

Summary
Liver disease remains a leading cause of mortality worldwide despite recent successes in the viral hepatitis field because increases in alcohol consumption and obesity are fueling epidemics of chronic fatty liver disease for which there are currently no effective medical therapies. About 20% of individuals with chronic liver injury ultimately develop end-stage liver disease due to cirrhosis and hence, treatments to prevent and reverse cirrhosis in individuals with ongoing liver injury are desperately needed. Success requires improved understanding of mechanisms that control liver disease progression. The liver responds to diverse insults with a conserved wound healing response, suggesting that it might be generally beneficial to optimize pathways that are crucial for effective liver repair. The Hedgehog pathway has emerged as a potential target based on compelling preclinical and clinical data which demonstrate that it critically regulates the liver's response to injury. This review will summarize evidence about the Hedgehog pathway's role in liver disease and discuss how modulating pathway activity might be applied to improve liver disease outcomes.
Keywords: Hedgehog pathway, liver disease, wound-healing response
Introduction
Liver disease is one of the leading causes of death worldwide1. Death from any type of acute or chronic liver injury results when sufficient healthy hepatic parenchyma cannot be regenerated to perform vital liver-specific functions. Although the regenerative capability of adult liver is legendary, 10-20% of individuals exhibit defective regenerative responses that progressively replace functional liver tissue with scar, placing them at risk for cirrhosis and primary liver cancer when confronted with chronic liver injury. The prognosis of cirrhosis is worse than that of many malignancies given that survival is merely 2-4 years once evidence of liver dysfunction becomes overt2. Further, after cirrhosis has developed, reversing it is an extremely difficult and lengthy process, even after eliminating the underlying cause of liver injury3. These dismal statistics underscore the importance of developing effective strategies to prevent and treat cirrhosis. Cirrhosis is an extreme consequence of recurrent futile efforts to repair liver injury. The liver has robust regenerative capability and typically responds to injury with precisely-coordinated wound healing responses that persist until healthy hepatic parenchyma is completely restored. Injured liver epithelial cells trigger wound healing by releasing signals that mobilize various types of cells that must collaborate to reconstruct healthy hepatic parenchyma. Immune cells are recruited to combat invading pathogens, remove dying epithelial cells, and help activate resident hepatic stellate cells and sinusoidal endothelial cells. These sinusoidal cells, in turn, generate signals that stimulate vasculogenesis and matrix remodeling. Together with epithelial and immune cell-derived factors, the changes in blood flow and matrix composition promote the viability of surviving epithelial cells while nurturing the outgrowth and eventual differentiation of progenitors needed to repopulate the damaged parenchyma. During these wound-healing responses the accumulation of immune cells, activated endothelial cells, myofibroblasts, and progenitors plus the resultant matrix and vascular remodeling (a.k.a., scarring) disrupt the normal hepatic architecture. However, as healthy hepatic parenchyma is regenerated, the signals driving repair dissipate, wound healing responses subside, and scarring gradually regresses. Wound healing “stalls” during the phase of active scarring when liver injury is recurrent or when the mechanisms that orchestrate the wound healing process itself become dysregulated. In order to optimize effective recovery from liver injury, hepatologists need to better understand how to regulate the signals that control liver repair.
Growing evidence indicates that the Hedgehog (Hh) pathway is a critical regulator of adult liver repair and hence, a potential diagnostic and/or therapeutic target in cirrhosis. Although the Nobel Prize laureates Wieschaus and Nussland-Volhart described the Hh pathway in 19804, its importance in dictating liver injury outcomes has emerged much more recently. Hh is a classic morphogen, i.e., it is secreted by ligand producing-cells, diffuses into the extracellular space and determines the fate of Hh-responsive target-cells according to its concentration and the duration of exposure5. Hh is crucial for embryogenesis and its name is based on evidence that genetic disruption of Hh production induces a spiculated appearance in fly larvae, causing them to resemble the homonymous mammal4. The first evidence that Hh might be involved in liver disease dates from the 2001 description of Hh pathway transcripts in a liver microarray analysis of liver samples from patients with cholangiopathies6. Since then, extensive evidence demonstrates that Hh pathway induction not merely associates with, but actually controls, liver disease progression, identifying this pathway as a potential therapeutic target to optimize liver repair. This review summarizes the preclinical and clinical data which show that Hh signaling regulates liver disease progression and discusses potential translational applications of this new knowledge.
The Hedgehog Signaling Pathway
The canonical Hh pathway is a conserved, highly complex signaling cascade, with many players and intricate regulation. However, it can be simplified into four fundamental components: i) the ligand Hedgehog, ii) the receptor Patched (Patch), iii) the signal transducer Smoothened (Smo), and iv) the effector transcription factor, Gli (Figure 1). Canonical Hh signaling occurs along a highly specialized organelle, the primary cilium7. Primary cilia (PC) are tubulin-polymerized immotile cilia that assemble from the centriole at the end of mitosis. Components of the Hh pathway concentrate in PC8,9 and a complex PC trafficking system regulates the interaction of Hh pathway components to enhance, or block, the Hh-initiated signal10,11.
Figure 1. The simplified representation of the Hedgehog signaling pathway.
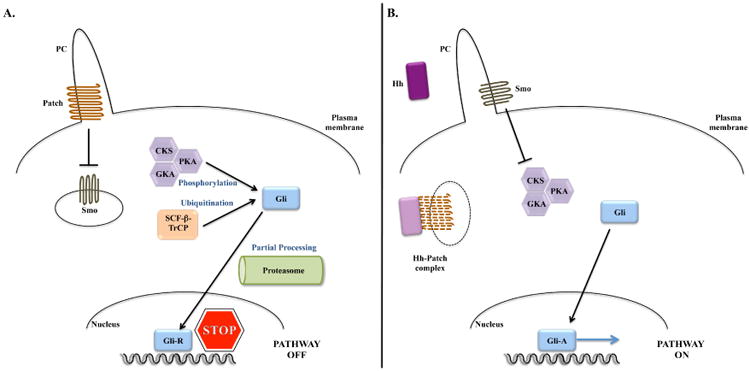
A. In the absence of Hedgehog ligand (Hh), Patched (Patch) prevents Smoothened (Smo) from entering the primary cilium (PC), repressing Smo activity. This allows the sequential phosphorylation of Gli by several kinases: protein kinase A (PKA), glycogen synthase-3β(GSK3β) and casein kinase-1 (CK1). Phosphorylated Gli is susceptible for ubiquitination by Skip-Cullin-F-box (SCF) protein/β-Transducing repeat Containing Protein (TrCP), which primes Gli to limited degradation in the proteasome. Truncated Gli (Gli-R) acts as a repressor of gene transcription.
B. When hedgehog binds to Patch, it removes Patch from the PC, allowing Smo to enter the PC. The complex Hh-Patch is degraded in vesicles in the cytoplasm. The entry of Smo into the PC allows Smo activation. Active Smo abrogates phosphorylation and subsequent degradation of Gli. Full length Gli translocates to the nucleus where it acts as a transcription factor for several target genes.
Of note, Shh, Ihh and Dhh ligands similarly activate the Hh pathway. Gli-1 does not undergo proteasomal degradation, and in the absence of ligand, Gli-2 is preferentially completely degraded in the proteasome while Gli-3 is partially degraded, and hence Gli-1 and Gli-2 act mostly as transcription promoters and Gli-3 can act as a transcription repressor.
Hh is a protein produced as a 45-kDa precursor that undergoes proteolytic processing in the endoplasmic reticulum12 and subsequent lipid modification to acquire cholesterol and palmitoyl groups13,14. Hh is secreted into the extracellular space, diffusing away from the ligand-producing cell to bind to other cells whereby it determines their fate according to the concentration and duration of exposure5. Extracellular matrix proteins, such as proteoglycans, modulate the diffusion of Hh through the extracellular space and thus, regulate the concentration of Hh to which target cells are exposed15,16. Mammals have 3 different Hh proteins: Sonic (Shh, named after the videogame personality), Indian (Ihh) and Desert (Dhh) hedgehog, (named after two mammalian hedgehog species)17. The 3 ligands similarly activate the Hh pathway in Hh-responsive cells, however their expression is differently regulated. While Shh and Ihh are widely expressed, Dhh is thought to be expressed mainly in the nervous system and testis10.
Patch, the Hh receptor, is a protein with 12 transmembrane domains. When Hh ligands are absent, Patch localizes to the PC and constitutively inhibits the Hh pathway by blocking Smo, the signal transducer protein, from entering the PC and being activated. When Hh ligand binds to Patch, it displaces Patch from the PC, allowing Smo to enter the PC and become active 9. The Hh-Patch complex is subsequently internalized and degraded18. Three Hh co-receptors, CAM-related down-regulated by oncogene (Cdo), brother of Cdo (Boc) and growth-arrest-specific (GAS)-2, 19. Conversely, Hhip, a soluble Hh receptor, inhibits Hh signaling by preventing Hh-Patch binding20.
