

SUMMARY
While microalgae are a promising feedstock for production of fuels and other chemicals, a challenge for the algal bioproducts industry is obtaining consistent, robust algae growth. Algal cultures include complex bacterial communities and can be difficult to manage because specific bacteria can promote or reduce algae growth. To overcome bacterial contamination, algae growers may use closed photobioreactors designed to reduce the number of contaminant organisms. Even with closed systems, bacteria are known to enter and cohabitate, but little is known about these communities. Therefore, the richness, structure, and composition of bacterial communities were characterized in closed photobioreactor cultivations of Nannochloropsis salina in F/2 medium at different scales, across nine months spanning late summer–early spring, and during a sequence of serially inoculated cultivations. Using 16S rRNA sequence data from 275 samples, bacterial communities in small, medium, and large cultures were shown to be significantly different. Larger systems contained richer bacterial communities compared to smaller systems. Relationships between bacterial communities and algae growth were complex. On one hand, blooms of a specific bacterial type were observed in three abnormal, poorly performing replicate cultivations, while on the other, notable changes in the bacterial community structures were observed in a series of serial large-scale batch cultivations that had similar growth rates. Bacteria common to the majority of samples were identified, including a single OTU within the class Saprospirae that was found in all samples. This study contributes important information for crop protection in algae systems, and demonstrates the complex ecosystems that need to be understood for consistent, successful industrial algae cultivation. This is the first study to profile bacterial communities during the scale-up process of industrial algae systems.
INTRODUCTION
Microalgae (herein, “algae”) are photosynthetic unicellular eukaryotes that grow in aquatic or marine environments. For reasons including rapid growth and high lipid content, certain varieties of algae are considered promising biofuels feedstocks (Chen et al., 2011; Mata et al., 2010). Algae may be cultivated on otherwise non-arable land in growth systems that use salt water or wastewater, so production of algae biomass does not necessarily divert land and fresh water from production of traditional agricultural crops (Shurin et al., 2013). Generally, large-scale industrial growth systems circulate algae, nutrients, and water around open ponds or within closed photobioreactors. Open ponds use a paddle wheel to circulate algae around a constantly exposed raceway. In closed systems, algae cultures are confined in bags or tubes that reduce exposure to the environment. Closed systems have higher capital costs but allow greater control over parameters such as CO2 and nutrient concentrations while limiting the potential for invasion by unwanted organisms (Grobbelaar, 2009; Slade and Bauen, 2013).
Growers typically desire to cultivate monocultures of algae selected or engineered for traits such as robust growth and accumulation of desired biochemical products (e.g., lipids or other high-value compounds) (Shurin et al., 2013). Following conventions used with traditional agricultural crops, these high performance algae varieties may be referred to as “elite”. For production of lipids, several commonly used elite strains are members of Nannochloropsis, a genus of marine algae with doubling times on the order of 30 h and lipid contents ranging from 30–60% (Griffiths et al., 2012; Rodolfi et al., 2009). Algae growth parameters are often studied and optimized using laboratory conditions including small-volume cultures, aseptic conditions, and precisely controlled light, temperature and nutrient regimes. Since elite algae have not historically been grown at the large volumes required by the biofuels industry (Fishman et al., 2010), a challenge is translating the productivity of elite strains optimized under highly controlled lab environments to consistent outdoor culture productivity at large scales.
Much like terrestrial crops, algae productivity may be modulated by biotic factors such as weeds, predators, and other microbes. For example, algae with low lipid content that contaminate elite cultures–and compete for resources such as light and nutrients–are considered weeds (Fulbright et al., 2014). Zooplankton grazers prey on small algae (Smith and Crews, 2014) such as Nannochloropsis. Fungi and bacteria also affect algae productivity (Smith and Crews, 2014; Lakaniemi et al., 2012); however, there is little understanding of the interactions among elite algae and co-resident microbes. The majority of algae pathogens and pests have not been identified, and industry pest management standards are at an early stage of development (Letcher et al., 2013, Fulbright et al., 2016).
Bacteria are abundant and dynamic in algae cultures, and bacterial counts commonly reach 1 × 109 cells/mL, outnumbering algae cells 10- to 100-fold (Wang et al., 2016). Although bacteria are often considered contaminants that can inhibit algae productivity or terminate algae populations, bacteria-algae interactions have a range of potential outcomes (Lakaniemi et al., 2012; Skerrat et al., 2002; Lee et al., 2000; Mayali and Azam, 2004). Algae support bacterial growth by releasing 25% of the total organic carbon fixed by photosynthesis (Rooney-Varga et al., 2005; Lakaniemi et al., 2012). Reciprocally, of hundreds of algae varieties surveyed, over half do not endogenously produce vitamin B12 and therefore require bacteria-produced vitamin B12 for growth (Croft et al., 2005). Additionally, specific bacteria may stimulate algae growth through activities including regulation of the amount of available nutrients such as iron, nitrogen, and phosphates (Amin et al., 2009; Foster et al., 2011; Reijnders, 2008), or by releasing phytohormones such as indole-3-acetic acid into the growth environment (De-Bashan et al., 2008). In some instances, bacteria reduce algae productivity by competing for these same nutrients (Cole, 1982; Kazamia et al., 2012). In addition to nutrient competition, non-lethal bacterial pathogens may inhibit algae productivity by diverting algal cellular resources from growth to defense. Finally, some bacteria can directly kill algae, causing cultures to collapse (Wang et al., 2012; Lewin, 1997). Much of this knowledge of algae-bacteria interactions derives from ecological studies of harmful algal blooms in natural environments, with the general aims of identifying bacteria or specific bacterial functions that promote or inhibit such blooms. Of immediate need for the algae bioproducts industry is an understanding of the relationships among elite algae and co-resident bacteria in engineered cultivation systems containing high concentrations of cells and nutrients.
In this study, bacterial communities were monitored during industrial algae production at Solix Biosystems (Fort Collins, CO). At this facility, production involves scale-up from 5-mL algae cultures grown under aseptic conditions to 200-L cultures grown in closed, but not aseptic, photobioreactors. Smaller cultures are used to inoculate larger ones until the 200-L scale is reached. Small cultures of 4 L or less are kept under aseptic laboratory conditions, including sterilized glassware and media, with all handling of open containers occurring in a laminar flow hood. These small cultures are grown under artificial light sources in shaking incubators or on shaking platforms. Medium cultures (20 to 60 L) are grown in flat-panel bioreactors under ambient light in a greenhouse, whereas large cultures (200 L) are grown in closed photobioreactors in an outdoor water basin under natural light. Though medium and large cultivations are grown in closed systems, handling of these cultures involves system components that are not sterile. In addition to opportunities for microbe entry during culture handling, the medium and large closed growth systems are technically more difficult to isolate from microbes in their environment.
It was hypothesized that bacterial communities would differ across growth system scales, across seasonal changes in environmental conditions, and in algae cultivations exhibiting different algae growth rates. To monitor bacterial communities in these N. salina cultivation systems, 275 samples were collected from small, medium, and large cultivations over 244 days. From these samples, a region of bacterial 16S rRNA was amplified and sequenced, and the composition, structure, and richness of bacterial communities associated with N. salina were determined. Although different growth systems contained distinct bacterial communities, 16 bacterial OTU were identified in 90% of N. salina cultivations, including a single OTU found in all samples. Differences in community composition were observed across N. salina growth systems, across the duration of the experiment, and among replicate large-scale cultivations supporting different algae growth rates. Relationships between bacterial community structure and algae growth rates were evaluated.
