

Abstract
Dissimilatory perchlorate reduction is an anaerobic respiratory pathway that in communities might be influenced by metabolic interactions. Because the genes for perchlorate reduction are horizontally transferred, previous studies have been unable to identify uncultivated perchlorate-reducing populations. Here we recovered metagenome-assembled genomes from perchlorate-reducing sediment enrichments and employed a manual scaffolding approach to reconstruct gene clusters for perchlorate reduction found within mobile genetic elements. De novo assembly and binning of four enriched communities yielded 48 total draft genomes. In addition to canonical perchlorate reduction gene clusters and taxa, a new type of gene cluster with an alternative perchlorate reductase was identified. Phylogenetic analysis indicated past exchange between these gene clusters, and the presence of plasmids with either gene cluster shows that the potential for gene transfer via plasmid persisted throughout enrichment. However, a majority of genomes in each community lacked perchlorate reduction genes. Putative chlorate-reducing or sulfur-reducing populations were dominant in most communities, supporting the hypothesis that metabolic interactions might result from perchlorate reduction intermediates and byproducts. Other populations included a novel phylum-level lineage (Ca. Muirbacteria) and epibiotic prokaryotes with no known role in perchlorate reduction. These results reveal unexpected genetic diversity, suggest that perchlorate-reducing communities involve substantial metabolic interactions, and encourage expanded strategies to further understand the evolution and ecology of this metabolism.
Introduction
Perchlorate (ClO4−), the most oxidized chlorine oxyanion, is produced naturally through successive oxidation of chloride by photochemical reactions or electrical discharge [1, 2]. Natural concentrations of perchlorate are low in most environments but appreciable in many desert soils and in regolith on Mars [1, 3]. Perchlorate is utilized as an anaerobic respiratory electron acceptor by dissimilatory perchlorate-reducing microorganisms (DPRM), which obtain energy by coupling the reduction of perchlorate to the oxidation of various electron donors [4]. In the canonical perchlorate reduction pathway, perchlorate reductase (PcrAB) reduces perchlorate to chlorate (ClO3−) and then chlorite (ClO2−), chlorite dismutase (Cld) dismutates chlorite to chloride (Cl−) and oxygen (O2), and a terminal oxygen reductase reduces oxygen to water (Supplementary Figure 1) [4]. The reactive chlorine species hypochlorite (ClO−), a spurious byproduct of chlorite dismutation, damages cells through oxidizing reactions, and in some DPRM a periplasmic methionine sulfoxide reductase (YedYZ) in combination with a methionine-rich peptide (MRP) subverts oxidative damage to proteins [5].
The genes for canonical perchlorate reduction are found within a horizontally transferred gene cluster referred to as the perchlorate reduction genomic island (PRI) [6]. PRI always contain chlorite dismutase (cld) and perchlorate reductase (pcr) and generally include genes for transcriptional regulation, oxidative stress response, molybdenum cofactor biosynthesis, and genetic mobility [6, 7]. The perchlorate reductase operon consists of catalytic subunits (pcrAB), a maturation protein (pcrD), and sometimes electron transfer subunits (pcrCO) and a quinol-oxidizing subunit (pcrQ) [7, 8]. Contemporary horizontal transfer of PRI complicates the identification of perchlorate-reducing bacteria in communities, though transfer seems to be restricted within specific families in the Alpha-, Beta-, Gamma-, and Epsilonproteobacteria [7]. In contrast, the genes involved in the closely related dissimilatory chlorate respiration, chlorate reductase (clrABDC) and cld, were recently exchanged between different Protebacteria classes [9]. Although a single ancestral PRI had been suggested based on monophyly in both PcrA and Cld [7], a broader extent of genetic exchange in chlorine oxyanion metabolism is hinted at by the polyphyletic nature of ClrA and the transfer of the cld gene from DPRM to dissimilatory chlorate-reducing microorganisms (DCRM) [9]. Further, the bias imposed by isolation techniques may mean that communities host a more diverse set of PRMs than previously identified.
Preliminary evidence suggests that the intermediates and byproducts of perchlorate reduction could potentially support other metabolisms in communities. For example, the intermediate chlorate transiently accumulated during perchlorate reduction by Dechlorosoma sp. HCAP-C, but was consumed when a DCRM was added in co-culture [10]. Similarly, chlorite, a toxic intermediate, was consumed by non-DPRM cells expressing Cld in an engineered mutualistic interaction with cells that reduced the perchlorate to chlorite [11] (denoted “symbiotic perchlorate reduction” [4]). An alternative mechanism of mutualism involves the sulfur redox cycle. Elemental sulfur (So) and polysulfide (Sn2-) are produced as byproducts of perchlorate reduction through enzymatic oxidation of sulfide by PcrAB, and abiotic reaction with chlorite, hypochlorite, and oxygen (Supplementary Figure 1) [12, 13]. This process, named perchlorate-dependent sulfide oxidation (PSOX), has been suggested to support elemental sulfur-reducing bacteria in multiple perchlorate-reducing communities based on the enrichment of taxa affiliated with isolated sulfur-reducing bacteria [14, 15]. In turn, the concurrent reduction of sulfur species to sulfide allows some organisms to grow by perchlorate reduction without chlorite dismutase [16, 17] (denoted “cryptic perchlorate reduction” [4]). None of the proposed metabolic interactions, however, have been demonstrated to occur in naturally enriched co-cultures or communities.
The goals of this study were to assess the genetic and phylogenetic diversity of uncultivated DPRM and to identify the possible extent of metabolic interactions occurring in perchlorate-reducing communities. Recently developed techniques to recover genomes from large metagenomic data sets enable the prediction of metabolic functions and interactions in complex communities [18–20]. A previous study using 16S rRNA gene sequencing showed that perchlorate-reducing communities from marine sediment enriched with acetate and perchlorate consisted mostly of phyla that do not contain known DPRM, such as Deltaproteobacteria and Bacteroidetes [14]. We re-evaluated these same enrichments by using genomic DNA sequencing, de novo assembly, and binning to link perchlorate reduction genes to the genomes of specific populations. The high sequence similarity of perchlorate reduction genes led to incomplete assembly and binning, a common problem with genes subject to mobility or strain variation. We overcame this limitation by employing contig assembly graphs to manually construct genomic regions containing pcr and cld sequences. While most diversity in DPRM matched that of isolated organisms, by using cld as a biomarker for novel (per)chlorate reductase genes we identified a gene cluster containing cld and a putative alternative perchlorate reductase. That numerous genomes lacked any perchlorate reduction genes led us to conclude that much of the community may be supported by metabolic interactions with DPRM.
