

SUMMARY
Regulated antimicrobial peptide expression in the intestinal epithelium is key to defense against infection and to microbiota homeostasis. Understanding the mechanisms that regulate such expression is necessary for understanding immune homeostasis and inflammatory disease, and for developing safe and effective therapies. We used Caenorhabditis elegans in a preclinical approach to discover mechanisms of antimicrobial gene expression control in the intestinal epithelium. We found an unexpected role for the cholinergic nervous system. Infection-induced acetylcholine release from neurons stimulated muscarinic signaling in the epithelium, driving downstream induction of Wnt expression in the same tissue. Wnt induction activated the epithelial canonical Wnt pathway, resulting in the expression of C-type lectin and lysozyme genes that enhanced host defense. Furthermore, the muscarinic and Wnt pathways are linked by conserved transcription factors. These results reveal a tight connection between the nervous system and the intestinal epithelium, with important implications for host defense, immune homeostasis, and cancer.
eTOC blurb
How intestinal epithelial antimicrobial defense functions are regulated is poorly understood. Using Caenorhabditis elegans, Labed et al. found infection-triggered neuronal acetylcholine release, driving intestinal defense gene expression via conserved muscarinic and Wnt pathways. This newly identified pathway may be relevant to the microbiota-gut-brain axis and immune homeostasis in humans.
INTRODUCTION
Innate host defenses, including antimicrobial peptides from the intestinal epithelium, are essential for defense against infection and for maintenance of the healthy microbiota (Hooper, 2015; Peterson and Artis, 2014). Although much is known about innate defenses in myeloid and lymphoid cells, much less is known about the regulation of antimicrobial peptide expression in the intestinal epithelium. Nonetheless, the intestinal epithelium is the most conserved host defense system (Bosch et al., 2009).
Caenorhabditis elegans gene clec-60 encodes a secreted C-type lectin related to mammalian antimicrobial lectin RegIIIγ (Vaishnava et al., 2011). clec-60 is upregulated in the intestinal epithelium during infection with Gram-positive pathogens, such as Enterococcus faecalis, Microbacterium nematophilum, and Staphylococcus aureus (O’rourke et al., 2006; Wong et al., 2007; Irazoqui et al., 2010), as well as Gram-negative Yersinia pestis (Bolz et al., 2010). In contrast, clec-60 is repressed by Pseudomonas aeruginosa and Salmonella enterica (Irazoqui et al., 2010; Head and Aballay, 2014). Overexpression of clec-60 results in enhanced host survival of infection (Irazoqui et al., 2010). Although differential expression of clec-60 suggests that it is controlled by pathogen-specific signal transduction pathways, the identity of such pathways is unknown. Thus, the dynamic and pathogen-specific expression profile of clec-60 makes an attractive model to investigate pathways that control the host response to infection.
Mammals and most invertebrates share a dependence on Toll-Like Receptors (TLRs) in the detection of Gram-positive bacterial infection (Akira et al., 2006). However, nematodes lack important components of canonical TLR pathways (Pujol et al., 2001). They also lack Nacht and Leucine-Rich domain containing proteins (NLRs). Despite the absence of both pattern recognition receptor (PRR) pathways, nematodes mount pathogen-specific transcriptional host responses to S. aureus (Irazoqui et al., 2010; Wong et al., 2007). Therefore, nematodes must possess a mechanism to detect S. aureus infection. In mammals, N-formyl peptides produced by bacteria are triggers of innate immune responses through the G-protein-coupled receptors (GPCRs), formyl peptide receptors 1 and 2 (Bloes et al., 2015). In C. elegans, the GPCR superfamily is greatly expanded (Rubin et al., 2000). GPCRs NPR-1 and FSHR-1 are involved in the C. elegans transcriptional and behavioral response to Gram-negative P. aeruginosa (Aballay et al., 2008; Powell et al., 2009). Because of this precedent, we hypothesized that a GPCR might function in the detection of S. aureus.
Instead, our studies revealed an unexpected connection between the canonical Wnt pathway and the neurotransmitter acetylcholine (ACh). Canonical Wnt signaling is important for intestinal development, homeostasis, and carcinogenesis in humans (Clevers and Nusse, 2012). In addition, it plays important roles in the function and activation of cells of the immune system (Silva-García et al., 2014). Disentangling the direct involvement of the canonical pathway in innate immunity from indirect roles through cellular differentiation remains challenging in mammalian models. In contrast to mammals, the nematode intestinal epithelium comprises just 20 fully differentiated cells (McGhee, 2007). These cells do not undergo anoikis, but remain functional for the entire life of the animal. Thus, canonical Wnt signaling in C. elegans is not involved in epithelial tissue renewal as it is in mammals, which greatly facilitates the study of its role in antimicrobial peptide production.
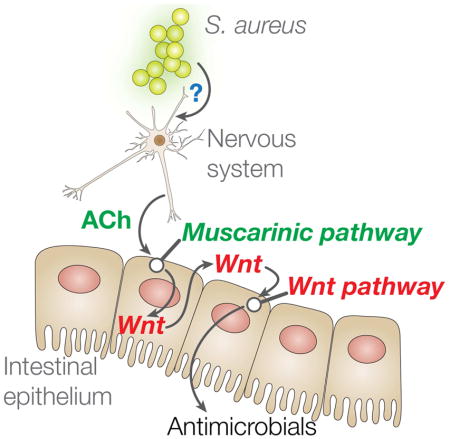
ACh secreted by cholinergic neurons engages ACh receptors on postsynaptic cells, including neurons and muscle cells. ACh receptors can be either muscarinic or nicotinic, which are GPCRs and ligand-gated ion channels, respectively (Rand, 2007). Muscarinic receptors engage downstream signaling pathways involving Gq, G11, Gi, or G0 heterotrimeric G proteins, depending on muscarinic receptor and cell type, to produce changes in gene expression and cellular function (Wess et al., 2007). In the central nervous system, muscarinic signaling is important for several behavioral and sensory processes, and has been implicated in Alzheimer’s disease and Parkinson’s disease. Peripheral muscarinic signaling controls cardiac rhythm, smooth-muscle contraction, and glandular secretion (Wess et al., 2007). In this work, we expanded the known roles of the muscarinic pathway to that of controlling host defense gene expression in the intestinal epithelium via the Wnt pathway. Because direct epithelial-neuronal synapses have not been described in this or any other organism (White et al., 1986; Yoo and Mazmanian, 2017), in C. elegans infection ACh appears to function in an endocrine fashion to engage muscarinic receptors in the intestinal epithelium and induce the expression of host defense genes through the activation of Wnt pathway.
RESULTS
Muscarinic signaling controls host defense
In addition to infection, induction of clec-60 was reported under conditions of nutritional deprivation (Melo and Ruvkun, 2012). To compare the induction of clec-60 under both conditions, we examined the expression of transcriptional reporter clec-60p::gfp (Irazoqui et al., 2008). In control animals fed ad libitum with Escherichia coli strain OP50, the standard laboratory diet, GFP expression was minimal (Fig. 1A). We observed similarly low GFP expression in animals that were starved for 8 h on the medium used in S. aureus infection assays (Fig. 1A, B). In contrast, animals that had been infected by feeding with S. aureus for 8 h exhibited greatly enhanced GFP expression in the intestinal epithelium (Fig. 1A, B). Thus, nutritional deprivation did not induce clec-60p::gfp under conditions used in S. aureus infection assays.