Smo is a 7-pass transmembrane G-protein coupled receptor that mediates activation of Gli transcription factors in various types of Hh-responsive cells. Gli proteins promote transcription of several genes important in the regenerative/repair process. For example, in endothelial cells Gli promotes the transcription of vascular endothelial growth factors (VEGF), angiopoietin-1 and -2; in fibroblasts, it stimulates transcription of snail, twist-2, α-smooth muscle actin (α-SMA) and vimentin; and in progenitor cells, Gli promotes expression of nanog, sox-2 and -910,21. In the absence of Hh, Smo activity is repressed by Patch and Gli binds to fused kinase (Fu), suppressor of fused (Sufu) and Costal-2, to form a suppressor protein complex which prevents Gli from entering the nucleus22. Arrested in the cytoplasm by the suppressor protein complex, Gli is vulnerable to sequential phosphorylation by protein kinase A (PKA), glycogen synthase kinase-3 (GSK3) and calmodulin kinase-1 (CK1). Phosphorylation of Gli enables Gli binding to β-transducin repeat containing protein (βTrCp) and the Gli-βTrCp complex is targeted to the proteasome for ubiquitination. In the proteasome, Gli can be either degraded entirely or processed to generate a truncated transcription repressor (Gli-R)23,24. When Hh binds to Patch, Smo is de-repressed and activated Smo dissociates Gli from the suppressor protein complex, preventing Gli phosphorylation and subsequent degradation. Full-length Gli moves to the nucleus where it acts as a transcription factor.
Mammals have 3 known 3 Gli proteins: Gli-1, -2 and -3. Gli-1 does not undergo proteosomal degradation and hence, remains untruncated and always acts as a transcription promoter. Gli-1 is an important target gene for Gli-225. Full-length Gli-2 accumulates when Smo is activated because activated Smo protects Gli-2 from proteosomal degradation. When Smo is inactive, both Gli-2 and Gli-3 are targeted to the proteasome; Gli-2 is usually fully degraded but Gli-3 is frequently partially processed to a truncated form that acts as a transcriptional repressor26. As such, Gli-3 can act as either a transcriptional repressor (when Smo is inactive) or as an activator of transcription (when activated Smo protects it from proteosomal degradation). In contrast, Gli-1 and Gli-2 act predominantly as transcription promoters.
Besides the canonical Hh pathway, there are also two known types of non-canonical Hh signaling. Type 1 non-canonical Hh signaling depends on Patch but is Smo-independent. In the absence of Hh, Patch has direct pro-apoptotic and anti-proliferative effects, by activating caspase-327 and preventing nuclear localization of cyclin D28, respectively. Both effects of Patch are lost when Hh binds to Patch. Type 2 non-canonical Hh signaling depends on Smo but it does not require PC29. This non-canonical signaling depends on the Gαi activity of Smo that directly regulates metabolism (for example it promotes a Warburg-like effect promoting glycolysis in muscle, adipose tissue and myofibroblasts30,31), proliferation, calcium flux and migration (in myofibroblasts and endothelial cells32-34), in a Gli-independent mechanism. Additionally, Gli signaling can occur in the absence of Hh via a process that also appears to be Patch and Smo-independent, as demonstrated by evidence that Gli induction is a direct downstream consequence of transforming growth factor (TGF) beta and RAS signaling35-37.
The Hedgehog Pathway In The Liver
Hepatic Development
The role of Hh in the embryogenesis of the liver is not fully understood. Shh is strongly expressed in the ventral foregut endoderm which gives rise to the liver, pancreas and lung buds. Shh expression disappears as the liver bud forms38,39 but it is transiently induced in hepatoblasts later in development. As hepatoblasts differentiate into hepatocytes, Shh expression is reduced again38, suggesting that Shh is necessary to generate, maintain, and expand certain populations of liver progenitors, but must be inhibited for these cells to differentiate into mature liver epithelial cells.
Healthy Adult Liver
In healthy adult liver, the Hh pathway is relatively dormant due to both very low production of ligands by liver-resident cells (e.g., occasional immature-appearing cholangiocytes), and robust expression of Hh inhibitors, such as Hhip, by quiescent HSC40. Interestingly, emerging evidence suggests that this low level of pathway activity may fluctuate in a circadian fashion and help to regulate zonal differences in hepatic metabolism by modulating the relative levels of various Gli factors in hepatocytes41,42. These recent observations raise the intriguing possibility that mammalian liver may be exposed (and respond) to Hh ligands derived from extra-hepatic sources. In flies, for example, Hh ligand is produced by intestinal epithelial cells and carried in lipoproteins to the fat body (an organ with dual adipose- and liver-like functions) where it has metabolic activity43. Hh inhibits lipogenesis in both flies and mammals22,44. Hh ligands have been demonstrated in human lipoproteins45 but the source(s) of lipoprotein-associated Hh ligands and their function in man are unknown. This issue merits further study given recent evidence that inherited Smoothened defects which abrogate Hh signaling in humans lead to hepatic steatosis46.
Injured Adult Liver
Hh ligand expression is induced in liver-resident cells and robust Hh pathway activity reemerges in adults in response to situations that trigger acute liver regeneration (e.g., after an acute liver insult with hepatic necrosis or after partial hepatectomy) or chronic liver regeneration/repair (e.g., all types of chronic liver injury). Indeed, the level of Hh pathway activation generally correlates with the severity and duration of the liver injury, regardless of etiology47. The fact that Hh pathway activity closely parallels the intensity of the regenerative stimulus probably reflects the fact that the production of Hh ligands is stimulated by several factors that accumulate in injured livers, including platelet derived growth factor (PDGF), TGF-β, and epidermal growth factor (EGF)48-50. During liver wound healing, various liver-resident cell types produce Shh and/or Ihh ligands, including injured/dying hepatocytes (e.g., ballooned hepatocytes in steatohepatitis), injured/activated cholangiocytes (e.g., in ductular reactions and cholangiopathies), myofibroblastic stellate cells (e.g., during fibrogenesis), sinusoidal endothelial cells (e.g., during capillarization), and immune cells (e.g., macrophages and NKT cells during fibrogenesis)40,48,51-58. Further, local production of Hh inhibitors is reduced as hepatic stellate cells quickly suppress their production of Hhip when they are becoming myofibroblastic53,54. These reciprocal changes in local production of Hh ligands and Hh inhibitors generate a microenvironment that promotes Hh pathway activation in Hh-responsive target cells.
There has been some debate about which types of liver cells might be able to activate canonical Hh signaling during liver injury. These uncertainties reflect technical challenges imposed by imperfect and inconsistent reagent specificity and the nature of the signaling process itself, which is quite dynamic and regulated at multiple levels. These challenges are compounded by the fact that PC are thought be necessary for cells to activate canonical Hh signaling in response to Hh ligand exposure. Cells possess a single 0.25 μm diameter PC and this structure forms and regresses as cells exit and enter the cell cycle59,60, making it quite difficult to visualize PC on any given cell in intact liver tissue, even with the best available approach (i.e., confocal microscopy with antibodies to acetylated tubulin61. This task is particularly daunting during the various phases of an active wound healing response. Indeed, it is conceivable that regeneration-related changes in PC contribute to the striking differences in Hh pathway activity in healthy and diseased livers.
Current dogma posits that all healthy adult hepatocytes are devoid of PC61, and hence cannot activate the canonical Hh pathway62. This assumption merits renewed scrutiny in light of the aforementioned evidence suggesting that Hh signaling regulates hepatocyte metabolism during health42,63. In addition, other reports in patients and animal models with liver disease have demonstrated Gli2 nuclear staining in periportal hepatocytes47,62,64-68. This suggests that some hepatocytes may be able to acquire PC and become responsive to Hh, since the activation of the transcription factor Gli2 is the end result of the Hh pathway. On the other hand, Gli2 activation in such cells could be Hh- and Smo independent, since Gli2 can be activated by other signaling pathways such as TGF-β35 (which increases after fibrogenic insults) and FOXC169 (which is upregulated in hepatocellular carcinoma70). It is also possible that hepatocytes exhibit Hh-dependent, PC-independent, Smo-dependent or Smo-independent activation of Gli71,72. In any case, several groups have demonstrated Smo activity in hepatocytes65,72.
Similar to healthy mature hepatocytes, liver sinusoidal endothelial cells are not thought to express PC in general. However, endothelial cells are known to become ciliated when exposed to increased hydrostatic pressure73,74. The resultant Hh pathway activation induces a vasoconstrictive phenotype and loss of fenestration in liver sinusoidal endothelial cells, promoting angiogenesis, capillarization and vascular remodeling that contributes to portal hypertension55,56,75,76.
In contrast to hepatocytes and liver sinusoidal endothelial cells, cholangiocytes and progenitor cells in healthy livers seem to express PC fairly consistently62,77 and thus, heritable ciliopathies which alter Hedgehog signaling along PC (e.g., Bardet Biedl syndrome, adult polycystic kidney disease, Caroli's disease) exhibit an aberrant biliary phenotype78. Hh activation in bipotent liver epithelial progenitors induces proliferation, inhibits apoptosis and blocks differentiation along the mature biliary lineage38,40,62. Such dysregulated Hh signaling has been implicated in the pathogenesis of “acquired” cholangiopathies, including biliary atresia79-81 and the ductular reaction that develops during many types of liver injury49,51,82. The ductular reaction is believed to reflect accumulation of immature liver epithelial cells83. Hh pathway activation in liver progenitors expands the pools of cells available to replace epithelial cells that are dying after an acute or chronic insult. However, full recuperation of liver-specific functions requires precise modulation of Hh signaling since complete differentiation of progenitors into mature liver epithelial cells seems to require repression of pathway activity38,39.