MATERIALS AND METHODS
Algae growth systems
All samples were collected from cultivations of Nannochloropsis salina at a single growth facility operated by Solix Biosystems (Fort Collins, CO). N. salina was originally obtained from the Provasoli-Guillard National Center for Marine Algae and Microbiota (formerly, Center for Culture of Marine Phytoplankton, CCMP) (Bigelow Laboratory for Ocean Sciences, East Boothbay, ME). All algae cultures were grown in F/2 medium (Quinn et al., 2012). To scale up the culture volume (Fig. 1A), a single N. salina colony was isolated from an F/2 agar plate and grown to high density in 5 mL liquid culture. Cultures were primarily grown in a serial batch mode with a portion of each harvest used to inoculate the subsequent cultivations in the same-volume system, or used to start a new cultivation in larger systems. For this study, culture volumes of 5 mL, 1 L, 2 L, and 4 L are all designated as “small”. Sterile technique was used with all small cultures, including growth in sterilized containers and F/2 medium, as well as use of a laminar flow hood during culture handling. Small cultures were maintained on a shaker table rotating at 200 rpm and supplemented under 24-hour artificial light at 50 μE. Cultivations designated as “medium” were grown in variable volume (20–60 L) flat-panel bioreactors aerated with 2% CO2 at 2.5 vvm (volume gas per volume liquid per minute) in a greenhouse under ambient light. Cultivations designated as “large” were approximately 200 L and grown in enclosed photobioreactors located outdoors in a water basin in which the temperature was maintained between 19 and 26 °C, and pH was maintained at approximately 7.3. System specifics are provided elsewhere (Fulbright et al., 2014). Flow cytometry was used to evaluate the purity of the algal population, and specifically the presence of a Tetraselmis sp. that had previously been observed at this site. This analysis revealed that the cultivations contained only low levels of this weedy species: 89.9% of the samples had less than 1% of Tetraselmis, 95.3% contained less than 2% of Tetraselmis, and 98.9% (3 samples) contained less than 5% of Tetraselmis (data not shown).
Figure 1. Overview of bacterial communities in N. salina growth systems.
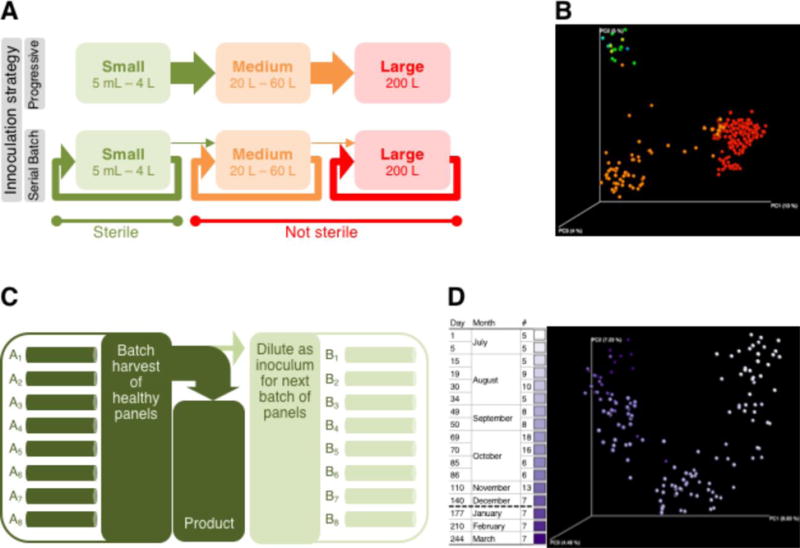
(A & B) Bacterial communities differ across growth scales
(A) Serial batch inoculation strategy of N. salina cultivation systems contrasted with a progressive inoculation strategy. Growth systems are categorized as small, medium or large as illustrated and further described in the text. Shading of system sizes corresponds to coloring in Fig. 1B. Arrow intensities indicate relative movement of inoculum biomass within and between systems.
(B) Principal coordinates analysis plot showing relationships among bacterial communities isolated from algae growth systems. Each point represents the bacterial community isolated in a single sample. Colors indicate samples from small (multi-color, blue–yellow spectrum), medium (orange), and large (red) cultivations.
(C & D) Relationships among bacterial communities in replicate 200-L N. salina cultivations grown in outdoor systems using a serial batch inoculation strategy
(C) Schematic representation of serial batch inoculation strategy for 200-L cultivations in large growth systems. Algae were grown to maturity in individual 200-L cultivations before being harvested in batch. The majority of the harvest was used for product extraction. A portion of the batch harvest was diluted and used to inoculate subsequent replicate panels of algae cultivation.
(D) Principal coordinates analysis plot showing 135 bacterial communities isolated from large N. salina growth systems during a 9-month period. Each point represents the bacterial community in a single sample. Day, day relative to start of experiment; Month, month of sampling day; #, number of large system samples analyzed
Algae cultivation sampling and growth monitoring
A total of 17, 81, and 177 samples were obtained from small, medium and large cultures, respectively. The frequency of sampling varied and not all systems were sampled on every sampling date, but the overall system was sampled at least once per calendar month from July, 2011 to March, 2012. Whenever a particular system scale was sampled, between 2–16 samples were isolated from cultivations within that growth scale. For samples from small cultures, an adjustable pipette was used to transfer 1 mL culture to a microcentrifuge tube in a laminar flow hood. Samples from medium and large systems were drawn using a sterile 10-mL needleless syringe through a non-sterile plastic sample line connected to sample ports at one end of the photobioreactor. To ensure that sample lines and ports were clear of waste material, a 20-mL volume of culture was drawn and discarded. Subsequently, 10 mL of culture were drawn and mixed by inversion, and 1 mL of mixed sample was transferred to a microcentrifuge tube. Sample biomass was pelleted using centrifugation at 15,000 × g. The supernatant was discarded, and the biomass was stored at −80 °C.
Algae culture density was monitored by optical density measured at 750 nm (OD750) using a Hach DR5000 spectrophotometer. Algae growth was estimated using Δ(OD750nm) = OD750(t2) – OD750(t1), where t1 and t2 represent adjacent time points. Additionally, a Guava easyCyte HT+ flow cytometer (EMD Millipore) equipped with an argon laser (488 nm) and 680/30 nm bandpass filter was used to directly count cells in a given volume, identifying algae cells based on size and chlorophyll fluorescence (Fulbright et al., 2014).
Extraction and sequencing of nucleic acids
DNA extractions and 16S rRNA amplification were done according to protocols standardized for the Earth Microbiome Project (EMP; http://www.earthmicrobiome.org/emp-standard-protocols/) (Caporaso et al., 2010). Briefly, community DNA (including algae and bacteria DNA) was extracted from archived biomass using PowerSoil®-htp 96 Well Soil DNA Isolation Kits (MoBio; Carlsbad, CA), and 300–350-bp amplicons from the V3–V4 regions of included 16S rRNA genes were generated by PCR using primers 515f and 806r. Amplification reactions were done in triplicate, and PCR reaction products were pooled prior to sequencing at the BioFrontiers Institute (University of Colorado, Boulder) using an Illumina MiSeq, resulting in 10.9 million 150-bp reads derived from the V3 region of amplicons.
Data processing
QIIME version 1.8.0 was used for all sequence analyses (Caporaso et al., 2010). Sequences were quality filtered and demultiplexed using default settings of the split_libraries_fastq.py QIIME script. Greengenes version 13_5 was used as the reference database for all OTU picking steps (McDonald et al., 2012). Since community DNA extracted from archived samples included significant amounts of algae DNA, sequences were filtered to eliminate reads of chloroplast or mitochondrial origin in two steps: one prior to the main OTU picking step, and one following. For the first filtering procedure, a subset of the Greengenes reference was generated that contained representatives from only mitochondrial and chloroplast clusters (using the 97% similarity Greengenes clusters and associated taxonomy assignments); all 10.9 million sample-derived sequences were assigned to OTUs at 97% similarity using the closed-reference protocol with this reduced reference database of chloroplast and mitochondria sequences; 5.6 million sample sequences that hit were assumed to be derived from algae chloroplasts or mitochondria and were eliminated from analysis. The main OTU picking step used the subsampling open-reference protocol to assign approximately 3.1 million of the remaining 5.3 million sequences to OTUs, using Greengenes 97% clusters and 97% similarity threshold. Approximately 200,000 sequences belonged to OTUs containing fewer than two sequences and were eliminated from further analyses, and a further 2 million sequences that did not align to reference 16S rRNA sequences using PyNAST was used [27]. Some of the new (i.e., non-reference) OTUs were assigned chloroplast or mitochondrial taxonomy; the second filtering step eliminated these OTUs, reducing the sequence for downstream analyses to 2 million out of the initial 10.9 million. An additional filtering step eliminated low abundance OTUs comprising less than 0.005% of the total sequence count.