Figure 1. Muscarinic receptors control host defense against infection.

(A) Epifluorescence micrographs of animals expressing GFP from the clec-60 promoter (clec-60p::gfp) after 8 h of feeding on E. coli, starvation, or infection with S. aureus. (B) Quantitative analysis of (A). Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). ** p ≤ 0.01 (see STAR★Methods for description of statistical methods). ns, not significant. (C) Representative epifluorescence micrographs of clec-60p::gfp animals that were reared on E. coli carrying empty vector (top row) or gar-2, gar-3 double RNAi (bottom row), and subsequently infected 16 h with S. aureus (left column) or treated for 16 h with 5 mM arecoline (right column). (D) Quantitative analysis of (C). Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). (E) clec-60p::gfp expression after 16 h incubation of uninfected animals with 5 mM arecoline. Data are mean ± SEM (at least two independent biological replicates, n ≥ 50 per condition). (F) qRT-PCR of clec-60, ilys-3, and lys-5 in wild type animals incubated with 5 mM arecoline for 8 h, normalized to vehicle treated animals. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). * p ≤ 0.05. (G) Representative epifluorescence micrographs of clec-60p::gfp animals that were treated with vehicle (top row) or 5 mM scopolamine (bottom row), during 16 h infection with S. aureus (left column) or treated with 5 mM arecoline for 16 h (right column). (H) Quantitative analysis of infection in (G). (I) Quantitative analysis of scopolamine treatment in (G). Data are mean ± SEM (at least two independent biological replicates, n ≥ 50 per condition). (J) qRT-PCR of clec-60 in wild type animals treated with 1 mM oxotremorine for 8 h. Results are normalized to vehicle-treated animals. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (K) Survival of wild type animals, treated with vehicle or 1 mM arecoline for 24 h prior to infection. Results are representative of 2 independent biological replicates. *** p ≤ 0.001. (L) Lifespan of wild type animals treated with vehicle or 1 mM arecoline for 24 h before transfer to E. coli OP50. Results are representative of 2 independent biological replicates. ns, p > 0.05. (M) Survival of wild type animals, treated with vehicle or 1 mM scopolamine during the entire course of infection. Results are representative of 2 independent biological replicates. (N) Lifespan of wild type animals on E. coli OP50, treated with vehicle or 1 mM scopolamine. Results are representative of 2 independent biological replicates. (O) Survival of wild type and unc-17 mutant animals infected with S. aureus. Results are representative of 2 independent biological replicates. (P) Aldicarb paralysis assays of infected and uninfected animals. Results are representative of at least 3 independent biological replicates. (Q) Schematic summary of results. Scale bars, 0.6 mm. Also see Figure S1.
To identify GPCRs involved in pathogen detection upstream of clec-60, we performed a screen, in which we examined the induction of clec-60p::gfp by S. aureus in animals that had been treated with individual RNAi against 890 GPCR genes. Silencing of gar-2 resulted in defective GFP expression (Fig. S1A). Mutants defective in gar-2 function lived slightly longer than wild type on nonpathogenic E. coli (Fig. S1B) but exhibited a mild defect in survival of infection (Fig. S1C), suggesting that their response defect was biologically important. gar-2 encodes a muscarinic receptor (Hobert, 2013). The C. elegans genome contains two other genes for muscarinic receptors (gar-1 and gar-3), raising the possibility of redundancy. Simultaneous silencing of gar-2 and gar-3 synergistically reduced clec-60p::gfp induction (Fig. 1C, D), suggesting that gar-2 and gar-3 share redundant functions.
To determine if the activation of muscarinic receptors can elicit a similar response, we used the ACh-mimic arecoline (Gilani et al., 2004). Administration of arecoline to uninfected (i.e. E. coli-fed) animals was sufficient to induce clec-60p::gfp expression similar to that in infected animals (Fig. 1C, E). Arecoline also induced the endogenous expression of host defense genes clec-60, ilys-3, and lys-5 (Irazoqui et al., 2010). Furthermore, silencing of gar-2 and gar-3 abrogated arecoline-induced clec-60 expression (Fig. 1C, E). Muscarinic antagonist scopolamine (Hwang et al., 1999) impaired clec-60p::gfp induction by either S. aureus or arecoline (Fig. 1G–I). Oxotremorine, a specific muscarinic agonist, also induced clec-60 (Fig. 1J). Collectively, these results showed that muscarinic activation is necessary and sufficient for the induction of the host response to S. aureus.
Treatment with arecoline 24 h prior to infection led to significantly enhanced survival of infection (Fig. 1K) with no effect on aging (Fig. 1L). In contrast, scopolamine enhanced susceptibility to S. aureus (Fig. 1M). However, scopolamine also caused reduced lifespan on nonpathogenic E. coli (Fig. 1N). Mutants defective in gene unc-17, which encodes the vesicular ACh transporter (vAChT) necessary for loading ACh in synaptic vesicles (Alfonso et al., 1993), have impaired ACh release. unc-17 (vAChT) mutants were significantly susceptible to infection (Fig. 1O) with a minor aging defect (Fig. S1D). Aldicarb is a cholinesterase inhibitor that prevents the hydrolysis of ACh in neuromuscular junctions, resulting in paralysis. Aldicarb paralysis assays are useful for estimating the endogenous concentration of ACh in C. elegans (Mahoney et al., 2006). We used aldicarb paralysis assays to estimate the endogenous amount of ACh in animals that were infected with S. aureus for 20 min. Infected animals paralyzed 30% faster than control animals fed E. coli (Fig. 1P), indicating that endogenous ACh is released during infection.
Taken together, these observations supported a hypothesis that S. aureus, and cholinergic agonists oxotremorine and arecoline, trigger muscarinic receptors GAR-2 and GAR-3, resulting in the induction of downstream host defense genes (Fig. 1Q).
Gαq, phospholipase Cβ, and protein kinase Cη function downstream of muscarinic receptors to promote host defense
The muscarinic receptor pathway has been delineated partially in C. elegans. Muscarinic receptor GAR-3 signals through the α subunit of heterotrimeric G protein q (Gαq, encoded by gene egl-30), which activates phospholipase Cβ (PLCβ, encoded by egl-8) (Lackner et al., 1999; Miller et al., 1999; Steger and Avery, 2004). Silencing of either egl-30 or egl-8 abrogated clec-60p::gfp induction by infection (Fig. 2A, B) and mutants lacking egl-8 were unable to induce clec-60 (Fig. 2C). RNAi of egl-30 or egl-8 also prevented clec-60p::gfp induction by arecoline (Fig. 2D, E). Taken together, these results suggested that ACh may signal through a pathway controlled by GAR-2 and -3, which includes EGL-30 (Gαq) and EGL-8 (PLCβ). In support of this idea, we recently showed that EGL-30 is important during host defense (Najibi et al., 2016). In addition, we found that egl-8 mutants were unable to induce clec-60 in response to arecoline (Fig. 2F) and that, while the longevity of egl-8 mutants was slightly extended (Fig. 2H), their survival of infection was severely compromised (Fig. 2G). These results indicated that the EGL-30-EGL-8 axis plays an important and specific role for host defense against S. aureus downstream of ACh.
