

Abstract
Background
Severe IgE-mediated, food-induced anaphylactic reactions are characterized by pulmonary venous vasodilatation and fluid extravasation, which is thought to lead to the life-threatening anaphylactic phenotype. The underlying immunological and cellular processes involved in driving fluid extravasation and the severe anaphylactic phenotype are not fully elucidated.
Objective
To define the interaction and requirement of IL-4 and vascular endothelial (VE) IL-4Rα chain signaling in histamine-ABL1–mediated VE dysfunction and fluid extravasation in the severity of IgE-mediated anaphylactic reactions in mice.
Methods
Mice deficient in VE IL-4Rα and models of passive and active oral antigen– and IgE-induced anaphylaxis were employed to define the requirements of VE IL-4Rα and ABL1 pathway in severe anaphylactic reactions. Human VE cell line (EA.hy926 cells) and pharmacologic (imatinib) and genetic approaches (shRNA knockdown of IL4RA and ABL1) were used to define the requirement of this pathway in VE barrier dysfunction.
Results
IL-4 exacerbation of histamine-induced hypovolemic shock in mice was dependent on VE expression of the IL-4Rα. IL-4 and histamine induced ABL1 activation in human VE cells and VE barrier dysfunction was ABL1 dependent. Development of severe IgE-mediated hypovolemia and shock required VE-restricted ABL1 expression. Treatment of mice with a history of food-induced anaphylaxis with the ABL kinase inhibitor imatinib protected the mice from developing severe IgE-mediated anaphylaxis.
Conclusion
IL-4 amplifies IgE- and histamine-induced VE dysfunction, fluid extravasation, and severity of anaphylaxis via a VE IL-4Rα-ABL1–dependent mechanism. These studies implicate an important contribution by the VE compartment in the severity of anaphylaxis and identify a new pathway for therapeutic intervention of IgE-mediated reactions.
Keywords: IL-4Rα, chain, hypovolemic shock, IgE and Mast cells, food-induced anaphylaxis, histamine, vascular endothelial barrier dysfunction, ABL1 kinase
Graphical Abstract
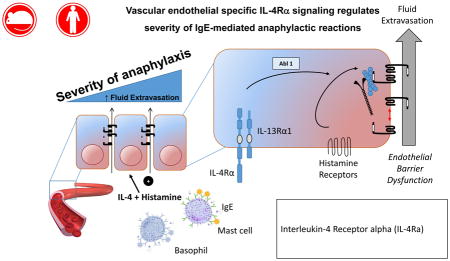
Introduction
Anaphylaxis is a severe, life-threatening allergic reaction that affects both children and adults and males and females in the United States 1. The most common inciting agents (33.2% of reactions) are foods, particularly peanuts and tree nuts, and food-induced anaphylaxis (FIA) hospitalization rates for children in US have more than doubled from 2000 to 2009 2.
A food-induced anaphylactic reaction encompasses a variety of symptoms that may affect one or more target organs including those of the gastrointestinal (GI), cutaneous, respiratory, and cardiovascular systems 3–5. In humans, compromise of either the cardiovascular or respiratory system defines a severe reaction 3, 4, and it is postulated that basophil- and mast cell (MC)-derived mediators, through inducing pulmonary venous vasodilatation and fluid extravasation, cause the respiratory and cardiovascular collapse that leads to the severe, life-threatening anaphylactic phenotype 6. Fluid extravasation in anaphylaxis is thought to be consequence of capillary fluid leak due to loss of the vascular endothelial (VE) barrier integrity, leading to the movement of fluids, electrolytes, and proteins from the vascular compartment into the interstitial spaces 7–9.
The VE barrier is maintained by adherens junction (AJ) and tight junction (TJ) proteins 10. The AJ proteins are the most ubiquitously expressed endothelial cell-cell junctional proteins and act as mechanical anchoring points that promote endothelial TJ protein-protein interactions and interjunctional integrity 11. The TJ proteins are tethered to the actin cytoskeleton and seal the intercellular space, establishing the dense “fence” barrier preventing the bilateral apical-basolateral passage of ions, proteins, and lipids 11. VE-cadherin is one of the first endothelial cell–specific molecules expressed and required for endothelial survival, blood vessel assembly, and stabilization 12, 13. VE-cadherin forms Ca2+- dependent homophilic interactions with adjacent endothelial cells via actin-linking catenin family proteins and the actin cytoskeleton, establishing the vascular barrier integrity 11. The stability of the VE-cadherin–catenin–cytoskeleton complex is essential to maintaining endothelial barrier function 14 and disruption of these processes via receptor-signaling pathways including non-receptor kinases, including SRC, ABL1 and ARG and myosin light chain kinase (MLCK) leads to VE-cadherin–mediated AJ disorganization or VE-cadherin internalization and loss of endothelial barrier integrity 14–20.
The cellular and molecular pathways that directly contribute to the severe anaphylaxis phenotype are unclear. Clinical studies have reported increased levels of IL-4 and histamine in the serum of human patients with severe anaphylaxis 21, and histamine, but not IL-4, levels were associated with severe disease 22 suggesting a possible role for these molecules in expression of the severe disease phenotype. We and others have demonstrated that symptoms of food-induced anaphylaxis in mice are dependent on IgE-MCs and histamine type I receptor signaling and that the severity of the reaction positively correlates with an increase in hemoconcentration (an indication for fluid extravasation and hypovolemic shock) 23–26. In vitro experimental evidence suggests that IL-4 modulates VE barrier properties 27–29 and we have demonstrated that IL-4 can interact with vasoactive mediators to increase hemoconcentration and the severity of anaphylaxis 30, 31. However, the cellular target of these IL-4-mediated effects and the underlying IL-4Rα-dependent signaling processes involved in the amplification of histamine-induced VE barrier dysfunction and fluid extravasation in IgE-mediated reactions is not yet fully understood.
Herein, we examine the relationship between IL-4 and histamine in IgE-mediated vascular endothelial leak and hypovolemic shock. We show that IL-4 amplifies histamine-induced hypovolemic shock via VE IL-4Rα chain–dependent process. Furthermore, we show that IL-4 and histamine stimulate ABL1 kinase activity in vascular endothelial cells and VE barrier dysfunction that was inhibited by pharmacologic and genetic ablation of ABL1 activity. Importantly, employing both passive and active models of food-induced anaphylaxis, treating mice with the ABL kinase inhibitor imatinib protected the mice from developing severe IgE-mediated anaphylactic phenotype after allergen exposure. These studies implicate an important contribution by the IL-4Rα-ABL1-signaling pathway in the VE compartment in the severity of IgE- and histamine-induced anaphylaxis.
Material and Methods
Animals
Intestinal IL-9 transgenic (iIL9Tg) mice were generated as previously described 32. WT BALB/c mice were originally provided by Charles River laboratories, (Wilmington, MA, USA) and bred in-house at Cincinnati Children’s Hospital Medical Center (CCHMC) (Cincinnati, OH, USA). IL-4RαY709F mice were obtained from Fred Finkelman, CCHMC 33. Cadherin-5Cre mice (purchased from Jackson Laboratory) (Bar Harbor, ME, USA) and IL-4Rαfl/fl mice (generously provided by Frank Brombacher, University of Cape Town, South Africa) and iIL9Tg were used to generate mice lacking IL-4Rα in VE cells 34. Tie2Cre mice (generously provided by Joseph E. Qualls, CCHMC) and Abl1fl/fl mice35 and iIL-9Tg mice were used to generate mice lacking ABL1 in VE cells 36. Age-, sex-, weight-matched littermates were used as controls in all experiments. The mice were maintained and bred in a clean barrier facility and were handled under an approved Institutional Animal Care and Use Committee protocols at CCHMC animal facility.
Oral Antigen–Induced Anaphylaxis
Six- to 8-week-old mice were sensitized and challenged as previously described 23. In some experiments, mice received imatinib mesylate (Santa Cruz Biotechnology, TX, USA) intraperitoneally (i.p.) (1.25 or 1.75 mg/100 μL distilled water per mouse) 2 h prior to OVA challenge.
Passive Anaphylaxis
Mice were injected intravenously (i.v.) with 20 μg/200 μL of anti-IgE (IgG2a mAb to mouse IgE; EM-95) (Obtained from Fred Finkelman, CCHMC) 37. EM95 cross-linking to FcεR1 lead to MC and basophil activation and degranulation 38.