Comparisons of communities across system scales included 17, 81, and 177 samples from small, medium and large cultures, respectively. The QIIME pipeline was used to identify and count OTUs, to compare relative abundances across scales, and to contrast phylogenetic composition of samples. To ensure even representation across system scales, 1000 amplicon sequences were randomly selected and analyzed for every sample. Using PYNAST, amplicon sequences that met a 97% similarity threshold were clustered together as an OTU (Caporaso, 2010), total numbers of OTUs were quantified, and relative abundance of each OTU was determined for each sample. To summarize the data by system size, relative abundances were averaged for all samples within each scale, resulting in a single relative abundance for each system. To calculate phylogenetic diversity represented within each sample, Faith’s Phylogentic Diversity was used (Faith et al. 1992, Peiffer et al., 2013). Essentially, this measures diversity by adding up all the branch lengths of OTUs found in samples. UniFrac was used to further clarify relationships between samples and systems. UniFrac takes taxa in each sample and places them on a phylogenetic tree. The phylogenetic trees produced from each sample are compared in a pairwise fashion. Taxa found in both samples are considered “shared”, whereas taxa found only in one sample are considered “unshared” (Lozupone & Knight, 2005). The fraction of unshared branch lengths relative to total branch lengths is used as a summary statistic for comparisons (Lozupone et al. 2007). To compare samples with principal coordinates analysis (PCoA), a multivariate statistical test principle coordinate analysis was used (Lozupone et al. 2011, Kuczynski et al., 2011). Computations were done on the Pando supercomputer. Data were deposited in the European Bioinformatics Institute with accession number ERP010414.
For comparisons that only involved samples from large-scale cultivations, 2740 amplicon sequences were randomly selected and analyzed for each sample. Further aspects of these community analyses were done as described above. To compare communities in outdoor, large-scale cultivations across nine months (Fig. 1D), 177 samples were used. Analysis of five large-scale cultivations inoculated using the serial batch strategy (Fig. 2) included five biological replicates (derived from a single inoculum source) per sampling date, with two slower growing cultivations being sampled on two dates each, resulting in comparison of 35 samples. Finally, the comparison of communities in healthy and stagnant large-scale cultivations (Fig. 3) included 13 healthy replicates and 3 stagnant replicates, all derived from the same batch inoculum.
Figure 2. Analysis of five serially inoculated batches of replicate N. salina 200-L cultivations.

Each batch was cultivated between 13 and 21 days before being harvested and used to inoculate the next batch of panels. In total, these batches spanned 77 days. For each sample day, data are presented for five replicate cultivations.
(A) Average algae growth. N. salina density was monitored using OD750. Green line indicates OD750 averaged across five replicate cultivations in each batch; light gray bars indicate one standard deviation. Day, day of batch harvest/inoculation relative to start of experiment; Batch, batch number; Time, length of batch cultivation (in days). Dotted orange vertical lines indicate sampling days; batches 938 and 949 were each sampled twice. Intensity of green shading near base of orange lines represents relative algae density at sampling, as in Fig. 2C. Error bars represent ± 1 standard deviation.
(B) Consensus taxonomy for 16 OTUs conserved among at least 90% of the 275 samples in this study, listed in order of relative abundance in the 35 samples in Fig. 2C. ID, indicates relative rank of abundance and color-coding for bar chart and heat map in Fig. 2C. Ph, Phylum (see Table 1). %, indicates OTUs found in 90%, 95% or 100% of the 275 samples in the entire study; gray and dark gray shading highlight OTUs conserved among 95% and 100% of communities, respectively.
(C) Relative bacterial abundance in replicates of sequentially inoculated large cultivations. Batches are labeled at the top and bottom, and are separated by dotted light-green vertical lines. Order and color-coding of OTU corresponds to Fig. 2B. Top, “Cultivations”: Growth metrics for cultivations in sampled panels. Batch Day, days since inoculation; [Bacteria], cells/mL of bacteria on sampling date, as measured using flow cytometry; [Algae], OD750 at sampling; Growth/d, ΔOD750 averaged across 5 panels and normalized to days of batch growth (Fig. 2A); Total Increase, percent increase in OD750 for each cultivation at harvest relative to inoculation; 24h Increase, percent increase in ΔOD750 for each panel during approximately 24 h preceding sampling. Heat map for “24h Increase” is formatted separately within each batch; other heat maps are formatted across all batches. Middle, “OTU Relative Abundance (%)”: stacked bar graph showing relative abundances; OTU order and color-coding correspond to Fig. 2B; white bars represent the category “Other” (top of stack, regardless of abundance). Bottom, “OTU Abundance, Normalized to Average”: ID, rank and color-coding correspond to Fig. 2B; [OTU], average OTU abundance across the 35 represented cultivations; a heat map is formatted within the [OTU] column (white = 0; orange = maximum). Remaining columns indicate OTU abundance in the sampled cultivation relative to the average abundance (i.e., [OTU]). To highlight OTU variability across cultivations and batches, heat maps are formatted separately for each OTU (blue = minimum; white = 1; yellow = maximum). Asterisk denotes community profiling data from a replicate of Batch 938 for which the statistics likely result from a sample handling or processing error and do not reflect the actual bacterial abundances in this cultivation.
Figure 3. Bacterial communities in healthy and stagnant replicate large cultivations of N. salina.

(A) Abundance of bacterial OTU in 16 replicate 200-L N. salina cultivations. Panels are labeled at far left. “Δ[Algae]”: algae growth rates (ΔOD750nm) for replicate cultivations are ranked from highest to lowest (left to right); light-green dashed vertical bar separates 13 healthy replicates from 3 replicates with stagnant growth; dark-green dashed vertical bar separates values for individual replicates from average values for healthy (+) and stagnant (−) cultivations. OTU Relative Abundance: stacked bar chart showing relative abundances of the 16 OTU present in at least 90% of samples in this study; these OTU are stacked based on average abundance in the 13 healthy replicates, from most to least abundant; all other OTU are represented at the top of the bar chart as single category (Other, O); consensus taxonomy and color-coding is given in Fig. 3B. “OTU Abundance, Normalized to (+)”: abundances were normalized to average abundances in the 13 healthy (+) replicate cultivations included in this figure; a single heat map is formatted across this panel (blue = minimum value, white = 1, yellow = highest value); column to left of heat map corresponds to consensus taxonomy in Fig. 3B. “OTU Abundance, Normalized to Serial Batches”: abundances were normalized to average abundances in the 35 communities shown in Fig. 2; heat map formatted as above.
(B) Comparison of OTU abundances in replicate healthy and stagnant cultivations in Fig. 3A with average abundances in serial batch cultivations in Fig. 2. Consensus taxonomy is given for 16 OTU conserved in at least 90% of samples in this study; OTU are listed by average abundance in healthy (+) cultivations shown in Fig. 3A. Ph, phylum (see Table 1); Cl, class: A, Alphaproteobacteria; C, Cytophagia; D, Deltaproteobacteria; G, Gammaproteobacteria; Sp, Sphingobacteria; S, [Saprospirae]. ID: rank abundance for each of the 16 OTU in healthy cultivations shown in Fig. 3A (+), and in 35 communities included in Fig. 2 (SB); color-coding for (+) corresponds to Fig. 3A. %: Indicates OTU found in 90%, 95% or 100% of the 275 samples in this study; gray and dark gray shading indicate OTU conserved among 95% and 100% of communities, respectively. [OTU]AVG: Average relative abundance for OTU in 13 healthy (+) and 3 stagnant (−) replicate cultivations in Fig. 3A, plus 35 communities analyzed in Fig. 2 (SB); heat maps are formatted separately within each column (lowest value = white; highest value = orange).