Like cholangiocytes and progenitors, some hepatic stellate cells (HSC) in healthy adult livers also appear to have PC. This conclusion is based on evidence that HSC in healthy livers are marked by both Patch- and Gli-reporter activity in transgenic mice84-86, similar to tissue-resident perivascular mesenchymal stem cell-like cells in multiple other tissues87. Hh activation appears to promote differentiation of such cells into proliferative myofibroblasts84,88-90. The mechanisms mediating this transdifferentiation have been delineated in HSC and shown to involve a epithelial-to-mesenchymal transition-like process whereby the cells transiently repress their expression of genes that favor a more quiescent, epithelial phenotype while inducing various factors that promote the transcription of mesenchymal genes and enhance the stability/translation of the respective mRNAs to promote proliferation, survival, migration, contractility, and angiogenesis91-94. Interestingly, although ultrastructure studies of HSC and studies with confocal microscopy described PC in only 2.5% of HSC of healthy livers59,68,95, in vitro studies show that HSC are highly responsive to both Hh ligand and antibodies that neutralize Hh84,90. HSC also appear to be responsive to Hh ligands in vivo as indicated by evidence that the pool of hepatic myofibroblasts expands in parallel with Hh ligand accumulation following various liver insults, whereas during recovery, a decline in Hh associates with involution of that pool of cells49. Further, in animal models neutralizing antibodies to Hh ligands promote myofibroblast inactivation, apoptosis and senescence96. Treatments that reduced injury-related production of Hh ligands by hepatocytes also caused regression of myofibroblast populations in patients97. Given evidence that HSC are highly responsive to direct antagonists and agonists of Smo in vivo and in vitro84,91, and reports that direct manipulation of Gli factors also regulates HSC fate98. It is conceivable that various canonical (i.e., PC-dependent) and noncanonical (i.e., PC-independent) pathways interact to modulate Hh signaling in HSC99.
Lastly, certain populations of immune cells may be Hh responsive. Although PC have not been demonstrated in healthy liver-resident macrophages or lymphocytes, macrophages in injured livers have been shown to produce Hh ligands58,100,101 and treating liver-derived macrophages with neutralizing antibodies to Hh ligands inhibits them from becoming M2 polarized in vitro58,101. Similarly, NKT cells (the predominant liver lymphocyte sub-population in healthy adult livers) are highly responsive to Hh, which promotes NKT cell viability, proliferation and skews their differentiation towards a phenotype that enhances both immune tolerance and fibrogenesis57,102.
In summary, while Hh signaling is generally relatively dormant in the healthy adult liver, the pathway becomes very active when the liver is injured and orchestrates a dialogue between different cell types to assure an effectively integrated repair response. After the insult subsides, the Hh pathway must be shut-down so that the liver can recover its mature structure and function.
Hedgehog In Liver Regeneration
A growing body of evidence demonstrates that activation of the Hh pathway is crucial for liver regeneration. The best-studied animal model for evaluating acute regeneration is surgical removal of 70% of the healthy liver (partial hepatectomy, PH) in adult rodents. Hh ligand expression increases transiently but significantly following PH in rodents65. Further, inhibiting Hh pathway induction with a direct pharmacologic antagonist of Smo decreased both recovery of liver mass and overall survival54,65. Interestingly, similar results were found when a targeted molecular approach was used to inhibit Smo and block Hh pathway activity in liver pericytes54, identifying Hh-responsive HSC as central players in liver regeneration.
Patients undergoing extensive liver resection to de-bulk metastatic cancer are at risk of liver failure due to massive loss of functional hepatic mass. To prevent this potentially-fatal outcome, a two-stage hepatectomy is often performed, with the first step being segmental portal vein ligation, followed by a partial hepatectomy (PH) to stimulate compensatory growth of the non-occluded liver section. To expedite the regenerative response, a modified two-step approach (dubbed, associating liver partition and portal vein ligation for staged hepatectomy, ALPPS) has been developed in which transection along the demarcation between occluded and non-occluded liver replaces the PH step. Compensatory liver growth is much faster after ALPSS than portal vein ligation alone, and it was recently reported that Ihh is massively up-regulated in the liver and blood of patients and mice subjected to ALPPS103. Interestingly, mice subjected to portal vein ligation with simultaneous administration of systemic Ihh, performed as well as mice submitted to ALPPS103, supporting the other preclinical evidence that Hh signaling plays a major role in promoting acute liver regeneration. Consistent with this concept, not only does the production of Hh ligands increase after PH, but the bioavailability of those ligands also changes. In the extracellular matrix of the healthy adult liver, the proteoglycan glypican-3 binds normally to Ihh to prevent Ihh from binding to Patch in order to constrain activation of the Hh pathway. After PH, the binding of Ihh to glypican-3 dramatically decreases, returning back to its baseline levels only when the liver recovers its initial size67. Thus, the bioavailability of Hh ligands increases rapidly and remains elevated for a period of time following PH, and this is accompanied by striking changes in hepatic Hh pathway activity. The kinetics of this process have been mapped after PH in rodents54,65,104.
Briefly, hepatic expression of Shh and Ihh ligands and Gli1/Gli2 transcription factors (the down-stream effectors of the canonical Hh signaling pathway) increases transiently after PH, with peak Hh pathway activity corresponding to the period of active hepatocyte replication. Interestingly, Ihh induction seems to occur slightly before, and slightly after, hepatocyte DNA proliferative activity peaks, while maximal Shh expression coincides with the time window during which hepatocyte replication and accumulation of α-SMA producing myofibroblasts are maximal65. Sinusoidal lining cells, particularly activated endothelial cells and inflammatory cells, appear to be major sources of Hh ligands, but hepatocytes isolated from mice after PH also produce these factors65, suggesting that PH may evoke transient stress and/or de-differentiation in residual mature hepatocytes105. While more research is necessary to clarify the latter issue, available data indicate that Smo-dependent nuclear accumulation of Gli-2 protein occurs initially in hepatocytes after PH, followed 24 hours later by nuclear accumulation of Gli-2 in cells that co-express progenitor markers65. Further, conditional disruption of Smo in α-SMA-expressing cells revealed that Hh pathway activity in myofibroblastic cells is required for post-PH matrix remodeling, progenitor cell expansion, and proliferative responses in hepatocytes and ductular cells54. Thus, multiple converging lines of evidence demonstrate that coordinated transient activation of the Hh pathway is critically important to re-construct healthy liver tissue following partial liver resection54. One appealing translation of this knowledge to improve current clinical practice might be to treat patients with Hh stimulants (for example recombinant Ihh) before extended hepatectomy or after transplantation of small-for-size liver grafts to optimize liver regeneration and avoid post-surgical liver failure. However, because maturation of liver epithelial cells is inhibited when Hh pathway activity is high, further study is essential to define safe doses and durations of pathway activation in these contexts.
Hedgehog In Chronic Liver Disease
The liver responds to different chronic insults with a highly conserved wound healing response during which different cell types must communicate to re-construct fully functional, healthy liver parenchyma. The inter-cellular dialogue that orchestrates effective liver repair is accomplished by diverse factors (e.g., cytokines, growth factors, and morphogens, such as Hh ligands) that interact to appropriately modulate the fates of surviving liver cells. Cumulative data from humans and animal models indicates that Hh signaling is a pivotal regulator of the wound healing response in chronic liver disease. This discovery identifies the Hh pathway as a common target for therapeutic manipulation, regardless of the primary insult perpetuating liver injury.
Diverse insults (e.g., alcohol, toxic drugs, metabolically active fat, autoimmune attack, viral and parasitic infections) induce stress and cell death in liver epithelial cells (hepatocytes or cholangiocytes). The injured/dying cells generate signals to recruit help and promote healing, including alarmins, damage-associated pattern molecules (DAMPs), cytokines, and morphogens. Because mortally-wounded liver epithelial cells cannot replicate, their alarm signals are configured to elicit a proliferative response in residual cells that survived, including less mature epithelial cells that can be incited to differentiate into replacements for the dead mature epithelia once the microenvironment becomes less noxious. The cellular source(s) of such immature liver epithelial cells has become a matter for debate, but morphologically the process is identified as a ductular reaction, i.e., the accumulation of immature-appearing ductal cells that aggregate to form nascent duct-like structures embedded in variable amounts of fibrous matrix with accompanying stromal cells (e.g., small oval-shaped cells with a high nuclear:cytoplasmic ratio, immune cells, activated endothelial cells and myofibroblasts)83,106.