Figure 2. Gαq, phospholipase Cβ, and protein kinase Cη function downstream of muscarinic receptors.

(A) Epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against egl-8, pkc-1, or egl-30, and subsequently infected with S. aureus for 16 h. (B) Quantitative analysis of (A). Data are mean ± SEM (at least 2 independent biological replicates, n ≥ 50 per condition). (C) qRT-PCR of clec-60 in wild type animals and egl-8 mutants after 8 h S. aureus infection. Results are normalized to non-infected wild type. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (D) Representative epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against egl-30 or pkc-1, and subsequently treated with 5 mM arecoline for 16 h. (E) Quantitative analysis of (D). Data are mean ± SEM (at least two independent biological replicates, n ≥ 50 per condition) (F) qRT-PCR of clec-60 in wild type and egl-8 mutant animals after 8 h treatment with 1 mM arecoline. Results are normalized to vehicle treated wild type. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (G) Survival of wild type and egl-8 mutant animals infected with S. aureus. Results are representative of 2 independent biological replicates. (H) Lifespan of wild type and egl-8 mutant animals on E. coli OP50. Results are representative of 2 independent biological replicates. (I) Survival of wild type and pkc-1 mutant animals infected with S. aureus. Results are representative of 2 independent biological replicates. (J) Lifespan of wild type and pkc-1 mutant animals on E. coli OP50. Results are representative of 2 independent biological replicates. ns, p > 0.05. (K) Schematic summary of results. Scale bars, 0.6 mm.
PLCβ is a major signaling hub (Rebecchi and Pentyala, 2000). It catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacyl glycerol (DAG) and inositol trisphosphate (IP3), second messengers that control a plethora of downstream signaling pathways. To identify signaling components that may function downstream of EGL-8 (PLCβ) during infection, we performed a screen to identify RNAi clones from a kinome library that prevented clec-60p::gfp induction by S. aureus. This screen identified pkc-1 (Fig. 2A, B), which encodes protein kinase Cη (PKCη) that is activated by diacyl glycerol (Stahelin et al., 2004). pkc-1 RNAi also abrogated the induction of clec-60p::gfp by arecoline (Fig. 2D, E). Partial loss-of-function pkc-1 mutants exhibited mildly defective host survival of infection (Fig. 2I), while their aging phenotype was unaffected (Fig. 2J). Whether such mild phenotype was a consequence of incomplete expressivity of the allele, or of partial redundancy with additional protein kinases, is unclear. Nonetheless, these results suggested that PKC-1 (PKCη) is an important signaling component of the pathway that links muscarinic receptors with host defense gene induction (Fig. 2K).
Wnt ligand is induced in response to infection and to acetylcholine
clec-60 induction by S. aureus is dependent on the β-catenin homolog bar-1, which functions within the canonical Wnt pathway (Irazoqui et al., 2008). To examine the role of the canonical Wnt pathway, we measured expression of key components during infection. Genes cwn-2, which encodes a Wnt ligand, and mig-1, which encodes Wnt receptor Frizzled, were highly induced, while Wnt gene mom-2 and Frizzled gene mom-5 were repressed (Fig. 3A). Furthermore, we found marked infection-induced expression of a translational reporter for CWN-2, in the intestinal epithelium (Fig. 3B, C). A transcriptional reporter for mig-1 was similarly induced (Fig. 3D, E). Thus, Wnt and its receptor were induced by S. aureus in the tissue where infection took place.
Figure 3. Infection and acetylcholine induce Wnt ligand in the intestinal epithelium.

(A) qRT-PCR of Wnt pathway genes in wild type animals after 16 h S. aureus infection. Results are normalized to wild type animals fed on E. coli OP50. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (B) Representative epifluorescence micrographs of animals expressing Venus-tagged CWN-2 protein from the cwn-2 promoter (cwn-2p::cwn-2::venus) after 24 h of feeding on E. coli or infection with S. aureus. (C) Quantitative analysis of (B). Results are normalized to E. coli controls. Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). (D) Representative epifluorescence micrographs of animals expressing GFP from the mig-1 promoter (mig-1p::gfp) after 24 h of feeding on E. coli or infection with S. aureus. (E) Quantitative analysis of (D). Results are normalized to E. coli controls. Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). (F) qRT-PCR of cwn-2 in wild type animals after 16 h treatment with 5 mM arecoline. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (G) Same as in (F), but after 16 h treatment with 1 mM oxotremorine. (H) Representative epifluorescence micrographs of cwn-2p::cwn-2::venus animals after 24 h treatment with 5 mM arecoline. (I) Quantitative analysis of (H). Results are normalized to vehicle controls. Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). (J) qRT-PCR of cwn-2 in wild type animals and unc-17 mutants infected with S. aureus for 16 h. Results are normalized to E. coli wild type control. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (K) CWN-2::Venus expression after 24 h S. aureus infection, in animals reared on E. coli carrying empty vector or RNAi against gar-2 and gar-3, egl-30, egl-8, or pkc-1. Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). Results are normalized to E. coli empty vector control. (L) qRT-PCR of cwn-2 in wild type and egl-8 mutant animals after 16 h treatment with 5 mM arecoline. Results are normalized to vehicle-treated wild type. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (M) Time course of aldicarb paralysis. Data are the ratio of paralysis frequencies of infected animals v. E. coli controls at each time point. Results are representative of 3 independent biological replicates. (N) Time course of cwn-2 and clec-60 mRNA induction. Data are cwn-2 or clec-60 expression in infected animals relative to E. coli controls. Mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (O) Schematic summary of results. Scale bars, 0.6 mm. Scale bars with *, 0.2 mm.
Systemic administration of arecoline or oxotremorine was sufficient to induce cwn-2 expression in uninfected animals (Fig. 3F, G). The CWN-2 reporter showed specific induction in the intestinal epithelium (Fig. 3H, I), as with infection. unc-17 mutants were unable to induce cwn-2 and clec-60 during infection (Fig. 3J, Fig. S1E), demonstrating that endogenous ACh was necessary. Furthermore, silencing of gar-2 and -3, egl-30, egl-8, and pkc-1, prevented induction of the CWN-2 translational reporter by infection (Fig. 3K). egl-8 mutation abrogated cwn-2 induction by arecoline in uninfected animals (Fig. 3L). These results showed that Wnt ligand was induced in the intestinal epithelium by the muscarinic pathway, and that muscarinic activation was both necessary and sufficient for epithelial induction of cwn-2.