RESULTS AND DISCUSSION
Bacterial communities differed across cultivation scales
Community DNA was extracted from archived biomass samples collected over an 8-month period from small, medium, and large industrial algae cultivation systems at Solix Biosystems (Fort Collins, CO) (Fig. 1A). In total, 275 samples were processed. The V3 region of 16S rRNA genes was amplified and sequenced, generating 10.9 million sequenced amplicons. Following filtering steps that removed algae-derived chloroplast and mitochondrial sequences along with extremely rare sequences and other potential sources of error, 2 million bacterial reads were used for further analyses.
Bacterial communities were characterized in small, medium, and large industrial cultivations of N. salina algae. The composition of these bacterial communities was compared across all samples using unweighted UniFrac as a distance metric. In the PCoA plot in Fig. 1B, the distance separating sample points represents differences among bacterial communities, measured as the fraction of evolutionary history in a phylogenetic tree that is unique to one of the samples (Peiffer et al, 2013). Three primary clusters were observed, corresponding to samples from small, medium, and large growth scales (Fig. 1B). Thus, algae cultivations at different scales contained bacterial communities that were distinct in terms of phylogenetic structure.
Since manipulations of small-scale cultures were done under sterile conditions, it is probable that bacterial communities in these cultures represent bacteria that were associated with the initial algal inoculum or introduced to stock cultivations in an early stage of sub-culturing. Beyond those initial cultures, there are numerous environmental differences during cultivation at small, medium, and large scales that might affect bacterial populations and cause distinct communities to dominate different growth systems. Some of these factors would directly influence bacteria (e.g., temperature management), while others (e.g., the ratio of surface area to volume, light source intensity, illumination period) have impacts on the growth of N. salina, which in turn would influence bacterial growth.
In addition to differences in environmental parameters, the serial batch strategy used for these cultivations may affect bacterial community composition across different scales. In the serial batch mode used here, biomass from dense N. salina cultures of a particular scale was harvested and additional cultures at that scale were inoculated using a portion of this harvest (Figs. 1A & 1C); occasionally, biomass harvested at one scale was used to inoculate cultivations in a larger growth system. Because culture communities (N. salina, bacteria, and other constituents) were repeatedly reused for cultivation at a particular scale, this inoculation strategy provides additional generations within which communities may have been affected by the conditions of that system scale and therefore became increasingly distinct from communities grown at different scales. It is conceivable that the community structure associated with productive N. salina cultivations at one growth scale could be less optimal at other scales.
The bacterial community structures in different algae growth systems were analyzed. At the phylum level, Bacteroidetes and Proteobacteria dominated communities from all system scales (Table 1). The total abundance of Bacteroidetes and Proteobacteria was constant across all systems, respectively comprising 91.8%, 89.9%, and 90.6% of bacteria in small, medium, and large cultivations. Considering all samples, Bacteroidetes increased in relative abundance as system scale increased, from 48.5% abundance in small-scale cultivations to 63.3% in medium-scale and 70.7% in large-scale growth environments (Table 1). Proteobacteria became less prevalent as the system size increased, having relative abundances of 43.6%, 28.6%, and 25.7% in cultivations grown at small, medium, and large scales, respectively. Bacteroidetes and Proteobacteria previously have been shown to be the most abundant bacteria in marine environments, with Alphaproteobacteria and Gammaproteobacteria typically dominating the Proteobacteria in marine systems (Kazamia et al., 2012). This finding is also consistent with results of previous studies of Nannochloropsis laboratory cultivations (Wang et al., 2016). Within both Bacteroidetes and Proteobacteria, the total number of distinct taxa identified at the class and order levels (and comprising at least 0.1% relative abundance) increased as culture scale increased (Tables 1 and S1).
Table 1. Bacteroidetes and Proteobacteria dominate communities across N. salina growth systems.
Relative abundances of bacterial classes identified in N. salina growth systems are represented. Phylum abbreviations given herein are used in subsequent Figures and Tables. cd, Candidate division. Heat maps are formatted separately for each scale growth system, ranging from zero to the maximum value in that system (white to black); for contrast, relative abundances above 18 are listed in white font. Since OTU counts are rounded to the nearest tenth, zero values represent relative abundances less than 0.05%. Σ (bottom) is the sum of all values, and reveals rounding errors; Σ(B+Pr) (bottom) indicates total abundance of Bacteroidetes and Proteobacteria in each system.
| Phylum | Class | Abundance | |||
|---|---|---|---|---|---|
| Sml | Med | Lrg | |||
| B | Bacteroidetes | Bacteroidia | 0.0 | 0.0 | 0.1 |
| Cytophagia | 5.0 | 18.9 | 22.3 | ||
| Flavobacteriia | 20.1 | 4.6 | 3.4 | ||
| Sphingobacteriia | 0.0 | 5.6 | 3.0 | ||
| [Rhodothermi] | 0.0 | 0.2 | 0.1 | ||
| [Saprospirae] | 23.2 | 32.0 | 37.3 | ||
| Pr | Proteobacteria | Alphaproteobacteria | 35.0 | 18.6 | 21.7 |
| Betaproteobacteria | 0.0 | 1.4 | 0.4 | ||
| Deltaproteobacteria | 0.0 | 5.1 | 2.1 | ||
| Epsilonproteobacteria | 0.0 | 0.0 | 0.1 | ||
| Gammaproteobacteria | 8.5 | 3.5 | 0.1 | ||
| A | Acidobacteria | Solibacteres | 0.0 | 0.1 | 0.1 |
| Ac | Actinobacteria | Acidimicrobiia | 0.2 | 0.1 | 0.0 |
| Actinobacteria | 0.2 | 0.1 | 0.0 | ||
| Ar | Armatimonadetes | [Fimbriimonadia] | 0.0 | 0.1 | 0.0 |
| Ch | Chloroflexi | Anaerolineae | 0.0 | 0.0 | 0.1 |
| Cy | Cyanobacteria | 4C0d-2 | 0.0 | 0.2 | 0.0 |
| ML635J-21 | 2.8 | 0.3 | 0.1 | ||
| Synechococcophycideae | 0.0 | 2.2 | 0.0 | ||
| F | Firmicutes | Clostridia | 0.0 | 0.0 | 0.2 |
| P | Planctomycetes | Planctomycetia | 1.4 | 1.8 | 0.9 |
| T | TM7 | SC3 | 0.0 | 0.3 | 0.0 |
| V | Verrucomicrobia | Opitutae | 0.0 | 0.6 | 0.9 |
| Verrucomicrobiae | 1.1 | 0.3 | 1.2 | ||
| cd | BRC1 | PRR-11 | 0.0 | 0.0 | 0.1 |
| OD1 | ZB2 | 0.0 | 0.2 | 0.2 | |
| Unassigned | 2.4 | 3.4 | 4.4 | ||
| Σ = | 99.9 | 99.6 | 98.8 | ||
| Σ(B+Pr) = | 91.8 | 89.9 | 90.6 | ||
Within each cultivation system size, bacteria were ranked by relative abundance and rankings were compared across systems (Tables 2 and S2). The 10 orders most abundant in small systems accounted for 94.9% of the bacterial communities (Table 2A). All ten of these orders were also identified in medium and large systems, although they totaled only 74.0% and 75.4% of the respective bacterial populations at these larger scales (Table 2A). In large systems, the ten most abundant bacterial orders represented, on average, 87.2% of the bacterial community (Table 2B). All these large-system OTUs were identified in medium systems, but four were not identified in small systems, indicating these OTUs may have entered the growth environment during non-sterile handling. With respect to specific orders, small systems had much higher abundance of Flavobacteriales and Rhizobiales than was observed at larger scales. Conversely, medium and large systems contained a higher relative abundance of Cytophagales than small systems.
Table 2. Bacteria predominant in small and large N. salina cultivation systems.