The cells in the ductular reaction have multiple duties that are essential for effective tissue repair, including nurturing progenitors to replace the dying epithelia, clearing dead cell debris, pathogen defense, vasculogenesis to optimize blood flow, and matrix remodeling to provide an appropriate scaffold for tissue reconstruction. Thus, it is intriguing that the ductular reaction and active scar tissue have many features in common given that the former is believed to be a potentially beneficial response, while the latter is thought to reflect failed regeneration. This apparent paradox might be reconciled if future research confirms our hypothesis that the microenvironment of the ductular reaction is dynamic and when optimally regulated, engenders an incubator-like niche that initially promotes the generation and expansion of epithelial precursors and then gradually morphs to instruct their appropriate differentiation into mature epithelial replacements. Viewed from this perspective, progressive scarring that leads to cirrhosis might simply reflect futile/stalled regeneration, i.e., inability to move beyond the initial phases of the wound healing response. Arrest during early wound healing might be appropriate if the liver is repetitively re-challenged with noxious insults (as occurs during metabolic liver disease or chronic viral infection). Alternatively, it could occur despite resolution of the initial insult if key mechanisms that control the normal evolution of the ductular reaction become dysregulated due to inherited traits or environmental factors.
Dysregulated wound healing itself promotes progressive tissue damage: persistent accumulation of myofibroblasts causes progressive liver fibrosis, ultimately resulting in cirrhosis; persistent accumulation of inflammatory immune cells perpetuates liver injury, leading to chronic hepatitis; and persistent expansion of progenitor cells and their arrest in immature forms favors carcinogenesis. The inability to shut down the wound healing response appropriately seems to more related to patient characteristics (e.g., inheritance, age, gender, environmental exposures) that convey susceptibility to defective tissue repair107 than to the specific etiology of the liver disease itself given that very different insults (e.g., alcoholic and nonalcoholic fatty liver disease108,109, chronic viral hepatitis110, schistosomiasis111) result in progressive damage and end-organ failure in a similar proportion of afflicted individuals.
The first suggestion that the Hh pathway might be involved in the pathogenesis of chronic liver disease was reported at the beginning of this millennium by Schakel et al who noted that Patch and Gli were upregulated in their microarray analysis of liver tissues from patients with primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC)6. Since that initial description, the Hh pathway has consistently been shown to be aberrantly activated in different human cholangiopathies and animal models of these diseases. For example, immunohistochemistry studies have demonstrated increased Hh pathway activity in human PBC and PSC relative to healthy age-matched controls68,112,113, with the main sources of Hh ligands being ductular cells in PBC and peribiliary gland progenitor cells in large bile ducts of patients with PSC112,113. Progenitor cells and myofibroblasts are the main Hh-responsive cell types in both diseases112,113. Importantly, the level of Hh activation strongly correlated with the degree of liver fibrosis in both PBC and PSC. Furthermore, genome wide association studies showed that genetic variants in the Hh pathway confer susceptibility for PBC114. In pediatric cholestatic diseases such as biliary atresia, Alagille's syndrome and progressive familial intrahepatic cholestasis, there is known activation of the Hh pathway. Further, high levels of Hh pathway activity correlate with dismal prognosis79,115. In pediatric cholangiopathies, the Hh pathway not only promotes fibrogenesis, but also induces epithelial to mesenchymal transition in biliary progenitors, which arrests them in an immature phenotype and abrogates ductular morphogenesis51,79. Studies in animal models of cholangiopathies not only replicated the evidence for excessive activation of the Hh pathway that was noted in the human diseases, but demonstrated a causal association. For example, a zebrafish model of biliary atresia caused by genetic deletion of glypican-1 could be reverted to a normal phenotype by administering a Hh inhibitor and replicated in wild type zebrafish embryos simply by administering recombinant Shh80. Rodent models of chronic bile duct ligation provides proof-of-concept that Hh signaling is also critical in the pathogenesis of cholangiopathies that are induced in adulthood. Bile duct ligation (BDL) in rats induces an exuberant fibroductular response that reproducibly culminates in biliary cirrhosis with accompanying high levels of Hh ligand production, profound downregulation of the Hh inhibitor Hhip, and striking nuclear accumulation of Gli, a Hh-regulated transcription factor. This liver phenotype can be reverted by submitting BDL rats to Roux-en-Y hepaticojejunostomy, which decompresses the obstructed biliary tree. Such biliary diversion progressively silences the Hh pathway, causes regression of biliary fibrosis, and ultimately normalizes the hepatic architecture 48,49,51. Inhibiting the Hh pathway, either pharmacologically or through conditional disruption of the Smo gene abrogates liver injury and fibrosis in BDL mice53,116. The reverse is also true, that is, genetically modified mice with an overactive Hh pathway demonstrate a more exuberant fibroductular response to BDL48.
Extensive data also demonstrate a role of the Hh pathway in the pathogenesis of alcoholic and nonalcoholic fatty liver disease. Studies of patients with alcoholic/nonalcoholic fatty liver disease consistently show that the level of Hh pathway activation correlates with the severity of liver cell injury/death, hepatic inflammation, liver fibrosis, and the risk for worse liver-related morbidity and mortality50,64,66,68,83,117. Various animal models of nonalcoholic fatty liver disease (NAFLD) have confirmed that the level of Hh pathway activity increases in parallel with the severity of steatohepatitis and liver fibrosis47,64,118-120. In contrast, the Hh pathway seems to protect the liver from steatosis in several preclinical NAFLD models. For example, obese ob/ob mice develop massive steatosis with age but are relatively protected from steatohepatitis and progressive liver fibrosis. This ob/ob phenotype results from monogenic deficiency of leptin; leptin has been proven to stimulate Hh signaling121; and Hh signaling is severely reduced in ob/ob mice96. Interestingly, a recent report indicates that the prevalence of NAFLD is higher in patients with germline mutations disrupting Smo than in the general population. This enrichment appears to be independent of obesity and seems to be driven predominately by the fact that reduced Smo function associates with hepatic steatosis. Indeed, expression of proinflammatory and profibrotic genes is generally low in such patients46. Similarly, genetically modified mice with reduced Hh pathway activity caused by global haploinsufficiency of Gli-2 develop more steatosis but less inflammation than wild type mice when exposed to steatogenic diets46. These findings in the livers of humans and mice with genetic inhibition of the Hh pathway are consistent with earlier publications proving that genetic activation of the Hh pathway directly inhibits lipogenesis in flies and mammals22,44, as well as more recent papers showing that targeted genetic/pharmacological activation of Hh signaling reverses steatosis in fatty hepatocytes harvested from obesity-related mice models of NAFLD42. Evidence linking reduced Hh activity with hepatic steatosis and excessive Hh signaling with steatohepatitis and fibrosis continues to accumulate as results of Hh pathway manipulation in additional animal models of NAFLD are published53,57,64,118,122-124.
Though studied less extensively than in cholangiopathies and fatty liver diseases, the Hh pathway is active in other forms of human liver disease, such as Schistosomiasis58,101 and chronic viral hepatitis100,125, and in several animal models of liver disease, including drug-induced liver injury62,126, radiotherapy-induced liver injury127,128 and liver injury caused by ischemia-reperfusion129. The aggregate data identify the Hh signaling pathway as a promising therapeutic target to prevent various types of fibrogenic liver disease. However, much more must to be learned before the available information can be applied in a clinical setting. For example, it will be important to clarify if Hh pathway inhibition is safe in patients with ongoing liver injury since Hh signaling is crucial for liver regeneration. Improved understanding of the relative contributions of canonical versus non-canonical activation of the Hh pathway in liver disease pathogenesis may also help to guide strategies to modulate Hh pathway activity during chronic liver disease. This is particularly important because the Hh pathway is highly regulated and involves both self-inhibitory and self-enhancing loops. Hence non-canonical signaling might not be blocked if the pathway is inhibited too far upstream. Conversely, blocking terminal activation of pathway targets is likely to have multiple off-target consequences because this approach would entirely abrogate much of the regulation that controls signaling initiated via either pathway.
Hedgehog In Liver Cancer
The Hh pathway has been implicated in the pathogenesis of different liver cancers, namely hepatocellular carcinoma, cholangiocarcinoma34,130-134, infantile hepatoblastoma135,136 and gallbladder cancer137-141. The most extensively studied primary liver cancer is hepatocellular carcinoma. Different hepatoma cell lines demonstrate constitutive activation of the Hh pathway, with upregulation of the expression of several ligands and effector proteins and downregulation of the Hh inhibitor Hhip142-144. A frequent mechanism of Hhip downregulation in hepatoma cell lines is the hypermethylation of its promoter145. Hepatoma cell lines with higher upregulation of the Hh pathway tend to be more undifferentiated, with a more mesenchymal and invasive phenotype, and resistant to chemo and radiotherapy146,147. Furthermore, manipulation of the Hh pathway in hepatoma cell lines confirms that Hh inhibits apoptosis, promoting viability, proliferation, migration and invasiveness142-144,148-150.