IL-4 and Histamine-Induced Anaphylaxis
Twenty-four hours before the experiments, mice were injected i.v. with IL-4C (recombinant, IL-4–neutralizing, anti–IL-4 monoclonal antibody [mAb] complex, 1:5 weight [1 μg of IL-4 + 5 μg anti-IL-4 mAb]) (anti–IL-4 mAb was obtained from Fred Finkelman, CCHMC) followed by histamine biphosphate monohydrate (Sigma–Aldrich, St. Louis, MO, USA) (0.4 or 2 mg/200 μL saline per mouse) 31, 39.
Passive Oral Antigen-Induced Anaphylaxis
Mice were primed and challenged as previously described 23. Mice received imatinib mesylate (Santa Cruz Biotechnology, TX, USA) i.p.(1.25–1.75 mg/100 μL distilled water per mouse) 2 h prior to OVA–2, 4, 6- trinitrophenyl hapten (TNP) challenge.
Anaphylaxis Assessments
Hypothermia (significant loss of body temperature) and hypovolemia (increased in hemoconcentration) were used as an indication of anaphylactic reaction. Rectal temperature was taken with a rectal probe and a digital thermocouple thermometer (Model BAT-12; Physitemp Instruments, Clifton, NJ, USA). Temperature was taken prior to the challenge and then every 15 min for 30 or 60 min. For hemoconcentration assessment, blood was drawn by retro-orbital bleeding into heparinized capillary tubes and centrifuged for 5 min at 10,000 rpm. Hematocrit (percentage of packed red blood cell [RBC] volume) was calculated as previously described 31.
Mast Cell Quantification
Jejunum and ileum were collected and processed for MC quantification by chloroacetate esterase (CAE) staining, as described previously 23.
ELISA Measurements
Mouse MC protease 1 (mMCPT-1) serum levels were measured by ELISA according to the manufacturer’s instructions (eBioscience, San Diego, CA, USA).
Cell Lines and Culture
EA.hy926 cell line (human VE cell line) (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (Atlanta Biological), nonessential amino acids (0.1 mM), sodium pyruvate (1 mM), HEPES (10 mM), and 1X penicillin and streptomycin (50 μg/mL) in a humidified incubator (5% CO2, 37°C) 40, 41. All reagents were obtained from Gibco, Life Technologies (Waltham, MA, USA) unless stated otherwise.
ABL Kinase Activity Assay
3×106 cells/well were plated in a 6-well plate and grown for 24 h. Cells were pretreated with IL-4 (10 ng for 24 h; Peprotech, Rocky Hill, NJ, USA) before imatinib treatment (10–100 μM for 3 h; Santa Cruz Biotechnology, TX, USA) followed by histamine (10 μM for 30 min; Sigma–Aldrich, St. Louis, MO, USA) stimulation. To assess for ABL1 kinase activity, ABL1 was immunopreciptated from treated EA.hy926 cells for a subsequent measurement of CRK-GST phosphorylation. The ABL1 activation assay was performed as describe by Dudek et al. 42.
In Vitro Permeability
EA.hy926 were plated on transwells (12-mm dimeter, 0.4-μm pore; Polyester Membrane Insert, Corning, NY, USA), cells were seeded and cultured until a monolayer formed. Transendothelial resistance (TER) was monitored with an EVOM/Endohm (WPI Inc, Sarasota, FL, USA), and endothelial monolayers with a TER >100 Ω/cm2 were used for all experiments. Monolayers were mounted between the hemi-chambers of an Ussing apparatus (P2300 Ussing chamber, Physiologic instruments, CA, USA), and 0.33 cm2 of cell monolayer was exposed to 6 mL of Krebs buffer at 37°C. The transendothelial potential difference was detected with two paired electrodes that contain 4% agar in 3M KCl. The electrodes were connected to a voltage clamp amplifier (VCC MC8, multichannel voltage/current clamp, Physiologic instruments, CA, USA). The electrode potential difference and fluid resistance were compensated before mounting the monolayer onto the chamber. To establish equilibrium, the potential difference was continuously monitored under open-circuit conditions for 15 min. Thereafter, the monolayers were voltage-clamped at 0 mV while continuously measuring the short circuit current (Isc). Voltage pulses (10-mV square waves sustained for 200 ms) were delivered every 20 s to yield a current response for calculating resistance across a mucosa (resistance) using Ohm’s law. IL-4– (100 ng/mL; 24 h), histamine- (100 mM; 30 min), and vehicle-stimulated endothelial monolayers were placed in Ussing chambers, where TER was measured as described previously 43. To measure paracellular flux, horse radish peroxidase (HRP) solution was left for 30 min at room temperature (RT) before the assay. The assay was started by adding 4 μg/mL of HRP to the apical side of the monolayers to measure flux from the apical to the basolateral direction. Afterward, aliquots of 250 μL from the basolateral side were collected at 30, 60, 90, 120, 150, and 180 min. HRP was assayed colorimetrically (BD OptEIA™, CA, USA) according to the manufacturer’s instructions.
Lentiviral Transduction
Bacterial glycerol stock (Sigma–Aldrich, St. Louis, MO, USA) containing shRNA plasmid DNA was used to generate lentiviral particles containing target shRNA. In brief, to expand the bacterial glycerol stock 1 μL was incubated at 37°C for 30 min in terrific broth (TB) without antibiotic then cultured in Luria broth (LB) agar with ampicillin (100 μg/ml). After an overnight incubation, a single colony was expanded by incubating with 3 mL of LB plus ampicillin for 8–10 h and then moved to 300 mL LB with ampicillin and incubated overnight. ShRNA plasmid DNA was extracted using QIAGEN Plasmid Maxi Kit (Hilden, Germany). Afterward, the lentivirus partials were produced by CCHMC’s Viral Vector Core. Cells were transduced with 5 μL of concentrated lentiviral particles or control shRNA lentiviral particles containing an empty vector (PLKO.1). Transduction was carried out with media in the presence of 10 μg/mL of polybrene (Sigma–Aldrich, St. Louis, MO, USA). The cells were incubated for 24 h at 37°C, and then the media was replaced with fresh media. Cells were selected with 10 μg/mL puromycin (Gibco, Grand Island, NY, USA) two days after transduction.
Flow Cytometry
Mice were injected with IL-4C (1 μg of IL-4 + 5 μg anti–IL-4 mAb) or vehicle. Twenty-four hours later, mice were sacrificed, and the spleen was harvested. To prepare a single-cell suspension, the spleen was homogenized, and then the suspension was filtered with a 40-μm pore cell strainer. Afterward, the cell solution was centrifuged (5 min/1200 rpm). Red blood cells (RBCs) were lysed with RBC lysing buffer (Sigma-Aldrich, St. Louis, MO, USA) and incubated for 2 min at 37°C; the reaction was stopped using 30 mL 1X HBSS buffer (Gibco, Grand Island, NY, USA). Cells were centrifuge and re-suspended with 200 μL FACS buffer (0.1% sodium azide, 2 % fetal calf serum, and 1X PBS) at 2×106 cells/mL and stained with anti-mouse MHC II (I-A) FITC and anti-mouse/human CD45R/B220 PerCP/Cy5.5 antibodies (BioLegend, San Diego, CA, USA). Following fixation with 1% paraformaldehyde (30 min at 4°C), the cells were re-suspended in 200 μL FACS buffer. Flow cytometry data acquisition and analysis were performed on BD LSR Fortessa using FACS Diva software (BD Biosciences, Mountain View, CA, USA).
Immunofluorescent Assay
Antibodies and concentrations used were as follows: anti–IL-4Rα at 1:400 (Santa Cruz, # sc-28361), anti-MECA32 at 1:100 (Novous Biologicals, # NB100-77668). Immunofluorescence was done as previously described 44, except that the antigen retrieval step was done using a decloaking chamber for 30 s at 125°C.
Statistical Analysis
Data are expressed as mean ± standard deviation (SD), unless otherwise stated. In experiments comparing multiple experimental groups, statistical differences between groups were analyzed using the one/two-way ANOVA parametric test. In experiments comparing two experimental groups, statistical differences between groups were determined using a Student’s t-test. A P < 0.05 was considered significant. Correlation analysis was performed using the Spearman’s rank correlation coefficient. All analyses were performed using Prism 7.0 software (GraphPad Software Inc., San Diego, CA, USA).