A) Ten most abundant bacterial orders identified in small growth systems. B) Ten most abundant bacterial orders identified in large growth systems. Zero values represent relative abundances less than 0.05%. Ph, Phylum (for phylum abbreviations, see Table 1). Heat maps are formatted separately for each growth system as in Table 1. Σ, total abundance of these ten orders in each system. Σ(Saprospirales + Cytophagales), combined abundance of the orders Saprospirales and Cytophagales in each system. Σ(Cytophagales + Flavobacteriales), combined abundance of the orders Cytophagales and Flavobacteriales in each system.
| Ph | Class | Order | Abundance | ||
|---|---|---|---|---|---|
| Sml | Med | Lrg | |||
| B | [Saprospirae] | [Saprospirales] | 23.2 | 32.0 | 37.3 |
| B | Flavobacteria | Flavobacteriales | 20.1 | 4.6 | 3.4 |
| Pr | Alphaproteobacteria | Rhizobiales | 19.7 | 8.0 | 6.2 |
| Pr | Alphaproteobacteria | Rhodobacterales | 8.7 | 3.6 | 2.9 |
| Pr | Gammaproteobacteria | Alteromonadales | 6.9 | 2.2 | 1.0 |
| Pr | Alphaproteobacteria | Sphingomonadales | 5.5 | 2.1 | 1.4 |
| B | Cytophagia | Cytophagales | 5.0 | 18.9 | 22.3 |
| Cy | ML635J-21 | 2.8 | 0.3 | 0.1 | |
| Pr | Gammaproteobacteria | Oceanospirillales | 1.6 | 0.8 | 0.1 |
| Pl | Planctomycetia | Pirellulales | 1.4 | 1.5 | 0.7 |
| Σ = | 94.9 | 74.0 | 75.4 | ||
| Ph | Class | Order | Abundance | ||
|---|---|---|---|---|---|
| Sml | Med | Lrg | |||
| B | [Saprospirae] | [Saprospirales] | 23.2 | 32.0 | 37.3 |
| B | Cytophagia | Cytophagales | 5.0 | 18.9 | 22.3 |
| Pr | Alphaproteobacteria | Kiloniellales | 0.0 | 1.1 | 7.1 |
| Pr | Alphaproteobacteria | Rhizobiales | 19.7 | 8.0 | 6.2 |
| B | Flavobacteria | Flavobacteriales | 20.1 | 4.6 | 3.4 |
| B | Sphingobacteriia | Sphingobacteriales | 0.0 | 5.6 | 3.0 |
| Pr | Alphaproteobacteria | Rhodobacterales | 8.7 | 3.6 | 2.9 |
| Pr | Alphaproteobacteria | Rhodospirillales | 1.1 | 1.3 | 2.0 |
| Pr | Deltaproteobacteria | Spirobacillales | 0.0 | 5.0 | 1.5 |
| Pr | Alphaproteobacteria | BD7-3 | 0.0 | 1.8 | 1.5 |
| Σ = | 77.8 | 81.9 | 87.2 | ||
| Σ (Saprospirales + Cytophagales) = | 28.2 | 50.9 | 59.6 | ||
| Σ (Cytophagales + Flavobacteriales) = | 25.1 | 23.5 | 25.7 | ||
Since the handling of cultures at medium and large scales was not sterile, each handling was an opportunity for bacteria and other microbes to enter the community and increase species richness and phylogenetic diversity. Average species richness within each growth scale was compared using OTU counts (Fig. S1). Overall, species richness increased as the size of the system increased. Small cultures averaged 88.0 ± 8.1 OTUs (N = 17), medium cultures contained 108 ± 22.8 OTUs (N = 81), and large cultures contained 132 ± 19 OTUs (N = 177). Furthermore, an increase in diversity across growth scales was also observed when assessed using phylogenetic distance (Fig. S2), which quantifies diversity based on total branch length of bacterial 16S rRNA phylogeny represented in a sample (Peiffer et al., 2013).
Bacteria prevalent in N. salina cultivations
To determine which bacteria were associated with N. salina across the majority of culture conditions, OTUs were identified that were present in 90%, 95%, or 100% of all samples (at least 0.01% abundance). There were 16 OTUs detected in at least 90% of cultivations (Table 3). These OTUs together averaged 63% of the relative abundance of bacterial communities across all systems. Of these, seven OTUs were identified in at least 95% of samples. Together, these seven OTUs were present at 47% relative abundance across all samples. A single OTU was present in 100% of samples (Table 3). This OTU is of the phylum Bacteroidetes, and is identified as Saprospiraceae at the family level. However, its classification at the Class and Order levels ([Saprospirae] and [Saprospirales]) is disputed within the Greengenes reference database (DeSantis et al., 2006). In addition to being in every sample, Saprospiraceae was the most abundant OTU on average, comprising 34.7% ± 14.3% of bacterial communities, and its average abundance increased in larger growth systems (Table 2B). Saprospiraceae abundance varied in individual samples from 0.3–67.0%, with the lowest and highest abundances both observed in large cultivations. Of 275 cultivations profiled, only 16 bacterial communities contained less than 5% Saprospiraceae. No correlation was observed between Saprospiraceae abundance and N. salina growth performance. Nonetheless, the presence of Saprospiraceae in every sample suggests that there are important interactions between this bacterium and N. salina, and makes Saprospiraceae a clear candidate for further study. While the activity of Saprospiraceae in this system is unknown, a strain of Saprospirales was shown to be capable of lysing the microalgae diatom Chaetoceros ceratosporum (Gou et al., 2003).
Table 3. Bacterial communities identified in greater than 90% of samples.
Bacteria identified in 100%, 95% and 90% of the 275 samples included in this study are indicated. Ph, Phylum (for phylum abbreviations, see Table 1).
| Community | Consensus Taxonomy* | |||||||
|---|---|---|---|---|---|---|---|---|
| 100% | 95% | 90% | Ph | Class | Order | Family | Genus | |
| B | [Saprospirae] | [Saprospirales] | Saprospiraceae | |||||
| Unassigned (95a) | ||||||||
| Unassigned (95b) | ||||||||
| B | Cytophagia | Cytophagales | Cytophagaceae | Leadbetterella | ||||
| Pr | Alphaproteobacteria | Rhizobiales | Phyllobacteriaceae | |||||
| Hyphomicrobiaceae | Parvibaculum | |||||||
| Hyphomicrobiaceae (95a) | ||||||||
| Unassigned (90) | ||||||||
| B | Cytophagia | Cytophagales | Cyclobacteriaceae | |||||
| [Saprospirae] | [Saprospirales] | Chitinophagaceae | ||||||
| Sphingobacteriia | Sphingobacteriales | |||||||
| Pr | Alphaproteobacteria | Sphingomonadales | Erythrobacteraceae | |||||
| Rhodobacterales | Hyphomonadaceae (90a) | |||||||
| Hyphomonadaceae (90b) | ||||||||
| Gammaproteobacteria | Alteromonadales | Alteromonadaceae | Marinobacter | |||||
| Deltaproteobacteria | Spirobacillales | |||||||
Bold text indicates taxonomy shared with bacteria isolated from N. oceanica cultures by Wang, et al. (2012). For more information, see Supplemental Table S3A)
In a previous study of bacteria associated with Nannochloropsis oceanica algae (Wang et al., 2012), several bacteria were isolated with taxonomy similar to bacteria prevalent in N. salina cultivations (Table S3). Members of the genus Marinobacter; the families Cytophagaceae, Phyllobacteriaceae, Hyphomonadaceae, and Erythrobacteraceae; and the orders Flavobacteriales, Oceanospirillales, Planctomycetales, and Pseudomonadales were identified in N. salina and N. oceanica cultures (Tables 3 and S3). Association of these bacteria with both N. salina and N. oceanica in distinct environments suggests these bacterial types may have specific relationships with Nannochloropsis species in general.
