

Abstract
Despite advances in cancer biology and therapeutics, drug resistance remains problematic. Resistance is often multifactorial, heterogeneous, and prone to undersampling. Nonetheless, many individual mechanisms of targeted therapy resistance may coalesce into a smaller number of convergences, including pathway reactivation (downstream re-engagement of original effectors), pathway bypass (recruitment of a parallel pathway converging on the same downstream output), and pathway indifference (development of a cellular state independent of the initial therapeutic target). Similar convergences may also underpin immunotherapy resistance. Such parsimonious, convergence-based frameworks may help explain resistance across tumor types and therapeutic categories and may also suggest strategies to overcome it.
Introduction
Most patients with advanced cancer die because their cancer exhibits or develops resistance to available therapies. This challenge remains pervasive despite many remarkable advances in cancer biology and treatment and has motivated renewed efforts to understand and combat cancer drug resistance. For example, addressing cancer drug resistance comprised one of the key recommendations from the Blue Ribbon Panel that advised the Beau Biden Cancer Moonshot initiative (Jacks et al., 2016).
Studies of cancer drug resistance yield several types of insights. First, defining mechanisms of resistance to a specific anticancer regimen may identify future therapeutic strategies against such resistance. Such knowledge might guide salvage treatment after resistance has already occurred or inform new up-front strategies that prevent its emergence. For example, identification of MEK reactivation as a mechanism of resistance to RAF inhibitor monotherapy in BRAF-mutant melanoma (Johannessen et al., 2010; Nazarian et al., 2010), or of EGFRT790M as a mechanism of resistance to first-generation EGFR inhibitors in EGFR-mutant lung cancer (Kobayashi et al., 2005; Pao et al., 2005), led to the development of new therapies with clinical efficacy against these resistance mechanisms (Mok et al., 2017; Robert et al., 2015).
Systematic characterization of drug resistance across tumor types and therapeutic categories may also enable new insights into cancer biology—regarding, for example, networks that differentiate the malignant state from normal, the cell-intrinsic and extrinsic factors regulating them, and how these pathways evolve to promote tumor survival during therapy. Investigating resistance to MAP kinase pathway inhibition revealed new aspects of feedback regulation within this pathway (Lito et al., 2014; Pratilas et al., 2009). Likewise, studies of EGFR inhibitor resistance in lung cancer highlighted the importance of intratumoral heterogeneity (Piotrowska et al., 2015).
Despite the potential benefits of such knowledge, systematic efforts to define the full spectrum of resistance mechanisms for any given therapeutic remain the exception rather than the rule in cancer research. In fact, the skeptic might even question whether studies of resistance in general offer the most efficient or incisive means to discover either novel therapeutic targets or disease-relevant biology. However, a synthesis of emerging genomic and functional portraits of cancer drug resistance begins to reveal overarching frameworks which both offer new insights into resistant states and suggest possible therapeutic approaches to overcome them.
Challenges in cancer drug resistance
As noted in earlier reviews (for example, Garraway and Janne, 2012), research into cancer drug resistance has produced many discoveries with both basic and clinical significance. As knowledge of resistance mechanisms has expanded, one question has become how best to organize and understand them collectively. At times, individual drug resistance effectors have been almost reflexively aggregated into “pie chart” representations that imply a “one patient, one mechanism” distribution of resistance to a given agent. Such representations, however, are likely to be overly simplistic.
One major challenge is that resistance is multifactorial. Just as initial catalogues of high-frequency somatic alterations in the cancer genome subsequently expanded into a “long tail” of driver alterations (Garraway and Lander, 2013), so too has the diversity of resistance mechanisms begun to increase considerably (Van Allen et al., 2014). While some mechanisms function through well-understood effectors, many others do not. Moreover, although many resistance mechanisms are genomically encoded (e.g., somatic mutations or amplifications), non-genetic resistance mechanisms (e.g., protein phosphorylation or gene expression changes) have become increasingly recognized. Furthermore, the tumor microenvironment can also modify drug exposure and response (Straussman et al., 2012). Thus, both the number of resistance effectors and the ways in which they become dysregulated are likely quite large for any given therapeutic context.
A second challenge is that resistance is heterogeneous. Heterogeneity between and within patients, or even within a single drug-resistant tumor focus (Sequist et al., 2011; Wagle et al., 2014), has been well described (reviewed in Meric-Bernstam and Mills, 2012). This heterogeneity can be spatial (both within a single tumor and among multiple metastases (Van Allen et al., 2014; Cai et al., 2015; Cooper et al., 2015; Gerlinger et al., 2012; Patel et al., 2014; Romano et al., 2013; Sanborn et al., 2015; Yates et al., 2015)) as well as temporal (e.g., adaptation as a result of the selective pressure induced by therapy (Bhang et al., 2015; Juric et al., 2015; Kwak et al., 2015; Menzies et al., 2014; Shi et al., 2014; Waclaw et al., 2015)). For example, varied response—or lack thereof—to a targeted therapeutic regimen across multiple metastatic lesions (Carlino et al., 2013; Menzies et al., 2014; Russo et al., 2016) implies underlying biological heterogeneity between those lesions. The simultaneous acquisition of resistance by multiple metastases underscores the fact that such resistant sub-populations may exist even prior to tumor dissemination (Sanborn et al., 2015; Wagle et al., 2011). Conversely, separate tumor foci can acquire similar resistance mechanisms independently through convergent evolution (Gundem et al., 2015; Juric et al., 2015).
A third challenge, arising out of the first two, is that resistance is prone to undersampling in translational investigation. One source of undersampling is technical: many studies have employed only one modality to query a small number of putative resistance mechanisms previously nominated by preclinical investigation. Other efforts have used large-scale genomic approaches, but such techniques also have limitations: a whole-exome sequencing study cannot identify transcriptional or post-translational resistance mechanisms, and vice versa. Moreover, clinical identification of resistance-associated alterations does not by itself demonstrate causality. Rather, experimental studies are necessary to evaluate their functional significance (Van Allen et al., 2014). The limited availability of paired pre-treatment (drug-sensitive) and post-treatment (drug-resistant) clinical samples has also constrained many of the above approaches. Finally, the aforementioned spatial and temporal heterogeneity may further compound undersampling.
Overall, therefore, cancer drug resistance is determined not by singular, mutually exclusive alterations, but rather by a multifactorial, heterogeneous landscape that is prone to undersampling (Figure 1). New approaches are needed to account for this complexity and facilitate future biological and therapeutic insights.
Figure 1. The landscape of cancer therapeutics and resistance.
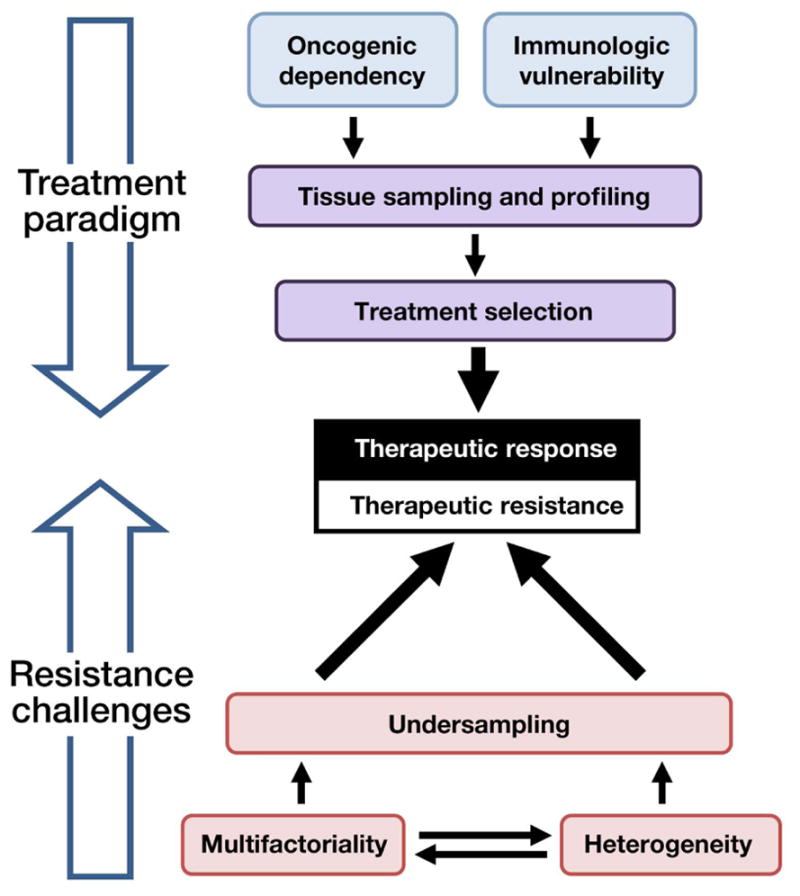
Recent research has more fully elucidated the landscape of cancer’s oncogenic dependencies and immunologic vulnerabilities. Clinical interrogation of these features can guide treatment selection, leading to therapeutic response (top). However, drug resistance often limits cure or long-term control of advanced cancer. Resistance poses several challenges (bottom), in that it is multifactorial, heterogeneous, and therefore prone to undersampling. If resistance is to be overcome, new frameworks are needed to understand and address these challenges.