Hh signaling activation has been consistently described in different animal models of hepatocellular carcinoma, such as chronic alcohol feeding, MDR2 deficient mice with spontaneous fibrotic cholangiopathy and hepatocellular carcinoma, and xenograft models of primary liver cancer151-154. Genetically inducing activation of Hh in only 2-5% of hepatocytes was able to enhance oncogene-induced hepatocarcinogenesis in mice155. Importantly, pharmacological treatment with Smo inhibitors was able to decrease tumor size, angiogenesis and metastasis, as well as increase radiosensitivity, in those models152-154,156,157. Studies in patients with hepatocellular carcinoma showed Hh activation in more than half the cases and higher levels of pathway activity tend to associate with higher tumor burden, invasion, metastatic disease, chemoresistance and worse prognosis with decreased overall survival and recurrence after liver transplant142,143,147,158-161. Phase I studies on Smo inhibitors are currently ongoing (NCT0215864,162). Smo inhibitors have been successfully used to treat other solid cancers such as basal cell carcinoma. However, the acquired resistance due to de novo mutations as well as the non-canonical Smo-independent Hh activation has generally challenged the treatment of malignancies with Smo inhibitors. Further, it is possible that Hh targets might be activated down-stream of Smo in at least some HCC given the recent discovery of a chromosomal translocation that constitutively activates Gli-1 in a subset of hepatic adenomas163.
Conclusion
Liver disease is a major cause of morbidity and mortality worldwide, reflecting the desperate need for effective treatments to prevent cirrhosis and primary liver cancers, the leading causes of fatal chronic liver disease. The liver responds to different insults with a similar wound-healing response. When this repair process cannot be shut down, scarring and neoplasia result instead of healing, increasing the risk for cirrhosis and primary liver cancer. This insight might be exploited in the future to develop novel therapies to prevent and treat cirrhosis and liver cancer. Therefore, growing evidence that the Hedgehog pathway critically regulates various facets of the wound healing response is particularly exciting because it identifies this morphogenic signaling pathway as a potential therapeutic target to prevent bad outcomes of liver injury. Further research is necessary to clarify how Hedgehog pathway activity might be safely manipulated to optimize effective regeneration of injured liver to thwart the evolution of liver cirrhosis and primary liver cancer. This is an exciting field of research that gives liver disease patients and their doctors new hope for a brighter future.
Figure 2. Summary of the hedgehog pathway in the different hepatic cell types.
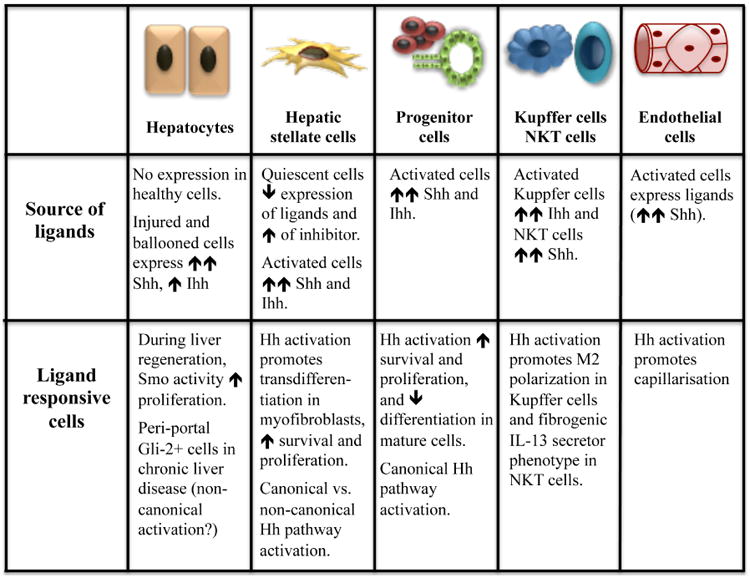
Table 1.
The hedgehog pathway in liver disease.
| Liver Regeneration | Chronic liver disease | Liver Cancer |
|---|---|---|
|
| ||
|
|
|
Hh, hedgehog; Ihh, Indian hedgehog; ALPPS, associating liver partition and portal vein ligation for staged hepatectomy; NAFLD, nonalcoholic fatty liver disease; ALD, alcoholic liver disease; HCC, hepatocellular carcinoma.
Keypoints.
The Hedgehog pathway is a complex and tightly-regulated signal transduction pathway that can be simplified into 4 main components: the ligand Hedgehog, the receptor Patched, the signal transducer Smoothened, and the effector transcription factor, Gli.
Hedgehog is a morphogen that is crucial for embryogenesis. In healthy adult liver, the Hedgehog pathway is dormant, but it reemerges after liver injury and is pivotal in orchestrating liver repair responses.
The Hedgehog pathway is critical for liver regeneration but if persistently activated, Hedgehog signaling induces misrepair and scarring that promote cirrhosis and hepatocellular carcinoma.
Upregulation of the Hedgehog pathway has been demonstrated consistently in chronic liver disease and liver cancer in humans, as well as in animal models. In all species examined to date, the level of pathway activity generally correlates with the severity of liver disease.
Preclinical studies indicate that pharmacological manipulation of the Hedgehog pathway has therapeutic potential in liver disease.
Abbreviations
- ALPPS
associating liver partition and portal vein ligation for staged hepatectomy
- Boc
brother of Cdo
- βTrCp
β-transducin repeat containing protein
- Cdo
CAM-related down-regulated by oncogene
- CK-1
calmodulin kinase-1
- Dhh
Desert hedgehog
- EGF
epidermal growth factor
- Fu
fused kinase
- GAS
growth-arrest-specific
- GSK3
glycogen synthase kinase-3
- HSC
hepatic stellate cells
- Hh
hedgehog
- Ihh
Indian hedgehog
- Patch
patched
- PBC
primary biliary cholangitis
- PC
primary cilia
- PDGF
platelet derived growth factor
- PKA
protein kinase A
- PSC
primary sclerosing cholangitis
- Shh
Sonic hedgehog
- SMA
smooth muscle actin
- Smo
Soothened
- Sufu
suppressor of fused
- TGF
transforming growth factor
- VEGF
vascular endothelial growth factors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mathers CD, Boerma T, Ma Fat D. Global and regional causes of death. Br Med Bull. 2009;92:7–32. doi: 10.1093/bmb/ldp028. [DOI] [PubMed] [Google Scholar]
- 2.D'Amico G, Morabito A, D'Amico M, et al. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol Int. 2017 doi: 10.1007/s12072-017-9808-z. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 3.Bedossa P. Reversibility of hepatitis B virus cirrhosis after therapy: who and why? Liver International. 2015;35(1):78–81. doi: 10.1111/liv.12710. [DOI] [PubMed] [Google Scholar]
- 4.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 5.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nature Reviews Molecular Cell Biology. 2013;14:416–29. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 6.Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–76. doi: 10.1136/gut.49.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bangs F, Anderson KV. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb Perspect Biol. 2017;9 doi: 10.1101/cshperspect.a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy S. Cilia and Hedgehog: when and how was their marriage solemnized? Differentiation; Research in Biological Diversity. 2012;83:S43–8. doi: 10.1016/j.diff.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Ramsbottom SA, Pownall ME. Regulation of Hedgehog Signalling Inside and Outside the Cell. J Dev Biol. 2016;4:23. doi: 10.3390/jdb4030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merchant JL, Saqui-Salces M. Inhibition of Hedgehog signaling in the gastrointestinal tract: targeting the cancer microenvironment. Cancer Treatment Reviews. 2014;40:12–21. doi: 10.1016/j.ctrv.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Sasai N, Ma G, et al. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011;9:e1001083. doi: 10.1371/journal.pbio.1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–9. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 14.Pepinsky RB, Zeng C, Wen D, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. The Journal of Biological Chemistry. 1998;273:14037–45. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 15.Ayers KL, Gallet A, Staccini-Lavenant L, Therond PP. The long-range activity of Hedgehog is regulated in the apical extracellular space by the glypican Dally and the hydrolase Notum. Dev Cell. 2010;18:605–20. doi: 10.1016/j.devcel.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Magistri P, Leonard SY, Tang CM, Chan JC, Lee TE, Sicklick JK. The glypican 3 hepatocellular carcinoma marker regulates human hepatic stellate cells via Hedgehog signaling. The Journal of Surgical Research. 2014;187:377–85. doi: 10.1016/j.jss.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Echelard Y, Epstein DJ, St-Jacques B, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–30. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 18.Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102:521–31. doi: 10.1016/s0092-8674(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 19.Izzi L, Levesque M, Morin S, et al. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–21. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- 21.Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15:24. doi: 10.1186/s12943-016-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teperino R, Aberger F, Esterbauer H, Riobo N, Pospisilik JA. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Seminars in Cell & Developmental Biology. 2014;33:81–92. doi: 10.1016/j.semcdb.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B, Li Y. Evidence for the direct involvement of {beta} TrCP in Gli3 protein processing. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:33–8. doi: 10.1073/pnas.0509927103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137:2001–9. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikram MS, Neill GW, Regl G, et al. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. The Journal of Investigative Dermatology. 2004;122:1503–9. doi: 10.1111/j.0022-202X.2004.22612.x. [DOI] [PubMed] [Google Scholar]
- 26.Pan Y, Wang B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. The Journal of Biological Chemistry. 2007;282:10846–52. doi: 10.1074/jbc.M608599200. [DOI] [PubMed] [Google Scholar]
- 27.Chinchilla P, Xiao L, Kazanietz MG, Riobo NA. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. 2010;9:570–79. doi: 10.4161/cc.9.3.10591. [DOI] [PubMed] [Google Scholar]