In experiments involving a third species of Nannochloropsis algae, bacteria from laboratory N. gaditana cultures were plated on marine agar, and a representative of the family Phyllobacteriaceae was recovered (SPF, KFR, unpublished). Since Phyllobacteriaceae was also identified in 95% of samples in this study and two Phyllobacteriaceae members were isolated from N. oceanica cultures (Wang, et al. 2012), there may be an intimate association of this bacterial family with several species of Nannochloropsis. In fact, members of the family Phyllobacteriaceae have been identified as supporting algae growth in additional studies. Mesorhizobium loti (of the Phyllobacteriaceae) was found to supply vitamin B to the alga Lobomonas rostrata, with this interaction optimized at a 1:30 (algae:bacteria) cellular ratio under the examined conditions (Grant et al., 2014). Separately, Mesorhizobium was shown to be one of several nitrogen-fixing species associated with growth promotion of four different green algae (Kim et al., 2014).
Although there are limited studies related to influences of bacteria on N. salina health, general ecological activities are known for some of the bacteria that were common across systems. The second most abundant bacterial order in larger systems was Cytophagales, of the class Cytophagia. “Cytophagia” roughly translates to “eats cells” and, like Saprospirales, members of Cytophagia are capable of lysing a variety of microalgae (Cole, 1982). The combined abundance of Saprospirales and Cytophagales averaged 50.9% and 59.6% in medium and large systems, respectively, whereas they totaled only 28.2% of the average bacterial communities in small cultivations (Table 2B). Given their common association with healthy N. salina cultivations, potential lytic activities of these bacterial orders may relate to desired processes like nutrient recycling. As such, they might represent “neutral” community members rather than pathogenic or otherwise negative organisms. Considering bacteria that might positively contribute to algal growth, four distinct bacterial families within the order Rhizobiales were conserved across 95% of sampled N. salina cultivations. Members of Rhizobiales are known to fix nitrogen and increase the growth of algae (Carney et al., 2014; Kim et al., 2014). In one instance, a Rhizobium sp. increased the growth of the algae Botryococcus brauni by 50% compared an axenic algal culture (Rivas et al., 2010). In addition, a Mesorizobium sp., a type of Rhizobiales, was found to provide vitamin B12 to algae (Grant et al., 2014). As such, members of the order Rhizobiales represent bacteria that algae producers might use to supplement growth media in order to maximize algal productivity. Another way to improve algal productivity would be to minimize the level of bacteria that have strictly negative impacts on algae. Bacteria of the order Sphingobacteriales can cause flocculation of some microalgae (Lee et al., 2000).
Sphingobacteriales was found in over 90% of all samples in this study and was one of the ten most abundant Orders in medium and large cultivations (Tables S1 and 2B), but was not detected in small-scale cultivations. Since small-scale cultivations typically lacked Sphingobacteriales, the bacteria could be added to laboratory-scale N. salina cultures to determine whether it has a direct negative impact on algal productivity.
Relationships among bacterial communities in replicate 200-L N. salina cultivations grown using the serial batch inoculation strategy
In large cultivations grown using a serial batch inoculation strategy (Fig. 1C), a series of replicate 200-L cultivations are simultaneously inoculated from a single source. Once cultivations are mature, biomass from healthy panels is harvested in a batch. The majority of the harvest is used toward product, while a fraction is diluted as inoculum for the next batch of replicate 200-L panels. In addition to algal biomass, this inoculum includes bacteria present in the preceding batch harvest.
To reveal relationships among bacterial communities in large cultivations spanning 9 calendar months (July 2011–March 2012), unconstrained, unweighted UniFrac was used as a distance metric. In the resulting PCoA plot (Fig. 1D), points representing bacterial communities sampled on a single day form a cluster; this is expected since such points represent bacterial communities from replicate algal cultivations. Furthermore, the overall arrangement of communities within the plot roughly corresponds with time from the start of monitoring (white data points, see legend) to the conclusion (purple data points). Presumably, a major factor governing this progression of data points relates directly to the serial batch inoculation strategy, specifically the inherent relatedness between the bacterial community in one batch at harvest and the community in the subsequent batch at inoculation. Since these large cultivations were grown outdoors, additional factors (e.g., day length) inherently varied along with the progression of time.
To further examine the relationship among bacterial communities in serially inoculated large-scale cultivations, five consecutive N. salina batches were characterized in more detail. These cultivations spanned 77 days from July–October 2011. Five replicate 200-L cultivations from each batch were characterized. These large-scale batches were grown outdoors under sunlight, and later batches grew more slowly than earlier ones as the day length shortened (Fig. 2A & 2C). For this reason, replicates in the first three batches were sampled only once, while the final batches were each sampled on two dates, meaning a total of 35 bacterial communities are represented. All batches in this analysis exhibited generally healthy growth, assessed visually by density and green color, and confirmed by OD750 (Fig. 2A). Relative abundances of the 16 OTU identified in at least 90% of all 275 samples in this study (Fig. 2B and Table 3) are shown as a stacked bar graph (Fig. 2C, middle panel), while their normalized abundances are represented in a heat map (Fig. 2C, bottom panel). Generally, bacterial communities from the five replicates sampled on a particular day appear similar to each other, even though individual replicates may have had different rates of increase in algal density over the entire batch growth period (Fig. 2C, “Total Increase”) or in the approximately 24 h period prior to sampling (Fig. 2C, “24h Increase”). An exception to this generalization is data from the second sampling date of N. salina batch 938 (Fig. 2C, “938-B”). The bacterial communities in these five replicates appear distinct from one another when compared by relative or normalized OTU abundances. However, the algal growth rates of the replicates are broadly similar to one another, whether calculated across the entire batch growth period or for the 24 h preceding sampling. Four of these replicates have elevated levels of the Sphingobacteriales, which is known to induce flocculation of some algae (Zhou et al., 2015). The data from the fifth replicate (denoted with an asterisk in Fig. 2C) indicate an extremely high amount of Saprospirales and a complete absence any of the other OTU conserved across 90% of samples in this study. As this culture replicate had no aberrant growth phenotypes and grew at a rate similar to the other four replicates presented, these statistics likely result from a sample handling or processing error that resulted in incorrect data and do not accurately indicate bacterial abundances in this cultivation.
As noted above, algae in the batches and individual replicates represented in Fig. 2 appeared generally healthy and algal growth rates were within the producer’s expectations. Nonetheless, average growth rates of batches varied approximately twofold across the course of the experiment (Fig. 2C, “Growth/d”). Since those batches spanned July–October, external factors such as day length presumably strongly influenced growth rates and, therefore, limited the ability to identify relationships between bacterial communities and algal productivity. To maximize the potential to identify bacterial community members that affect productivity, 16 replicate 200-L N. salina cultivations were analyzed (Fig. 3). In the algae batch presented in Fig. 3, three replicates exhibited stagnant growth, while the remaining 13 were generally healthy, though the growth rates of the healthy replicates varied twofold (Fig 3A, top). When comparing average OTU abundances in the 13 healthy and 3 stagnant cultivations, stagnant cultivations have elevated relative abundances of Spirobacillales (Fig 3A, upper heat map). Abundances of OTUs in the 13 healthy and 3 stagnant replicates were also normalized to their abundances in the 35 healthy serial batch communities in Fig. 2A–2C (Fig. 3A, lower heat map). This reveals that in the healthy and stagnant cultivations in Fig. 3, levels of Spirobacillales are, on average, respectively elevated to 11.3× and 133× their abundance in the serial batches. Therefore, it is possible that the batch of inoculum used to start these 16 replicate cultivations already contained an elevated level of Spirobacillales and that conditions in the stagnant cultivations lead to its dominance of those communities. Spirobacillales is generally uncharacterized, and it is unknown whether the higher Spirobacillales abundance limited N. salina growth, or itself was a result of culture stagnation. Similarly high levels of Spirobacillales were not observed in other large cultivations with slow or stagnant growth.
Implications for industrial algal cultivations
As demonstrated in this study, bacteria were abundant in closed phototrophic algal production systems, and differences in community composition were found across growth conditions. Since all samples characterized in this study were obtained from a single facility, some members of the associated bacterial communities may be unique to this particular growth environment or geographic location. Growers at different locations might observe distinct populations of common bacteria. To identify bacteria or bacterial functions required for (or detrimental to) efficient Nannochloropsis growth across multiple environments, future studies could include cultivations grown in different cultivation systems and from around the world.