Aldicarb assays showed that endogenous ACh amounts rose very quickly during infection, peaking at 10 min, and then diminishing and plateauing after 4 h (Fig. 3M). In contrast, endogenous cwn-2 and clec-60 mRNA concentrations showed highest expression after 16 h of infection (Fig. 3N). Together, these data supported a hypothesis that S. aureus induced ACh release, which drove the muscarinic pathway, whose activation induced Wnt ligand and Frizzled receptor in the intestinal epithelium (Fig. 3O).
Canonical Wnt signaling is necessary and sufficient for defense gene induction
Silencing of positive regulators in the canonical Wnt pathway (Sawa and Korswagen, 2013), including homologs of Wnt, Frizzled, Disheveled, β-catenin, and TCF(LEF), abrogated induction of clec-60p::gfp by S. aureus (Fig. 4A, B). Silencing of Frizzled homolog mig-1 impaired induction of a set of host response genes (Fig. 4C). Additionally, silencing of the Wnt signaling genes impaired induction of clec-60p::gfp by arecoline (Fig. 4D, E). Silencing of negative regulators in the canonical Wnt pathway, including homologs of Axin, Casein kinase I, and β-TrCP, resulted in constitutive induction of clec-60p::gfp in uninfected animals (Fig. 4F, G) in a bar-1 (β-catenin)-dependent manner (Fig. 4G). Silencing of negative regulator gene gsk-3, homologous to mammalian glycogen synthase kinase 3β (GSK-3β), resulted in constitutively high expression of several host defense genes in uninfected animals (Fig. 4H). Taken together, these results showed that activation of the canonical Wnt signaling pathway was necessary and sufficient to induce host defense gene expression by infection and by ACh.
Figure 4. Canonical Wnt signaling is necessary and sufficient for host defense gene induction.

(A) Representative epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against cwn-2, mig-1, mig-5, bar-1, or pop-1, and subsequently infected with S. aureus for 16 h. (B) Quantitative analysis of (A). Data are mean ± SEM (at least two independent biological replicates, n ≥ 50 per condition). (C) qRT-PCR of clec-60, cpr-2, ilys-2, and ilys-3 in wild type and mig-1 mutant animals after 8 h S. aureus infection. Results are normalized to E. coli-fed wild type. Data are mean ± SEM (three independent biological replicates, n ≥ 3,000 per condition). (D) Representative epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against cwn-2, mig-1, mig-5, bar-1, or pop-1, and subsequently treated with 5 mM arecoline for 16 h. (E) Quantitative analysis of (D). Results are normalized to non-treated wild type animals. Data are mean ± SEM (at least 2 independent biological replicates, n ≥ 50 per condition). (F) Representative epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against gsk-3, pry-1, kin-19, or lin-23. (G) Quantitative analysis of (F). Results are normalized to wild type animals on empty vector. Data are mean ± SEM (two independent biological replicates, n ≥ 50 per condition). (H) qRT-PCR of antimicrobial genes in wild type animals on gsk-3 RNAi. Results are normalized to empty vector controls. Data are mean ± SEM (three independent biological replicates, n ≥ 3000 per condition). (I) Survival of wild type and cwn-2 mutant animals infected with S. aureus. Results are representative of 2 independent biological replicates. (J) Lifespan of wild type and cwn-2 mutant animals fed E. coli OP50. Results are representative of 2 independent biological replicates. (K) Survival of wild type and mig-1 mutant animals infected with S. aureus. Results are representative of 2 independent biological replicates. (L) Lifespan of wild type and mig-1 mutant animals fed E. coli OP50. Results are representative of 2 independent biological replicates. (M) Survival of wild type animals grown on empty vector or gsk-3 RNAi, and subsequently infected with S. aureus. Results are representative of 2 independent biological replicates. (N) Lifespan of animals treated as in (M), but transferred to E. coli OP50 after RNAi. Results are representative of 2 independent biological replicates. (O) Survival of bar-1 mutant animals treated as in (M). Results are representative of 2 independent biological replicates. (P) Survival of wild type animals treated with vehicle, 1 mM arecoline, or 1 mM scopolamine. Arecoline treatment was 24 h prior to infection and scopolamine was applied during infection. Results are representative of 2 independent biological replicates (Q) Survival of mig-1 mutant animals, treated as in (P). Results are representative of 2 independent biological replicates. (R) Schematic summary of results. Scale bars, 0.6 mm. Also see Figure S2.
Mutants defective in cwn-2 or mig-1 exhibited impaired infection survival (Fig. 4I, K) with no effect on aging (Fig. 4J, L). gsk-3 silencing resulted in enhanced host survival (Fig. 4M), even when it negatively affected aging (Fig. 4N). In contrast, gsk-3 silencing had no effect in animals lacking bar-1 function (Fig. 4O). These results demonstrated how activation of the canonical Wnt pathway enhanced host defense in a bar-1 (β-catenin)-dependent manner.
Arecoline administration, which activates muscarinic signaling, also resulted in enhanced survival of infection, while administration of scopolamine, which inhibited muscarinic signaling, impaired survival (Fig. 4P). Deletion of mig-1 (Frizzled) abrogated such effects (Fig. 4Q), again placing the Wnt pathway genetically downstream of muscarinic signaling. Muscarinic and Wnt pathway mutants showed no defects in aldicarb paralysis assays, ruling out the possibility that such mutations decrease ACh release (Fig. S2C to H). RNAi of egl-8 (PLCβ) or double RNAi of gar-2 and gar-3 did not enhance susceptibility in bar-1 (β-catenin) mutants, suggesting by epistasis analysis that gar-2, gar-3, egl-8 (PLCβ), and bar-1 (β-catenin) functioned in the same pathway (Fig. S2A). Additionally, unc-17, egl-8, and bar-1 null mutants showed similar susceptibilities to infection (Fig. S2B). Collectively, these observations supported a genetic pathway (Fig. 4R) in which S. aureus stimulated the release of ACh, which activated the muscarinic pathway, driving transcription of cwn-2 and mig-1 in the intestinal epithelium. Such activation of Wnt engaged the canonical Wnt pathway, resulting in the transcription of downstream host defense genes.
Canonical Wnt signaling functions post-developmentally
A key prediction of the pathway proposed in Fig. 4R is that the canonical Wnt pathway is activated in adult animals during infection, after its known roles in development (Sawa and Korswagen, 2013). The induction of cwn-2 and mig-1 in the intestinal epithelium strongly supports this prediction. Post-embryonic RNAi-mediated silencing has been used to circumvent developmental effects of key conserved genes, revealing phenotypes that would otherwise be masked by developmental defects or inviability (Curran, 2007). Adult-specific silencing of cwn-2, mig-1, and bar-1 resulted in defective host survival of infection (Fig. 5A, C, E), with little to no effect on aging (Fig. 5B, D, F), suggesting that the Wnt pathway functions during adulthood for host defense. Inorganic lithium compounds are known GSK-3β chemical inhibitors in clinical use. Brief administration of LiCl to adult uninfected animals resulted in high induction of clec-60p::gfp in a bar-1-dependent manner (Fig. 5G, H). Together, these results showed that post-developmental activation of canonical Wnt signaling in adult animals was necessary and sufficient for induction of host defense genes in the intestinal epithelium.