- 28.Barnes EA, Kong M, Ollendorff V, Donoghue DJ. Patched1 interacts with cyclin B1 to regulate cell cycle progression. The EMBO journal. 2001;20:2214–23. doi: 10.1093/emboj/20.9.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan X, Cao J, He X, et al. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nature Communications. 2016;7:11024. doi: 10.1038/ncomms11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teperino R, Amann S, Bayer M, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414–26. doi: 10.1016/j.cell.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29 e1. doi: 10.1053/j.gastro.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polizio AH, Chinchilla P, Chen X, Manning DR, Riobo NA. Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Science Signaling. 2011;4:pt7. doi: 10.1126/scisignal.2002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bijlsma MF, Borensztajn KS, Roelink H, Peppelenbosch MP, Spek CA. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cellular Signalling. 2007;19:2596–604. doi: 10.1016/j.cellsig.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Razumilava N, Gradilone SA, Smoot RL, et al. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. Journal of Hepatology. 2014;60:599–605. doi: 10.1016/j.jhep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson RW, Nguyen MP, Padalecki SS, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Research. 2011;71:822–31. doi: 10.1158/0008-5472.CAN-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dennler S, Andre J, Verrecchia F, Mauviel A. Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. The Journal of Biological Chemistry. 2009;284:31523–31. doi: 10.1074/jbc.M109.059964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nolan-Stevaux O, Lau J, Truitt ML, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes & Development. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirose Y, Itoh T, Miyajima A. Hedgehog signal activation coordinates proliferation and differentiation of fetal liver progenitor cells. Experimental Cell Research. 2009;315:2648–57. doi: 10.1016/j.yexcr.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 39.Zhao R, Duncan SA. Embryonic development of the liver. Hepatology. 2005;41:956–67. doi: 10.1002/hep.20691. [DOI] [PubMed] [Google Scholar]
- 40.Sicklick JK, Li YX, Melhem A, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. American Journal of Physiology Gastrointestinal and Liver Physiology. 2006;290:G859–70. doi: 10.1152/ajpgi.00456.2005. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt-Heck W, Matz-Soja M, Aleithe S, Marbach E, Guthke R, Gebhardt R. Fuzzy modeling reveals a dynamic self-sustaining network of the GLI transcription factors controlling important metabolic regulators in adult mouse hepatocytes. Mol Biosyst. 2015;11:2190–7. doi: 10.1039/c5mb00129c. [DOI] [PubMed] [Google Scholar]
- 42.Matz-Soja M, Rennert C, Schonefeld K, et al. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. Elife. 2016;5 doi: 10.7554/eLife.13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodenfels J, Lavrynenko O, Ayciriex S, et al. Production of systemically circulating Hedgehog by the intestine couples nutrition to growth and development. Genes & Development. 2014;28:2636–51. doi: 10.1101/gad.249763.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pospisilik JA, Schramek D, Schnidar H, et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–60. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 45.Panakova D, Sprong H, Marois E, Thiele C, Eaton S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature. 2005;435:58–65. doi: 10.1038/nature03504. [DOI] [PubMed] [Google Scholar]
- 46.Sacoto MJG, Martinez AF, Abe Y, et al. Human Germline Hedgehog Pathway Mutations Predispose to Fatty Liver. Journal of Hepatology. 2017;67:809–17. doi: 10.1016/j.jhep.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleig SV, Choi SS, Yang L, et al. Hepatic accumulation of Hedgehog-reactive progenitors increases with severity of fatty liver damage in mice. Laboratory Investigation. 2007;87:1227–39. doi: 10.1038/labinvest.3700689. [DOI] [PubMed] [Google Scholar]
- 48.Omenetti A, Yang L, Li YX, et al. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Laboratory Investigation. 2007;87:499–514. doi: 10.1038/labinvest.3700537. [DOI] [PubMed] [Google Scholar]
- 49.Omenetti A, Popov Y, Jung Y, et al. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut. 2008;57:1275–82. doi: 10.1136/gut.2008.148619. [DOI] [PubMed] [Google Scholar]
- 50.Jung Y, Brown KD, Witek RP, et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology. 2008;134:1532–43. doi: 10.1053/j.gastro.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. The Journal of Clinical Investigation. 2008;118:3331–42. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guy CD, Suzuki A, Zdanowicz M, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–21. doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michelotti GA, Xie G, Swiderska M, et al. Smoothened is a master regulator of adult liver repair. The Journal of Clinical Investigation. 2013;123:2380–94. doi: 10.1172/JCI66904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swiderska-Syn M, Syn WK, Xie G, et al. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut. 2014;63:1333–44. doi: 10.1136/gutjnl-2013-305962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie G, Choi SS, Syn WK, et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut. 2013;62:299–309. doi: 10.1136/gutjnl-2011-301494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xing Y, Zhao T, Gao X, Wu Y. Liver X receptor alpha is essential for the capillarization of liver sinusoidal endothelial cells in liver injury. Scientific Reports. 2016;6:21309. doi: 10.1038/srep21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Syn WK, Witek RP, Curbishley SM, et al. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. European Journal of Immunology. 2009;39:1879–92. doi: 10.1002/eji.200838890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pereira TA, Xie G, Choi SS, et al. Macrophage-derived Hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver International. 2013;33:149–61. doi: 10.1111/liv.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tobe K, Tsuchiya T, Itoshima T, Nagashima H, Kobayashi T. Electron microscopy of fat-storing cells in liver diseases with special reference to cilia and cytoplasmic cholesterol crystals. Arch Histol Jpn. 1985;48:435–41. doi: 10.1679/aohc.48.435. [DOI] [PubMed] [Google Scholar]
- 60.Quarmby LM, Parker JD. Cilia and the cell cycle? The Journal of cell biology. 2005;169:707–10. doi: 10.1083/jcb.200503053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biology International. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- 62.Grzelak CA, Martelotto LG, Sigglekow ND, et al. The intrahepatic signalling niche of hedgehog is defined by primary cilia positive cells during chronic liver injury. Journal of Hepatology. 2014;60:143–51. doi: 10.1016/j.jhep.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 63.Rennert C, Eplinius F, Hofmann U, et al. Conditional loss of hepatocellular Hedgehog signaling in female mice leads to the persistence of hepatic steroidogenesis, androgenization and infertility. Arch Toxicol. 2017 doi: 10.1007/s00204-017-1999-5. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 64.Syn WK, Jung Y, Omenetti A, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–88 e8. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ochoa B, Syn WK, Delgado I, et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology. 2010;51:1712–23. doi: 10.1002/hep.23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swiderska-Syn M, Suzuki A, Guy CD, et al. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology. 2013;57:1814–25. doi: 10.1002/hep.26230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhave VS, Mars W, Donthamsetty S, et al. Regulation of liver growth by glypican 3, CD81, hedgehog, and Hhex. The American Journal of Pathology. 2013;183:153–9. doi: 10.1016/j.ajpath.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grzelak CA, Sigglekow ND, Tirnitz-Parker JE, et al. Widespread GLI expression but limited canonical hedgehog signaling restricted to the ductular reaction in human chronic liver disease. PloS One. 2017;12:e0171480. doi: 10.1371/journal.pone.0171480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han B, Qu Y, Jin Y, et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015;13:1046–58. doi: 10.1016/j.celrep.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia L, Huang W, Tian D, et al. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology. 2013;57:610–24. doi: 10.1002/hep.26029. [DOI] [PubMed] [Google Scholar]
- 71.Chen MH, Wilson CW, Li YJ, et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes & Development. 2009;23:1910–28. doi: 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matz-Soja M, Aleithe S, Marbach E, et al. Hepatic Hedgehog signaling contributes to the regulation of IGF1 and IGFBP1 serum levels. Cell Commun Signal. 2014;12:11. doi: 10.1186/1478-811X-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mohieldin AM, Zubayer HS, Al Omran AJ, et al. Vascular Endothelial Primary Cilia: Mechanosensation and Hypertension. Curr Hypertens Rev. 2016;12:57–67. doi: 10.2174/1573402111666150630140615. [DOI] [PubMed] [Google Scholar]
- 74.Ke YN, Yang WX. Primary cilium: an elaborate structure that blocks cell division? Gene. 2014;547:175–85. doi: 10.1016/j.gene.2014.06.050. [DOI] [PubMed] [Google Scholar]