Materials and methods
Enrichment and sampling
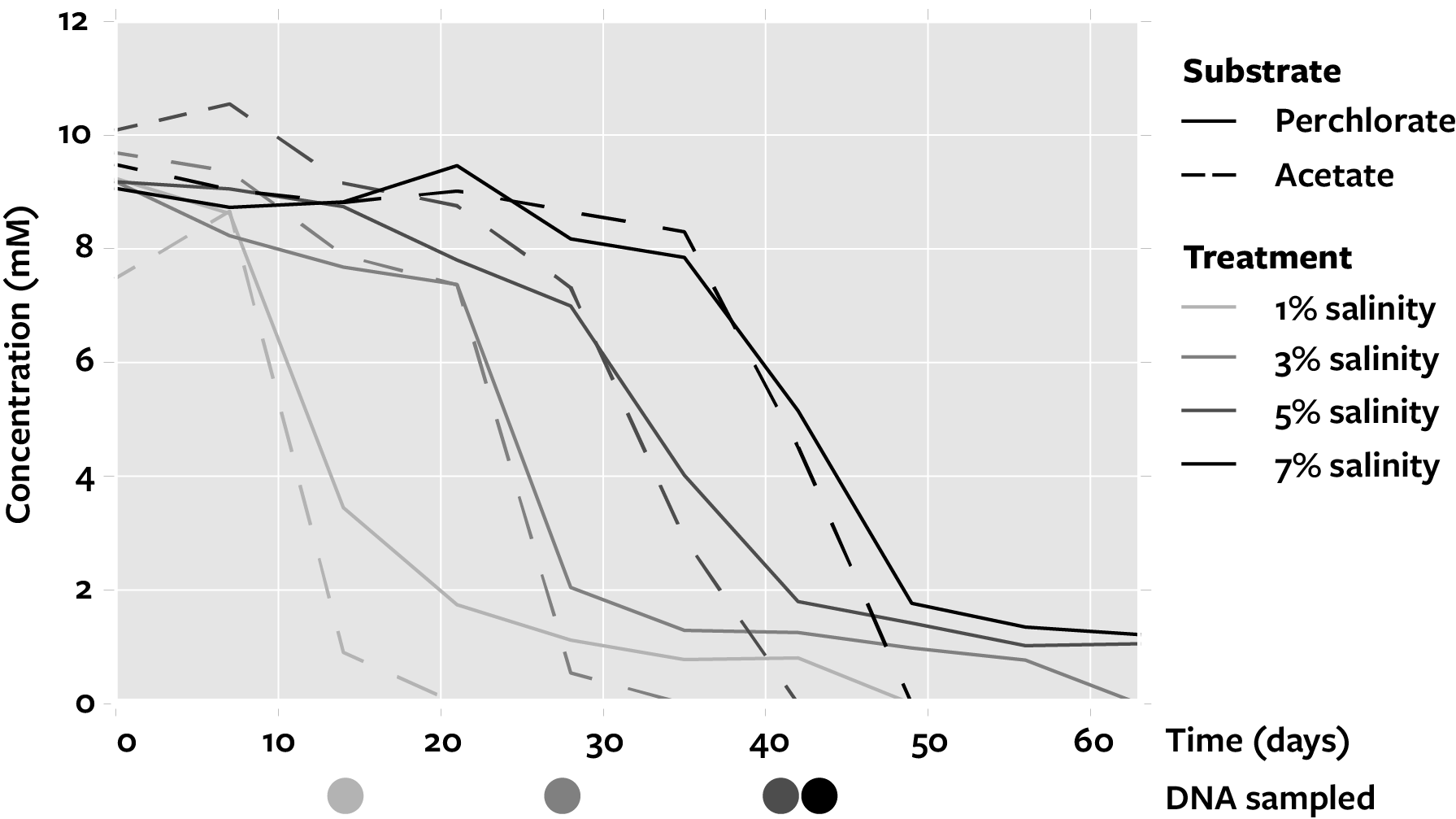
Samples were selected from enrichments described in ref. [14]. Briefly, those enrichments utilized a sterile, anoxic, defined medium at pH 6.8 (bicarbonate-buffered) with 10 mM perchlorate (electron acceptor) and 10 mM acetate (non-fermentable electron donor and carbon source) [14, 21]. Inoculum consisted of sulfidogenic marine sediment collected from the San Francisco Estuary near the outlet of a freshwater stream (37.8629° N, 122.3132° W) in Berkeley, California, USA. Bottles with defined media and 5% (v/v) sediment inoculum were incubated at 30 °C for 63 days, and biomass samples (20 mL) were collected weekly using 0.22-µm filters (MoBio, Carlsbad, CA) and preserved at −80 °C. Partial removal of perchlorate (2.1 ± 0.4 mM) and complete removal of acetate occurred in the no-donor and no-acceptor controls, respectively, indicating the presence of alternative electron donors and acceptors from the sediment inoculum, although no sulfate was removed [22]. For this study, to obtain distinct perchlorate-reducing communities a single sample was chosen to represent each condition at a time point following substantial perchlorate removal: 14 days at 1% salinity (NaCl, w/v), 28 days at 3% salinity, 42 days at 5% salinity, and 42 days at 7% salinity (Supplementary Figure 2).
Metagenome sequencing and assembly
DNA was extracted using the MoBioRNAPowerSoil Kit with the DNA Elution Accessory Kit (MoBio Laboratories, Inc., Carlsbad, CA) as previously described [14]. DNA library preparation and sequencing were performed by the Vincent J. Coates Genomics Sequencing Laboratory at the California Institute of Quantitative Biosciences (QB3−Berkeley, Berkeley, CA) using an Illumina HiSeq 2000 platform with 100-bp paired-end reads. Prior to assembly, reads were trimmed using Sickle v. 1.33 with default parameters [23]. Reads from all samples were then co-assembled using MEGAHIT v. 1.0.2 with “meta-sensitive” presets [24]. Following assembly, reads were dereplicated by mapping reads to contigs using the Burrows-Wheeler Alignment Tool v. 0.7.10 (BWA) BWA-MEM algorithm [25] and selecting unique reads using Picard Tools v. 2.4.1 (http://picard.sourceforge.net/). Automated scaffolding of contigs was performed using SSPACE v. 3.0, which uses paired-end read information to join contigs by filling the length of the gap with the character “N” [26], without scaffold extension and with 25% variation from the estimated median insert size (275 bp) allowed. All computation was performed on the computer cluster at the QB3 Computational Genomics Resource Laboratory (CGRL) (UC Berkeley, Berkeley, CA).
Metagenome-assembled genome binning
Binning of scaffolds was performed using the default pipeline in Anvi’o v. 2.0.2 [27] as follows. Dereplicated reads from each sample were mapped to scaffolds using BWA-MEM and sorted and indexed using Samtools v. 0.1.19 [28] to provide differential abundance estimates across samples. Scaffolds greater than 2000 bp were binned using CONCOCT [29], which uses abundance, nucleotide composition, and read-pairing for automated binning, and the resulting bins were manually refined using abundance, GC content, taxonomic markers, and single-copy gene counts to remove incorrectly binned genomic and viral sequences. Plasmid sequences, which sometimes can be maintained at a higher copy number than chromosomal sequences, were assigned to bins based on similar patterns of coverage between samples and the taxonomic identity of plasmid genes. Coverage for scaffolds and bins was calculated as follows: coverage = (number reads mapped to sequence) × (read length)/(total scaffold length).
The quality of each bin was assessed using CheckM v. 1.0.1 [30], which uses lineage-specific sets of single-copy genes (SCGs) to estimate completeness and contamination and assigns contamination to strain heterogeneity if amino acid identity is great than 90%. Further analysis used only high-quality draft genomes, defined as bins that are near-complete (>90% SCGs present) with low contamination (<5% redundant SCGs), as well as medium-quality draft genomes that were more than moderately complete (>70% SCGs present) with medium contamination (<10% redundant SCGs) or less. The quality of bins belonging to candidate phyla radiation (CPR) bacteria, a broad lineage missing SCGs found in other bacteria [31], was assessed using a set of CPR-specific SCGs provided by the developers of Anvi’o.
Activity estimates were calculated using the index of replication (iRep), which measures genome replication as the ratio of coverage between the replication origin and terminus that are predicted from the distribution of coverage across a genome [32].
Resolution of sequences through individual assembly and manual scaffolding
Key genes for perchlorate reduction were subject to poor assembly and automated scaffolding and were not binned. Therefore, we applied contig graphs, which are visualizations of assemblies where unresolved contigs are displayed as branches between contigs, to identify, scaffold, and bin sequences containing perchlorate reduction genes. To reduce sequence complexity, each sample was assembled individually using MEGAHIT as above. Contig graphs (FASTG format) were generated using the “contig2fastg” tool in MEGAHIT. Bandage (“a Bioinformatics Application for Navigating De novo Assembly Graphs Easily”) v. 0.8.0 [33] was used to visualize contigs, identify BLAST hits to specific gene sequences, manually merge contigs, and relate merged contigs to a genome bin. Criteria for merging contigs were that contigs (1) shared a similar abundance and (2) lay in the same contiguous path. Merged contigs were binned when contiguity with multiple contigs from the same genome bin was certain. This strategy was also used to recover some 16S rRNA genes.
Phylogenetic analysis
The taxonomic position of metagenome-assembled genomes was determined using multi-locus sequence analysis (MLSA) as performed in PhyloPhlAn v. 0.99 [34], which constructs a phylogenetic tree based on 400 conserved protein sequences. Following initial placement of recovered genomes among 2013 IMG genomes [35], additional published genomes were added (Supplementary Table 1) to provide refined placement. As contamination in bins may lead to an aberrant MLSA phylogeny, taxonomic novelty was confirmed by comparing the identity of ribosomal protein amino acid sequences to the NCBI database and, when available, 16S rRNA gene sequences to the NCBI and SILVA databases [36, 37].