A convergence-based framework for cancer drug resistance
Given these challenges, the task of understanding and addressing cancer drug resistance might at first seem hopeless. A deeper view of this same complexity, however, makes it clear that resistance effectors are not randomly distributed. Rather, they appear to converge into recognizable patterns on the basis of their relationship to the drug target and its downstream pathways—an observation that in turn suggests the possibility of principled and broadly applicable strategies against resistance (Figure 2). This convergence-based framework, described in detail below, has two key properties. First, it reduces many individual resistance effectors into a much smaller set of resistance principles. Second, it may prove generalizable across many tumor types and therapeutic settings. In defining parsimonious classes of resistance mechanisms, this convergence-based approach may therefore assist in understanding and combatting the complex phenomenon of cancer drug resistance.
Figure 2. A convergence-based framework for cancer drug resistance.
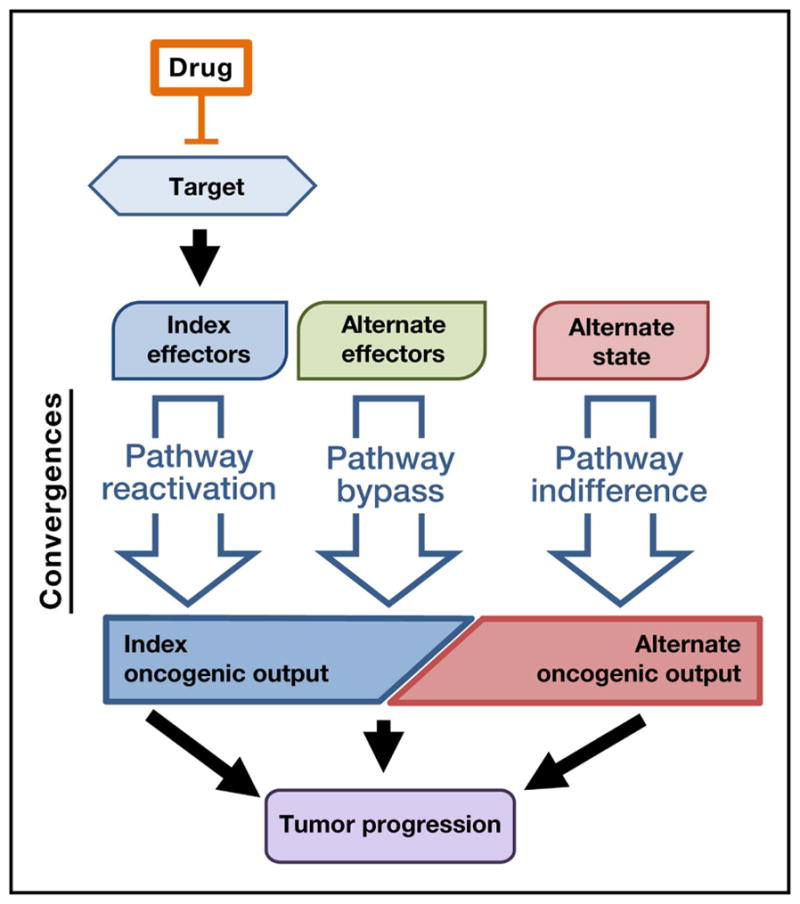
Mechanisms of resistance to targeted therapeutics converge into patterns on the basis of their relationship to the drug target and its downstream pathways. In a “pathway reactivation” convergence, resistance mechanisms re-engage the index effector pathway downstream of the drug target. “Pathway bypass” resistance uses alternate effectors to bypass the inhibited index effectors and re-engage the original downstream oncogenic output (e.g., transcriptional or translational state). “Pathway indifference,” in contrast, is characterized by an alternative cell state that is indifferent to inhibition of the drug target as well as its index effectors and oncogenic output. In each case, resistance mechanisms support an index or alternate oncogenic output that drives continued tumor progression.
Convergences in resistance to targeted therapy
Convergence 1: Pathway reactivation
Targeted anticancer agents act against specific cellular dependencies, often proteins or pathways that have undergone oncogenic dysregulation. Correspondingly, many individual mechanisms of resistance to a given targeted therapy converge towards maintenance of the index oncogenic signaling output by re-engaging core pathway effectors downstream of the drug target (Figure 3). Arguably, pathway reactivation represents the most common resistance convergence described to date, as discussed in detail below.
Figure 3. Pathway reactivation as a resistance convergence.

Pathway reactivation confers drug resistance by re-engaging index effectors downstream of the drug target, thereby maintaining the original oncogenic signaling output.
- In BRAF-mutant melanoma, MAPK pathway inhibitors target BRAF and its index effectors in the MAPK pathway. Pathway reactivation mechanisms include target alterations (e.g., amplification or alternate splicing), which render BRAF insensitive to drug inhibition, as well as recruitment of upstream, parallel, and downstream effectors, which re-activate index effectors in a BRAF-independent fashion. These mechanisms reactivate the index MAPK pathway effectors downstream of BRAF, conferring resistance to BRAF inhibition.
- In prostate cancer, oncogenic signaling through the androgen receptor (AR) is targeted by androgen-deprivation therapy and anti-androgen therapy. Resistance to AR-directed therapy can be mediated by pathway reactivation mechanisms including target alterations, which render AR itself resistant to drug inhibition, and recruitment of upstream effectors (non-gonadal androgens or alternate AR ligands), which reactivate drug-inhibited signaling through AR. In either case, the index oncogenic signaling downstream of AR is re-activated, conferring drug resistance.
Drug target alterations
Among the simplest ways to re-activate the pathway downstream of a drug-inhibited oncoprotein is to render the protein target itself insensitive to the drug. This effect is most commonly achieved through second-site mutations that arise in cis with the original oncogenic mutation. A now-classic example of this mechanism involves secondary mutations in the BCR-ABL fusion kinase that confer acquired imatinib resistance in chronic myeloid leukemia (CML) (Gorre et al., 2001). Since this discovery, secondary drug resistance mutations have been described in many kinase drug targets (reviewed in Daver et al., 2015; Lito et al., 2013; Zhang et al., 2009).
One of the best known kinase inhibitor resistance mutations affects the conserved “gatekeeper” residue, altering the accessibility of a hydrophobic pocket critical for the binding of ATP-competitive kinase inhibitors. Gatekeeper mutations identified clinically in kinase oncoprotein drug targets include BCR-ABLT315I (in CML (Gorre et al., 2001)), EGFRT790M (in non-small cell lung cancer (NSCLC) (Kobayashi et al., 2005; Pao et al., 2005)), ALKL1196M (in NSCLC (Choi et al., 2010; Katayama et al., 2012)), and KITT670I (in gastrointestinal stromal tumors (Heinrich et al., 2006)). Third-generation tyrosine kinase inhibitors that retain efficacy against the EGFRT790M mutation represent an important advance in the clinical management of this resistance mechanism in EGFR-mutant lung cancer (Mok et al., 2017), although they remain vulnerable to other resistance mutations in the drug target (Thress et al., 2015).