- 75.Witek RP, Yang L, Liu R, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–30 e2. doi: 10.1053/j.gastro.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao S, Zhang Z, Yao Z, et al. Tetramethylpyrazine attenuates sinusoidal angiogenesis via inhibition of hedgehog signaling in liver fibrosis. IUBMB Life. 2017;69:115–27. doi: 10.1002/iub.1598. [DOI] [PubMed] [Google Scholar]
- 77.Venkatesh D. Primary cilia. J Oral Maxillofac Pathol. 2017;21:8–10. doi: 10.4103/jomfp.JOMFP_48_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rock N, McLin V. Liver involvement in children with ciliopathies. Clin Res Hepatol Gastroenterol. 2014;38:407–14. doi: 10.1016/j.clinre.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 79.Omenetti A, Bass LM, Anders RA, et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology. 2011;53:1246–58. doi: 10.1002/hep.24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cui S, Leyva-Vega M, Tsai EA, et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology. 2013;144:1107–15 e3. doi: 10.1053/j.gastro.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang V, Cofer ZC, Cui S, Sapp V, Loomes KM, Matthews RP. Loss of a Candidate Biliary Atresia Susceptibility Gene, add3a, Causes Biliary Developmental Defects in Zebrafish. J Pediatr Gastroenterol Nutr. 2016;63:524–30. doi: 10.1097/MPG.0000000000001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jalan-Sakrikar N, De Assuncao TM, Lu J, et al. Hedgehog Signaling Overcomes an EZH2-Dependent Epigenetic Barrier to Promote Cholangiocyte Expansion. PloS One. 2016;11:e0168266. doi: 10.1371/journal.pone.0168266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Machado MV, Michelotti GA, Pereira TA, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. Journal of Hepatology. 2015;63:962–70. doi: 10.1016/j.jhep.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sicklick JK, Li YX, Choi SS, et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Laboratory Investigation. 2005;85:1368–80. doi: 10.1038/labinvest.3700349. [DOI] [PubMed] [Google Scholar]
- 85.Kramann R, Wongboonsin J, Chang-Panesso M, Machado FG, Humphreys BD. Gli1+ Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J Am Soc Nephrol. 2017;28:776–84. doi: 10.1681/ASN.2016030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kramann R, Goettsch C, Wongboonsin J, et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell. 2016;19:628–42. doi: 10.1016/j.stem.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kramann R, Schneider RK, DiRocco DP, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan XR, Jing YY, Liu WT, et al. Lipopolysaccharide induces the differentiation of hepatic progenitor cells into myofibroblasts via activation of the Hedgehog signaling pathway. Cell cycle. 2017;16:1357–65. doi: 10.1080/15384101.2017.1325976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin N, Tang Z, Deng M, et al. Hedgehog-mediated paracrine interaction between hepatic stellate cells and marrow-derived mesenchymal stem cells. Biochemical and Biophysical Research Communications. 2008;372:260–5. doi: 10.1016/j.bbrc.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 90.Yang L, Wang Y, Mao H, et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. Journal of Hepatology. 2008;48:98–106. doi: 10.1016/j.jhep.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Choi SS, Omenetti A, Witek RP, et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. American Journal of Physiology Gastrointestinal and Liver Physiology. 2009;297:G1093–106. doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li T, Leng XS, Zhu JY, Wang G. Suppression of hedgehog signaling regulates hepatic stellate cell activation and collagen secretion. Int J Clin Exp Pathol. 2015;8:14574–9. [PMC free article] [PubMed] [Google Scholar]
- 93.Kim J, Hyun J, Wang S, et al. Thymosin beta-4 regulates activation of hepatic stellate cells via hedgehog signaling. Scientific Reports. 2017;7:3815. doi: 10.1038/s41598-017-03782-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang F, Hao M, Jin H, et al. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br J Pharmacol. 2017;174:409–23. doi: 10.1111/bph.13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Geerts A, Bouwens L, Wisse E. Ultrastructure and function of hepatic fat-storing and pit cells. J Electron Microsc Tech. 1990;14:247–56. doi: 10.1002/jemt.1060140306. [DOI] [PubMed] [Google Scholar]
- 96.Xie G, Swiderska-Syn M, Jewell ML, et al. Loss of pericyte smoothened activity in mice with genetic deficiency of leptin. BMC Cell Biol. 2017;18:20. doi: 10.1186/s12860-017-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guy CD, Suzuki A, Abdelmalek MF, Burchette JL, Diehl AM, for the NC Treatment response in the PIVENS trial is associated with decreased hedgehog pathway activity. Hepatology. 2015;61:98–107. doi: 10.1002/hep.27235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu F, Zheng Y, Hong W, Chen B, Dong P, Zheng J. MicroRNA200a suppresses epithelialtomesenchymal transition in rat hepatic stellate cells via GLI family zinc finger 2. Mol Med Rep. 2015;12:8121–8. doi: 10.3892/mmr.2015.4452. [DOI] [PubMed] [Google Scholar]
- 99.Uschner FE, Ranabhat G, Choi SS, et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Scientific Reports. 2015;5:14573. doi: 10.1038/srep14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pereira Tde A, Witek RP, Syn WK, et al. Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Laboratory Investigation. 2010;90:1690–703. doi: 10.1038/labinvest.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.de Almeida Pereira T, Borthwick L, Xie G, et al. Crosstalk between IL13 and Hedgehog pathways contributes to Schistosomiasis mansoni fibrosis. Journal of Hepatology. 2016;62:S204–S. [Google Scholar]
- 102.Kling JC, Blumenthal A. Roles of WNT, NOTCH, and Hedgehog signaling in the differentiation and function of innate and innate-like lymphocytes. J Leukoc Biol. 2017;101:827–40. doi: 10.1189/jlb.1MR0616-272R. [DOI] [PubMed] [Google Scholar]
- 103.Langiewicz M, Schlegel A, Saponara E, et al. Hedgehog pathway mediates early acceleration of liver regeneration induced by a novel two-staged hepatectomy in mice. Journal of Hepatology. 2017;66:560–70. doi: 10.1016/j.jhep.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 104.Hanaoka J, Shimada M, Utsunomiya T, et al. Significance of sonic hedgehog signaling after massive hepatectomy in a rat. Surgery Today. 2013;43:300–7. doi: 10.1007/s00595-012-0248-z. [DOI] [PubMed] [Google Scholar]
- 105.Wang Z, Li W, Li C, et al. Small hepatocyte-like progenitor cells may be a Hedgehog signaling pathway-controlled subgroup of liver stem cells. Exp Ther Med. 2016;12:2423–30. doi: 10.3892/etm.2016.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Richardson MM, Jonsson JR, Powell EE, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133:80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 107.Moylan CA, Pang H, Dellinger A, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology. 2014;59:471–82. doi: 10.1002/hep.26661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. Journal of Hepatology. 2015;62:S38–46. doi: 10.1016/j.jhep.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Machado MV, Diehl AM. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology. 2016;150:1769–77. doi: 10.1053/j.gastro.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nguyen DL, Hu KQ. Clinical Monitoring of Chronic Hepatitis C Based on its Natural History and Therapy. N Am J Med Sci. 2014;7:21–7. [PMC free article] [PubMed] [Google Scholar]
- 111.Lambertucci JR. Revisiting the concept of hepatosplenic schistosomiasis and its challenges using traditional and new tools. Rev Soc Bras Med Trop. 2014;47:130–6. doi: 10.1590/0037-8682-0186-2013. [DOI] [PubMed] [Google Scholar]
- 112.Jung Y, McCall SJ, Li YX, Diehl AM. Bile ductules and stromal cells express hedgehog ligands and/or hedgehog target genes in primary biliary cirrhosis. Hepatology. 2007;45:1091–6. doi: 10.1002/hep.21660. [DOI] [PubMed] [Google Scholar]
- 113.Carpino G, Cardinale V, Renzi A, et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. Journal of Hepatology. 2015;63:1220–8. doi: 10.1016/j.jhep.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 114.Kar SP, Seldin MF, Chen W, et al. Pathway-based analysis of primary biliary cirrhosis genome-wide association studies. Genes and Immunity. 2013;14:179–86. doi: 10.1038/gene.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jung HY, Jing J, Lee KB, Jang JJ. Sonic hedgehog (SHH) and glioblastoma-2 (Gli-2) expressions are associated with poor jaundice-free survival in biliary atresia. J Pediatr Surg. 2015;50:371–6. doi: 10.1016/j.jpedsurg.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 116.Kumar V, Mundra V, Mahato RI. Nanomedicines of Hedgehog Inhibitor and PPAR-gamma Agonist for Treating Liver Fibrosis. Pharmaceutical Research. 2014;31:1158–69. doi: 10.1007/s11095-013-1239-5. [DOI] [PubMed] [Google Scholar]