Functional annotation of metagenome-assembled genomes
Functional annotation of each genome was performed using Prokka v. 1.11 [38], which employs a suite of sequence prediction software to annotate RNA and protein gene sequences [39–46]. Gene loci were plotted using Annotation Sketch as implemented by genometools v. 1.5.9 [47]. Metabolic functions were determined by the presence or absence of key genes using criteria defined in Supplementary Table 2. Specific genes were confirmed using a combination of BLAST searches, sequence alignment, motif analysis, and phylogenetic trees (Supplementary Figure 3). For phylogenetic tree construction, protein sequences were downloaded from protein families on the Pfam databases (Uniprot and NCBI), selected using custom HMM profiles, and supplemented with the top 100 IMG homology search results [35] and top 250 BLASTP search results for a representative protein. A reduced set of proteins was obtained by clustering proteins at either 80 or 100% identity using UCLUST v. 5.2.32 [48]. Novel metagenomic sequences were added to these sequences and aligned in MUSCLE v. 3.8.31 [49]. Alignments were edited in AliView v. 1.18 [50] to remove N-terminal and C-terminal overhangs and positions consisting of greater than 95% gaps. Phylogenetic trees were generated in PhyML v. 3.0 [51] using 100 bootstraps and the appropriate evolutionary model as determined by ProtTest v. 3.0 [52]. Phylogenetics trees were visualized using the FigTree v. 1.4.3 software package (http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
All reads and genome sequences are available through the NCBI Bioproject accession PRJNA387015.
Results and discussion
Community overview
Previously performed perchlorate-reducing enrichments [14] were subjected to metagenomic sequencing to identify perchlorate reduction genes, taxa, and potential interactions in communities. Samples selected from the marine sediment inoculum and the endpoint of four enrichments (1, 3, 5, and 7% salinity) provided a baseline community and four distinct communities enriched for PRMs. The five samples were sequenced, yielding 303 million total 100-bp reads (Supplementary Table 3), then co-assembled and scaffolded to produce a metagenomic data set of 603 Mbp of scaffold sequences (Supplementary Table 4). Automatic and manual binning yielded 36 high-quality and 12 medium-quality metagenome-assembled genomes totaling 145 Mbp (Table 1). Enrichment enabled a large proportion of communities to be recovered: an average of 68.8% of reads from enriched communities mapped to binned sequences, compared to only 3.2% of reads from the sediment inoculum (Supplementary Figure 4).
Table 1.
Summary of metagenome-assembled genome bins
| Phylum | Genome Bin | 16Sa | GC (%) | Size (Mbp) CDS | Compl. (%)b | Contam. (%)b | Scaffolds (#) N50 (bp)c | Coveraged | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteroidetes | Marinilabiliales BM514 | – | 39.0 | 3.14 | 2677 | 91.49 | 4.43/50 | 478 | 8468 | 15 |
| Marinilabiliales BM518 | – | 39.7 | 3.35 | 2758 | 95.61 | 2.15/25 | 259 | 20,566 | 13 | |
| Marinilabiliales BM520 | – | 41.5 | 3.49 | 2732 | 97.85 | 1.08/0 | 348 | 13,857 | 12 | |
| Marinilabiliales BM523 | – | 35.4 | 3.92 | 3206 | 79.8 | 3.9/0 | 751 | 5963 | 7 | |
| Marinilabiliales BM524 | – | 34.4 | 3.60 | 2910 | 93.23 | 1.08/0 | 223 | 30,104 | 15 | |
| Marinilabiliales BM703 | 32.9 | 4.24 | 4035 | 96.15 | 3.49/0 | 27 | 265,205 | 113 | ||
| Marinilabiliales BM718 | – | 32.4 | 3.57 | 3157 | 91.08 | 1.61/20 | 544 | 7945 | 10 | |
| Marinilabiliales BM719 | – | 33.9 | 3.29 | 2740 | 94.3 | 1.14/0 | 324 | 15,276 | 10 | |
| Salinivirgaceae BM715 | – | 35.1 | 4.32 | 3493 | 94.23 | 3.33/25 | 364 | 18,792 | 11 | |
| Ignavibacteria | Ignavibacteria BM516 | + | 56.7 | 3.93 | 3264 | 92.35 | 3.01/33.33 | 436 | 13,823 | 22 |
| Ca. Muirbacteriae | Ca. Muirbacterium halophilum BM706 | (+) | 32.9 | 2.92 | 2669 | 96.56 | 3.5/20 | 147 | 33,603 | 56 |
| Ca. Parcubacteria | Parcubacteria BM507 | + | 35.4 | 1.27 | 1204 | 71.78 | 0.99/0 | 49 | 76,005 | 21 |
| Parcubacteria BM519 | + | 37.2 | 0.97 | 948 | 72.94 | 2.09/0 | 59 | 28,615 | 9 | |
| Parcubacteria BM521 | + | 39.0 | 1.06 | 983 | 83.15 | 1.12/0 | 67 | 30,243 | 10 | |
| Parcubacteria BM705 | + | 33.5 | 1.43 | 1424 | 76.81 | 0/NA | 15 | 156,199 | 46 | |
| Ca. Woesearchaeotaf | Woesearchaeota BM511 | + | 31.1 | 1.59 | 1702 | 85.05 | 1.87/66.67 | 70 | 31,020 | 16 |
| Deferribacteres | Denitrovibrio BM701 | + | 41.1 | 2.88 | 2811 | 95.35 | 0/NA | 29 | 155,259 | 324 |
| Denitrovibrio BM704 | – | 38.8 | 2.78 | 2700 | 93.97 | 0/NA | 13 | 410,371 | 44 | |
| Firmicutes | Clostridiales BM714 | – | 44.6 | 1.93 | 1911 | 92.13 | 3.9/75 | 370 | 5971 | 13 |
| Alphaproteobacteria | Alphaproteobacteria BM001 | + | 42.8 | 1.13 | 1175 | 97.85 | 0 | 10 | 189,894 | 28 |
| Rhizobiales BM303 | – | 61.4 | 3.50 | 3308 | 96.12 | 2.17/44.44 | 428 | 11,363 | 24 | |
| Rhizobiales BM505 | + | 63.6 | 4.04 | 3609 | 98.58 | 0.79/0 | 7 | 859,782 | 31 | |
| Betaproteobacteria | Azoarcus BM101 | + | 62.9 | 4.90 | 4690 | 98.86 | 3.63/20 | 160 | 59,964 | 515 |
| Deltaproteobacteria | Deltaproteobacteria BM512 | – | 54.2 | 2.53 | 2404 | 92.11 | 1.68/50 | 67 | 65,871 | 15 |
| Deltaproteobacteria BM525 | – | 54.5 | 2.60 | 2497 | 94.21 | 2.52/0 | 192 | 22,954 | 9 | |
| Desulfobacteraceae BM002 | – | 50.0 | 3.80 | 3595 | 94.35 | 3.25/33.33 | 638 | 7626 | 11 | |
| Desulfobacteraceae BM005 | – | 43.3 | 3.05 | 2788 | 84.52 | 1.81/0 | 577 | 6310 | 9 | |
| Desulfobulbaceae BM004 | – | 47.5 | 2.66 | 2487 | 98.13 | 1.64/25 | 189 | 21,028 | 12 | |
| Desulfobulbaceae BM506 | + | 56.3 | 3.71 | 3194 | 98.81 | 0/NA | 83 | 74,437 | 31 | |
| Desulfuromonas BM103 | – | 55.9 | 2.93 | 2860 | 90.07 | 6.13/7.69 | 426 | 9781 | 9 | |
| Desulfuromonas BM302 | – | 55.8 | 2.57 | 2473 | 94.03 | 1.51/85.71 | 216 | 19,147 | 52 | |
| Desulfuromonas BM304 | – | 56.7 | 3.30 | 3091 | 100 | 0.32/0 | 84 | 56,769 | 15 | |
| Desulfuromonas BM501 | – | 62.4 | 3.70 | 3348 | 98.06 | 0.65/0 | 56 | 94,881 | 437 | |
| Desulfuromonas BM508 | – | 55.0 | 2.66 | 2434 | 97.74 | 1.64/25 | 109 | 43,575 | 31 | |
| Desulfuromonas BM509 | – | 54.7 | 3.29 | 3141 | 96.02 | 0.65/100 | 219 | 24,493 | 40 | |
| Desulfuromonas BM513 | – | 52.5 | 3.12 | 2851 | 95.38 | 1.94/33.33 | 186 | 26,287 | 20 | |
| Desulfuromonas BM515 | – | 56.1 | 2.39 | 2258 | 74.23 | 1.61/33.33 | 138 | 24,721 | 11 | |
| Desulfuromonas BM522 | – | 56.3 | 3.12 | 2941 | 90 | 2.1/0 | 203 | 22,928 | 10 | |
| Desulfuromonas BM707 | – | 53.3 | 3.08 | 2941 | 99.35 | 0.65/0 | 121 | 56,077 | 23 | |
| Desulfuromonas BM709 | – | 58.3 | 3.03 | 2916 | 93.44 | 7.74/42.86 | 226 | 20,005 | 20 | |
| Epsilonproteobacteria | Arcobacter BM102 | + | 28.1 | 3.48 | 3530 | 98.78 | 1.56/0 | 42 | 139,512 | 49 |
| Arcobacter BM504 | – | 28.9 | 2.67 | 2847 | 97.76 | 0.95/0 | 12 | 347,029 | 58 | |
| Sulfurimonas BM502 | – | 36.6 | 2.38 | 2554 | 95.03 | 0.2/0 | 46 | 90,685 | 96 | |
| Sulfurimonas BM702 | – | 34.4 | 1.88 | 1999 | 97.14 | 3.22/60 | 122 | 26,298 | 291 | |
| Gammaproteobacteria | Chromatiales BM003 | – | 60.6 | 3.62 | 3404 | 93.35 | 5.53/25.93 | 337 | 14,686 | 15 |
| S. selenatireducens BM301 | – | 56.4 | 4.26 | 4076 | 98.72 | 1.46/0 | 31 | 238,438 | 675 | |
| Sedimenticola BM503 | – | 53.9 | 3.86 | 3805 | 99.65 | 0.64/0 | 109 | 74,804 | 89 | |
| Vibrio alginolyticus BM517 | – | 44.9 | 3.94 | 3460 | 84.18 | 2.94/73.33 | 725 | 6657 | 16 | |
a16S rRNA gene presence in genome bin: +, presence; -, absence; and (+), recovered through manual assembly using contig graphs
bCheckM estimates of completeness (Compl.), contamination (Contam.; left), and fraction of contamination from related strains (Contam.; right)
cAssembly contiguity: 50% of the total genome length is accounted for by contigs of this length or greater