Results
IL-4 enhances histamine-induced hypovolemic shock via vascular endothelium–dependent IL-4Rα chain signaling
We first examined the effect of IL-4 on histamine-induced increase in hemoconcentration and hypothermia (as an indication for anaphylactic shock severity). WT mice were i.v. injected with IL-4C (a long-acting formula IL-4 complexed with anti–IL-4 39) or vehicle and 24 h later received an i.v. challenge of histamine. Treatment of mice with IL-4C alone did not impact hemoconcentration nor impact body temperature (Figure 1). In contrast, histamine alone induced an increase in hemoconcentration that was associated with hypothermia (Figure 1). Notably, this response was potentiated by pretreatment with IL-4C, suggesting that IL-4 amplifies histamine-induced hypovolemic shock (Figure 1). To determine whether IL-4 amplification of the histamine response was via direct modulation of VE barrier function, we examined the effect of histamine and IL-4 on the human VE cell line EA.hy926 40, 41. We show that TER decreased and flux of HRP (40 kDa) increased in histamine-stimulated compared to unstimulated EA.Hy926 cells (Figure 2A–B). Importantly, the histamine-induced increase in endothelial cell permeability was enhanced by pretreatment with IL-4 (Figure 2A–B). Collectively, these data suggest that IL-4 amplifies histamine-induced VE permeability through a direct activation of VE cells.
Figure 1. IL-4 enhancement of histamine-induced shock negatively correlates with fluid extravasation.

VEIL-4Rα WT (iIL-9WT Cadherin-5WT IL-4Rαfl/fl) mice were injected i.v. with (0.2 mL) of vehicle or IL-4C (1 μg of IL-4 + 5 μg anti–IL-4 mAb) and then challenged i.v. 24 h later with vehicle or histamine (0.4 mg/200 μL). Shown is Spearman’s rank correlation coefficient between hematocrit percentage and the maximum temperature change from 0 to 30 minutes. Each circle represents an individual mouse, n = 3 – 11 per group.
Figure 2. IL-4 amplifies histamine-induced vascular endothelial barrier dysfunction.

Percent reduction in transendothelial resistance (TER) (A) and horseradish peroxidase (HRP) flux (B) of vehicle- or IL-4-treated EA.hy926 cells following histamine stimulation. EA.hy926 monolayers were pretreated with vehicle or IL-4 (100 ng/mL) for 24 hrs and stimulated with histamine (100 μM for 30 min) or vehicle and TER and HRP flux were measured as described in methods. Data are represented as the mean ± SD; n = 3 wells per group from n = 2 independent experiments (A, B). **** P < 0.0001, *** P < 0.0001, ** P < 0.01, ns > 0.05.
To identify the requirement of IL-4 signaling on VE cells in the exacerbation of histamine-induced hypovolemic shock in vivo, we took a transgenic approach using the VE-specific promoter of the junction protein cadherin-5 (also called VE-cadherin) 34. Cadherin-5Cre IL-4Rαfl/fl mice (referred to as VEΔIL-4Rα) lack the expression of the IL-4Rα chain on the VE compartment, whereas Cadherin-5WT IL-4Rαfl/fl mice (referred to as VEIL-4Rα WT) were used as a WT control (Supplemental Figure S1). Mice received an i.v. injection of either vehicle or IL-4C and 24 h later received an i.v. injection of histamine; evidence of hypovolemic shock and hypothermia were evaluated (Figure 3A). I.v. administration of histamine induced hypothermia and increased hemoconcentration in both VEIL-4Rα WT and VEΔIL-4Rα (Figure 3B–C). Pretreating VEIL-4Rα WT with IL-4C enhanced histamine-induced hypothermia and hemoconcentration (Figure 3B–C). However, the IL-4C amplification of the histamine-induced hypovolemic shock was ablated in VEΔIL-4Rα mice (Figure 3B–C). Consistent with previous studies, we found that IL-4C alone had no effect on body temperature nor on hemoconcentration 31 (Figure 3B–C). To confirm IL-4C activity in VEΔIL-4Rα mice, we evaluated the expression of MHC II on B cells (B220+) after IL-4C treatment. We showed that the expression of MHC II on B cells increased in IL-4C–treated compared to vehicle-treated mice (Supplemental Figure S2). Notably, IL-4C treatment induced a comparable increase in MHC II expression in the VE IL-4RαWT and VEΔIL-4Rα mice (Supplemental Figure S2). These studies indicate that IL-4 amplification of histamine-induced hypovolemic shock was dependent on IL-4 signaling on the VE compartment.
Figure 3. Loss and amplification of vascular endothelial IL-4Rα signaling alters IL-4 enhancement of histamine-induced hypovolemic shock.

Experimental regimen (A and D), Maximum Temperature Change (B and E) and Hemacrit (%) (C and F) in iIL-9Tg VEΔIL-4Rα and iIL-9Tg VEIL-4Rα WT mice (A – C) and IL-4RαY709F and IL-4RαWT mice (D – F) following i.v. injection with vehicle or IL-4C and i.v. challenged with histamine (0.4 mg). Data are represented as the mean ± SD; n = 3 – 10 (B) and n = 3 – 6 mice (C) and n = 9 – 13 mice (E and F) per group from n = 3 – 4 independent experiments. **** P < 0.0001, ** P < 0.001, * P < 0.05, ns > 0.05.
Interestingly, clinical and experimental studies indicate that gain-of-function mutations in the IL-4Rα chain have been associated with increased susceptibility to atopic disease and enhanced allergic inflammatory responses 45–50. Given the observation that IL-4/IL-4Rα signaling plays an important role in histamine-induced hypovolemic shock, we hypothesized that mice with a gain-of-function in IL-4 receptor signaling would have increased severity of histamine-induced hypovolemic shock. To test this, we employed IL-4RαY709F mice33, which have an activating variant IL-4Rα chain (Figure 3D–F). Histamine challenge of IL-4RαY709F mice induced a hypovolemic shock response equivalent to that observed in WT mice (Figure 3E and F). IL-4C treatment amplified histamine-induced hypothermia and hypovolemic shock in both IL-4RαY709F and WT BALB/c mice (Figure 3E and F). Notably, we observed a significantly greater IL-4C–induced amplification of the histamine-induced hypothermic response in the IL-4RαY709F mice than BALB/c mice, and this increased hypothermic response was associated with increased hemoconcentration (Figure 3E and F). These data indicate that enhanced IL-4Rα signaling can exacerbate histamine- and IL-4–induced hypovolemic shock.
Vascular endothelial IL-4Rα chain deletion attenuated hypovolemic shock induced by passive oral antigen–induced anaphylaxis
To confirm the requirement of IL-4Rα chain expression on VE in IgE/MC-mediated anaphylaxis, we used a passive oral antigen–triggered IgE-mediated anaphylaxis model 51. Cadherin-5Cre IL-4Rαfl/fl mice were backcrossed on the iIL-9Tg background (referred to as iIL-9Tg VEΔIL-4Rα). iIL-9Tg VEΔIL-4Rα mice and iIL-9Tg VEIL-4Rα WT mice were injected with anti-TNP IgE with IL-4C and then administered TNP-OVA 24 h later (Figure 4A). Oral gavage of TNP-OVA increased hemoconcentration and induced shock in both iIL-9Tg VEIL-4Rα WT and iIL-9Tg VEΔIL-4Rα mice treated with anti-TNP IgE (Figure 4B–C). Notably, pretreatment of the iIL-9Tg VEIL-4Rα WT mice with IL-4C exacerbated hypothermia and hemoconcentration; however, the IL-4 enhancement was absent in iIL-9Tg VEΔIL-4Rα mice (Figure 4B–C). The reduced disease in iIL-9Tg VEΔIL-4Rα mice could not be explained by decreased MC activation or levels or immunoglobulin levels, as serum mMCPT-1 levels, intestinal MC levels, and immunoglobulin (Ig) (IgE and IgG1) were comparable between groups (Figure 4D; Supplemental Figure S3A–E). Collectively, these data indicate that IL-4 enhancement of passive oral antigen–triggered anaphylaxis is dependent on the VE IL-4Rα chain.
Figure 4. Vascular endothelial IL-4Rα chain deletion attenuated hypovolemic shock in a passive oral antigen anaphylaxis model.