For allosteric (Type IIB), non-ATP competitive kinase inhibitors, resistance mutations outside of the gatekeeper residue can alter the intrinsic activity of the target kinase, as seen in resistance to MEK inhibitors (Emery et al., 2009), or interfere with drug binding, as observed with resistance to the BTK inhibitor ibrutinib (Woyach et al., 2014). In other cases, such as BRAF-mutant melanoma (Figure 3A), other genomic alterations of the target kinase such as amplification (Van Allen et al., 2014; Long et al., 2014; Shi et al., 2012; 2014) and alternative splicing (Poulikakos et al., 2011) contribute to resistance.
The importance of drug target alterations as resistance effectors is not limited to kinase inhibitors. In prostate cancer, for example, where signaling through the androgen receptor (AR) is central to oncogenesis (Figure 3B), the efficacy of AR-directed therapeutics can be thwarted by AR mutation (Taplin et al., 1995), amplification (Visakorpi et al., 1995), or overexpression (Chen et al., 2004). These genomic events restore AR activity by converting first-generation AR antagonists into partial agonists or by rendering AR sensitive to alternative steroid hormone ligands. Second-generation anti-androgen therapies (e.g., abiraterone and enzalutamide) are also susceptible to target-based resistance mechanisms. For example, abiraterone resistance can be driven by AR amplification or the ART878A mutation, which renders AR responsive to progesterone instead (Chen et al., 2015; Romanel et al., 2015). Similarly, the ARF876L mutation converts enzalutamide to a partial AR agonist, conferring enzalutamide resistance (Balbas et al., 2013; Joseph et al., 2013; Korpal et al., 2013). The AR-V7 splice variant, which retains transcriptional activity but may abrogate enzalutamide binding, has also in some reports been associated with drug resistance (Antonarakis et al., 2014; Miyamoto et al., 2015). Analogously, in estrogen receptor (ER)-positive breast cancer, mutations in ESR1 encoding ERα result in constitutive, estrogen-independent ERα activation and are associated with resistance to anti-estrogen therapy (Robinson et al., 2013; Toy et al., 2013).
Upstream effectors
Downstream pathway activation as a resistance mechanism can also be achieved by effectors that act upstream of the drug target. In BRAF-mutant melanoma, for example, resistance can be conferred by mutations that activate NRAS (Van Allen et al., 2014; Nazarian et al., 2010; Romano et al., 2013) or by loss of the NF1 tumor suppressor, which negatively regulates Ras proteins (Maertens et al., 2013; Shalem et al., 2014; Whittaker et al., 2013). Further upstream, RTKs including MET (Straussman et al., 2012; Wilson et al., 2012) and EGFR (Girotti et al., 2013) can override single-agent RAF inhibition by activating both Ras and C-RAF, restoring downstream MEK/ERK signaling. RTKs may also contribute to resistance to RAF or PI3 kinase inhibition through relief of feedback inhibitory mechanisms engaged by oncogenic mutations (Chandarlapaty, 2012; Corcoran et al., 2012; Le et al., 2016; Prahallad et al., 2012). Autocrine or paracrine activation of RTKs (Montero-Conde et al., 2013) represents another upstream resistance mechanism, one that can in some cases originate from the tumor microenvironment (Straussman et al., 2012; Wilson et al., 2012).
Parallel effectors
Downstream pathway reactivation and drug resistance can also be achieved by effector proteins operating in parallel to the target (onco)protein. For example, resistance to anti-EGFR monoclonal antibodies in colorectal cancer (Bertotti et al., 2015; Siravegna et al., 2015) and to EGFR TKIs in EGFR-mutant NSCLC can be driven by engagement of alternative RTKs including MET (Bardelli et al., 2013; Engelman et al., 2007), ERBB2 (Takezawa et al., 2012; Yonesaka et al., 2011), and IGF1R (Guix et al., 2008). Likewise, EGFR (Katayama et al., 2012; Sasaki et al., 2011), ERBB3 (Wilson et al., 2015), and KIT (Katayama et al., 2012) can rescue inhibition of ALK in ALK-rearranged NSCLC (reviewed in Niederst and Engelman, 2013). In BRAF-mutant melanoma (Figure 3A), single-agent RAF inhibition can be rescued by alternative MAP3Ks, including COT/MAP3K8 (Johannessen et al., 2010; 2013), which reactivates MEK and ERK signaling.
Downstream effectors
Reactivation of downstream effectors, independent of upstream signaling, can also confer pathway re-engagement. In BRAF-mutant melanoma (Figure 3A), for example, activating MEK mutations drive resistance to RAF and MEK inhibition (Van Allen et al., 2014; Carlino et al., 2015; Emery et al., 2009; Oddo et al., 2016; Wagle et al., 2011). Similarly, for EGFR-directed therapies in colorectal cancer, activating mutations in the downstream effectors KRAS (Amado et al., 2008; Diaz et al., 2012; Douillard et al., 2013; Van Emburgh et al., 2016; Misale et al., 2012), BRAF (Di Nicolantonio et al., 2008), and MEK (Bertotti et al., 2015; Russo et al., 2016; Siravegna et al., 2015) are associated with intrinsic and acquired resistance. In CLL, gain-of-function mutations in downstream PLCγ2 confer BTK-independent B cell receptor activity and resistance to the BTK inhibitor ibrutinib (Woyach et al., 2014).
Thus, many individual resistance mechanisms converge on reactivation of the index drug-inhibited oncogenic signaling pathway (Johannessen et al., 2010; 2013; Wilson et al., 2015). The same general principle appears to hold true in the clinical setting; for example, around 70% of BRAF-mutant melanomas clinically progressing on RAF/MEK inhibition have, via one mechanism or another, reactivated ERK phosphorylation (Van Allen et al., 2014; Kwong et al., 2015; Rizos et al., 2014; Shi et al., 2014; Wagle et al., 2014). The frequency of pathway reactivation as a mechanism of resistance may reflect biological parsimony, in that minimal re-wiring of the malignant cell is needed to escape pharmacologic inhibition.
Convergence 2: Pathway bypass
While many mechanisms of resistance to targeted therapy converge on reactivation of the index intermediary signaling pathway, other convergences may also contribute. In the BRAF-mutant melanoma example above, a significant minority of drug-resistant cases do not seem to re-establish ERK phosphorylation (Van Allen et al., 2014; Kwong et al., 2015; Rizos et al., 2014; Shi et al., 2014; Wagle et al., 2014). At least some such instances may harbor resistance mechanisms that bypass index signaling pathways, finding alternative ways to re-engage the more downstream oncogenic output (transcriptional, translational, or otherwise) required by the malignant cell state (Hsieh et al., 2015; Pratilas et al., 2009). We denote this effect as a “pathway bypass” convergence (Figure 4). Of note, others have used the term “bypass” to describe resistance mechanisms that substitute for an individual protein drug target, thus reactivating the index intermediary signaling effectors (for example, Niederst and Engelman, 2013). We, however, consider such mechanisms part of the “pathway reactivation” continuum, because they restore the original drug-inhibited signaling pathway. Here, we use the term “bypass” to describe mechanisms that circumvent the index signaling pathway as a whole, conferring resistance without reactivation of the original intermediary signaling effectors.
Figure 4. Pathway bypass as a resistance convergence.

Pathway bypass confers drug resistance by recruiting alternate effector pathways to sustain the index downstream oncogenic output (for example, transcriptional or translational state).
- In BRAF-mutant melanoma, pathway bypass mechanisms reactivate the core MITF transcriptional output in a manner that is independent of upstream MAPK input. For example, a GPCR-cAMP-CREB signaling axis can substitute for MAPK signaling to sustain an MITF-driven transcriptional program. Likewise, MITF amplification can render MITF itself independent of MAPK signaling.