dSum of mean read depth from each sample
eProposed candidate phylum from this study
fMisassigned to Parvarchaeota in ref. [14]
Among the recovered genomes were taxa related to previously isolated canonical DPRM as well as unrelated taxa, including multiple uncultivated lineages (Table 1; Supplementary Figure 5). While the enrichments contained considerable phylogenetic diversity, only a minority of genomes recovered from each community encoded both perchlorate reductase (pcr or pnar; see below) and chlorite dismutase (Fig. 1a). Distinct sets of these DPRM were abundant (×>5 coverage) at each salinity: bin Arcobacter BM102 at 1% salinity; bins Sedimenticola BM301, Sedimenticola BM503, and Rhizobiales BM303 at 3% salinity; bins Sulfurimonas BM502, Sedimenticola BM503, Arcobacter BM504, Rhizobiales BM505, Sulfurimonas BM702, and Rhizobiales BM303 at 5% salinity; and bin Sulfurimonas BM702 at 7% salinity (Fig. 1b; Supplementary Table 5). Activity, as measured by the iRep, varies within DPRM: Arcobacter BM102 (iRep = 1.42) and Rhizobiales BM303 (iRep = 1.33, 1.49) were actively replicating, whereas Sulfurimonas BM502 (iRep = 1.17), Arcobacter BM504 (iRep = 1.18), and Rhizobiales BM505 (iRep = 1.23) were likely not replicating and values for genomes Sedimenticola BM301, Sedimenticola BM503, and Sulfurimonas BM702 could not be determined (Fig. 1c; Supplementary Table 5).
Fig. 1.

Summary of energy metabolism genes, abundance, and activity of metagenome-assembled genomes from perchlorate-reducing enrichments (1, 3, 5, and 7% salinity) and sediment inoculum. Genomes predicted to be dissimilatory perchlorate-reducing bacteria are shown in red. a Gene content based on presence (filled squares) or absence (empty squares) of key genes involved different metabolisms. Genes in the same vertical division perform the same function. Gene abbreviations: pcr perchlorate reductase, pnar periplasmic Nar nitrate reductase, cld chlorite dismutase, yedYZ periplasmic methionine sulfoxide reductase, clr chlorate reductase, nap periplasmic nitrate reductase, cbb3 cbb3-type cytochrome c oxidase, aa3 aa3-type cytochrome c oxidase or cytochrome bo(3) ubiquinol oxidase, bd cytochrome bd quinol oxidase, psr polysulfide reductase, pMHC periplasmic multi-heme cytochrome c, ack-pta acetate kinase and phosphotransacetylase, acs acetyl-CoA synthase, nqo proton-translocating NADH:quinol oxidoreductase, nqr sodium-translocating NADH:quinol oxidoreductase. b Relative abundance of metagenome-assembled genomes within samples (coverage/coverage of most abundant community member). c Relative activity across samples as measured by index of replication (iRep). Slash indicates the iRep value could not be determined
Enrichment proved sufficient and necessary to recover DPRM genomes from marine sediment. All perchlorate reductase genes recovered could be associated with binned genomes. Furthermore, the sediment inoculum data set contained effectively none of the enriched DPRM genomes (×<0.3 coverage) or scaffolds containing perchlorate reduction genes (×<1 coverage; Supplementary Figure 6), and perchlorate reduction genes were not present in an individual assembly of the sediment inoculum. Similarly, perchlorate reduction genes are largely absent from published metagenomic sequences on the IMG database (results not shown), emphasizing the apparent low relative abundance of DPRM in natural ecosystems.
Expanded diversity of perchlorate reduction genes and taxa in communities
Perchlorate reduction genes and taxa in the enriched communities mostly resemble those of previously isolated DPRM, suggesting that studied isolates are representative of active environmental perchlorate reducers. Phylogenetic analysis of pcrA and cld from the enriched populations indicated that most were closely related to pcrA and cld from characterized marine isolates (Fig. 2). Correspondingly, these genomes were closely related to previously isolated DPRM (Table 1; Supplementary Figure 5). All recovered pcrA sequences were co-located in a perchlorate reduction genomic island (PRI) with cld and other perchlorate reductase subunits, and gene order within related PRI were conserved (Supplementary Figure 7): Sedimenticola genomes possessed the gene cluster pcrABCDQ-cld, and Arcobacter and Sulfurimonas genomes possessed pcrABD-cld. Our database search also detected pcrA and cld in previously sequenced strains Sulfurospirillum sp. MES and Sulfurospirillum cavolei NBRC 104982, which possessed a distinct PRI with cld-pcrABDQ (Supplementary Figure 7). The ability of Sulfurospirillum spp. to use perchlorate as an electron acceptor has not been tested [53, 54]. These results expand characterized or putative DPRM to four genera in the Epsilonproteobacteria: Arcobacter, Wolinella, Sulfurimonas, and Sulfurospirillum [22, 55]. Additionally, Rhizobiales BM303 and Rhizobiales BM505 constituted a putative novel family in the order Rhizobiales, whereas all other alphaproteobacterial DPRM have been found in the order Rhodospirillales [4]. The pcrA and cld sequences of DPRM in the Sulfurimonas, Sulfurospirillum, and Rhizobiales differ somewhat from previously characterized sequences but generally cluster together with other marine organisms, a pattern that could be caused by unique selection pressures in the marine ecosystems, by decreased gene transfer between environments, or simply by sampling bias.
Fig. 2.

Maximum-likelihood tree of perchlorate and chlorate reduction enzymes obtained from metagenome-assembled genomes (bold) and the NCBI and IMG databases. Trees were constructed using MUSCLE and PhyML(100 bootstraps) and visualized with FigTree. Branch length and local support values provided by bootstrapping are indicated. Blue text indicates taxa isolated from marine environments. a Perchlorate reductase alpha subunit. b Chlorite dismutase; colored squares indicate the closest perchlorate or chlorate reductase gene to the chlorite dismutase gene. c Chlorate reductase alpha subunit
In addition to observing the canonical PRI, genomic analysis identified a novel perchlorate reduction gene cluster distinguished by an alternative putative perchlorate reductase. In bins Rhizobiales BM303 and Rhizobiales BM505 and three previously sequenced genomes, cld is adjacent to the gene for a periplasmic Nar-like molybdopterin reductase (pnarGHIJ) (Fig. 3a). Available evidence suggests that the pNar enzyme could function as an alternative perchlorate reductase: pNar from thermophilic bacteria and archaea reduce perchlorate to chlorite, and no pNar tested has lacked perchlorate reductase activity [16, 17, 56–58]. Additionally, in sequence alignments pNarG, like cNarG, had structurally similar amino acids in the key residues (positions α, β, and γ) that shape the PcrA active site and influence its affinity for perchlorate (Fig. 3b) [59]. Whereas the lack of chlorite dismutase in other pnar-containing organisms suggests their perchlorate reduction is inadvertent, the conserved association of pnar with cld in this gene cluster strongly suggests it is specialized for perchlorate reduction. The gene clusters also resembled PRI by encoding, variously, genes for reactive chlorine stress response, signaling, plasmid replication, transposon mobility, and phage proteins. Furthermore, pNarG proteins from the pnar-cld gene cluster formed a deep-branching monophyletic clade basal to pNarG proteins from other organisms (Fig. 3c), which supports a specialized function distinct from other pNar proteins. Available genomic and phylogenetic evidence support that pNar enzymes in this clade primarily function as perchlorate reductases, henceforth referred to as group 2 perchlorate reductases, although biochemical and genetic evidence are ultimately needed to confirm their function.