Figure 5. Wnt signaling functions post-developmentally for host defense.

(A, B) Wild type animals were subjected to RNAi against cwn-2, starting at the late L4 or young adult stage. Following RNAi, animals were infected with S. aureus (A), or transferred to E. coli OP50 plates (B). Results are representative of 2 independent biological replicates. (C, D) Same as in (A, B), but performing RNAi against mig-1. Results are representative of 2 independent biological replicates. (E, F) Same as in (A, B), but performing RNAi against bar-1. Results are representative of 2 independent biological replicates. (G) Representative epifluorescence micrographs of clec-60p::gfp animals reared on E. coli carrying empty vector or RNAi against bar-1, and subsequently treated with 100 mM LiCl for 16 h. (H) Quantitative analysis of (G). Results are normalized to vehicle-treated wild type animals. Data are mean ± SEM (2–3 independent biological replicates, n ≥ 50 per condition). Scale bars, 0.6 mm.
Muscarinic and Wnt signaling take place in the intestinal epithelium
Because intestinal epithelial clec-60 induction is under control of the canonical Wnt pathway, we examined whether the Wnt pathway functions in the intestinal epithelium. We adopted a method to produce RNAi-mediated silencing specifically in the intestinal epithelium (Luallen et al., 2015; Qadota et al., 2007). Intestine-specific silencing of genes mig-1 or bar-1 greatly impaired host survival of infection (Fig. 6A, B) with minor detrimental effects on aging. As mentioned, cwn-2 mutants exhibited defective survival of infection (Fig. 4I), but not defective aging (Fig. 4J). Re-expression of wild type cwn-2 only in the intestinal epithelium partially restored survival of infection in these mutants (Fig. 6D), whereas muscle-specific expression did not (Fig. 6E). Similarly, intestinal expression of mig-1 restored survival of infection in mig-1 mutants almost completely (Fig. 6F). These results showed that the canonical Wnt pathway functioned in the intestinal epithelium for host defense.
Figure 6. Muscarinic and Wnt signaling function in the intestinal epithelium.

(A) Survival of MGH167 animals subjected to RNAi against mig-1 until young adulthood, and subsequently infected with S. aureus. Results are representative of 2 independent biological replicates. (B) Same as in (A), but performing RNAi against bar-1. (C) Lifespan on E. coli OP50 of animals treated as in (A, B). Results are representative of 2 independent biological replicates. (D) Survival of wild type, cwn-2(ok895), and cwn-2(ok895) mutants expressing wild type cwn-2 in the intestinal epithelium. Results are representative of 2 independent biological replicates. (E) Survival of wild type, cwn-2(ok895) mutants, and cwn-2 mutants expressing wild type cwn-2 in the body wall muscle. (F) Survival of wild type, mig-1(e1787) mutants, and mig-1(e1787) mutants expressing wild type mig-1 in the intestinal epithelium. Results are representative of 2 independent biological replicates. (G) Survival of MGH167 animals subjected to RNAi against gar-2 and gar-3, egl-30, and egl-8 until young adulthood, and subsequently infected with S. aureus. Results are representative of 2 independent biological replicates. (H, I) Lifespan on E. coli OP50 of animals treated with RNAi as in (G). Results are representative of 2 independent biological replicates. Also see Figure S6.
Because both unc-17 (the vesicular ACh transporter) and cha-1 (choline acetyltransferase) are expressed exclusively in the nervous system of C. elegans, it is thought that ACh is produced and released only by neurons in nematodes (Alfonso et al., 1993; Duerr et al., 2008; Pereira et al., 2015). We wondered if the muscarinic pathway might also function in the intestinal epithelium. Intestinal silencing of gar-2 or gar-3 caused mild infection survival defects (Fig. S3A, B), whereas simultaneous gar-2 and gar-3 silencing caused a strong defect (Fig. 6G), suggesting that these two receptors share redundant functions in the intestine. Simultaneous gar-2, gar-3 intestinal silencing greatly extended the lifespan of non-infected animals (Fig. 1I), ruling out that the enhanced susceptibility to infection was due to a defect in viability or aging. In addition, colony forming unit (cfu) assays did not show a difference in bacterial load between treated and control animals, indicating that the enhanced susceptibility was not caused by enhanced bacterial colonization of, or residence in, the intestine (Fig. S3C). Intestinal RNAi of egl-30 (Gαq) or of egl-8 (PLCβ) also strongly impaired host survival (Fig. 6G), but resulted in minor reductions of lifespan on E. coli, suggesting a possible role in aging (Fig. 6H). These results showed that the muscarinic pathway performed important functions for host defense in the intestinal epithelium.
Identification of transcription factors that link muscarinic and Wnt signaling
The results so far indicated that the muscarinic pathway and the downstream Wnt pathway functioned in the intestinal epithelium to control host defense gene induction. However, the mechanism of the connection between these two pathways was unknown. We determined that muscarinic signaling caused induction of cwn-2 (Fig. 3F–L), implying the existence of transcription factors that mediate muscarinic-induced Wnt induction. Therefore, we performed a reverse genetic screen to identify such transcription factors. Following gene silencing, we administered arecoline and measured clec-60p::gfp. Thus, we identified 10 transcription factor genes that were required for full clec-60p::gfp induction by arecoline (Fig. 7A). Because these transcription factors could be involved upstream or downstream of Wnt signaling, we measured cwn-2 induction after arecoline treatment. We expected genes that function as a link between muscarinic and Wnt signaling to disrupt cwn-2 induction by arecoline. Out of the 10 candidate genes, only 4 satisfied this criterion (Fig. 7B). Because of the availability of genetic tools and its evolutionary conservation, we decided to focus on one such gene, lin-1, which is homologous to human ELK1-3 transcription factor genes with known roles in inflammation (Oettgen, 2006).
Figure 7. Transcription factors that link muscarinic and Wnt signaling.

(A) Quantification of GFP expression in clec-60p::gfp animals subjected to RNAi against the indicated transcription factor genes, and subsequently treated with 5 mM arecoline for 16 h. Data are mean ± SEM (2 independent biological replicates, n ≥ 50 per condition). Results are normalized to vehicle-treated empty vector control. (B) Quantification of Venus expression in cwn-2p::cwn-2::venus animals treated with RNAi as in (A) and subsequently with 5 mM arecoline for 24 h. Data are mean ± SEM (2 independent biological replicates, n ≥ 50 per condition). Results are normalized to vehicle-treated empty vector control. (C) qRT-PCR of clec-60 in wild type and lin-1 null mutant animals treated with 5 mM arecoline for 8 h. Results are normalized to vehicle-treated wild type control. Data are mean ± SEM (3 independent biological replicates, n ≥ 3,000 per condition). (D) qRT-PCR of cwn-2 in the same animals as in (C), treated with 5 mM arecoline for 16 h. (E) qRT-PCR of clec-60 in E. coli-fed wild type and lin-1 gain of function (GF) mutant animals. Results are normalized to wild type. Data are mean ± SEM (3 independent biological replicates, n ≥ 3,000 per condition). (F) qRT-PCR of cwn-2 in the same animals as in (E). (G) MGH167 animals subjected to RNAi against lin-1 until young adulthood, and subsequently infected with S. aureus. Results are representative of 2 independent biological replicates. (H) Lifespan of MGH167 animals treated as in (G), but placed on E. coli OP50. Also see Figure S5.