- 117.Guy CD, Suzuki A, Burchette JL, et al. Costaining for keratins 8/18 plus ubiquitin improves detection of hepatocyte injury in nonalcoholic fatty liver disease. Human Pathology. 2012;43:790–800. doi: 10.1016/j.humpath.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PloS One. 2013;8:e70599. doi: 10.1371/journal.pone.0070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PloS One. 2015;10:e0127991. doi: 10.1371/journal.pone.0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang X, Zheng Z, Caviglia JM, et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016;24:848–62. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Choi SS, Syn WK, Karaca GF, et al. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. The Journal of Biological Chemistry. 2010;285:36551–60. doi: 10.1074/jbc.M110.168542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Syn WK, Choi SS, Liaskou E, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–15. doi: 10.1002/hep.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Syn WK, Oo YH, Pereira TA, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kwon H, Song K, Han C, et al. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease (NAFLD) Hepatology. 2016;63 doi: 10.1002/hep.28289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Granato M, Zompetta C, Vescarelli E, et al. HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI2. Scientific reports. 2016;6:30649. doi: 10.1038/srep30649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.El-Agroudy NN, El-Naga RN, El-Razeq RA, El-Demerdash E. Forskolin, a hedgehog signalling inhibitor, attenuates carbon tetrachloride-induced liver fibrosis in rats. Br J Pharmacol. 2016;173:3248–60. doi: 10.1111/bph.13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang S, Hyun J, Youn B, Jung Y. Hedgehog signaling regulates the repair response in mouse liver damaged by irradiation. Radiation Research. 2013;179:69–75. doi: 10.1667/RR3091.1. [DOI] [PubMed] [Google Scholar]
- 128.Kim J, Wang S, Hyun J, Guy CD, Jung Y. Hedgehog Signaling is Associated with Liver Response to Fractionated Irradiation in Mice. Cellular Physiology and Biochemistry. 2016;40:263–76. doi: 10.1159/000452543. [DOI] [PubMed] [Google Scholar]
- 129.Pratap A, Panakanti R, Yang N, Eason JD, Mahato RI. Inhibition of endogenous hedgehog signaling protects against acute liver injury after ischemia reperfusion. Pharmaceutical Research. 2010;27:2492–504. doi: 10.1007/s11095-010-0246-z. [DOI] [PubMed] [Google Scholar]
- 130.Jinawath A, Akiyama Y, Sripa B, Yuasa Y. Dual blockade of the Hedgehog and ERK1/2 pathways coordinately decreases proliferation and survival of cholangiocarcinoma cells. Journal of Cancer Research and Clinical Oncology. 2007;133:271–8. doi: 10.1007/s00432-006-0166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fingas CD, Bronk SF, Werneburg NW, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–88. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.El Khatib M, Kalnytska A, Palagani V, et al. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology. 2013;57:1035–45. doi: 10.1002/hep.26147. [DOI] [PubMed] [Google Scholar]
- 133.Tang L, Tan YX, Jiang BG, et al. The prognostic significance and therapeutic potential of hedgehog signaling in intrahepatic cholangiocellular carcinoma. Clinical Cancer Research. 2013;19:2014–24. doi: 10.1158/1078-0432.CCR-12-0349. [DOI] [PubMed] [Google Scholar]
- 134.Kim Y, Kim MO, Shin JS, et al. Hedgehog signaling between cancer cells and hepatic stellate cells in promoting cholangiocarcinoma. Annals of Surgical Oncology. 2014;21:2684–98. doi: 10.1245/s10434-014-3531-y. [DOI] [PubMed] [Google Scholar]
- 135.Eichenmuller M, Gruner I, Hagl B, et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology. 2009;49:482–90. doi: 10.1002/hep.22649. [DOI] [PubMed] [Google Scholar]
- 136.Li YC, Deng YH, Guo ZH, et al. Prognostic value of hedgehog signal component expressions in hepatoblastoma patients. European Journal of Medical Research. 2010;15:468–74. doi: 10.1186/2047-783X-15-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li J, Wu T, Lu J, et al. Immunohistochemical evidence of the prognostic value of hedgehog pathway components in primary gallbladder carcinoma. Surgery Today. 2012;42:770–5. doi: 10.1007/s00595-012-0157-1. [DOI] [PubMed] [Google Scholar]
- 138.Matsushita S, Onishi H, Nakano K, et al. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014;105:272–80. doi: 10.1111/cas.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xie F, Xu X, Xu A, et al. Aberrant activation of Sonic hedgehog signaling in chronic cholecystitis and gallbladder carcinoma. Human Pathology. 2014;45:513–21. doi: 10.1016/j.humpath.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 140.Takebe N, Yang SX. Sonic hedgehog signaling pathway and gallbladder cancer: targeting with precision medicine approach. Chin Clin Oncol. 2016;5:1. doi: 10.3978/j.issn.2304-3865.2016.01.02. [DOI] [PubMed] [Google Scholar]
- 141.Dixit R, Pandey M, Tripathi SK, Dwivedi AN, Shukla VK. Comparative Analysis of Mutational Profile of Sonic hedgehog Gene in Gallbladder Cancer. Dig Dis Sci. 2017;62:708–14. doi: 10.1007/s10620-016-4438-1. [DOI] [PubMed] [Google Scholar]
- 142.Sicklick JK, Li YX, Jayaraman A, et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis. 2006;27:748–57. doi: 10.1093/carcin/bgi292. [DOI] [PubMed] [Google Scholar]
- 143.Huang S, He J, Zhang X, et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis. 2006;27:1334–40. doi: 10.1093/carcin/bgi378. [DOI] [PubMed] [Google Scholar]
- 144.Patil MA, Zhang J, Ho C, Cheung ST, Fan ST, Chen X. Hedgehog signaling in human hepatocellular carcinoma. Cancer Biology & Therapy. 2006;5:111–7. doi: 10.4161/cbt.5.1.2379. [DOI] [PubMed] [Google Scholar]
- 145.Tada M, Kanai F, Tanaka Y, et al. Down-regulation of hedgehog-interacting protein through genetic and epigenetic alterations in human hepatocellular carcinoma. Clinical Cancer Research. 2008;14:3768–76. doi: 10.1158/1078-0432.CCR-07-1181. [DOI] [PubMed] [Google Scholar]
- 146.Chen YJ, Lin CP, Hsu ML, Shieh HR, Chao NK, Chao KS. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. International Journal of Radiation Oncology, Biology, Physics. 2011;80:851–9. doi: 10.1016/j.ijrobp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 147.Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. Journal of Hepatology. 2011;55:838–45. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen JS, Li HS, Huang JQ, et al. Down-regulation of Gli-1 inhibits hepatocellular carcinoma cell migration and invasion. Mol Cell Biochem. 2014;393:283–91. doi: 10.1007/s11010-014-2071-x. [DOI] [PubMed] [Google Scholar]
- 149.Byrne S, Dlamini N, Lumsden D, et al. SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder. Neuromuscul Disord. 2015;25:585–8. doi: 10.1016/j.nmd.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 150.Fan YH, Ding J, Nguyen S, et al. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene. 2016;35:116–24. doi: 10.1038/onc.2015.67. [DOI] [PubMed] [Google Scholar]
- 151.Chan IS, Guy CD, Machado MV, et al. Alcohol activates the hedgehog pathway and induces related procarcinogenic processes in the alcohol-preferring rat model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2014;38:787–800. doi: 10.1111/acer.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Philips GM, Chan IS, Swiderska M, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PloS One. 2011;6:e23943. doi: 10.1371/journal.pone.0023943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jeng KS, Sheen IS, Jeng WJ, et al. Blockade of the sonic hedgehog pathway effectively inhibits the growth of hepatoma in mice: An in vivo study. Oncology Letters. 2012;4:1158–62. doi: 10.3892/ol.2012.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pinter M, Sieghart W, Schmid M, et al. Hedgehog inhibition reduces angiogenesis by downregulation of tumoral VEGF-A expression in hepatocellular carcinoma. United European Gastroenterol J. 2013;1:265–75. doi: 10.1177/2050640613496605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chung SI, Moon H, Ju HL, et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. Journal of hepatology. 2016;64:618–27. doi: 10.1016/j.jhep.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 156.Tsai CL, Hsu FM, Tzen KY, Liu WL, Cheng AL, Cheng JC. Sonic Hedgehog inhibition as a strategy to augment radiosensitivity of hepatocellular carcinoma. Journal of Gastroenterology and Hepatology. 2015;30:1317–24. doi: 10.1111/jgh.12931. [DOI] [PubMed] [Google Scholar]
- 157.Jeng KS, Jeng CJ, Jeng WJ, et al. Sonic hedgehog pathway inhibitor mitigates mouse hepatocellular carcinoma. Am J Surg. 2015;210:554–60. doi: 10.1016/j.amjsurg.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 158.Dugum M, Hanouneh I, McIntyre T, et al. Sonic hedgehog signaling in hepatocellular carcinoma: A pilot study. Mol Clin Oncol. 2016;4:369–74. doi: 10.3892/mco.2016.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Che L, Yuan YH, Jia J, Ren J. Activation of sonic hedgehog signaling pathway is an independent potential prognosis predictor in human hepatocellular carcinoma patients. Chinese Journal of Cancer Research. 2012;24:323–31. doi: 10.3978/j.issn.1000-9604.2012.10.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cheng WT, Xu K, Tian DY, Zhang ZG, Liu LJ, Chen Y. Role of Hedgehog signaling pathway in proliferation and invasiveness of hepatocellular carcinoma cells. International Journal of Oncology. 2009;34:829–36. doi: 10.3892/ijo_00000209. [DOI] [PubMed] [Google Scholar]
- 161.Lu JT, Zhao WD, He W, Wei W. Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacologica Sinica. 2012;33:691–700. doi: 10.1038/aps.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Abou-Alfa GK, Lewis LD, LoRusso P, et al. Pharmacokinetics and safety of vismodegib in patients with advanced solid malignancies and hepatic impairment. Cancer Chemother Pharmacol. 2017;80:29–36. doi: 10.1007/s00280-017-3315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nault JC, Couchy G, Balabaud C, et al. Molecular Classification of Hepatocellular Adenoma Associates With Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology. 2017;152:880–94 e6. doi: 10.1053/j.gastro.2016.11.042. [DOI] [PubMed] [Google Scholar]