Experimental regimen (A), Maximum Temperature Change (B) Hemacrit (%) (C) and serum mMCPT-1 levels (D) in iIL-9Tg VEΔIL-4Rα and iIL-9Tg VEIL-4RαWT mice injected i.v. with IL-4C mixed with anti-TNP IgE and then oral gavaged the next day with TNP-OVA. Data are represented as the mean ± SD; n = 3 – 13 mice per group from n = 4 independent experiments. **** P < 0.0001, *** P < 0.001, ** P < 0.01, * P < 0.05, ns > 0.05.
Histamine and IL-4 modulation of paracellular leakage in the human vascular endothelial cell line (EA.hy926) is ABL1 kinase-dependent
We were next interested in defining the signaling pathways involved in IL-4C amplification of the histamine-induced VE dysfunction. Recent studies have identified roles for members of the ABL family of non-receptor tyrosine kinases (ABL1 and ARG) and RhoA and ROCK in histamine-induced VE leakage; therefore, we examined histamine and IL-4C induction of ABL1 kinase activity 15, 52. Employing the immortalized human endothelial cell line (EA.hy926), we firstly examined ABL1 kinase activity after histamine and IL-4C stimulation. We found that histamine or IL-4C alone stimulated ABL1 kinase activity as indicated by GST-CRK (a known ABL1 substrate) phosphorylation (Figure 5A). Notably, EA.hy926 cell costimulation with both histamine and IL-4C amplified ABL1 kinase activity (Figure 5A). To determine the requirement of ABL1 kinase in the histamine/IL-4C–induced barrier function, we used a pharmacologic (imatinib) and shRNA ABL1 kinase knockdown approach. Histamine stimulation of EA.hy926 cells reduced the TER in a dose dependent manner (Supplemental Figure S5A). Pretreatment of EA.hy926 cells with Imatinib antagonized histamine-induced reduction in TER and paracellular HRP-flux (Supplemental Figure S4B and C). Pretreatment of EA.hy926 cells with imatinib blocked IL-4 amplification of histamine-induced VE barrier dysfunction in a dose-dependent manner (Figure 5C–D). Notably, the decreased VE response in the presence of imatinib was associated with decrease of ABL1 kinase activity (Figure 5B). Consistent with these observations, pretreating shRNA ABL1–transduced EA.hy926 cells with histamine or histamine and IL-4 did not induce VE barrier dysfunction (Figure 5F–G). Taken together, these findings suggest that histamine-induced, IL-4–enhanced paracellular leakage is dependent on the VE ABL kinase, ABL1 kinase.
Figure 5. Histamine and IL-4 induced-vascular endothelial barrier dysfunction is ABL1 kinase dependent.

ABL1 kinase activity in vehicle or IL-4 stimulated EA.hy926 cells following (A) histamine stimulation and (B) in the presence of Imatinib. TER (C and F) and HRP-flux (D and G) of EA.hy926 cells and EA.hy926 cells transduced with ABL1 shRNA or empty vector (PLKO-1) pretreated with IL-4 (10 ng/mL) and stimulated with histamine in the presence and absence of imatinib (100 μM). (E) ABL1 levels in EA.hy926 transduced with ABL1 shRNA lentiviral particles or an empty vector (PLKO-1) detected by western blot. (C and D; F and G) Data are represented as the mean ± SD; n = 3 wells per group from two representative experiments. **** P < 0.0001, *** P < 0.001, * P < 0.05, ns > 0.05.
Vascular endothelial ABL1 kinase involvement in passive anaphylaxis in iIL-9Tg VEΔABL1 mice
We next examined the requirement of the VE ABL1 kinase in passive anaphylaxis by employing mice lacking ABL1 expression in the VE compartment. Tie2Cre transgenic mice have been used as a genetic tool for the analyses of endothelial cell–specific gene targeting 36. Therefore, we employed Tie2Cre ABL1fl/fl mice on an iIL9Tg background (referred to as iIL-9Tg VEΔABL1) and WT control iIL9Tg Tie2WT ABL1fl/fl (referred to as iIL-9Tg VEABL1 WT). iIL-9Tg VEΔABL1 mice and iIL-9Tg VEABL1 WT mice received an i.p. injection with vehicle or imatinib 30 min before i.v. injection with anti-IgE mAb (EM95) (Supplemental Figure S5A). In iIL-9Tg VEABL1 WT mice, i.v. anti-IgE mAb induced hypothermia and increased hemoconcentration, with these changes being sensitive to imatinib inhibition (Supplemental Figure S5B–C). Importantly, iIL-9Tg VEΔABL1 mice showed an attenuated hypothermia that was comparable to the hypothermia observed in iIL-9Tg VEABL1 WT treated with imatinib (Supplemental Figure 5B). Importantly, levels of MC activation and degranulation were similar between groups (Supplemental Figure S5D). Collectively, these data indicate a requirement for VE ABL1 in hypovolemic shock induced by IgE.
ABL kinase inhibitor attenuated passive oral antigen–induced anaphylaxis
To determine whether the ABL kinase inhibitors could suppress the development of symptoms of oral antigen–induced IgE/MC-mediated reactions in mice, we examined the effect of imatinib in a passive oral antigen–triggered, IgE-mediated anaphylaxis model 23. Evidence of anaphylaxis was evaluated in iIL-9Tg mice who received an i.v. injection of anti-TNP IgE and 24 h later received either vehicle or imatinib (16–70 μg/kg per mice; 2 h) and subsequently received oral gavage with TNP-OVA (Figure 6A). In vehicle-treated mice, oral challenge of TNP-OVA induced shock, which was associated with an increase in hemoconcentration (Figure 6B–C). Pretreating mice with imatinib prior to TNP-OVA administration protected the mice from hypothermia and hemoconcentration (Figure 6B–C). Importantly, imatinib-mediated protection was not associated with reduced MC degranulation as levels of the mMCPT-1 were comparable between all groups (Figure 6D).
Figure 6. Pharmacologic inhibition of ABL kinases attenuated IgE-mediated hypovolemic shock in a passive oral antigen anaphylaxis model.

Experimental regimen (A), Maximum Temperature Change (B), Hemacrit (%) (C), and serum mMCPT-1 levels (D) in iIL-9Tg mice injected i.v. with anti-TNP IgE (20 μg) and o.g. with TNP-OVA. (E) mMCPT-1 values verse temperature change following oral challenge and (F) Fisher’s exact test analysis. (G) Serum histamine levels in iIL-9Tg mice following anti-TNP IgE and o.g. with TNP-OVA in the presence and absence of Imatinib. Data are represented as the mean ± SD; n = 3 – 9 mice per group (B–D) from n = 3 experiments and n = 5 – 6 mice per group (G). **** P < 0.0001, *** P < 0.001, ** P < 0.01, * P < 0.05, ns > 0.05.
Imatinib is known to target c-KIT, which is required for optimum MC degranulation 53, 54. Though mMCPT-1 levels were not significantly different between groups, the levels of mMCPT-1 in the Imatinib-treated mice were lower than vehicle-treated mice (Figure 6D). To confirm that the imatinib-mediated protective effects were not due to decreased MC degranulation, we examined the relationship between mMCPT-1 levels and the onset of hypothermic response (Figure 6E). We show that 10 out of 11 mice in the vehicle-treated, TNP-OVA–challenged group with mMCPT-1 >10,000 ng/mL were hypothermic (≥1.5°C temperature loss) and that no mice (0/6) with <10,000 ng/mL mMCPT-1 developed hypothermia (Figure 6E–F). Therefore, if we set a cut-off threshold of ≥10,000 ng/mL mMCPT-1 as a level of MC degranulation sufficient to induce symptoms of anaphylaxis, this provides us with ≥90% confidence that mice with ≥10,000 ng/mL mMCPT-1 will develop evidence of anaphylaxis. Examining mMCPT-1 levels from the imatinib-treated, TNP-OVA–challenged group revealed that 10/13 mice had ≥10,000 ng/mL mMCPT-1 and were protected from hypothermic response (≥1.5°C temperature loss) (Figure 6E–F). Therefore, the protection from hypothermia in those mice is unlikely due to an effect on MC degranulation. mMCPT-1 is a surrogate for MC degranulation and the hypothermic response is histamine-dependent; we therefore examined histamine levels after TNP-OVA administration in anti-TNP IgE–primed mice treated with vehicle or imatinib. TNP-OVA administration induced a significant increase in histamine levels in the vehicle-treated group, which was comparable with histamine levels in the imatinib-treated group (Figure 6G). These studies suggest that imatinib protection is mainly via inhibiting ABL kinase activity independent of MC degranulation. Collectively, these data indicate that imatinib can reduce the severity of IgE/MC-driven anaphylaxis and that this effect is independent of MC degranulation.