- In prostate cancer, signaling through the glucocorticoid receptor (GR) drives a transcriptional program similar to that of the index AR signaling pathway, rendering cells resistant to inhibition of the AR axis.
BRAF-mutant melanoma provides an instructive example of one possible pathway-bypass convergence. Here, RAF/MEK inhibitor resistance can be driven by a MAP kinase-independent module that begins at the plasma membrane with G protein coupled receptor (GPCR) signaling. This GPCR activity triggers a cAMP/protein kinase A signal which in turn engages the CREB transcriptional regulator to harness several transcription factors that confer RAF/MEK inhibitor resistance (Johannessen et al., 2013) (Figure 4A). In wild-type melanocytes, cAMP/CREB signaling downstream of the GPCR MC1R both provides an essential pro-survival signal and, via PKA, suppresses RAF-mediated MAPK activation (Dumaz and Marais, 2003). In the setting of oncogenic BRAF mutations, cAMP/CREB signaling becomes dispensable and its inhibitory effect on MAPK signaling is lost (Dumaz et al., 2006; Garraway et al., 2005). Following RAF/MEK inhibition, loss of MAPK signaling can then be rescued by restoration of GPCR/cAMP/CREB signaling (Johannessen et al., 2013). Crucially, both MAPK and cAMP/CREB converge on a core set of transcriptional outputs, including ETV1 (Jane-Valbuena et al., 2010; Wang et al., 2017) and the melanoma oncogene and master lineage regulator MITF (Bertolotto et al., 1998; Huber et al., 2003; Johannessen et al., 2013; Khaled et al., 2010; Price et al., 1998). Thus, the GPCR/cAMP/CREB module effects resistance by sustaining a key transcriptional output of the oncogenic MAPK pathway, but in a manner that bypasses the index RAF/MEK/ERK module.
An analogous situation occurs in prostate cancer, in which blockade of AR by enzalutamide can be bypassed by signaling through the glucocorticoid receptor (GR) (Arora et al., 2013) (Figure 4B). GR and AR, similar to CREB and MAPK in melanoma, appear to converge on a similar gene expression program that sustains tumor survival. Thus, following therapeutic inhibition of AR signaling, activation of GR may rescue the original oncogenic transcriptional output in an AR-independent fashion. Re-activation of an “AR-on” transcriptional phenotype probably also contributes to abiraterone resistance (Miyamoto et al., 2012), although the distinctive contributions of AR bypass versus AR-dependent mechanisms in this setting are less clearly resolved at present.
Pathway bypass mechanisms may also contribute to resistance in several other settings. In resistance to ALK inhibition in NSCLC, a putative bypass module operates independently of both MAP kinase and PI3 kinase signaling by engaging members of the P2Y purinergic receptor GPCR family to transduce a signal through protein kinase C (Wilson et al., 2015). Pathway bypass resistance to PI3 kinase inhibition in ER+ breast cancer can engage PIM kinases or other effectors and may converge not on a specific transcriptional program, but rather on a TOR-dependent translational output (Le et al., 2016). Likewise, PARP inhibition is synthetic lethal with BRCA1/2 deficiency in human tumors, and PARP-inhibitor resistance can arise by pathway bypass mechanisms including loss of 53BP1 (Jaspers et al., 2013) or REV7 (Xu et al., 2015), which restore function of the index drug-inhibited homologous recombination pathway (without restoration of PARP activity itself).
Together, these examples suggest that pathway bypass mechanisms can reactivate fundamental oncogenic transcriptional or translational outputs through engagement of alternative intermediary effectors. As with pathway-dependent convergences (above), we speculate that for any given therapeutic regimen, multiple pathway-independent resistance mechanisms may converge onto a much smaller number of bypass modules which in turn re-engage the core transcriptional or translational programs that underpin the oncogenic dependency being targeted.
Convergence 3: Pathway indifference
Both pathway reactivation and pathway bypass resistance convergences re-engage downstream oncogenic transcriptional or translational outputs disrupted by drug therapy. In contrast, a growing body of evidence supports the existence of an additional, fundamentally distinct type of convergence centered on alternative malignant cell states that are independent of the index oncogenic dependency. This “pathway indifference” may arise despite the continued presence of a driver alteration that conferred a therapeutic vulnerability in the treatment-naive setting. Here, the term “pathway indifference” is not intended as a catch-all classification for unknown mechanisms of resistance. Rather, it is proposed to describe a specific phenotype: an alternative cell state (transcriptional or otherwise) that is independent of the index oncogenic pathway and that confers drug resistance despite continued inhibition of the index drug target and its downstream outputs.
One well-known example of pathway-indifferent resistance occurs when EGFR-mutant NSCLC is treated with EGFR inhibitors. Biopsies obtained at the time of drug resistance reveal that some of these cancers show markers of a mesenchymal-like (Sequist et al., 2011; Yauch et al., 2005) or small cell lung cancerlike (SCLC-like) state (Piotrowska et al., 2015; Sequist et al., 2011; Yu et al., 2013). Importantly, the activating EGFR mutation that conferred initial sensitivity to EGFR inhibition is preserved in the relapsing SCLC-like tumor, implying a common origin of both populations. Thus, the pathway-indifference phenomenon appears to represent either outgrowth of an SCLC-like subpopulation originally present within the EGFR-mutant NSCLC tumor population or an adaptive cell state switch that arises within NSCLC cells that survive drug exposure (Hata et al., 2016, reviewed in Oser et al., 2015). Of note, these possibilities (clonal outgrowth versus de novo acquisition) apply in principle to the emergence of any resistance mechanism.
A related phenomenon has been observed in melanoma (Figure 5A). Around 20% of BRAF-mutant melanomas exhibit intrinsic resistance to RAF/MEK inhibition (Chapman et al., 2011; Robert et al., 2015; Konieczkowski et al., 2014; Larkin et al., 2014). Sensitivity to RAF/MEK inhibition correlates strongly with a transcriptional profile governed by the aforementioned MITF transcription factor, which requires MAPK input for its activity. In contrast, melanomas that are intrinsically resistant to RAF/MEK inhibition display lower expression of MITF and its target genes, but instead elaborate a transcriptional profile characterized by NF-κB activation and expression of the AXL receptor tyrosine kinase (Konieczkowski et al., 2014; Muller et al., 2014; Tap et al., 2010; Zipser et al., 2011). Although either AXL expression or NF-κB activation can confer MAPK resistance, neither appears required to maintain the pathway-indifferent resistant state (Anastas et al., 2014; Konieczkowski et al., 2014; Wood et al., 2012). In other words, these factors may be sufficient but not necessary for intrinsic resistance to RAF/MEK inhibition in melanoma. Thus, particularly in the intrinsic resistance setting, an alternative transcriptional state can confer pathway-indifferent resistance to RAF/MEK inhibition in BRAF-mutant melanoma.
Figure 5. Pathway indifference as a resistance convergence.

Pathway indifference confers drug resistance via an alternate cellular state that is independent of the index drug-inhibited pathway and its original oncogenic effectors.
- In BRAF-mutant melanoma, the index drug-sensitive transcriptional state is characterized by MAPK-dependent activity of the MITF transcription factor. Resistance to MAPK pathway inhibition can be achieved by transition to an NF-κB-driven, low-MITF transcriptional state that does not require MAPK input for its maintenance.
- In prostate cancer, alternative AR-independent cellular states are characterized by Wnt5A-driven and neuroendocrine-like transcriptional programs and are indifferent to inhibition of the AR signaling axis.
An alternative cell state also appears relevant to pathway-indifferent drug resistance in prostate cancer (Figure 5B). During escape from androgen receptor blockade (i.e., development of castration resistance), many prostate cancers transition into a so-called “neuroendocrine-like” state that exhibits reduced AR expression (Beltran et al., 2011; 2012; 2016). This neuroendocrine state, which also lacks expression of AR-driven markers such as PSA and is resistant to further anti-androgen therapy, appears indifferent to signaling through the AR axis. Instead, this state appears to be governed by an alternative transcriptional program enacted, at least in part, by SOX2 (Bishop et al., 2017; Ku et al., 2017; Mu et al., 2017). Other studies raise the possibility of a distinct Wnt5A-driven state that arises during acquisition of androgen independence (Miyamoto et al., 2015). Hence, this phenomenon resembles the aforementioned cell state transitions in EGFR-mutant NSCLC and BRAF-mutant melanoma.