Fig. 3.

Characterization of periplasmic nitrate reductase (pNar) genes with possible perchlorate reductase activity. a Gene clusters containing both pnarGHJI and cld. b Alignment of selected Nar-type enzymes constructed using MUSCLE and edited using AliView. Perchlorate reductase residue positions α, β, and γ are structurally conserved and functionally significant [59]. Substrate utilization:+, reduced substrate in vivo or in vitro; −, did not reduce substrate; (+), predicted to reduce substrate. Tree: position in the phylogenetic tree at right. c Maximum-likelihood phylogeny of Nar-type molybdoenzymes constructed using MUSCLE and PhyML with 100 bootstraps. Four lineages are found in gene clusters with chlorite dismutase (blue background): group 1 PcrA, group 1 ClrA (SerA-like), group 2 ClrA (DdhA-like), and group 2 PcrA (pNarG-like). Genome bins encoding proteins from each group are indicated
The physiological difference between group 2 perchlorate reductases and the previously characterized group 1 perchlorate reductases is uncertain. Two gene clusters encoded both reductases. Additionally, strains encoding only the alternative perchlorate reductase, Magnetospirillum magnetotacticum MS-1 and Tropicimonas isoalkanivorans B51, did not grow using perchlorate or chlorate as an electron acceptor with acetate as the electron donor (results not shown) [60]. However, Rhizobiales BM303, which also encoded only a group 2 Pcr, was enriched in two communities, suggesting that group 2 Pcr alone may be sufficient for dissimilatory perchlorate reduction in some conditions. The similarity of pNarG and cNarG could explain the failure to cultivate organisms with group 2 Pcr: both NarG enzymes lack a key tryptophan residue (position γ; Fig. 3b) for the proposed gating mechanism in group 1 Pcr that limits the rate of perchlorate reduction at unnaturally high concentrations of perchlorate, such as those supplied in cultivation media [59]. Rapid reduction of perchlorate to chlorite by group 2 Pcr could be detrimental to cells unless chlorite is removed, as would occur by chemical reaction with sulfide present in the sediment inoculum but absent from most cultivation media.
Genetic mobility of perchlorate reduction genes
PRIs suffered from fragmentation into multiple contigs during co-assembly that initially prevented binning and characterization. We noticed that individually assembled samples had a higher average contig length than the co-assembled samples (Supplementary Table 4), which implied that co-assembly led to greater fragmentation. By assembling samples individually then using contigs graphs to manually combine contigs, we reconstructed complete PRI and linked them to specific genomes binned from the co-assembly. To our knowledge, this is the first study in which contig graphs have been used to associate mobile genetic elements with populations in microbial communities. Contig graphs revealed that even within individual samples, the same sequences (Fig. 4a) and perchlorate reduction genes (Fig. 4b) were associated with multiple genomes. In the 3% salinity community, a PRI was contiguous with both Sedimenticola BM301 and BM503, though pcrA and pcrD did not assemble as a consensus sequence. In the 5% salinity community, a PRI was contiguous with sequences from Arcobacter BM504 and Sulfurimonas BM502. Notably, the pcrAB sequence from that PRI was also contiguous with Sedimenticola BM503. In contig graphs, the presence of identical sequences in multiple metagenome-assembled genomes indicates either horizontal transfer during the enrichment or the assembly of slightly diverged sequences as a consensus sequence. The latter conclusion is supported by similarity of PcrA and Cld phylogenies between metagenome-assembled genomes and previously isolated strains (Fig. 2).
Fig. 4.

Use of contig graphs to manually assemble perchlorate reduction genes within mobile genetic elements. a Each sample was assembled individually and inspected for perchlorate reduction genes in contig graphs. Contig graphs depict possible connections between contigs produced by de Bruijn graph assembly. The real connections between contigs may be determined using information such as coverage (line weight) and gene content (line colors). PRI sequences including group 1 pcrA (red), group 2 pcrA (“pnarG”, green), and cld (blue) that were clearly connected to contigs from genome bins (arrows) were added to those bins. Mean coverage for particular plasmid sequences and bins is indicated in parenthesis. For simplicity, only contigs within a few connections of PRI sequences are shown, and some contigs are not shown in their entirety. b Reconstructed PRI from contig graphs. Shaded genes indicate sequences that were highly similar and assembled as a consensus sequence from multiple strains. Each genome was annotated independently using Prokka
Two plasmids encoding perchlorate reduction genes were identified in the metagenomic data set. A transposon containing key perchlorate reduction genes pcrABCDQ and cld was integrated into the chromosome of Sedimenticola BM301 in 3% salinity community but into a conjugative plasmid in the 3 and 5% salinity communities. We assigned this plasmid to Sedimenticola BM503 on the basis of similar abundance to the plasmid in the 3% salinity condition (×24 and ×29 coverage, respectively) (Fig. 4a) and the close taxonomic relationship of Sedimenticola BM503 to Sedimenticola BM301. The plasmid was four times more abundant than Sedimenticola BM503 in the 5% salinity community, however, in this community the plasmid was either maintained at high copy number or present in multiple organisms. A second plasmid, containing a PRI with group 2 pcr instead of group 1 pcr, was present in both the 3 and 5% salinity. While this plasmid initially binned with Sedimenticola BM503 (which is at two-fold greater coverage in one sample), we re-assigned the plasmid to Rhizobiales BM303 due to more similar patterns of abundance (×15:×28 and ×14:×49 coverage) (Fig. 4a) and the close taxonomic relationship of Rhizobiales BM303 to Rhizobiales BM505, which has a closely related group 2 pcr on a 326,675-bp chromosomal scaffold. The plasmid encodes group 2 pcrABD, cld, and genes for cytoplasmic methionine sulfoxide reductase (msrAB), a sensor histidine kinase system, and a transposase. Perchlorate reduction genes have previously only been associated with evidence of prior integration, such as bordering transposases and insertion scars, or potential excision, in the case of the integrative conjugative element in Dechloromonas aromatica RCB [7]. Thus Sedimenticola BM503 and Rhizobiales BM303 provide the first examples of DPRM with perchlorate reduction gene clusters on intact plasmids. However, the available data is not sufficient to determine whether or not these plasmids served as vectors for gene transfer during enrichment.
Implications for the evolution of perchlorate reduction
The perchlorate reduction genes obtained from these communities make evident a substantial reorganization of perchlorate reduction gene clusters throughout their evolutionary history. Protein sequences from recovered genomes showed a distinct phylogenetic grouping of Cld between Arcobacter-type and Sedimenticola-type proteins (Fig. 2b) that was not replicated for PcrA (Fig. 2a). For example, Sedimenticola BM301 and Sedimenticola BM503 encoded identical Cld (100% identity) but distinct PcrA (89% identity). These discordant phylogenies support the past independent exchange of cld or pcr between PRI. The most likely evolutionary history is that a group of Gammaproteobacteria (Sedimenticola BM503, Marinobacter sp. P4B1, and Sedimenticola sp. CUZ) acquired pcrA from one of a group of Epsilonproteobacteria (Sulfurospiriullum spp., Sulfurimonas spp., or Arcobacter spp.). These results challenge the previous hypotheses that pcrAB and cld were propagated together following the formation of a single ancestral PRI and that the exchange of perchlorate reduction genes between phyla is rare [7]. The exchange of these genes may result from the need to optimize perchlorate reductase and chlorite dismutase activity when perchlorate concentrations or other geochemical conditions change.