ABL kinase inhibitor attenuated pre-existing oral antigen–induced anaphylaxis
Next, we examined the effect of the ABL kinases pharmacologic antagonist, imatinib, on mice with established food allergy 23. BALB/c WT mice were primed i.p. with OVA-alum and subsequently challenged by repeated oral gavage with OVA (Figure 7A). By the 6th allergen challenge, mice developed symptoms of food induced-anaphylaxis, including hypothermia and diarrhea 51, 55, 56. Prior to the 7th challenge, mice were stratified into two groups, with some mice receiving vehicle while others were pretreated with imatinib (50–75 μg/kg i.p.; 2 h) and then subsequently challenged with OVA (Figure 7A). Oral antigen challenge induced hypothermia in vehicle-treated mice (Figure 7B–C). In contrast, pretreating mice with imatinib protected the mice from anaphylactic shock; the level of hypothermia and diarrhea were significantly reduced in the imatinib-treated mice compared to the vehicle-treated mice (Figure 7B–D). Notably, the protection from hypothermia was associated with reduced hemoconcentration (Figure 7E) in the presence of comparable MC degranulation (mMCPT-1 levels) (Figure 7F), suggesting that imatinib attenuates histamine-induced fluid extravasation. Collectively, these studies indicate that pretreating mice with imatinib can protect from the development of a severe food-induced reaction in mice with a previous history of severe reactions.
Figure 7. Established food-induced anaphylaxis is attenuated with ABL kinase inhibitor.

Experimental regimen (A), Maximum temperature change at the 6th and 7th challenge (± Imatinib) (B) the 7th challenge ± imatinib (C), percentage of mice with diarrhea (D), Hemacrit (%) (E) and serum mMCPT-1 levels (F) of OVA-sensitized WT BALB/c mice treated ± imatinib (1.27–1.75 mg/mice). Data are represented as the mean ± SD; n = 8 mice per group (D) and n = 7 – 8 per group except baseline n = 3 mice per group (E, F) from n = 2 experiments. (D) Ratio indicate number of mice with diarrhea: total number of mice. **** P < 0.0001, ** P < 0.01, * P < 0.05, ns > 0.05.
Discussion
Clinical studies suggest a relationship between increased levels of IL-4 and histamine and severity of anaphylaxis 22, 57, 58. This was supported animal-based studies indicating that IL-4 can exacerbate IgE-mediated reactions 31. IL-4 alone has been shown to induce vascular endothelial barrier dysfunction in vitro 27, 28. However, the cellular target of IL-4 action and the underlying IL-4Rα-dependent signaling processes involved in the amplification of the IgE-MC-histamine driven VE barrier dysfunction and fluid extravasation was not previously fully understood.
Herein, we show that: 1) IL-4 increased histamine-induced fluid extravasation, and this increase positively correlated with anaphylaxis severity (hypovolemic shock); 2) IL-4 enhancement of histamine- and IgE/MC-induced systemic anaphylaxis symptoms was dependent on signaling through the VE IL-4Rα chain; 3) IL-4 enhanced histamine-induced ABL1 activity and VE barrier dysfunction in human VE cells, which could be antagonized by ABL kinase inhibition; and 4) pharmacologic inhibition of ABL kinases by imatinib protected mice from passive and established oral antigen–induced anaphylaxis via suppressing VE barrier dysfunction and fluid extravasation. 59, 60
Clinical evidence supports possible involvement of hypovolemia and distributive and cardiogenic shock in human anaphylaxis 7, however mounting evidence identifies hypovolemic shock as an important contributor to the cardiovascular collapse that leads to the severe, life-threatening anaphylactic phenotype. 59–61. Consistent with these observations, aggressive fluid resuscitation has been shown to be an effective treatment for anaphylaxis occurring under anesthesia 8, 60. Similarly, In the mouse, anaphylaxis causes fluid extravasation, leading to severe hypovolemia and fatal tissue hypoxia 62, 63. IgE-mediated MC activation promotes the secretion of vasoactive amines (histamine and serotonin) and lipid mediators (leukotrienes, prostaglandins) that are thought to induce VE dysfunction and the hypovolemic shock 64, 65. Consistent with this observation, histamine treatment of mice increases hemoconcentration and hypovolemia-induced shock 31 and conversely, treating mice with H1R antagonists with or without concurrent H2R blockade attenuates IgE/MC-mediated hypovolemic shock 25, 66. Herein, we demonstrate that IL-4 can amplify the IgE-mediated, histamine-induced fluid extravasation through priming of the vascular endothelial compartment. Previous studies have revealed an important role for IL-4 and IL-4Rα signaling in IgE-mediated anaphylaxis; however, this was predominantly thought to be through its effect on the hematopoietic compartment driving IgE-isotype switch and the associated CD4+ Th2 and mast cell response in anaphylaxis 31, 67–69. We show that IL-4 also acts on the non-hematopoietic compartment, namely the VE to increase the sensitivity of the VE to histamine. This is supported by our observation, that levels of mMCPT-1 were comparable between iIL-9Tg VEΔIL4Rα and iIL-9Tg VEIL-4Rα WT even though the iIL-9Tg VEIL-4Rα WT mice experienced greater hypovolemic shock. Notably, the mice lacking the expression of the IL-4Rα chain on their VE developed histamine-induced anaphylactic shock; however, they were protected from the IL-4–induced amplification of anaphylaxis symptoms, suggesting that VE IL-4Rα chain signaling is not required for histamine-induced hypovolemia but exacerbates the histamine-dependent component.
IL-4 can signal through either the type I (IL-4Rα chain and γc chain) and type II (IL-4Rα chain and IL-13Rα1 chain) IL-4 receptor pathways. The previous demonstration of IL-4– enhanced anaphylaxis in RAG2/γc–deficient mice (which lack B cells, T cells, MCs, eosinophils, and NK cells) indicates the involvement of the IL-4 type II receptor and not the IL-4 type I receptor 31. Interestingly, mutations in the IL4RA gene have been associated with susceptibility to atopic disease 45–50. Importantly, gain-of-function mutations in the IL-4Rα chain (E375A, S478P, or Q551R) have been linked with enhanced allergic inflammatory responses including increased MCs and elevated IgE levels 45–50; Consistent with these observations, murine-based studies have revealed that mutations in the IL-4Rα that promote gain-of-function (IL-4RαY709F, IL-4RαQ576R, and IL-4RαY500F) led to enhanced allergic inflammatory phenotypes including asthma, IgE-mediated responses, and food allergy 33, 70–73. We show that pretreating IL-4RαY709F mice with IL-4 enhanced histamine-induced hypovolemic shock, suggesting that gain-of-function mutations can increase severity of food-induced anaphylaxis. Relevant to this study, Burton et al. and Mathias et al. demonstrated that IL-4RαY709F mice have an enhanced sensitivity to food allergens and that the enhanced IgE/MC-mediated systemic anaphylaxis in IL-4RαY709F mice is due to a direct effect on MCs 71, 74. Of note, Burton et al. showed that IL-4RαY709F mice had a similar responsiveness to histamine to that of WT mice, suggesting no role for gain-of-function mutations in the IL-4Rα chain in severity of anaphylaxis 74. However, these studies were performed in the absence of IL-4 treatment. We show enhanced histamine-induced anaphylaxis with exogenous IL-4 treatment in IL-4RαY709F mice, suggesting that gain-of- function mutations in IL-4Rα signaling can affect not only hematopoietic cell function but also non-hematopoietic cell function and severity of food-induced reactions.