Finally, resistance to PARP inhibition may offer a distinct example of pathway indifference, in this case effected by loss of the underlying “BRCA-like” cell state that conferred synthetic lethal dependency on PARP1. Thus, secondary mutations in BRCA1/2 that restore BRCA1/2 function abolish the synthetic lethal relationship with PARP1 and confer indifference to PARP1 inhibition (Edwards et al., 2008; Norquist et al., 2011). The shift from such a genome-unstable to genome-stable state may differ in some respects from the aforementioned transcriptional state transitions. Nonetheless, and while the mechanistic details of these pathway-indifferent cell states are still emerging, it seems that a convergence involving such cell state transitions may contribute to drug resistance in multiple contexts.
Convergence-based resistance to cancer immunotherapy
The foregoing sections summarize the growing evidence in support of a convergence-based framework for resistance to targeted therapy. Given the profound and growing impact of immunotherapy across multiple cancer types, an obvious question is whether convergences may also be evident in resistance to immunotherapy. Of note, the dimensionality of the problem increases markedly in the setting of a multicellular antitumor immune response and an assortment of immunomodulatory microenvironmental factors. Moreover, characterization of mechanisms of resistance to immunotherapy remains in its infancy. Nonetheless, it is possible that the tumor/immune interface may also harbor relevant convergences.
As others have described, clearing of tumor cells by T lymphocytes depends not on a single pathway but rather on a multi-step “cancer immunity cycle.” This process begins with (neo)antigen presentation to T cells, progresses to T cell priming, activation, and trafficking/expansion in tumor tissue, and concludes with cytotoxic T cell recognition of antigen/MHC complexes followed by killing of malignant cells (Figure 6A; reviewed in Chen and Mellman, 2013; Kim and Chen, 2016). The efficiency of this overall process is modified at each step by factors including the availability of immunogenic tumor antigens, immunomodulatory factors in the microenvironment, regulatory cells (such as myeloid suppressor cells and T-regs), and engagement of T cell checkpoint cascades. Immunotherapy, in turn, targets specific steps in the above process that may be rate-limiting for a particular tumor, allowing the cancer-immunity cycle to progress and generating a therapeutic anti-tumor immune response. Resistance to immunotherapy, then, can arise from further aberrations that affect the same fundamental steps, thereby bringing the tumor immunity cycle to a halt. As seen with targeted therapeutics, resistance to immunotherapy will undoubtedly prove both heterogeneous and multifactorial and so may perturb multiple of these steps simultaneously in the same tumor. Moreover, resistance to immunotherapy can manifest as both an intrinsic and an acquired phenomenon.
Figure 6. Convergences in resistance to immunotherapy.

- Anti-tumor immunity requires a multi-step cycle to achieve immune cell clearance of tumor cells.
- Many immunotherapy resistance mechanisms converge on modulation of neoantigen expression, processing, or presentation, thus restoring the index state of escape from anti-tumor immunity.
- Alternatively, induction of T cell dysfunction can bypass anti-tumor immunity, either directly (e.g., through alternative checkpoint ligands) or indirectly (e.g., through T regulatory cells or the tumor microenvironment).
- Finally, certain oncogenes or malignant cell states may confer an “immune indifferent” resistance state.
In the face of this complexity, the characterization of the clinical landscape immunotherapy resistance mechanisms remains in its infancy. Nonetheless, certain themes have begun to emerge. For example, resistance mechanisms affecting antigen presentation have become evident in the setting of both intrinsic and acquired resistance to immune checkpoint blockade (Figure 6B). Somatic alterations predicted to reduce antigen presentation (e.g., in HLA and beta-2 microglobulin (B2M) (Shukla et al., 2015, 2014)) or processing (Giannakis et al., 2016) demonstrate enrichment in several cancer types, suggesting that intrinsic resistance to immune-mediated killing contributes to the evolution of primary tumors. Low mutation or neoantigen burden (Van Allen et al., 2015; Le et al., 2015; Rizvi et al., 2015; Snyder et al., 2014) have also been identified in the setting of intrinsic resistance to immunotherapy. Moreover, B2M mutations are associated with acquired resistance to anti-PD1 therapy (Zaretsky et al., 2016). Thus, a growing catalogue of mechanisms implicates antigen presentation as a convergence for resistance to immune checkpoint blockade.
In addition, antigen-based mechanisms of resistance to chimeric antigen receptor (CAR) T cell based therapies have been reported. In this setting, genetic deletion of specific epitopes recognized by CAR T cell clones facilitates tumor escape (Sotillo et al., 2015). While not modifying antigen presentation per se, these mechanisms may represent a convergence on antigen recognition (or lack thereof) in resistance to cellular immunotherapy. Indeed, convergence on antigen presentation and recognition might tentatively be considered analogous to pathway-based resistance. In targeted therapeutics, pathway-based resistance reactivates the index drug-inhibited signaling pathway, restoring essential downstream oncogenic outputs; the above immunotherapy resistance mechanisms, likewise, re-establish suppression of an index “pathway” of antigen presentation and recognition, restoring immune escape.
A second emerging commonality involves T cell dysfunction or “exhaustion” (Figure 6C, reviewed in Wherry and Kurachi, 2015). Here, one set of mechanisms may involve up-regulation of alternate immune checkpoint ligands on malignant or other antigen presenting cells. Alternatively, induction of T cell dysfunction (e.g., by IDO, which diminishes the supply of tryptophan, an amino acid essential for T cell function (Holmgaard et al., 2013; Uyttenhove et al., 2003)) or down-regulation of T cell activating factors (e.g., OX40/OX40 ligand (Guo et al., 2014)) may achieve similar ends (reviewed in Mahoney et al., 2015), as may modulation by intratumoral Tregs (Overacre-Delgoffe et al., 2017). Likewise, disruption of interferon signaling via JAK mutation/deletion may render tumor cells non-responsive to immunotherapy (Garcia-Diaz et al., 2017; Manguso et al., 2017; Shin et al., 2017; Zaretsky et al., 2016). Akin to the pathway bypass convergence proposed for targeted therapeutics, these mechanisms restore the index oncogenic state (here, suppression of anti-tumor immunity) without directly re-engaging the index pathways (here, antigen presentation and immune checkpoint engagement) that originally determined it.
Other immunotherapy resistance mechanisms do not obviously alter either antigen presentation or T cell exhaustion. Some of these may represent states of indifference to anti-tumor immunity (Figure 6D). For example, certain oncoproteins may contribute to intrinsic resistance to immune checkpoint blockade. While immunotherapy offers significant clinical benefit in NSCLC as a whole, this effect appears attenuated in the setting of EGFR activating mutations (Gainor et al., 2016). On the one hand, EGFR-mutant NSCLC often harbors a distinct mutational spectrum (Shukla et al., 2015, 2014a), suggesting that the oncogene driver itself may not be entirely responsible for immunotherapy resistance. On the other hand, mutant EGFR regulates both PD-L1 expression and the immune microenvironment in mouse models (Akbay et al., 2013), raising the possibility that aberrant RTK signaling may directly contribute to immunotherapy resistance in some fashion. Conversely, concomitant MAP kinase pathway inhibition may enhance responses to anti-PD1 therapy in other oncogene-driven cancers, such as BRAF-mutant melanoma and KRAS-mutant colorectal cancer (Cooper et al., 2014; Frederick et al., 2013; Hu-Lieskovan et al., 2015), although this effect may be attenuated by contributions of MAP kinase signaling to T cell function (Callahan et al., 2014). Alternatively, an EMT-like alternative cell state may be associated with intrinsic resistance to immunotherapy in melanoma (Titz et al., 2016). Finally, transition to cell states in which critical (neo-)antigen expression is lost may provide a distinct pathway-indifferent resistance mechanism. Thus, for example, in CD19+ B-ALL, resistance to anti-CD19 CAR-T cell therapy can be mediated by transition to a myeloid-like phenotype that lacks CD19 expression (Gardner et al., 2016; Jacoby et al., 2016); in contrast to specific loss of expression of a single neoantigen described earlier, this resistance mechanisms appear to involve a more fundamental transition to a cell state independent of CD19 and therefore indifferent to immunotherapy against it.