Ultimately, algae producers will benefit from detailed molecular understanding of mechanisms underlying bacterial impacts on algal culture performance. In the near-term, however, culture management strategies may be best informed by determining associations between system constituents and algal culture performance. The profiling of 16S rRNA sequences presently allows a detailed systems-level characterization of bacterial communities during algal cultivation. The presence or absence of specific community members may be correlated with algal performance metrics such as growth rate and lipid productivity. Although complete evaluation of the influences of the bacteria in these cultures was beyond the scope of this study, a limited study demonstrated that at least one isolate had the potential to have a detrimental impact on N. salina. Whether such bacteria directly impact algal productivity or merely serve as predictors of culture performance, diagnostics may be developed to routinely monitor for presence or abundance of these specific community members. For example, 16 bacterial OTU were identified in 90% of all samples in this study, seven OTU were in 95% of samples, and a single OTU was found in every sample. To favor stable N. salina growth in large systems, potential sources of inoculum could be screened to confirm that they contain the bacterial community found in 90%, 95%, or 100% of samples in this study. In some instances, it may not be sufficient to monitor for the presence or absence of specific organisms. In this study, Spirobacillales was one of the OTUs observed in more than 90% of samples. This conservation across samples suggests it is beneficial to monitor for retention of Spirobacillales in cultivations and potential inoculum. However, of 16 replicate large system cultivations of N. salina, Spirobacillales was present at unusually high abundances in three cultures undergoing stagnant growth, but was found at lower levels in the remaining 13 cultures growing at normal rates (Fig. 3). Thus, it may be desirable to monitor for its abundance of Spirobacillales relative to some standard across cultivations (such as N. salina abundance or total bacterial abundance).
Small cultivations grown under sterile conditions contained less bacterial diversity than cultivations grown in medium and large systems. As a practical matter, experiments to determine optimal conditions for algae productivity often use small cultivation systems. The different bacterial community composition and reduced diversity of small cultivations may impact the ability of researchers and producers to translate N. salina productivity levels observed in small laboratory systems to performance in large systems.
This study revealed major shifts in the composition of bacterial communities in N. salina algae cultivation systems. Understanding bacterial functions in algae cultures is critical for successful large-scale algae cultivation. Bacteria that are detrimental to algae growth must be identified, tracked, and minimized. Bacterial communities that promote algae growth and stability could be included in a probiotic cultivation supplement (Kazamia et al., 2012). In addition to systems-level monitoring of community constituents, targeted experiments are necessary to determine specific bacterial functions that promote or inhibit algae productivity. Candidates for further characterization include bacteria associated with the majority of all cultivations, with specific growth scales, or with cultures exhibiting extreme growth rates. These targeted efforts will be facilitated by isolation and cultivation of highly conserved bacterial strains or, conversely, by removal of bacterial types from algal cultures through use of antibiotics or dilution strategies. Additional molecular procedures–such as monitoring algal growth rates, transcriptomes, and proteomes–can be used to define the effects of these bacteria on algal phenotypes.
Supplementary Material
Highlights.
Bacteria in N. salina cultivations were studied over time at different scales
10.9 million amplicons of 16S rRNA sequences from 275 samples were analyzed
Bacterial communities in small, medium, and large algae cultures were different
Larger algae cultivations contained richer bacterial communities than smaller ones
Acknowledgments
The authors thank Joel Butler and Ron Parsons of Solix Biosystems for assisting in sampling their algae systems. We also thank Mark Clark, Caleb Hulse, Evan Piland, and John Walden for assisting with collection and analysis of cultures, and Mike Desarro for assisting with flow cytometer analysis. We thank William Walters for developing strategies with primer selection. SPF was supported by a National Science Foundation (NSF) Graduate Research Fellowship ID#: 2011129113, and by a fellowship from the NSF-IGERT program Multidisciplinary Approaches to Sustainable Bioenergy at Colorado State University (DGE 0801707). ARP was supported by a fellowship from the NSF-IGERT of the Interdisciplinary Quantitative Program at the Biofrontiers Institute at the University of Colorado, Boulder (DGE 1144807). Compute was performed on the Pando supercomputer, which is funded by NIH grant 1S10OD012300.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information consists of additional data presented in three tables and two figures.
AUTHOR CONTRIBUTIONS
S.P.F., K.F.R., and S.T.C conceptualized the study; A.R.P. and S.P.F. performed the formal analysis; S.P.F., D.B.-L., and A.R.P. conducted the investigation; S.T.C, KFR, and R.K. provided resources; S.P.F. and A.R.P. performed data curation; S.P.F. wrote the original manuscript draft; K.F.R, S.T.C, R.K., and A.R.P reviewed and edited the manuscript; S.P.F. and S.T.C. developed the data visualization; and K.F.R., S.T.C., and R.K. supervised the project.
STATEMENTS REGARDING CONFLICTS, CONSENT, AND HUMAN/ANIMAL RIGHTS
No conflicts, informed consent, human or animal rights applicable.
References
- Amin SA, Green DH, Hart MC, Küpper FC, Sunda WG, Carrano CJ. Photolysis of iron–siderophore chelates promotes bacterial–algal mutualism. Proceedings of the National Academy of Sciences. 2009;106(40):17071–17076. doi: 10.1073/pnas.0905512106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JC, Bittinger K, Bushman FD, DeSantis TD, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney LT, Reinsch SS, Lane PD, Solberg OD, Jansen LS, Williams KP, Trent JD, Lane TW. Microbiome analysis of a microalgal mass culture growing in municipal wastewater in a prototype OMEGA photobioreactor. Algal Research. 2014;4:52–61. [Google Scholar]
- Chen CY, Yeh KL, Aisyah R, Lee DJ, Chang JS. Cultivation, photobioreactor design and harvesting of microalgae for biodiesel production: a critical review. Bioresource Technology. 2011;102(1):71–81. doi: 10.1016/j.biortech.2010.06.159. [DOI] [PubMed] [Google Scholar]
- Cole JJ. Interactions between bacteria and algae in aquatic ecosystems. Annual Review of Ecology and Systematics. 1982:291–314. [Google Scholar]
- Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature. 2005;438(7064):90–93. doi: 10.1038/nature04056. [DOI] [PubMed] [Google Scholar]
- De-Bashan LE, Antoun H, Bashan Y. Involvement of indole-3-acid produced by the growth-promoting bacterium Azospirillum spp. in promoting growth of Chlorella vulgaris. Journal of Phycology. 2008;44(4):938–947. doi: 10.1111/j.1529-8817.2008.00533.x. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Applied and Environmental Microbiology. 2006;72:5062–72. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biological Conservation. 1992;61(1):1–10. [Google Scholar]
- Fishman D, Majumdar R, Morello J, Pate R, Yang J. National algal biofuels technology roadmap, US Department of Energy, Office of Energy Efficiency and Renewable Energy. Biomass Program 2010 [Google Scholar]
- Foster RA, Kuypers MM, Vagner T, Paerl RW, Musat N, Zehr JP. Nitrogen fixation and transfer in open ocean diatom–cyanobacterial symbioses. The ISME journal. 2011;5(9):1484–1493. doi: 10.1038/ismej.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulbright SP, Dean MK, Wardle G, Lammers PJ, Chisholm ST. Molecular diagnostics for monitoring contaminants in algal cultivation. Algal Research. 2014;4:41–51. [Google Scholar]
