, Lowy DR. Papillomaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Lippincott Williams and Wilkins; Philadelphia: 2007. [Google Scholar]
- Hu J, Han R, Cladel NM, Pickel MD, Christensen ND. Intracutaneous DNA Vaccination with the E8 Gene of Cottontail Rabbit Papillomavirus Induces Protective Immunity against Virus Challenge in Rabbits. Journal of Virology. 2002;76:6453–6459. doi: 10.1128/JVI.76.13.6453-6459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SG, Lee D, Kim J, Seo T, Choe J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J Biol Chem. 2002;277:2923–2930. doi: 10.1074/jbc.M109113200. [DOI] [PubMed] [Google Scholar]
- Ilves I, Maemets K, Silla T, Janikson K, Ustav M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol. 2006;80:3660–3665. doi: 10.1128/JVI.80.7.3660-3665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Anderson DE, van Doorslaer K, McBride AA. A proteomic approach to discover and compare interacting partners of papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics. 2015;15:2038–2050. doi: 10.1002/pmic.201400613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Kwon D, McBride AA. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J Virol. 2009;83:2592–2600. doi: 10.1128/JVI.02275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jeckel S, Huber E, Stubenrauch F, Iftner T. A Transactivator Function of Cottontail Rabbit Papillomavirus E2 Is Essential for Tumor Induction in Rabbits. Journal of Virology. 2002;76:11209–11215. doi: 10.1128/JVI.76.22.11209-11215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck KM, Moquin SA, He A, Fernandez SG, Somberg JJ, Liu SM, Martinez DM, Miranda JL. Bromodomain and extraterminal inhibitors block the Epstein-Barr virus lytic cycle at two distinct steps. Journal of Biological Chemistry. 2017;292:13284–13295. doi: 10.1074/jbc.M116.751644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreider JW, Christensen ND, Christian CB, Pickel MD. Preclinical System for Evaluating Topical Podofilox Treatment of Papillomas: Dose-Response and Duration of Growth Prior to Treatment. Journal of Investigative Dermatology. 1992;99:813–818. doi: 10.1111/1523-1747.ep12614781. [DOI] [PubMed] [Google Scholar]
- Kreider JW, Howett MK, Leure-Dupree AE, Zaino RJ, Weber JA. Laboratory production in vivo of infectious human papillomavirus type 11. Journal of Virology. 1987;61:590–593. doi: 10.1128/jvi.61.2.590-593.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Chiang CM. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem. 2009;284:2778–2786. doi: 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, Jang MK. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses. 2013;5:1374–1394. doi: 10.3390/v5061374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, Romanczuk H, Howley PM. The papillomavirus E2 regulatory proteins. Journal of Biological Chemistry. 1991;266:18411–18414. [PubMed] [Google Scholar]
- McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Jacob Y, Jones L, Weiss A, Brino L, Chantier T, Lotteau V, Favre M, Demeret C. Large scale genotype comparison of human papillomavirus E2-host interaction networks provides new insights for e2 molecular functions. PLoS Pathog. 2012;8:e1002761. doi: 10.1371/journal.ppat.1002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo RD, Webb HM, West MJ. RNA polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein-Barr virus. PLoS Pathog. 2011;7:e1002334. doi: 10.1371/journal.ppat.1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Research. 2001;29:e45–e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Schofer H, Van Ophoven A, Henke U, Lenz T, Eul A. Randomized, comparative trial on the sustained efficacy of topical imiquimod 5% cream versus conventional ablative methods in external anogenital warts. European journal of dermatology : EJD. 2006;16:642–648. [PubMed] [Google Scholar]
- Schweiger MR, Ottinger M, You J, Howley PM. Brd4-independent transcriptional repression function of the papillomavirus e2 proteins. J Virol. 2007;81:9612–9622. doi: 10.1128/JVI.00447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology. 2007;358:10–17. doi: 10.1016/j.virol.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squiquera L, Taxman DJ, Brendle SA, Torres R, Sulley J, Hodge T, Christensen N, Sidransky D. Ranpirnase eradicates human papillomavirus in cultured cells and heals anogenital warts in a Phase I study. Antiviral Therapy. 2017;22:247–255. doi: 10.3851/IMP3133. [DOI] [PubMed] [Google Scholar]
- Stanley MA. Genital human papillomavirus infections: current and prospective therapies. J Gen Virol. 2012;93:681–691. doi: 10.1099/vir.0.039677-0. [DOI] [PubMed] [Google Scholar]
- Stanley MA, Browne HM, Appleby M, Minson AC. Properties of a non-tumorigenic human cervical keratinocyte cell line. International Journal of Cancer. 1989;43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- Stern PL, van der Burg SH, Hampson IN, Broker TR, Fiander A, Lacey CJ, Kitchener HC, Einstein MH. Therapy of human papillomavirus-related disease. Vaccine. 2012;30(Suppl 5):F71–82. doi: 10.1016/j.vaccine.2012.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrjanen K. Anogenital human papillomavirus and the problem of persistence. European Journal of Dermatology. 1998;8:5–7. [PubMed] [Google Scholar]
- Theodoulou NH, Tomkinson NC, Prinjha RK, Humphreys PG. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr Opin Chem Biol. 2016;33:58–66. doi: 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- Wang YW, Chang HS, Lin CH, Yu WC. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int J Biochem Cell Biol. 2007;39:402–412. doi: 10.1016/j.biocel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Weidner-Glunde M, Ottinger M, Schulz F. WHAT do viruses BET on? Front Biosci (Landmark Ed) 2010;15:537–549. doi: 10.2741/3632. [DOI] [PubMed] [Google Scholar]
- Wolfgang GH, Shibata R, Wang J, Ray AS, Wu S, Doerrfler E, Reiser H, Lee WA, Birkus G, Christensen ND, Andrei G, Snoeck R. GS-9191 is a novel topical prodrug of the nucleotide analog 9-(2-phosphonylmethoxyethyl)guanine with antiproliferative activity and possible utility in the treatment of human papillomavirus lesions. Antimicrob Agents Chemother. 2009;53:2777–2784. doi: 10.1128/AAC.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge TR, Laimins LA. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology. 2008;374:371–380. doi: 10.1016/j.virol.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, Nin DS, Lee AY, Simanski S, Kodadek T, Chiang C-M. BRD4 Phosphorylation Regulates HPV E2-Mediated Viral Transcription, Origin Replication, and Cellular MMP-9 Expression. Cell Reports. 2016;16:1733–1748. doi: 10.1016/j.celrep.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Li Q, Lievens S, Tavernier J, You J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J Virol. 2010;84:76–87. doi: 10.1128/JVI.01647-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- You J. Papillomavirus interaction with cellular chromatin. Biochim Biophys Acta. 2010;1799:192–199. doi: 10.1016/j.bbagrm.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the Bovine Papillomavirus E2 Protein with Brd4 Tethers the Viral DNA to Host Mitotic Chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- You J, Schweiger MR, Howley PM. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J Virol. 2005;79:14956–14961. doi: 10.1128/JVI.79.23.14956-14961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, Brass AL. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.