Genetic deletion of lin-1 (ELK) partially impaired arecoline induction of clec-60 (Fig. 7C) and strongly impaired induction of cwn-2 (Fig. 7D). A gain of function allele of lin-1 (ELK) was sufficient for high constitutive expression of clec-60 (Fig. 7E) and cwn-2 (Fig. 7F) in uninfected animals. These results indicated that lin-1 was sufficient and partially necessary for host defense gene induction downstream of ACh.
Intestine-specific silencing of lin-1 (ELK) strongly impaired host survival of infection (Fig. 7G), with no effect on aging (Fig. 7H). Furthermore, genetic epistasis experiments showed a lack of effect of lin-1 RNAi in the survival of infected bar-1 (β-catenin) mutants (Fig. S5), consistent with lin-1 (ELK) and bar-1 (β-catenin) participating in the same pathway. Thus, lin-1 (ELK), and potentially other genes identified in our screen, mediate control of Wnt signaling by the muscarinic pathway.
DISCUSSION
C. elegans possess a simple nervous system, comprising 302 neurons (White et al., 1986)). C. elegans neurons are divided into sensory, interneuron, and motor neuron classes. A majority of the neurons in C. elegans serve sensory functions and are present in the anterior head region, where they are organized into ganglia surrounding the pharynx. About 150 neurons are thought to synthesize and release ACh (Duerr et al., 2008; Pereira et al., 2015). Sensory, interneuron, and motor neurons can be cholinergic. Synapses connecting neurons and the intestinal epithelium have not been observed.
Our results suggested that ACh, released from the nervous system in response to infection with S. aureus, functioned in a neuroendocrine fashion to activate muscarinic receptors in the intestinal epithelium. This activation led to increased expression of Wnt and its receptor Frizzled in the intestinal epithelium. Wnt, presumably secreted to the basolateral side of the polarized epithelium, could signal to the intestinal epithelial cells in an autocrine and paracrine fashion. Activation of the canonical Wnt pathway led to the induction of host defense genes, including antimicrobials such as C-type lectin clec-60 and lysozymes. The genetic link between the muscarinic and Wnt pathways was mediated by at least four highly conserved transcription factors, including the Ets family transcription factor LIN-1, which is homologous to human transcription factors ELK1-3.
The nervous system may have evolved to stimulate cells distal from the site of activation, injury, or infection to help produce an integrated physiological response to insult. Also, this mechanism may allow the translation of a fast and short-lived signal (ACh) to a slow and longer-lived signal (Wnt ligand) to produce a robust response to infection. Furthermore, release of ACh increases C. elegans locomotion (Rand, 2007). Pathogen-triggered ACh release may thus provide an elegant way of coupling emergency evasive behaviors with the enhanced expression of host defense genes. This view is supported by previous work, which showed that cholinergic signaling is involved in the behavioral response to infection by Microbacterium nematophilum (McMullan et al., 2012).
Our studies highlight muscarinic signaling as a key mechanism by which the nervous system controls intestinal epithelial host defense. In mice and humans, the vagal nerve and the enteric nervous system project cholinergic fibers to the viscera and the intestinal epithelium (Erickson et al., 2014; Foong et al., 2014; Gautron et al., 2013; Jönsson et al., 2007; Matteoli and Boeckxstaens, 2013). Muscarinic receptors are broadly expressed in the mammalian intestinal epithelium (Harrington et al., 2010; Khan et al., 2013). In vitro, muscarinic stimulation of intestinal epithelial cells modulated ion secretion, cell proliferation, and barrier permeability and repair (Hirota and McKay, 2006; Khan et al., 2014; Peng et al., 2013). However, mammalian epithelial cells and other cells are capable of producing and secreting non-neuronal ACh (Grando et al., 2015). Therefore, muscarinic signaling at the mammalian intestinal epithelium is complex and its function is poorly understood.
Nonetheless, emerging clues hint that epithelial muscarinic signaling is important for immune homeostasis at several mucosal sites in mammals. For example, muscarinic stimulation of intestinal epithelial cells prevents barrier disruption under inflammatory conditions (Dhawan et al., 2015). OCTN1, which encodes an ACh exporter, is a susceptibility locus for Crohn’s disease (Pochini et al., 2012). Muscarinic stimulation causes IL-8 secretion by bronchial epithelial cells, driving inflammation (Profita et al., 2008). Muscarinic signaling is important during experimental sepsis (Amaral et al., 2016; Jeremias et al., 2016), and anticholinergic treatment is associated with community-acquired pneumonia in elderly adults (Chatterjee et al., 2016). Mice lacking M3 muscarinic receptor exhibit impaired pro-inflammatory signaling and clearance of bacterial and helminth infections (Darby et al., 2015; McLean et al., 2015). Secretion of ACh by mouse T cells enhances antimicrobial peptide production by the intestinal epithelium via muscarinic signaling (Dhawan et al., 2016). Anti-muscarinic therapies for the treatment of inflammation are being considered in diseases such as chronic obstructive pulmonary disease (COPD) or inflammatory bowel disease (IBD) (Matera et al., 2014; Sales, 2010; Verbout and Jacoby, 2012). However, the molecular mechanisms by which muscarinic signaling enhances mucosal host defense are not known.
The canonical Wnt pathway plays several important roles in the mammalian intestinal epithelium, including development, homeostasis, and repair after inflammation (Karin and Clevers, 2016). Wnt signaling within Paneth cells (crypt epithelial cells specialized for antimicrobial peptide secretion) is critical for antimicrobial gene expression (Clevers and Bevins, 2013; Clevers et al., 2007). Wnt signaling defects in Paneth cells are associated with IBD (Bevins and Salzman, 2011; Clevers et al., 2007; Kini et al., 2015; Koslowski et al., 2009, 2012). Thus, our delineation of a canonical Wnt pathway that is important for antimicrobial peptide expression in the nematode intestine highlights evolutionary conservation of its host defense function.
We identified four conserved transcription factor genes as genetic links between the muscarinic and Wnt pathways. hlh-26 has weak resemblance to human genes HEYL and ARNTL2, and hlh-34 is related to NPAS3 and HIF1A. ceh-6 and lin-1 are homologous to human genes POU3F1-4 and ELK1-3, respectively. lin-1 is under control of the ERK signaling cascade in C. elegans (Lackner and Kim, 1998), and had not been implicated in host defense previously. Human ELK1 is activated downstream of muscarinic-Gαq-PKC signaling (Blaukat et al., 2000; Rössler et al., 2008). Both ELK1 and ELK3 are known to be important for host defense induced by bacterial peptidoglycan and for phagocytosis of bacteria by macrophages (Tsoyi et al., 2015; Xu et al., 2001). Thus, our results placing lin-1 (ELK) as a link between upstream muscarinic signaling and downstream Wnt signaling are of great importance.