We have previously demonstrated that the onset of a severe food-induced anaphylaxis in mice requires increased type 2 cytokines, food-specific IgE, and an intestinal mastocytosis 23. Antigen sensitization and repeated food antigen challenge of sensitized mice promotes an intestinal antigen-specific CD4+ Th2 cell/ILC2 response, which is thought to drive IL-9 producing mucosal MCs (MMC9 cells) and lead to the intestinal mastocytosis 75, 76. We speculate that the repeated antigen challenge promotes heightened IL-4 levels, leading to the increased sensitivity of the VE compartment to MC-derived mediators. The cellular source of IL-4 during these allergic conditions is likely to be CD4+ T cells and basophils 77, 78. Repeated oral antigen exposure drives the antigen-specific CD4+ type 2 response and generation of IL-4–dependent, antigen-specific IgE and mastocytosis 75, 76, 79. Concurrently, oral antigen exposure will also lead to IgE-FcεRI ligation on basophils and MCs. The demonstration that IgE-mediated IL-4 responses are dependent on FcεRI and can occur independently of MCs but not basophils suggest that basophils are another likely source of IL-4 77, 78, 80. Studies of mediators in human anaphylaxis revealed that in patients with anaphylaxis, histamine levels peaked during emergency department arrival and that histamine levels correlated with reaction severity 22. From our observations, one would predict that IL-4 levels should also correlate with reaction severity; however, IL-4 has previously been reported to not correlate with reaction severity. Interestingly, the most sensitive known in vivo effect of IL-4 is upregulation of B cell MHC II expression 39, 81, and IL-4 doses barely capable of inducing an increase in B cell class II MHC expression have been shown to be sufficient to enhance anaphylaxis 31. These data suggest that incremental changes in IL-4 may be sufficient to enhance histamine-induced VE dysfunction and anaphylaxis severity and may explain the lack of association between IL-4 levels and severity of anaphylaxis in humans.
The mechanism by which IL-4 modulates histamine responses is not fully elucidated. We show that IL-4 amplifies histamine-induced ABL1 kinase activation and that blockade of ABL1 kinase genetically or pharmacologically abrogated histamine-induced VE dysfunction and leak, suggesting that IL-4 modulation of histamine-induced responses is likely through increased ABL1 activity. The ABL family of non-receptor tyrosine kinases, including both ABL1 and ARG, have been implicated in the regulation of endothelial barrier function 6. Notably, Chislock et al. demonstrated a requirement for activation of the ABL kinases in endothelial permeability induced by thrombin and Vascular endothelial growth factor (VEGF) in human microvascular endothelial cells (HMVECs) 15. VE-cadherin is a critical requirement for assembly and stability of the VE-cadherin–catenin–cytoskeleton complex and maintenance of endothelial barrier function 12, 13, 82. Histamine is thought to destabilize the VE-cadherin–catenin complex through activating protein kinase C (PKC) and Rho-associated protein kinase (ROCK) and subsequent downstream activation of myosin light chain (MLC), resulting in reduced endothelial cell adhesiveness and increased paracellular permeability 83. Pharmacologic and genetic blockade of ABL1 has been shown to reduce myosin light chain kinase (MLCK) phosphorylation and VE barrier dysfunction after VEGF and thrombin stimulation 15.
Collectively, our data suggest that blockade of ABL1 kinase-dependent processes does not ablate anaphylaxis but rather dramatically reduces the severity of the IgE-mediated reaction. Based upon these observations, we suggest that inhibiting ABL1 kinase-dependent processes may be a therapeutic target for diminishing or preventing severe IgE-mediated anaphylactic reactions and could be used in conjunction with desensitization protocols such as sublingual immunotherapy and oral immunotherapy to minimize the risk of possible severe adverse reactions. Recently, there has been significant development of second- and third-generation tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML), including second-generation TKIs (dasatinib, nilotinib, and bosutinib) and third-generation TKIs (Ponatinib and GNF-2, -5, and -7) that act as ATP-binding site competitive inhibitors and allosteric ABL inhibitors that have distinct efficacy, specificity, and long-term safety 84, 85. Given that TKIs such as Imatinib bind and inhibit c-KIT-dependent TK signaling which is essential for MC development and maturation, an added advantage of these class of inhibitors is that they can target both MC levels and the VE compartment to attenuate IgE/MC-mediated reactions 86–88. Consistent with this argument, long-term treatment with imatinib caused a profound MC deficiency in mice and patients with PH+ CML 89. Furthermore, a recent study examined the clinical utilization of Imatinib for treatment of severe refractory asthmatic patients and demonstrated that Imatinib had excellent efficacy, reducing mast cell counts and activation and asthma symptoms 90.
In conclusion, we show that IL-4 enhances histamine responses through the VE IL-4Rα chain by modulating VE function and amplifying vascular leakage and anaphylaxis phenotypes. We demonstrated that IgE-mediated fluid extravasation and hypovolemic shock in mice are mediated by a histamine-induced, VE ABL kinase–dependent mechanism, possibly via ABL1 kinase. These studies identify that targeting of the ABL1- sensitive VE compartment may be a new pathway for therapeutic intervention and prevention of food-induced severe and life-threatening anaphylaxis and that repurposing TKI inhibitors such as imatinib may prevent food-induced reactions.
Supplementary Material
Key Messages.
IL-4 amplifies IgE- and histamine-induced hypovolemic shock.
IL-4 amplification of the shock response is dependent on vascular endothelial expression of ABL1 and IL-4Rα.
Utilization of the ABL kinase inhibitor imatinib prevented the development of severe IgE-mediated anaphylaxis.
Acknowledgments
Grant support: This work was supported by NIH R01 AI073553 and DK090119 and Food Allergy Research & Education.
We thank members of the Divisions of Allergy and Immunology Cincinnati Children’s Hospital Medical Center for critical review of the manuscript and insightful conversations. We would also like to thank Shawna Hottinger for editorial assistance and manuscript preparation. This work was supported by NIH R01 AI073553, R01 DK090119, P30DK078392; U19A1070235 and Food Allergy Research Education Award (S.P.H).
Abbreviations
- HRP
Horseradish peroxidase
- iIL-9Tg
intestinal interleukin 9 transgenic mice
- IL-4C
Interleukin-4 complex
- IL-4Rα
Interlaken 4 receptor alpha chain
- mMCPT-1
Mouse mast cells protease-1
- OVA-TNP
Ovalbumin- 2,4,6-trinitrophenol
- shRNA
Short hairpin RNA
- TER
Transendotheilial resistance
- VE
Vascular endothelial
References
- 1.Sicherer SH, Sampson HA. Food allergy: Epidemiology, pathogenesis, diagnosis, and treatment. J Allergy Clin Immunol. 2014;133:291–307. doi: 10.1016/j.jaci.2013.11.020. quiz 8. [DOI] [PubMed] [Google Scholar]
- 2.Rudders SA, Arias SA, Camargo CA., Jr Trends in hospitalizations for food-induced anaphylaxis in US children, 2000–2009. J Allergy Clin Immunol. 2014;134:960–2. e3. doi: 10.1016/j.jaci.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 3.Sampson HA, Munoz-Furlong A, Bock SA, Schmitt C, Bass R, Chowdhury BA, et al. Symposium on the definition and management of anaphylaxis: summary report. J Allergy Clin Immunol. 2005;115:584–91. doi: 10.1016/j.jaci.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Sampson HA, Munoz-Furlong A, Campbell RL, Adkinson NF, Jr, Bock SA, Branum A, et al. Second symposium on the definition and management of anaphylaxis: summary report--second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med. 2006;47:373–80. doi: 10.1016/j.annemergmed.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Sampson HA. Food-induced anaphylaxis. Novartis Found Symp. 2004;257:161–71. discussion 71–6, 207–10, 76–85. [PubMed] [Google Scholar]
- 6.Silverman HJ, Van Hook C, Haponik EF. Hemodynamic changes in human anaphylaxis. Am J Med. 1984;77:341–4. doi: 10.1016/0002-9343(84)90717-4. [DOI] [PubMed] [Google Scholar]
- 7.Brown SG. The pathophysiology of shock in anaphylaxis. Immunol Allergy Clin North Am. 2007;27:165–75. doi: 10.1016/j.iac.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Fisher MM. Clinical observations on the pathophysiology and treatment of anaphylactic cardiovascular collapse. Anaesth Intensive Care. 1986;14:17–21. doi: 10.1177/0310057X8601400105. [DOI] [PubMed] [Google Scholar]