- Fulbright SP, Chisholm ST, Reardon KF. Growth inhibition of Nannochloropsis by Bacillus pumilus. Algal Research. 2016;20:70–76. [Google Scholar]
- Furusawa G, Yoshikawa T, Yasuda A, Sakata T. Algicidal activity and gliding motility of Saprospira sp. SS98-5. Canadian Journal of Microbiology. 2003;49(2):92–100. doi: 10.1139/w03-017. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T, Somerfield PJ, Huse S, Joint I. The seasonal structure of microbial communities in the Western English Channel. Environmental Microbiology. 2009;11(12):3132–3139. doi: 10.1111/j.1462-2920.2009.02017.x. [DOI] [PubMed] [Google Scholar]
- Grant MA, Kazamia E, Cicuta P, Smith AG. Direct exchange of vitamin B12 is demonstrated by modelling the growth dynamics of algal–bacterial cocultures. The ISME Journal. 2014;8(7):1418–1427. doi: 10.1038/ismej.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths MJ, van Hille RP, Harrison ST. Lipid productivity, settling potential and fatty acid profile of 11 microalgal species grown under nitrogen replete and limited conditions. Journal of Applied Phycology. 2012;24(5):989–1001. [Google Scholar]
- Grobbelaar JU. Factors governing algal growth in photobioreactors: the “open” versus “closed” debate. Journal of Applied Phycology. 2009;21:489–492. [Google Scholar]
- Grossart HP, Levold F, Allgaier M, Simon M, Brinkhoff T. Marine diatom species harbour distinct bacterial communities. Environmental Microbiology. 2005;7(6):860–873. doi: 10.1111/j.1462-2920.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- Kazamia E, Aldridge DC, Smith AG. Synthetic ecology–A way forward for sustainable algal biofuel production? Journal of Biotechnology. 2012;162(1):163–169. [Google Scholar]
- Kim BH, Ramanan R, Cho DH, Oh HM, Kim HS. Role of Rhizobium, a plant growth promoting bacterium, in enhancing algal biomass through mutualistic interaction. Biomass and Bioenergy. 2014;69:95–105. [Google Scholar]
- Krohn-Molt I, Wemheuer B, Alawi M, Poehlein A, Güllert S, Schmeisser C, Streit WR. Metagenome survey of a multispecies and alga-associated biofilm revealed key elements of bacterial-algal interactions in photobioreactors. Applied and Environmental Microbiology. 2013;79(20):6196–6206. doi: 10.1128/AEM.01641-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. Using QIIME to analyze 16S rRNA gene sequences from Micobial Communities. Current Protocols in Bioinformatics. 2011 doi: 10.1002/0471250953.bi1007s36. Chapter: Unit10.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakaniemi AM, Intihar VM, Tuovinen OH, Puhakka JA. Growth of Chlorella vulgaris and associated bacteria in photobioreactors. Microbial Biotechnology. 2012;5(1):69–78. doi: 10.1111/j.1751-7915.2011.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Kato J, Takiguchi N, Kuroda A, Ikeda T, Mitsutani A, Ohtake H. Involvement of an Extracellular Protease in Algicidal Activity of the Marine Bacterium Pseudoalteromonassp. Strain A28. Applied and Environmental Microbiology. 2000;66(10):4334–4339. doi: 10.1128/aem.66.10.4334-4339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letcher PM, Lopez S, Schmieder R, Lee PA, Behnke C, Powell MJ, McBride RC. Characterization of Amoeboaphelidium protococcarum, an algal parasite new to the cryptomycota isolated from an outdoor algal pond used for the production of biofuel. PloS One. 2013;8(2):e56232. doi: 10.1371/journal.pone.0056232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin RA. Saprospira grandis: A Flexibacterium That Can Catch Bacterial Prey by mixotrophy. Microbial Ecology. 1997;34(3):232–236. doi: 10.1007/s002489900052. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Hamady M, Kelly ST, Knight R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Applied and Environmental Microbiology. 2007;73(5):1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. The ISME Journal. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata TM, Martins AA, Caetano NS. Microalgae for biodiesel production and other applications: a review. Renewable and Sustainable Energy Reviews. 2010;14(1):217–232. [Google Scholar]
- Mayali X, Azam F. Algicidal bacteria in the sea and their impact on algal blooms. Journal of Eukaryotic Microbiology. 2004;51(2):139–144. doi: 10.1111/j.1550-7408.2004.tb00538.x. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME Journal. 2012;6(3):610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda LN, Hutchison K, Grossman AR, Brawley SH. Diversity and abundance of the bacterial community of the red macroalga Porphyra umbilicalis: did bacterial farmers produce macroalgae. PloS One. 2013;8(3):e58269. doi: 10.1371/journal.pone.0058269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler S, Ley RE. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Sciences. 2013;110(16):6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JC, Yates T, Douglas N, Weyer K, Butler J, Bradley TH, Lammers PJ. Nannochloropsis production metrics in a scalable outdoor photobioreactor for commercial applications. Bioresource Technology. 2012;117:164–171. doi: 10.1016/j.biortech.2012.04.073. [DOI] [PubMed] [Google Scholar]
- Shurin JB, Abbott RL, Deal MS, Kwan GT, Litchman E, McBride RC, Smith VH. Industrial‐ strength ecology: trade‐ offs and opportunities in algal biofuel production. Ecology Letters. 2013;16(11):1393–1404. doi: 10.1111/ele.12176. [DOI] [PubMed] [Google Scholar]
- Skerratt JH, Bowman JP, Hallegraeff G, James S, Nichols PD. Algicidal bacteria associated with blooms of a toxic dinoflagellate in a temperate Australian estuary. Marine Ecology Progress Series. 2002;244(1):15. [Google Scholar]
- Slade R, Bauen A. Micro-algae cultivation for biofuels: Cost, energy balance, environmental impacts and future prospects. Biomass and Bioenergy. 2013;5(3):29–38. [Google Scholar]
- Smith VH, Crews T. Applying ecological principles of crop cultivation in large-scale algal biomass production. Algal Research. 2014;4:23–34. [Google Scholar]
- Stevens H, Stübner M, Simon M, Brinkhoff T. Phylogeny of Proteobacteria and Bacteroidetes from oxic habitats of a tidal flat ecosystem. FEMS Microbiology Ecology. 2005;54(3):351–365. doi: 10.1016/j.femsec.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Reijnders L. Do biofuels from microalgae beat biofuels from terrestrial plants? Trends in Biotechnology. 2008;26(7):349–350. doi: 10.1016/j.tibtech.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Rivas MO, Vargas P, Riquelme CE. Interactions of Botryococcus braunii cultures with bacterial biofilms. Microbial Ecology. 2010;60(3):628–635. doi: 10.1007/s00248-010-9686-6. [DOI] [PubMed] [Google Scholar]
- Rodolfi L, Chini Zittelli G, Bassi N, Padovani G, Biondi N, Bonini G, Tredici MR. Microalgae for oil: Strain selection, induction of lipid synthesis and outdoor mass cultivation in a low‐ cost photobioreactor. Biotechnology and Bioengineering. 2009;102(1):100. doi: 10.1002/bit.22033. [DOI] [PubMed] [Google Scholar]
- Rooney-Varga JN, Giewat MW, Savin MC, Sood S, LeGresley M, Martin JL. Links between phytoplankton and bacterial community dynamics in a coastal marine environment. Microbial Ecology. 2005;49(1):163–175. doi: 10.1007/s00248-003-1057-0. [DOI] [PubMed] [Google Scholar]
- Wang H, Laughinghouse HD, Anderson MA, Chen F, Williams E, Place AR, Zmora O, Zohar Y, Zheng RT, Hill R. Novel bacterial isolate from Permian groundwater, capable of aggregating potential biofuel-producing microalga Nannochloropsis oceanica IMET1. Applied and Environmental Microbiology. 2012;78(5):1445–1453. doi: 10.1128/AEM.06474-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hill RT, Zheng T, Hu X, Wang B. Effects of bacterial communities on biofuel-producing microalgae: stimulation, inhibition and harvesting. Critical Reviews in Biotechnology. 2016;36(2):341–352. doi: 10.3109/07388551.2014.961402. [DOI] [PubMed] [Google Scholar]
- Zhou D, Li R, Yang Y. Granulation, control of bacterial contamination, and enhanced lipid accumulation by driving nutrient starvation in coupled wastewater treatment and Chlorella regularis cultivation. Applied Microbiology and Biotechnology. 2015;99(3):1531–1541. doi: 10.1007/s00253-014-6288-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.