Genetic and pharmacological inhibition of muscarinic signaling reduces intestinal epithelial cell proliferation and carcinogenesis in mice (Raufman et al., 2008). In humans, cholinergic fibers of the vagal nerve enhance gastric tumorigenesis, by enhanced Wnt3 expression driven by the M3 muscarinic receptor (Zhao et al., 2014). This observation provides a striking clinical parallel to our results, highlights their clinical relevance, and suggests that the implications of our findings may transcend host-pathogen interactions. Moreover, members of the human microbiota are able to produce physiologically relevant quantities of ACh (Sarkar et al., 2016), perhaps providing a direct mechanism of microbiota-epithelium interaction, and an avenue of therapeutic intervention for chronic inflammation. It seems likely that cholinergic induction of Wnt in the gut epithelium constitutes a fundamental mechanism of the so-called Gut-Brain-Microbiota axis of immune homeostasis (Dhawan et al., 2012; Tracey, 2014).
STAR★Methods
Contact for Reagents and Resources
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Javier E. Irazoqui (Javier.Irazoqui@umassmed.edu).
Experimental Model
C. elegans strains
C. elegans strains were grown on nematode-growth media (NGM) plates seeded with E. coli OP50 at 15 – 20 °C, according to standard procedures (Powell and Ausubel, 2008).
Method Details
Infection
S. aureus HA-MRSA USA100 (Carroll et al., 2013) was grown overnight in tryptic soy broth (TSB) containing 50 μg/ml kanamycin (KAN). 10 μl of overnight cultures was uniformly spread on the entire surface of 35 mm TSA plates containing 10 μg/ml kanamycin, and incubated 4–6 h at 37 °C. Animals we re treated with 80–100 μg/ml 5-fluoro-2′-deoxyuridine (FUDR) at L4 larval stage for 24 h at 15 °C before transfer to S. aureus plates. After FUDR treatment, 25 – 40 infertile animals were transferred to each of three replicate infection plates per strain. Survival was quantified as described (Powell and Ausubel, 2008). Animals that died of bursting vulva, matricidal hatching, or crawling off the agar were censored.
Starvation
Worms were supplemented with 80–100 μg/ml FUDR at L4 larval stage for 24 h at 15 °C before transfer to solid TSA plates without bacteria. After 8 h, animals were harvested for imaging.
Longevity (aging) assays
All assays were performed as described in (Powell and Ausubel, 2008). Animals were transferred to NGM plates seeded with E. coli OP50, supplemented with 80 – 100 μg/ml FUDR and incubated at 25 °C. Experiments were performed at least twice.
RNAi by feeding
RNAi was carried out using bacterial feeding RNAi (Timmons et al., 2001). Briefly, 8 to 12 gravid adult worms were spot-bleached in 20% alkaline hypochlorite solution directly on the plates (outside the bacterial lawn) to synchronize progeny. Progeny of bleached adults were treated with FUDR at the L4 larval stage for 24 h at 15 °C prior to use. The RNAi plates contain E. coli HT115 carrying vector L4440 (empty vector) or expressing dsRNA of selected genes. Adult RNAi treatment was done by incubating young adult animals for 2 days at 15 °C on RNAi plates. gar-2 and gar-3 double RNAi plates were prepared by adding a mixture of 150 μl of overnight culture of HT115 carrying gar-2 RNAi and 150 μl of HT115 carrying gar-3 RNAi. HT115 RNAi clones were obtained from the Ahringer genomic RNAi library, or the Vidal library when absent in the former. Clone identity was confirmed by sequencing, and absence of off target effects was verified against predictions by the C. elegans genomic database resource, WormBase (www.wormbase.org). RNAi gene silencing was confirmed by real-time RT-PCR (Fig. S4).
RNAi Screening
Briefly, 2,000 gravid adult, clec-60p::gfp worms were bleached with alkaline hypochlorite, followed by several washes with M9 buffer (Powell and Ausubel, 2008). Subsequently, the eggs were incubated in M9 buffer overnight at room temperature. RNAi was carried out using bacterial feeding on L1 stage animals in a semi-automated liquid setup until they reached the young adult stage at 15 °C (Lehner et al., 2006). Following RNAi treatment, animals were washed and transferred to solid medium in 96 well plates containing S. aureus for the induction of clec-60p::gfp. The RNAi sub-libraries used, assembled from the Ahringer and Vidal genomic libraries, were: GPCRs (1748 genes; assembled by Justine Melo, MGH), protein kinases (441 genes; assembled by Javier Irazoqui, MGH), and transcription factors (427 genes; assembled by Sean Curran and Dave Simon, MGH).
Aldicarb assays
Aldicarb paralysis after infection was performed using a previously described protocol (Mahoney et al., 2006) with minor changes. Briefly, 30 young adult animals were placed for 10 min to 12 h on infection plates. Next, animals were immediately transferred to TSA (Tryptic Soy Agar) plates containing 1 mM aldicarb (Sigma-Aldrich) and 100 μl of 10X overnight culture of OP50. Paralysis was quantified blindly at room temperature.
qRT-PCR
After treatment, C. elegans were collected in M9 buffer and lysed using TRI Reagent (Molecular Research Center) following manufacturer’s instructions. cDNA was obtained with iScript cDNA (Bio-Rad) and qRT-PCR was performed using iQ SYBR Green (Bio-Rad), as described previously (Irazoqui et al., 2010). Data analysis was performed using the Pfaffl method (Pfaffl, 2001).
Imaging
Animals expressing a fluorescent reporter gene were kept for 1 h at room temperature prior to imaging. Animals were next harvested by washing with M9 buffer (Powell and Ausubel, 2008), and paralyzed with 10 % NaN3 in half-well 96-well plates. Image acquisition was automatically performed using a Cytation 3 Imaging Plate Reader (Biotek).
Drug treatments
Arecoline hydrobromide (Sigma, 31593-250MG), 1 mM for killing assays and 5 mM for other experiments; Scopolamine (Sigma PHR1470-500MG), 1 mM for killing assays and 5 mM for other assays; Oxotremorine (sigma, O100-100MG) 1 mM, LiCl (Sigma 203637-10G) 100 mM). For killing assays and qRT-PCR, all drug treatments were performed on solid NGM plates supplemented with drug. Animals were treated at the young adult stage and incubated at 25 °C for 24 h. After 16 h, animals were harvested for killing assays or qRT-PCR. For reporter gene quantification, all drug treatments were performed in NGM liquid culture for 16 h. Subsequently, animals were washed with M9 buffer and prepared for imaging.
Quantification and Statistical Analysis
Statistical analyses were performed using Prism 6 software (GraphPad). Survival data were compared using the Log-Rank test. Data are represented as median survival (MS), as defined by Kaplan-Meier analysis, or Time to 50% Death (LT50), as defined by nonlinear regression. A p value ≤ 0.05 was considered significantly different from control. For qRT-PCR, two-sample, two-tailed t tests were performed to evaluate differences among pooled ΔCt values according to Pfaffl (Pfaffl, 2001) using Excel. A p value ≤ 0.05 was considered significant. For image quantification, two-sample, two-tailed t tests were performed. Prior to use of the t-test, all values were confirmed for normal distribution by the Agostino Pearson omnibus test.