Although clinical characterization of immunotherapy resistance remains fragmentary (reviewed in Restifo et al., 2016), these preliminary observations suggest that resistance convergences—ones rooted in foundational tumor immunology mechanisms—may also pertain to this class of therapeutics. Such knowledge may eventually facilitate the identification of molecular subsets intrinsically less likely to respond to specific immunotherapeutic approaches and may also help clarify mechanisms of acquired resistance to immunotherapy. These insights may ultimately guide strategies to overcome resistance to immunotherapy.
Therapeutic frameworks to overcome cancer drug resistance
Strategies to prevent the emergence of resistance or salvage the cancer patient after resistance has developed remain limited. Indeed, despite the aforementioned insights, the physician and patient are still very likely to run out of therapeutic options before the tumor runs out of resistance mechanisms (reviewed in Garraway and Janne, 2012). Therefore, a critical question is how insights into tumor dependencies, immunologic vulnerabilities, and resistance convergences might help address this clinical challenge.
One intuitively appealing approach—what might be termed a “dependency-based” combinatorial framework—proposes to simultaneously target multiple dependencies that lack shared mechanisms of resistance. Theoretically, such combinations should make it improbable for a tumor to acquire resistance to an entire multi-drug cocktail simultaneously. Indeed, a compelling precedent exists for this approach in the treatment of infectious diseases, particularly the development of highly active anti-retroviral therapy (HAART) for HIV. In both HIV and cancer, drug-resistant subclones arise via mutations or other stochastic changes and expand under the selective pressure imposed by therapy. Whereas this phenomenon thwarts effective disease control by monotherapies, it is countered by rational combinations that target parallel dependencies, thus enabling durable therapeutic control. One might even be tempted to argue that studies of drug resistance mechanisms—however biologically interesting—offer therapeutic value that is supplementary at best in comparison to this dependency-based combinatorial approach.
In fact, however, several difficulties constrain the application of this dependency-based framework to cancer patients. First, while most tumors do harbor multiple driver (epi)genomic alterations, many of these aberrancies remain poorly druggable—a fact that probably will not change quickly. Second, in HIV and other microbial settings, the biological dissimilarity between host and pathogen minimizes off-target toxicities. Thus, most antimicrobial drug combinations are safe as well as effective. In contrast, many “horizontal” combinations of targeted anti-cancer agents (i.e., those targeting multiple parallel dependencies) have proven unacceptably toxic (reviewed in Park et al., 2013; Yap et al., 2013). This phenomenon often reflects on-target toxicities since the targeted pathways are essential to the function of various normal tissues. Thus, it cannot be assumed that targeting multiple cancer dependencies in parallel will always be feasible. These challenges have given rise to recent efforts to combine targeted therapeutics with immunotherapy (Hu-Lieskovan et al., 2015; Ribas et al., 2013), as well as with other therapeutic classes such as chemotherapy and radiation therapy. Such studies will undoubtedly be followed with great interest, particularly in treatment-resistant settings.
In addition to the above dependency-based framework, an alternative “convergence-based” therapeutic approach might also be envisioned. This strategy would use the mechanistic convergences identified by the above resistance framework as “anchors” for combinatorial therapeutic intervention, simultaneously inhibiting both an index dependency and its major resistance convergences. In the context of such an approach, just as the convergence-based resistance framework suggests a parsimonious explanatory model for resistance, it may also offer predictive guidance for strategies to overcome resistance.
Pathway-based resistance mechanisms, for example, might be interdicted by “stacking” therapeutics against the target pathway. For instance, the same target can be inhibited with two drugs. This strategy has been exploited clinically in dual blockade of HER2 with both pertuzumab and trastuzumab (von Minckwitz et al., 2017; Swain et al., 2015) or lapatinib and trastuzumab (Blackwell et al., 2012) in HER2-amplified breast cancer; in BCR-ABL rearranged CML, an analogous strategy involving combined ATP-competitive and allosteric inhibitors of ABL has recently been proposed in the preclinical setting (Wylie et al., 2017). Of note, the individual drugs in such a dual-blockade approach must be non-competitive (e.g., have different target binding sites) in order to cooperate. Alternatively, multiple nodes in the same pathway might be targeted. For example, in prostate cancer, dual blockade of AR signaling with a “vertical” combination of ADT and abiraterone has demonstrated clinical superiority compared to ADT alone (Fizazi et al., 2017; James et al., 2017). Likewise, in BRAF-mutant melanoma, suppression of the MAPK pathway by combined RAF/MEK inhibition overcomes several mechanisms of resistance to, and is clinically superior to, single-agent RAF inhibition (Johannessen et al., 2013; Larkin et al., 2014; Robert et al., 2015). Conceivably, targeting a pathway-based convergence more distally (for example, ERK inhibitors in BRAF-mutant cancers) might be still more effective. Unfortunately, such approaches remain constrained by the inevitable emergence of resistance to single-pathway blockade (Larkin et al., 2014; Robert et al., 2015). Nonetheless, the prevalence of pathway-based resistance raises the possibility that additional optimization of pathway inhibition might further improve clinical outcomes in many contexts.
Pathway bypass resistance mechanisms could be targeted either at the level of the bypass itself or at the convergent downstream oncogenic output. Targeting bypass pathways with horizontal combinations has been proposed based on preclinical studies. For example, inhibition of the GR bypass pathway restores sensitivity to AR inhibition (Shah et al., 2017). Such approaches may, however, face the same in vivo toxicity challenges discussed above if the bypass module also comprises a normal tissue dependency. On the other hand, combination immunotherapy directed against parallel checkpoint proteins (e.g., CTLA-4 and PD-1) has shown significant clinical benefit with manageable toxicity (Larkin et al., 2015; Postow et al., 2015; Wolchok et al., 2013; 2017).
Targeting the convergent oncogenic output (further downstream) could theoretically have efficacy against both the index pathway (both as primary therapy and in the setting of pathway-based resistance) and pathway bypass mechanisms of resistance. One challenge to this approach is that many such downstream convergences are transcriptional programs, which are not readily druggable directly. Many transcription factors, however, require chromatin factors as co-regulators, some of which are already targeted by therapeutics in the clinic. For example, it has been proposed that histone deacetylase inhibitors (which indirectly suppress MITF expression) could be combined with MAP kinase pathway inhibitors to overcome MITF-driven resistance in melanoma (Johannessen et al., 2013; Yokoyama et al., 2008).
Resistance mediated by pathway indifference, in turn, suggests either disrupting the drug-indifferent state (thus returning the cell to the index drug-sensitive state) or targeting novel vulnerabilities in the alternative state itself. There is preclinical evidence for the former strategy in EGFR-mutant NSCLC and in neuroendocrine prostate cancer, in which targeting effectors of the EMT-associated or neuroendocrine-like resistance states, respectively, restores sensitivity to inhibition of EGFR or AR (Kim et al., 2013; Ku et al., 2017; Marin-Aguilera et al., 2014; Mu et al., 2017; Zhang et al., 2012). Clinical evidence for the latter strategy likewise comes from EGFR-mutant NSCLC, in which relapse under EGFR inhibition has been associated with a transition from NSCLC to an SCLC-like cell state and with sensitivity to a drug regimen (carboplatin/etoposide) used in SCLC (Piotrowska et al., 2015; Sequist et al., 2011, reviewed in Oser et al., 2015). The extent to which these state transitions are mediated by epigenetic alterations remains uncertain, but, in T-ALL, preclinical evidence suggests that an epigenetic state resistant to NOTCH1 targeting via gamma secretase inhibition is in turn sensitive to epigenetic drugging via bromodomain inhibitors (Knoechel et al., 2014). Similarly, convergence-based approaches might show efficacy in preventing or attacking the alternative cell states associated with indifference to immunotherapy. For example, in cases where indifference to immunotherapy is mediated by specific oncogenic pathways, targeting those pathways may represent a promising strategy against resistance (Akbay et al., 2013; Cooper et al., 2014; Frederick et al., 2013; Hu-Lieskovan et al., 2015). In the future, broader clinical application of strategies against indifferent cell states will require clinically robust markers of such state changes as well as identification and targeting of their vulnerabilities.