The identification of gene clusters with group 2 perchlorate reductase challenge the very notion of a single ancestral PRI. While group 1 and group 2 Pcr formed distinct monophyletic groups (Fig. 3c), the Cld proteins from the gene clusters showed no such phylogenetic clustering (Fig. 2b). Therefore, cld has been exchanged between gene clusters with phylogenetically distinct perchlorate reductases. Clusters that included both group 1 and group 2 pcr, as found in Dechlorobacter hydrogenophilus LT-1 and Rhizobiales BM505, may represent an intermediate point in this exchange. The exchange may be driven by environmental perchlorate concentrations: it was recently demonstrated that group 1 PcrAB is a specialized member of the DMSO reductase superfamily with a relatively high perchlorate affinity (KM = 6 μM), unlike the related cNarGH nitrate reductase which has a much lower affinity for perchlorate (KM = 1.1 mM) [59]. In contrast to supporting a single ancestral PRI, these data support the formation of two perchlorate reduction gene clusters through the independent acquisition of chlorite dismutase. A similar history was inferred for the gene cluster responsible for dissimilatory chlorate reduction, the chlorate reduction composite transposon interior (CRI) [9]. Thus this study adds to empirical support for the idea that enzymes in the NarG subfamily of molydopterin oxidoreductases possess reductase activity towards various oxyanions but have been repeatedly adopted for perchlorate or chlorate reduction through the acquisition of the downstream enzyme Cld.
Metabolic interactions within perchlorate-reducing communities
Metagenome-assembled genomes lacking perchlorate reduction genes accounted for 35 of 43 enriched genomes (Fig. 1a) and 10.5 to 91.5% of the recovered community (Fig. 5a). Although we cannot dismiss the idea that unknown mechanisms for perchlorate reduction exist, it is not necessary to explain the growth of organisms in these communities. One explanation is that organisms use alternative electron acceptors or fermentable substrates that were present in the sediment inoculum [14]. A non-conflicting possibility of particular interest is that perchlorate reduction intermediates and byproducts provide growth substrates for non-DPRM. We identified genes for energy metabolism in every genome (Fig. 1a) to determine whether interactions between metabolisms were possible in each community.
Fig. 5.

Metabolic potential of perchlorate-reducing communities. (a) The relative abundance, among high-quality bins, of genomes with each predicted metabolism: dissimilatory perchlorate-reducing bacteria (red), dissimilatory chlorate-reducing bacteria (orange), dissimilatory elemental sulfur-reducing bacteria (gray), free-living fermentative bacteria (dark blue), and epibiotic fermentative bacteria (light blue). Communities differ in salinity (% NaCl w/v), sampling time (days), and perchlorate removed (%). (b). Possible metabolic interactions and their commonality among the four enriched communities
Intermediates of perchlorate reduction may have supported the growth of non-DPRM in the 1% salinity community. The most abundant population in this community, Azoarcus BM101, lacked any perchlorate reductase yet encodes a complete chlorate reduction pathway consisting of genes for chlorate reductase (clrABDC), chlorite dismutase (cld), and terminal oxygen reductases (Fig. 1a). Azoarcus BM101 could therefore utilize any perchlorate reduction intermediate (ClO3−, ClO2−, or O2), similar to the symbiotic perchlorate reduction metabolism previously identified [4, 11]. Unexpectedly, although this would have made it dependent on DPRM, Azoarcus BM101 was found at 10-fold higher abundance than the perchlorate-reducing population Arcobacter BM102 (Fig. 5a). Specific growth characteristics of either strain that could explain this interaction are unknown. The most closely related isolated strain to BM102, Arcobacter sp. CAB, is not known to accumulate chlorate during perchlorate reduction, and though the chlorate reduction proteins in Azoarcus BM101 were nearly identical to those in previously characterized organisms (Fig. 2c), it is the first chlorate-reducing bacteria to be described in the betaproteobacterial order Rhodocyclales. The consumption of perchlorate reduction intermediates by Azoarcus BM101 would likely limit the growth of DPRM due to energy losses, although which intermediates were involved in this interaction is uncertain (Fig. 5b). While evidence for interaction between DPRM and chlorate-reducing microorganisms was only observed in one of four enriched communities, whether or not such interactions are a common feature of perchlorate-reducing communities is presently unknown.
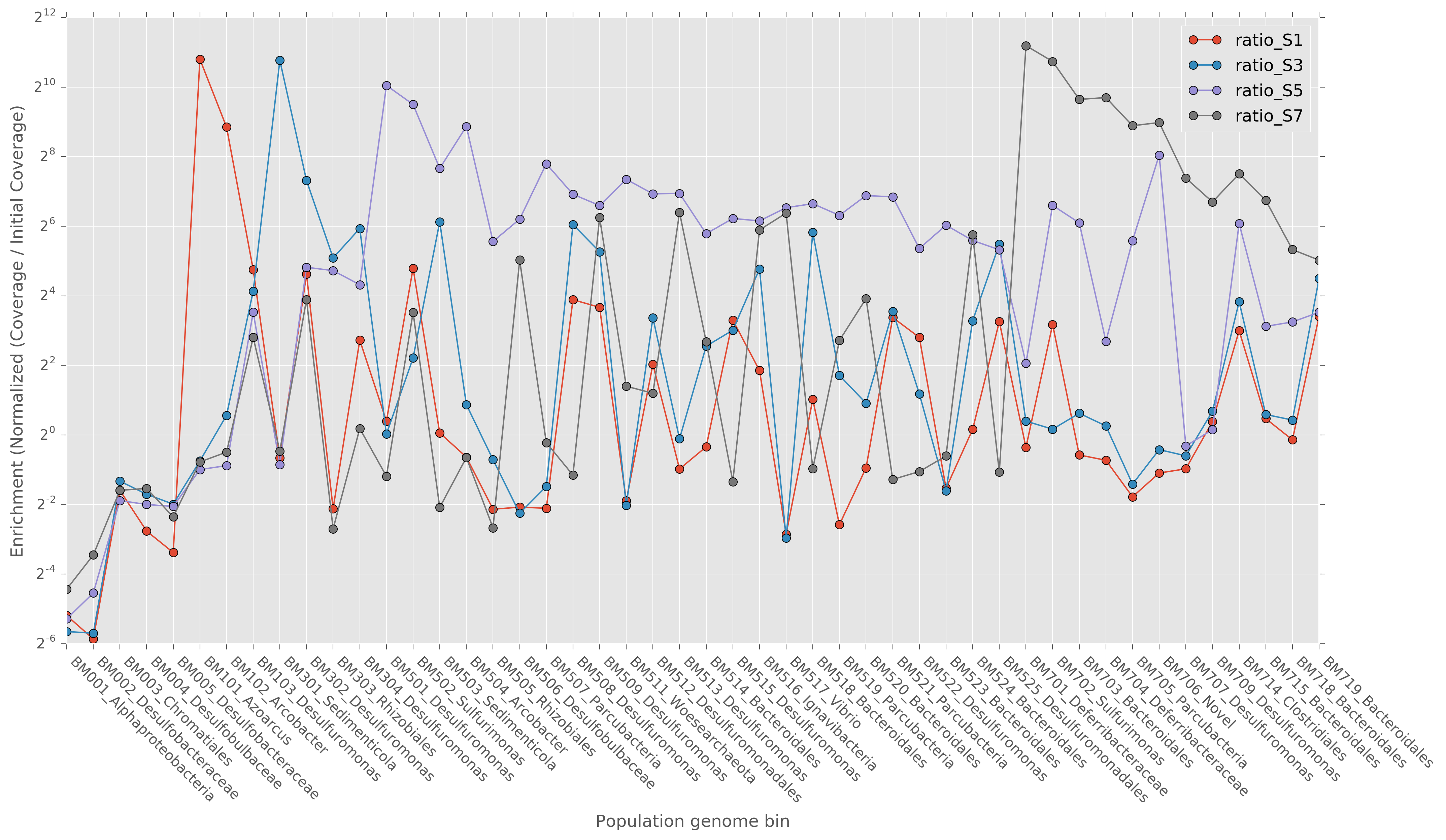
PSOX to elemental sulfur and polysulfide could have supported dissimilatory elemental sulfur-reducing microorganisms (DSRM) in every community at the expense of outgrowth of canonical DPRM [12, 13] (Fig. 5). Populations in every community encoded genes for enabling elemental sulfur reduction: polysulfide reductase (psr) and periplasmic high-molecular weight multi-heme cytochromes (Fig. 1a) [61, 62]. Putative DSRM included Denitrovibrio (BM701, BM704), Ignavibacteria BM516, Marinilabiliales (BM514, BM524, BM523, BM719), Desulfobulbaceae BM506, Deltaproteobacteria (BM512, BM525), and Desulfuromonas (BM103, BM302, BM304, BM501, BM508, BM509, BM513, BM515, BM522, BM707, BM709). At higher salinities DSRM dominated, as these populations accounted for 58.7 and 45.0% of the 5 and 7% salinity communities, respectively (Fig. 5a). Many of these populations were actively replicating (iRep > 1.3; Supplementary Table 5) and showed equal or greater enrichment than DPRM populations (Supplementary Figure 8), demonstrating that DSRM were not only persisting through enrichment but actively growing. Because the inoculum is marine sediment, reduced sulfur species including elemental sulfur were likely present. Any sulfide present or produced by DSRM would be re-oxidized to elemental sulfur by PSOX (Fig. 5b). The inhibition of DPRM by sulfide [12, 13] provides a possible mechanism by which elemental sulfur-reducing microorganisms could dominate communities without affecting the stoichiometric removal of acetate and perchlorate. Genomic evidence from these enrichments therefore supports the hypothesis that cryptic elemental sulfur cycling is a feature of perchlorate-reducing communities with sulfur-reducing microorganisms and reduced sulfur species.