- 9.Beaupre PN, Roizen MF, Cahalan MK, Alpert RA, Cassorla L, Schiller NB. Hemodynamic and two-dimensional transesophageal echocardiographic analysis of an anaphylactic reaction in a human. Anesthesiology. 1984;60:482–4. doi: 10.1097/00000542-198405000-00017. [DOI] [PubMed] [Google Scholar]
- 10.Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–41. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 11.Gavard J. Endothelial permeability and VE-cadherin: a wacky comradeship. Cell Adh Migr. 2014;8:158–64. doi: 10.4161/cam.29026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–57. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 13.Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, Dejana E, et al. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood. 2005;105:2771–6. doi: 10.1182/blood-2004-06-2244. [DOI] [PubMed] [Google Scholar]
- 14.Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87:272–80. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chislock EM, Pendergast AM. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS One. 2013;8:e85231. doi: 10.1371/journal.pone.0085231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, et al. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation. 2012;126:2728–38. doi: 10.1161/CIRCULATIONAHA.112.134304. [DOI] [PubMed] [Google Scholar]
- 17.Alcaide P, Martinelli R, Newton G, Williams MR, Adam A, Vincent PA, et al. p120-Catenin prevents neutrophil transmigration independently of RhoA inhibition by impairing Src dependent VE-cadherin phosphorylation. Am J Physiol Cell Physiol. 2012;303:C385–95. doi: 10.1152/ajpcell.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikezoe T, Yang J, Nishioka C, Umezawa K, Yokoyama A. Thrombomodulin blocks calcineurin inhibitor-induced vascular permeability via inhibition of Src/VE-cadherin axis. Bone Marrow Transplant. 2017;52:245–51. doi: 10.1038/bmt.2016.241. [DOI] [PubMed] [Google Scholar]
- 19.Zhang WJ, Li PX, Guo XH, Huang QB. Role of moesin, Src, and ROS in advanced glycation end product-induced vascular endothelial dysfunction. Microcirculation. 2017:24. doi: 10.1111/micc.12358. [DOI] [PubMed] [Google Scholar]
- 20.Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol (Oxf) 2017 doi: 10.1111/apha.12860. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa Y, Grant JA. Mediators of anaphylaxis. Immunol Allergy Clin North Am. 2007;27:249–60. vii. doi: 10.1016/j.iac.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Stone SF, Cotterell C, Isbister GK, Holdgate A, Brown SG Emergency Department Anaphylaxis I. Elevated serum cytokines during human anaphylaxis: Identification of potential mediators of acute allergic reactions. J Allergy Clin Immunol. 2009;124:786–92. e4. doi: 10.1016/j.jaci.2009.07.055. [DOI] [PubMed] [Google Scholar]
- 23.Ahrens R, Osterfeld H, Wu D, Chen CY, Arumugam M, Groschwitz K, et al. Intestinal mast cell levels control severity of oral antigen-induced anaphylaxis in mice. Am J Pathol. 2012;180:1535–46. doi: 10.1016/j.ajpath.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulton JD, Harris WE, Craft CE. Hematocrit chnage as indication of anaphyactic shock in the mouse. Proc Soc Exp Biol Med. 1957;95:625–7. doi: 10.3181/00379727-95-23309. [DOI] [PubMed] [Google Scholar]
- 25.Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol. 2007;120:506–15. doi: 10.1016/j.jaci.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 26.Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol. 2002;109:658–68. doi: 10.1067/mai.2002.123302. [DOI] [PubMed] [Google Scholar]
- 27.Skaria T, Burgener J, Bachli E, Schoedon G. IL-4 Causes Hyperpermeability of Vascular Endothelial Cells through Wnt5A Signaling. PLoS One. 2016;11:e0156002. doi: 10.1371/journal.pone.0156002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotowicz K, Callard RE, Klein NJ, Jacobs MG. Interleukin-4 increases the permeability of human endothelial cells in culture. Clin Exp Allergy. 2004;34:445–9. doi: 10.1111/j.1365-2222.2004.01902.x. [DOI] [PubMed] [Google Scholar]
- 29.Chalubinski M, Wojdan K, Luczak E, Gorzelak P, Borowiec M, Gajewski A, et al. IL-33 and IL-4 impair barrier functions of human vascular endothelium via different mechanisms. Vascul Pharmacol. 2015;73:57–63. doi: 10.1016/j.vph.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Strait R, Morrist SC, Finkelman FD. Cytokine enhancement of anaphylaxis. Novartis Found Symp. 2004;257:80–91. discussion -100, 276–85. [PubMed] [Google Scholar]
- 31.Strait RT, Morris SC, Smiley K, Urban JF, Jr, Finkelman FD. IL-4 exacerbates anaphylaxis. J Immunol. 2003;170:3835–42. doi: 10.4049/jimmunol.170.7.3835. [DOI] [PubMed] [Google Scholar]
- 32.Forbes EE, Groschwitz K, Abonia JP, Brandt EB, Cohen E, Blanchard C, et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J Exp Med. 2008;205:897–913. doi: 10.1084/jem.20071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tachdjian R, Al Khatib S, Schwinglshackl A, Kim HS, Chen A, Blasioli J, et al. In vivo regulation of the allergic response by the IL-4 receptor alpha chain immunoreceptor tyrosine-based inhibitory motif. J Allergy Clin Immunol. 2010;125:1128–36. e8. doi: 10.1016/j.jaci.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, et al. VE-Cadherin- Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–67. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 35.Moresco EM, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25:6105–18. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2- Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 37.Baniyash M, Eshhar Z. Inhibition of IgE binding to mast cells and basophils by monoclonal antibodies to murine IgE. Eur J Immunol. 1984;14:799–807. doi: 10.1002/eji.1830140907. [DOI] [PubMed] [Google Scholar]
- 38.Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol. 2002;109:658–68. doi: 10.1067/mai.2002.123302. [DOI] [PubMed] [Google Scholar]
- 39.Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, et al. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151:1235–44. [PubMed] [Google Scholar]
- 40.Bouis D, Hospers GA, Meijer C, Molema G, Mulder NH. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis. 2001;4:91–102. doi: 10.1023/a:1012259529167. [DOI] [PubMed] [Google Scholar]
- 41.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–7. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, et al. Abl tyrosine kinase phosphorylates nonmuscle Myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21:4042–56. doi: 10.1091/mbc.E09-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu D, Ahrens R, Osterfeld H, Noah TK, Groschwitz K, Foster PS, et al. Interleukin-13 (IL-13)/IL-13 receptor alpha1 (IL-13Ralpha1) signaling regulates intestinal epithelial cystic fibrosis transmembrane conductance regulator channel-dependent Cl- secretion. J Biol Chem. 2011;286:13357–69. doi: 10.1074/jbc.M110.214965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noah TK, Kazanjian A, Whitsett J, Shroyer NF. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp Cell Res. 2010;316:452–65. doi: 10.1016/j.yexcr.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandford AJ, Chagani T, Zhu S, Weir TD, Bai TR, Spinelli JJ, et al. Polymorphisms in the IL4, IL4RA, and FCERIB genes and asthma severity. J Allergy Clin Immunol. 2000;106:135–40. doi: 10.1067/mai.2000.107926. [DOI] [PubMed] [Google Scholar]
- 46.Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, et al. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genet. 2002;70:230–6. doi: 10.1086/338242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hershey GK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA. The association of atopy with a gain-of-function mutation in the alpha subunit of the interleukin-4 receptor. N Engl J Med. 1997;337:1720–5. doi: 10.1056/NEJM199712113372403. [DOI] [PubMed] [Google Scholar]
- 48.Faffe DS, Whitehead T, Moore PE, Baraldo S, Flynt L, Bourgeois K, et al. IL-13 and IL-4 promote TARC release in human airway smooth muscle cells: role of IL-4 receptor genotype. Am J Physiol Lung Cell Mol Physiol. 2003;285:L907–14. doi: 10.1152/ajplung.00120.2003. [DOI] [PubMed] [Google Scholar]
- 49.Risma KA, Wang N, Andrews RP, Cunningham CM, Ericksen MB, Bernstein JA, et al. V75R576 IL-4 receptor alpha is associated with allergic asthma and enhanced IL-4 receptor function. J Immunol. 2002;169:1604–10. doi: 10.4049/jimmunol.169.3.1604. [DOI] [PubMed] [Google Scholar]