Finally, certain strategies against resistance may be naive to the specific mechanism of resistance at work. For example, in the absence of drug, resistance mutations are frequently deleterious (Chmielecki et al., 2011; Das Thakur et al., 2013). Accordingly, following emergence of drug resistance, drug discontinuation can lead to drug-sensitive populations out-competing the drug-resistant populations, re-emergence of drug sensitivity, and clinical response upon drug rechallenge (Browning et al., 2013; Chmielecki et al., 2011; Hata et al., 2013; Riely et al., 2007; Romano et al., 2013; Schweizer et al., 2015; Seghers et al., 2012; Sequist et al., 2011; et al., 2015; Das Thakur et al., 2013, reviewed in Camidge et al., 2014). In the future, drug scheduling strategies based on these principles may prove helpful in forestalling the development of resistance or at least in modulating its clinical course.
Conclusion
Although the multifactorial and heterogeneous nature of cancer drug resistance remains a significant challenge, systematic experimental and clinical studies across many cancer types have revealed a wealth of resistance mechanisms. This review has proposed a convergence-based framework—pathway reactivation, pathway bypass, and pathway indifference—to organize the multiplicity of individual resistance mechanisms to targeted therapeutics into a parsimonious set of generalizable principles. Importantly, this framework may not prove comprehensive, nor is it intended to be proscriptive. Rather, it seems likely that additional as-yet unsuspected convergences will emerge. Nonetheless, the most fundamental lesson from such consideration may not be the identity of specific resistance convergences themselves, but rather that even highly complex landscapes of resistance can be understood through the paradigm of convergence. Going forward, such frameworks may provide both explanatory power to aid biological understanding and predictive power to guide future therapeutic approaches. Ultimately, these insights may help to achieve durable disease control in patients with advanced cancer.
Footnotes
Declaration of Interests
D.J.K. and C.M.J. declare no competing interests. L.A.G. is a founder of and equity holder in Foundation Medicine and Tango Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen E, Wagle N, Sucker A, Treacy D, Johannessen C, Goetz E, Place C, Taylor-Weiner A, Whittaker S, Kryukov G, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discovery. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHGH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado RG, Wolf M, Peeters M, Cutsem E, Van Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long GV, Euw EM, von Yang P-TT, Chen H-WW, Haydu L, Toroni RA, et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J Clin Invest. 2014;124:2877–90. doi: 10.1172/JCI70156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–73. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, Rubin MA, Nanus DM. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386–9. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Abbe P, Hemesath TJ, Bille K, Fisher DE, Ortonne JP, Ballotti R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998;142:827–35. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263–7. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhang HCE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21:440–8. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, Jama R, Nip KM, Angeles A, Johnson F, et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017;7:54–71. doi: 10.1158/2159-8290.CD-15-1263. [DOI] [PubMed] [Google Scholar]
- Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, Ellis C, Florance A, Vukelja S, Bischoff J, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol. 2012;30:2585–92. doi: 10.1200/JCO.2011.35.6725. [DOI] [PubMed] [Google Scholar]
- Browning ET, Weickhardt AJ, Camidge DR. Response to crizotinib rechallenge after initial progression and intervening chemotherapy in ALK lung cancer. J Thorac Oncol. 2013;8:e21. doi: 10.1097/JTO.0b013e31827a892c. [DOI] [PubMed] [Google Scholar]
- Cai W, Lin D, Wu C, Li X, Zhao C, Zheng L, Chuai S, Fei K, Zhou C, Hirsch FR. Intratumoral Heterogeneity of ALK-Rearranged and ALK/EGFR Coaltered Lung Adenocarcinoma. J Clin Oncol. 2015;33:3701–9. doi: 10.1200/JCO.2014.58.8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014;2:70–9. doi: 10.1158/2326-6066.CIR-13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- Carlino MS, Saunders CA, Haydu LE, Menzies AM, Martin Curtis C, Lebowitz PF, Kefford RF, Long GV. (18)F-labelled fluorodeoxyglucose-positron emission tomography (FDG-PET) heterogeneity of response is prognostic in dabrafenib treated BRAF mutant metastatic melanoma. Eur J Cancer. 2013;49:395–402. doi: 10.1016/j.ejca.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Carlino MS, Fung C, Shahheydari H, Todd JR, Boyd SC, Irvine M, Nagrial AM, Scolyer RA, Kefford RF, Long GV, et al. Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin Cancer Res. 2015;21:98–105. doi: 10.1158/1078-0432.CCR-14-0759. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012;2:311–9. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015;21:1273–80. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- Cooper CS, Eeles R, Wedge DC, Loo P, Van Gundem G, Alexandrov LB, Kremeyer B, Butler A, Lynch AG, Camacho N, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47:367–372. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2:643–54. doi: 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, Kantarjian H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood. 2015;125:3236–45. doi: 10.1182/blood-2014-10-605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douillard J-YY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem. 2003;278:29819–23. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66:9483–91. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, Mussolin B, Baldi F, Buscarino M, Bartolini A, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665. doi: 10.1038/ncomms13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CMM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, Ozguroğlu M, Ye D, Feyerabend S, Protheroe A, et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med. 2017;377:352–360. doi: 10.1056/NEJMoa1704174. [DOI] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res. 2016;22:4585–93. doi: 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Wu D, Cherian S, Fang M, Hanafi LAA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–10. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–26. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–67. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMCM, Papaemmanuil E, Brewer DS, Kallio HMLM, Hognas G, Annala M, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Wang X, Cheng D, Xia Z, Luan M, Zhang S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE. 2014;9:e89350. doi: 10.1371/journal.pone.0089350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Katakami N, Kaji R, Fujita S, Imai Y. Does T790M disappear? Successful gefitinib rechallenge after T790M disappearance in a patient with EGFR-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8:e27–9. doi: 10.1097/JTO.0b013e318282e047. [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HCE, Krishnamurthy Radhakrishna V, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–9. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, Mehren M, von Fletcher CD, Sandau K, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Nguyen HG, Wen L, Edlind MP, Carroll PR, Kim W, Ruggero D. Cell type-specific abundance of 4EBP1 primes prostate cancer sensitivity or resistance to PI3K pathway inhibitors. Sci Signal. 2015;8:ra116. doi: 10.1126/scisignal.aad5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber WE, Price ER, Widlund HR, Du J, Davis IJ, Wegner M, Fisher DE. A tissue-restricted cAMP transcriptional response: SOX10 modulates alpha-melanocyte-stimulating hormone-triggered expression of microphthalmia-associated transcription factor in melanocytes. J Biol Chem. 2003;278:45224–30. doi: 10.1074/jbc.M309036200. [DOI] [PubMed] [Google Scholar]
- Jacks T, Jaffee E, Singer D. Cancer Moonshot Blue Ribbon Panel Report 2016 2016 [Google Scholar]
- Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, Yang Y, Chien CD, Seif AE, Lei H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7:12320. doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James ND, Bono JS, de Spears MR, Clarke NW, Mason MD, Dearnaley DP, Ritchie AWSW, Amos CL, Gilson C, Jones RJ, et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med. 2017;377:338–351. doi: 10.1056/NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM, Baker AC, Nazarian RM, Vijayendran KG, Sellers WR, et al. An oncogenic role for ETV1 in melanoma. Cancer Res. 2010;70:2075–84. doi: 10.1158/0008-5472.CAN-09-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–42. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518:240–4. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaled M, Levy C, Fisher DE. Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit. Genes Dev. 2010;24:2276–81. doi: 10.1101/gad.1937710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann Oncol. 2016;27:1492–504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- Kim HR, Kim WS, Choi YJ, Choi CM, Rho JK, Lee JC. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol Oncol. 2013;7:1093–102. doi: 10.1016/j.molonc.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–70. doi: 10.1038/ng.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Ahronian LG, Siravegna G, Mussolin B, Godfrey JT, Clark JW, Blaszkowsky LS, Ryan DP, Lennerz JK, Iafrate AJ, et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015;5:1271–81. doi: 10.1158/2159-8290.CD-15-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest. 2015;125:1459–70. doi: 10.1172/JCI78954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, Castel P, Scaltriti M, Baselga J, Garraway LA. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov. 2016;6:1134–1147. doi: 10.1158/2159-8290.CD-16-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–49. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Aguilera M, Codony-Servat J, Reig O, Lozano JJ, Fernandez PL, Pereira MVV, Jimenez N, Donovan M, Puig P, Mengual L, et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol Cancer Ther. 2014;13:1270–84. doi: 10.1158/1535-7163.MCT-13-0775. [DOI] [PubMed] [Google Scholar]