Not all populations interacted directly with DPRM through perchlorate reduction intermediates or byproducts. Several populations from higher salinity communities lacked key genes for anaerobic respiration and likely obtained energy only through fermentation. Putatively fermentative populations include Clostridiales (BM714), Vibrio alginolyticus (BM517), and Marinilabiliales (BM518, BM520, BM703, BM715, BM718), Most of these genomes lacked NADH:quinone oxidoreductase, genes for acetate catabolism through the TCA cycle, and respiratory perchlorate, chlorate, oxygen, and sulfur reductases (Fig. 1a). Previously isolated Marinilabiliales organisms have indeed been specialized for the fermentation of dead cells present in hypersaline marine sediments [63]. In contrast, like previously described members of the CPR Bacteria and DPANN Archaea superphyla [31, 64–66], Ca. Woesearchaeota and Ca. Parcubacteria/OD1 bins have a small size and limited gene content and were likely obligate epibiotic symbionts of other populations. One putatively fermentative population, BM706, constituted a distinct phylum-level lineage in our MLSA (Supplementary Figure 5). BM706 is abundant in both the 5 and 7% salinity communities, totals 2.92 Mbp of sequence, and is predicted to be near-complete (96.6%) with low contamination (3.5%) (Table 1). BM706 is distinct from other genomes as determined by comparison ribosomal protein S3 (63% identity) and 16S rRNA gene sequences (84% identity) using BLAST. The only closely related (>84% identity) environmental 16S rRNA clones were found in hypersaline microbial mats in Guerrero Negro, Baja California Sur, Mexico, (all >94% identity) [67]. Indeed, the genome recovered from our enrichment appears to be specialized for high salinity environments, encoding a sodium/proton-translocating ATP synthetase, several sodium transporters, and numerous extracellular enzymes for the degradation of lysed biomass, and we suggest that the energy metabolism of BM706 relies on the production of a sodium motive force powered by the fermentation of dead cells (a graphical depiction of the metabolism can be found in Supplementary Figure 9). For this candidate taxon we propose a genus and species name Ca. Muirbacterium halophilum BM706 and the phylum name “Muirbacteria” in recognition of the contributions of naturalist John Muir (1838–1914) to the protection of natural areas in California. While the growth of Ca. Muirbacteria halophilum BM706 and other putative free-living fermenters may be attributable to fermentable substrates from the sediment, the reactive chlorine species produced by perchlorate-reducing bacteria could accelerate cell lysis and provide additional substrates for fermentative bacteria (Fig. 5).
Conclusions
The metagenomic data set obtained here resolves long-standing questions regarding the genetic and metabolic composition of perchlorate-reducing communities and opens exciting new questions about the evolution and ecology of this unique metabolism. The discovery of a second specialized perchlorate reductase, if confirmed, fundamentally reorders the evolution of this metabolism. Further attempts to cultivate and characterize the organisms with this gene cluster may identify ecologically important differences from the canonical perchlorate reductase. In these communities, gene clusters containing either perchlorate reductase could be found on plasmids, which may provide a genetic tool to study the exchange of perchlorate reduction genes between organisms. The high sequence identity of perchlorate reduction genes and their association with mobile genetic elements required manual assembly using contig graphs—a technique that may be widely applicable for binning key genes and identifying possible gene transfer in simple metagenomic data sets. In addition to DPRM, the typical perchlorate-reducing community appears to involve interactions with other organisms that could substantially influence ecosystem functions. Together, these results suggest a model of perchlorate reduction involving the combined activity of distinct perchlorate reductases and multiple interacting metabolisms. A tool for using contig graphs to study gene transfer in metagenomic data sets has been published [68] since our initial submission.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial support for work on perchlorate metabolism was provided through a grant from the Energy Biosciences Institute to J.D.C. Additional funding was provided through the NSF Graduate Research Fellowship Program to T.P.B. We thank Ouwei Wang and Matthew Olm for contributing comments to this manuscript; QB3 Computational Genomics Resource Laboratory Science Director Jason Huff and Bioinformatics Scientist Ke Bi for their assistance in using advanced computational resources; Ryan Wick for his assistance in using Bandage; Chris Brown for his assistance in using iRep; Alexander Probst for providing additional genomes for phylogenetic analysis; and Matthew Olm and Rose Kantor for their advice in the assembly and binning of metagenomic data sets.
Author contributions
JDC conceived of the study and guided the research. CC and AE designed the enrichments. CC and LL performed and sampled the enrichments and extracted DNA. TPB and IF developed sequencing, assembly, and binning strategies. TPB performed all sequencing, assembly, binning, phylogenetic, and functional analyses. TPB, IF, AE, and JDC contributed significantly to data interpretation. TPB and JDC wrote the manuscript. All authors reviewed the manuscript and approved of its publication.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0081-5) contains supplementary material, which is available to authorized users.
References
- 1.Catling DC, Claire MW, Zahnle KJ, Quinn RC, Clark BC, Hecht MH, et al. Atmospheric origins of perchlorate on Mars and in the Atacama. J Geophys Res. 2010;115:E00E11. doi: 10.1029/2009JE003425. [DOI] [Google Scholar]
- 2.Dasgupta PK, Martinelango PK, Jackson WA, Anderson TA, Tian K, Tock RW, et al. The origin of naturally occurring perchlorate: The role of atmospheric processes. Environ Sci Technol. 2005;39:1569–75. doi: 10.1021/es048612x. [DOI] [PubMed] [Google Scholar]
- 3.Rajagopalan S, Anderson T, Cox S, Harvey G, Cheng Q, Jackson WA. Perchlorate in wet deposition across North America. Environ Sci Technol. 2009;43:616–22. doi: 10.1021/es801737u. [DOI] [PubMed] [Google Scholar]
- 4.Youngblut MD, Wang O, Barnum TP, Coates JD. (Per)chlorate in biology on earth and beyond. Annu Rev Microbiol. 2016;70:435–59. doi: 10.1146/annurev-micro-102215-095406. [DOI] [PubMed] [Google Scholar]
- 5.Melnyk Ra, Youngblut MD, Clark IC, Carlson HK, Wetmore KM, Price MN, et al. Novel mechanism for scavenging of hypochlorite involving a periplasmic methionine-rich peptide and methionine sulfoxide reductase. MBio. 2015;6:e00233–15. doi: 10.1128/mBio.00233-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melnyk Ra, Engelbrektson A, Clark IC, Carlson HK, Byrne-Bailey K, Coates JD. Identification of a perchlorate reduction genomic island with novel regulatory and metabolic genes. Appl Environ Microbiol. 2011;77:7401–4. doi: 10.1128/AEM.05758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melnyk RA, Coates JD. The perchlorate reduction genomic island: mechanisms and pathways of evolution by horizontal gene transfer. BMC Genom. 2015;16:862. doi: 10.1186/s12864-015-2011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melnyk Ra, Clark IC, Liao A, Coates JD. Transposon and deletion mutagenesis of genes involved in perchlorate reduction in Azospira suillum PS. MBio. 2014;5:e00769–13. doi: 10.1128/mBio.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark IC, Melnyk Ra, Engelbrektson A, Coates JD. Structure and evolution of chlorate reduction composite transposons. MBio. 2013;4:e00379–13. doi: 10.1128/mBio.00379-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley M, Nerenberg R. Microbial ecology of perchlorate-reducing bacteria that accumulate high levels of chlorate. MSc Thesis, University of Notre Dame, Notre Dame, Indiana, 2007.