- 50.Wenzel SE, Balzar S, Ampleford E, Hawkins GA, Busse WW, Calhoun WJ, et al. IL4R alpha mutations are associated with asthma exacerbations and mast cell/IgE expression. Am J Respir Crit Care Med. 2007;175:570–6. doi: 10.1164/rccm.200607-909OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahrens R, Osterfeld H, Wu D, Chen C-Y, Groschwitz K, Arumugam M, et al. Intestinal mast cell levels control severity of oral antigen-induced anaphylaxis in mice. Am J Pathol. 2012;180:1535–46. doi: 10.1016/j.ajpath.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun. 2015;6:6725. doi: 10.1038/ncomms7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyseng-Williamson K, Jarvis B. Imatinib. Drugs. 2001;61:1765–74. doi: 10.2165/00003495-200161120-00007. discussion 75–6. [DOI] [PubMed] [Google Scholar]
- 54.Iwaki S, Tkaczyk C, Satterthwaite AB, Halcomb K, Beaven MA, Metcalfe DD, et al. Btk plays a crucial role in the amplification of Fc epsilonRI-mediated mast cell activation by kit. J Biol Chem. 2005;280:40261–70. doi: 10.1074/jbc.M506063200. [DOI] [PubMed] [Google Scholar]
- 55.Forbes EE, Groschwitz K, Abonia JP, Brandt EB, Cohen E, Blanchard C, et al. IL-9– and mast cell–mediated intestinal permeability predisposes to oral antigen hypersensitivity. J Exp Med. 2008;205:897–913. doi: 10.1084/jem.20071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osterfeld H, Ahrens R, Strait R, Finkelman FD, Renauld JC, Hogan SP. Differential roles for the IL-9/IL-9 receptor alpha-chain pathway in systemic and oral antigen-induced anaphylaxis. J Allergy Clin Immunol. 2010;125:469–76. e2. doi: 10.1016/j.jaci.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogawa Y, Grant JA. Mediators of anaphylaxis. Immunol Allergy Clin North Am. 2007;27:249–60. doi: 10.1016/j.iac.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 58.Brown SG, Stone SF, Fatovich DM, Burrows SA, Holdgate A, Celenza A, et al. Anaphylaxis: clinical patterns, mediator release, and severity. J Allergy Clin Immunol. 2013;132:1141–9. e5. doi: 10.1016/j.jaci.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 59.Sonin L, Grammer LC, Greenberger PA, Patterson R. Idiopathic anaphylaxis. A clinical Summary. Annals Intern Med. 1983;99:634–5. doi: 10.7326/0003-4819-99-5-634. [DOI] [PubMed] [Google Scholar]
- 60.Fisher M. Blood volume replacement in acute anaphylactic cardiovascular collapse related to anaesthesia. Br J Anaesth. 1977;49:1023–6. doi: 10.1093/bja/49.10.1023. [DOI] [PubMed] [Google Scholar]
- 61.Black JH, Kemp HA. Blood density in anaphylaxis in hay fever artificially induced. Am J Clin Pathol. 1937;7:300. [Google Scholar]
- 62.Munoz J, Bergman RK. MECHANISM OF ANAPHYLACTIC DEATH IN THE MOUSE. Nature. 1965;205:199–200. doi: 10.1038/205199b0. [DOI] [PubMed] [Google Scholar]
- 63.Bergmann RK, Munoz J. Circulatory chnages in anaphylaxis and histamine toxicity in mice. J Immunol. 1965;95:1–8. [PubMed] [Google Scholar]
- 64.Osher E, Weisinger G, Limor R, Tordjman K, Stern N. The 5 lipoxygenase system in the vasculature: emerging role in health and disease. Mol Cell Endocrinol. 2006;252:201–6. doi: 10.1016/j.mce.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 65.Williams TJ, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–7. doi: 10.1038/246215a0. [DOI] [PubMed] [Google Scholar]
- 66.Wechsler JB, Schroeder HA, Byrne AJ, Chien KB, Bryce PJ. Anaphylactic responses to histamine in mice utilize both histamine receptors 1 and 2. Allergy. 2013;68:1338–40. doi: 10.1111/all.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burton OT, Darling AR, Zhou JS, Noval-Rivas M, Jones TG, Gurish MF, et al. Direct effects of IL-4 on mast cells drive their intestinal expansion and increase susceptibility to anaphylaxis in a murine model of food allergy. Mucosal Immunol. 2013;6:740–50. doi: 10.1038/mi.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tordesillas L, Berin MC, Sampson HA. Immunology of Food Allergy. Immunity. 2017;47:32–50. doi: 10.1016/j.immuni.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 69.Sampath V, Tupa D, Graham MT, Chatila TA, Spergel JM, Nadeau KC. Deciphering the black box of food allergy mechanisms. Ann Allergy Asthma Immunol. 2017;118:21–7. doi: 10.1016/j.anai.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tachdjian R, Mathias C, Al Khatib S, Bryce PJ, Kim HS, Blaeser F, et al. Pathogenicity of a disease-associated human IL-4 receptor allele in experimental asthma. J Exp Med. 2009;206:2191–204. doi: 10.1084/jem.20091480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathias CB, Hobson SA, Garcia-Lloret M, Lawson G, Poddighe D, Freyschmidt EJ, et al. IgE-mediated systemic anaphylaxis and impaired tolerance to food antigens in mice with enhanced IL-4 receptor signaling. J Allergy Clin Immunol. 2011;127:795–805. e1–6. doi: 10.1016/j.jaci.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blaeser F, Bryce PJ, Ho N, Raman V, Dedeoglu F, Donaldson DD, et al. Targeted inactivation of the IL-4 receptor alpha chain I4R motif promotes allergic airway inflammation. J Exp Med. 2003;198:1189–200. doi: 10.1084/jem.20030471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sledd J, Wu D, Ahrens R, Lee J, Waggoner L, Tsai YT, et al. Loss of IL-4Ralpha-mediated PI3K signaling accelerates the progression of IgE/mast cell-mediated reactions. Immun Inflamm Dis. 2015;3:420–30. doi: 10.1002/iid3.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burton OT, Darling AR, Zhou JS, Noval-Rivas M, Jones TG, Gurish MF, et al. Direct effects of IL-4 on mast cells drive their intestinal expansion and increase susceptibility to anaphylaxis in a murine model of food allergy. Mucosal Immunol. 2012 doi: 10.1038/mi.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen CY, Lee JB, Liu B, Ohta S, Wang PY, Kartashov A, et al. Induction of Interleukin-9- producing Mucosal Mast cells Promotes Susceptibility to IgE-mediated Experimental Food Allergy. Immunity. 2015;43:788–802. doi: 10.1016/j.immuni.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee J, CC, Liu B, Mugge L, Angkasekwinai P, Facchinetti V, et al. IL-25 and CD4+ Th2 cells enhance ILC2-derived IL-13 production that promotes IgE-mediated experimental food allergy. JACI. 2015 doi: 10.1016/j.jaci.2015.09.019. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng LE, Sullivan BM, Retana LE, Allen CD, Liang HE, Locksley RM. IgE-activated basophils regulate eosinophil tissue entry by modulating endothelial function. J Exp Med. 2015;212:513–24. doi: 10.1084/jem.20141671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sullivan BM, Liang HE, Bando JK, Wu D, Cheng LE, McKerrow JK, et al. Genetic analysis of basophil function in vivo. Nat Immunol. 2011;12:527–35. doi: 10.1038/ni.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brandt EB, Munitz A, Orekov T, Mingler MK, McBride M, Finkelman FD, et al. Targeting IL-4/IL-13 signaling to alleviate oral allergen-induced diarrhea. J Allergy Clin Immunol. 2009;123:53–8. doi: 10.1016/j.jaci.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khodoun MV, Orekhova T, Potter C, Morris S, Finkelman FD. Basophils initiate IL-4 production during a memory T-dependent response. J Exp Med. 2004;200:857–70. doi: 10.1084/jem.20040598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noelle R, Krammer PH, Ohara J, Uhr JW, Vitetta ES. Increased expression of Ia antigens on resting B cells: an additional role for B-cell growth factor. Proc Natl Acad Sci U S A. 1984;81:6149–53. doi: 10.1073/pnas.81.19.6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhasin M, Yuan L, Keskin DB, Otu HH, Libermann TA, Oettgen P. Bioinformatic identification and characterization of human endothelial cell-restricted genes. BMC Genomics. 2010;11:342. doi: 10.1186/1471-2164-11-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–97. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 84.Jabbour E, Kantarjian H, Cortes J. Use of second- and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: an evolving treatment paradigm. Clin Lymphoma Myeloma Leuk. 2015;15:323–34. doi: 10.1016/j.clml.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–63. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 87.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925–32. [PubMed] [Google Scholar]
- 88.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–8. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 89.Cerny-Reiterer S, Rabenhorst A, Stefanzl G, Herndlhofer S, Hoermann G, Mullauer L, et al. Long-term treatment with imatinib results in profound mast cell deficiency in Ph+ chronic myeloid leukemia. Oncotarget. 2015;6:3071–84. doi: 10.18632/oncotarget.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A, et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N Engl J Med. 2017;376:1911–20. doi: 10.1056/NEJMoa1613125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.