- Menzies AM, Haydu LE, Carlino MS, Azer MW, Carr PJ, Kefford RF, Long GV. Inter- and intra-patient heterogeneity of response and progression to targeted therapy in metastatic melanoma. PLoS ONE. 2014;9:e85004. doi: 10.1371/journal.pone.0085004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F, Mills GB. Overcoming implementation challenges of personalized cancer therapy. Nat Rev Clin Oncol. 2012;9:542–8. doi: 10.1038/nrclinonc.2012.127. [DOI] [PubMed] [Google Scholar]
- Minckwitz G, von Procter M, Azambuja E, de Zardavas D, Benyunes M, Viale G, Suter T, Arahmani A, Rouchet N, Clark E, et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med. 2017;377:122–131. doi: 10.1056/NEJMoa1703643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M, Smas ME, Lord JB, Brannigan BW, Trautwein J, et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2012;2:995–1003. doi: 10.1158/2159-8290.CD-12-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351–6. doi: 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TS, Wu Y-L-L, Ahn M-J-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, Fagin JA. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–33. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CCC, Wongvipat J, Ku SYY, Gao D, Cao Z, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes Foppen MH, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. 2014;5:5712. doi: 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MKK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolantonio F, Di Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, Dosso S, De Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6:re6. doi: 10.1126/scisignal.2004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–15. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo D, Sennott EM, Barault L, Valtorta E, Arena S, Cassingena A, Filiciotto G, Marzolla G, Elez E, Geel RM, van JM, et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016;76:4504–15. doi: 10.1158/0008-5472.CAN-16-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165–72. doi: 10.1016/S1470-2045(14)71180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. Interferon-γ Drives Treg Fragility to Promote Antitumor Immunity. Cell. 2017;169:1130–1141. e11. doi: 10.1016/j.cell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Davis M, Doroshow JH, Kummar S. Safety and feasibility of targeted agent combinations in solid tumours. Nat Rev Clin Oncol. 2013;10:154–68. doi: 10.1038/nrclinonc.2012.245. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman EL, Kalsy A, et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015;5:713–22. doi: 10.1158/2159-8290.CD-15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009;106:4519–24. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ER, Horstmann MA, Wells AG, Weilbaecher KN, Takemoto CM, Landis MW, Fisher DE. alpha-Melanocyte-stimulating hormone signaling regulates expression of microphthalmia, a gene deficient in Waardenburg syndrome. J Biol Chem. 1998;273:33042–7. doi: 10.1074/jbc.273.49.33042. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–6. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- Riely GJ, Kris MG, Zhao B, Akhurst T, Milton DT, Moore E, Tyson L, Pao W, Rizvi NA, Schwartz LH, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13:5150–5. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–77. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Wu Y-MM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, Salvi S, Amadori D, Zafeiriou Z, Rescigno P, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7:312re10. doi: 10.1126/scitranslmed.aac9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano E, Pradervand S, Paillusson A, Weber J, Harshman K, Muehlethaler K, Speiser D, Peters S, Rimoldi D, Michielin O. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res. 2013;19:5749–57. doi: 10.1158/1078-0432.CCR-13-0661. [DOI] [PubMed] [Google Scholar]
- Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, Mussolin B, Kwak EL, Buscarino M, Lazzari L, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016;6:147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, Butler T, Thompson JF, Mann GJ, Haydu LE, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci USA. 2015;112:10995–1000. doi: 10.1073/pnas.1508074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, Luo J, Haffner MC, Yegnasubramanian S, Carducci MA, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med. 2015;7:269ra2. doi: 10.1126/scitranslmed.3010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghers ACC, Wilgenhof S, Lebbe C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22:466–72. doi: 10.1097/CMR.0b013e3283541541. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Wang P, Wongvipat J, Karthaus WR, Abida W, Armenia J, Rockowitz S, Drier Y, Bernstein BE, Long HW, et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife. 2017:6. doi: 10.7554/eLife.27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Moriceau G, Kong X, Lee MKK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–95. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SM, Baselga J, Kim S-BB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J-MM, Schneeweiss A, Heeson S, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–34. doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–33. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap WD, Gong KWW, Dering J, Tseng Y, Ginther C, Pauletti G, Glaspy JA, Essner R, Bollag G, Hirth P, et al. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia. 2010;12:637–49. doi: 10.1593/neo.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–2. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titz B, Lomova A, Le A, Hugo W, Kong X, Ten Hoeve J, Friedman M, Shi H, Moriceau G, Song C, et al. JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. Cell Discov. 2016;2:16028. doi: 10.1038/celldisc.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Eynde BJ, Van den J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525:261–4. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen E, Treacy D, Frederick D, Cooper Z, Taylor-Weiner A, Rosenberg M, Goetz E, Sullivan R, Farlow D, et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discovery. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, Sicinska E, Sulahian R, Goodale A, Cowley GS, et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017;18:1543–1557. doi: 10.1016/j.celrep.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker S, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley G, Schadendorf D, Root D, Garraway L. A Genome-Scale RNA Interference Screen Implicates NF1 Loss in Resistance to RAF Inhibition. Cancer Discovery. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, Corsello SM, Capelletti M, Calles A, Butaney M, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27:397–408. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon R-AA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KC, Konieczkowski DJ, Johannessen CM, Boehm JS, Tamayo P, Botvinnik OB, Mesirov JP, Hahn WC, Root DE, Garraway LA, et al. MicroSCALE screening reveals genetic modifiers of therapeutic response in melanoma. Sci Signal. 2012;5:rs4. doi: 10.1126/scisignal.2002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach JA, Furman RR, Liu T-MM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543:733–737. doi: 10.1038/nature21702. [DOI] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31:1592–605. doi: 10.1200/JCO.2011.37.6418. [DOI] [PubMed] [Google Scholar]
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Loo P, Van Aas T, Alexandrov LB, Larsimont D, Davies H, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–9. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Feige E, Poling LL, Levy C, Widlund HR, Khaled M, Kung AL, Fisher DE. Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment Cell Melanoma Res. 2008;21:457–63. doi: 10.1111/j.1755-148X.2008.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFRdirected therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–7. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipser MC, Eichhoff OM, Widmer DS, Schlegel NC, Schoenewolf NL, Stuart D, Liu W, Gardner H, Smith PD, Nuciforo P, et al. A proliferative melanoma cell phenotype is responsive to RAF/MEK inhibition independent of BRAF mutation status. Pigment Cell Melanoma Res. 2011;24:326–33. doi: 10.1111/j.1755-148X.2010.00823.x. [DOI] [PubMed] [Google Scholar]
- Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014a;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014b;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]