- 11.Clark IC, Youngblut M, Jacobsen G, Wetmore KM, Deutschbauer A, Lucas L, et al. Genetic dissection of chlorate respiration in Pseudomonas stutzeri PDA reveals syntrophic (per)chlorate reduction. Environ Microbiol. 2016;18:3342–54. doi: 10.1111/1462-2920.13068. [DOI] [PubMed] [Google Scholar]
- 12.Gregoire P, Engelbrektson A, Hubbard CG, Metlagel Z, Csencsits R, Auer M, et al. Control of sulfidogenesis through bio-oxidation of H2S coupled to (per)chlorate reduction. Environ Microbiol Rep. 2014;6:558–64. doi: 10.1111/1758-2229.12156. [DOI] [PubMed] [Google Scholar]
- 13.Mehta-Kolte MG, Loutey D, Wang O, Youngblut MD, Hubbard CG, Wetmore KM, et al. Mechanism of H2S oxidation by the dissimilatory perchlorate-reducing microorganism Azospira suillum PS. MBio. 2017;8:1–16. doi: 10.1128/mBio.02023-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlström C, Coates JD. Characterization of an anaerobic marine microbial community in response to perchlorate addition and varying salinity. Appl Microbiol Biotechnol. 2016;100:9719–32. doi: 10.1007/s00253-016-7780-5. [DOI] [PubMed] [Google Scholar]
- 15.Engelbrektson A, Hubbard CG, Tom LM, Boussina A, Jin YT, Wong H, et al. Inhibition of microbial sulfate reduction in a flow-through column system by (per)chlorate treatment. Front Microbiol. 2014;5:1–11. doi: 10.3389/fmicb.2014.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liebensteiner MG, Pinkse MWH, Nijsse B, Verhaert PDEM, Tsesmetzis N, Stams AJM, et al. Perchlorate and chlorate reduction by the Crenarchaeon Aeropyrum pernix and two thermophilic Firmicutes. Environ Microbiol Rep. 2015;7:936–45. doi: 10.1111/1758-2229.12335. [DOI] [PubMed] [Google Scholar]
- 17.Liebensteiner MG, Pinkse MWH, Schaap PJ, Stams AJM, Lomans BP. Archaeal (per)chlorate reduction at high temperature: an interplay of biotic and abiotic reactions. Science. 2013;340:85–87. doi: 10.1126/science.1233957. [DOI] [PubMed] [Google Scholar]
- 18.Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. doi: 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker BJ, Lazar CS, Teske AP, Dick GJ. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome. 2015;3:14. doi: 10.1186/s40168-015-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speth DR, in’t Zandt MH, Guerrero-Cruz S, Dutilh BE, Jetten MSM. Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nat Commun. 2016;7:11172. doi: 10.1038/ncomms11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coates JD, Woodward J, Allen J, Philp P, Lovley DR. Anaerobic degradation of polycyclic aromatic hydrocarbons and alkanes in petroleum-contaminated marine harbor sediments. Appl Environ Microbiol. 1997;63:3589–93. doi: 10.1128/aem.63.9.3589-3593.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlström CI, Wang O, Melnyk Ra, Bauer S, Lee J, Engelbrektson A, et al. Physiological and genetic description of dissimilatory perchlorate reduction by the novel marine bacterium Arcobacter sp. strain CAB. MBio. 2013;4:1–9. doi: 10.1128/mBio.00217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joshi N, Fass J. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33). 2011. https://github.com/najoshi/sickle.
- 24.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 27.Eren AM, Esen OumlC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 30.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–11. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 32.Brown CT, Olm MR, Thomas BC, Banfield JF. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol. 2016;34:1256–63. doi: 10.1038/nbt.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–2. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segata N, Börnigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun. 2013;4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen IMA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45:507–16. doi: 10.1093/nar/gkw929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST plus: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 39.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F et al. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–8. [DOI] [PMC free article] [PubMed]
- 41.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–3. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 45.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:61–65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–5. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10:645–56. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 48.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 49.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larsson A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014;30:3276–8. doi: 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 52.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest-HPC: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:177–84. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kodama Y, Ha LT, Watanabe K. Sulfurospirillum cavolei sp. nov., a facultatively anaerobic sulfur-reducing bacterium isolated from an underground crude oil storage cavity. Int J Syst Evol Microbiol. 2007;57:827–31. doi: 10.1099/ijs.0.64823-0. [DOI] [PubMed] [Google Scholar]
- 54.Ross DE, Marshall CW, May HD, Norman RS. Draft genome sequence of Sulfurospirillum sp. strain MES, reconstructed from the metagenome of a microbial electrosynthesis system. Genome Announc. 2015;3:e01336–14. doi: 10.1128/genomeA.01336-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace W, Ward T, Breen A, Attaway H. Identification of an anaerobic bacterium which reduces perchlorate and chlorate as Wolinella succinogenes. J Ind Microbiol. 1996;16:68–72. doi: 10.1007/BF01569924. [DOI] [Google Scholar]
- 56.Marangon J, Paes De Sousa PM, Moura I, Brondino CD, Moura JJG, González PJ. Substrate-dependent modulation of the enzymatic catalytic activity: reduction of nitrate, chlorate and perchlorate by respiratory nitrate reductase from Marinobacter hydrocarbonoclasticus 617. Biochim Biophys Acta. 2012;1817:1072–82. doi: 10.1016/j.bbabio.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 57.Martínez-Espinosa RM, Richardson DJ, Bonete MJ. Characterisation of chlorate reduction in the haloarchaeon Haloferax mediterranei. Biochim Biophys Acta. 2015;1850:587–94. doi: 10.1016/j.bbagen.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 58.Oren A, Elevi Bardavid R, Mana L. Perchlorate and halophilic prokaryotes: implications for possible halophilic life on Mars. Extremophiles. 2014;18:75–80. doi: 10.1007/s00792-013-0594-9. [DOI] [PubMed] [Google Scholar]
- 59.Youngblut MD, Tsai CL, Clark IC, Carlson HK, Maglaqui AP, Gau-Pan PS, et al. Perchlorate reductase is distinguished by active site aromatic gate residues. J Biol Chem. 2016;291:9190–202. doi: 10.1074/jbc.M116.714618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Achenbach LA, Coates JD. Disparity between bacterial phylogeny and physiology. ASM News. 2000;66:714–5. [Google Scholar]
- 61.Burns JL, DiChristina TJ. Anaerobic respiration of elemental sulfur and thiosulfate by Shewanella oneidensis MR-1 requires psrA, a homolog of the phsA gene of Salmonella enterica Serovar Typhimurium LT2. Appl Environ Microbiol. 2009;75:5209–17. doi: 10.1128/AEM.00888-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banci L, Bertini I, Bruschi M, Sompornpisut P, Turano P. NMR characterization and solution structure determination of the oxidized cytochrome c7 from Desulfuromonas acetoxidans. Proc Natl Acad Sci USA. 1996;93:14396–14400. doi: 10.1073/pnas.93.25.14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hania WBen, Joseph M, Bunk B, Spröer C, Klenk HP, Fardeau ML, et al. Characterization of the first cultured representative of a Bacteroidetes clade specialized on the scavenging of cyanobacteria. Environ Microbiol. 2016;19:1134–48. doi: 10.1111/1462-2920.13639. [DOI] [PubMed] [Google Scholar]
- 64.Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol. 2015;25:690–701. doi: 10.1016/j.cub.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 65.Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD, et al. Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci USA. 2010;107:8806–11. doi: 10.1073/pnas.0914470107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luef B, Frischkorn KR, Wrighton KC, Holman HYN, Birarda G, Thomas BC, et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun. 2015;6:6372. doi: 10.1038/ncomms7372. [DOI] [PubMed] [Google Scholar]
- 67.Harris JK, Caporaso JG, Walker JJ, Spear JR, Gold NJ, Robertson CE, et al. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J. 2013;7:50–60. doi: 10.1038/ismej.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Olekhnovich Evgenii I, Vasilyev Artem T, Ulyantsev Vladimir I, Kostryukova Elena S, Tyakht Alexander V. MetaCherchant: analyzing genomic context of antibiotic resistance genes in gut microbiota. Bioinformatics. 2018;34:434–444. doi: 10.1093/bioinformatics/btx681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All reads and genome sequences are available through the NCBI Bioproject accession PRJNA387015.