Supplementary Material
Highlights.
Acetylcholine regulates transcriptional host defense in intestinal epithelial cells
Acetylcholine activates muscarinic and Wnt signaling in the intestine
Transcription factor lin-1 (ELK) links muscarinic and Wnt pathways by Wnt induction
Acknowledgments
Dr. Frederick Ausubel and his laboratory provided generous intellectual input and shared laboratory space and equipment. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number GM101056, and by the National Science Foundation under award number NSF1457055 (to J.E.I.). Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
S.L. performed, analyzed, and reported most experiments. K.W. performed aldicarb assays and specific experiments. S.J. and A.H. helped with the RNAi screens. M.N. contributed to specific experiments. J.E.I. provided key intellectual input and overall guidance for experimental design, execution, interpretation, and reporting. All authors contributed to writing the manuscript.
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aballay A, Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI. Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Sci N Y NY. 2008;322:460–464. doi: 10.1126/science.1163673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alfonso A, Grundahl K, Duerr J, Han H, Rand J. The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Sci N Y NY. 1993;261:617–619. doi: 10.1126/science.8342028. [DOI] [PubMed] [Google Scholar]
- Amaral FA, Fagundes CT, Miranda AS, Costa VV, Resende L, Gloria de Souza D, da Prado VF, Teixeira MM, Maximo Prado MA, Teixeira AL. Endogenous Acetylcholine Controls the Severity of Polymicrobial Sepsis-associated Inflammatory Response in Mice. Curr Neurovasc Res. 2016;13:4–9. doi: 10.2174/1567202612666151026105915. [DOI] [PubMed] [Google Scholar]
- Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G Protein-Coupled Receptor-Mediated Mitogen-Activated Protein Kinase Activation through Cooperation of Gαq and Gαi Signals. Mol Cell Biol. 2000;20:6837–6848. doi: 10.1128/mcb.20.18.6837-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloes DA, Kretschmer D, Peschel A. Enemy attraction: bacterial agonists for leukocyte chemotaxis receptors. Nat Rev Microbiol. 2015;13:95–104. doi: 10.1038/nrmicro3390. [DOI] [PubMed] [Google Scholar]
- Bolz DD, Tenor JL, Aballay A. A conserved PMK-1/p38 MAPK is required in caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J Biol Chem. 2010;285:10832–10840. doi: 10.1074/jbc.M109.091629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch TCG, Augustin R, Anton-Erxleben F, Fraune S, Hemmrich G, Zill H, Rosenstiel P, Jacobs G, Schreiber S, Leippe M, et al. Uncovering the evolutionary history of innate immunity: the simple metazoan Hydra uses epithelial cells for host defence. Dev Comp Immunol. 2009;33:559–569. doi: 10.1016/j.dci.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Carroll RK, Burda WN, Roberts JC, Peak KK, Cannons AC, Shaw LN. Draft Genome Sequence of Strain CBD-635, a Methicillin-Resistant Staphylococcus aureus USA100 Isolate. Genome Announc. 2013:1. doi: 10.1128/genomeA.00491-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Carnahan RM, Chen H, Holmes HM, Johnson ML, Aparasu RR. Anticholinergic Medication Use and Risk of Pneumonia in Elderly Adults: A Nested Case-Control Study. J Am Geriatr Soc. 2016;64:394–400. doi: 10.1111/jgs.13932. [DOI] [PubMed] [Google Scholar]
- Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75:289–311. doi: 10.1146/annurev-physiol-030212-183744. [DOI] [PubMed] [Google Scholar]
- Clevers HC, Nusse R. Wnt/β-Catenin Signaling and Disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Clevers HC, Wehkamp J, Wang G, Kübler I, Nuding S, Gregorieff A, Schnabel A, Kays RJ, Fellermann K, Burk O, et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J Immunol Baltim Md 1950. 2007;179:3109–3118. doi: 10.4049/jimmunol.179.5.3109. [DOI] [PubMed] [Google Scholar]
- Curran SP. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby M, Schnoeller C, Vira A, Culley F, Bobat S, Logan E, Kirstein F, Wess J, Cunningham AF, Brombacher F, et al. The M3 Muscarinic Receptor Is Required for Optimal Adaptive Immunity to Helminth and Bacterial Infection. PLOS Pathog. 2015;11:e1004636. doi: 10.1371/journal.ppat.1004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Cailotto C, Harthoorn LF, de Jonge WJ. Cholinergic signalling in gut immunity. Life Sci. 2012;91:1038–1042. doi: 10.1016/j.lfs.2012.04.042. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Hiemstra IH, Verseijden C, Hilbers FW, Te Velde AA, Willemsen LEM, Stap J, den Haan JM, de Jonge WJ. Cholinergic receptor activation on epithelia protects against cytokine-induced barrier dysfunction. Acta Physiol Oxf Engl. 2015;213:846–859. doi: 10.1111/apha.12469. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Palma GD, Willemze RA, Hilbers FW, Verseijden C, Luyer MD, Nuding S, Wehkamp J, Souwer Y, de Jong EC, et al. Acetylcholine-producing T cells in the intestine regulate antimicrobial peptide expression and microbial diversity. Am J Physiol - Gastrointest Liver Physiol. 2016;311:G920–G933. doi: 10.1152/ajpgi.00114.2016. [DOI] [PubMed] [Google Scholar]
- Duerr JS, Han H-P, Fields SD, Rand JB. Identification of major classes of cholinergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol. 2008;506:398–408. doi: 10.1002/cne.21551. [DOI] [PubMed] [Google Scholar]
- Erickson CS, Lee SJ, Barlow-Anacker AJ, Druckenbrod NR, Epstein ML, Gosain A. Appearance of cholinergic myenteric neurons during enteric nervous system development: comparison of different ChAT fluorescent mouse reporter lines. Neurogastroenterol Motil Off J Eur Gastrointest Motil Soc. 2014;26:874–884. doi: 10.1111/nmo.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foong JPP, Tough IR, Cox HM, Bornstein JC. Properties of cholinergic and non-cholinergic submucosal neurons along the mouse colon. J Physiol. 2014;592:777–793. doi: 10.1113/jphysiol.2013.265686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautron L, Rutkowski JM, Burton MD, Wei W, Wan Y, Elmquist JK. Neuronal and nonneuronal cholinergic structures in the mouse gastrointestinal tract and spleen. J Comp Neurol. 2013;521:3741–3767. doi: 10.1002/cne.23376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilani AH, Ghayur MN, Saify ZS, Ahmed SP, Choudhary MI, Khalid A. Presence of cholinomimetic and acetylcholinesterase inhibitory constituents in betel nut. Life Sci. 2004;75:2377–2389. doi: 10.1016/j.lfs.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Grando SA, Kawashima K, Kirkpatrick CJ, Kummer W, Wessler I. Recent progress in revealing the biological and medical significance of the non-neuronal cholinergic system. Int Immunopharmacol. 2015;29:1–7. doi: 10.1016/j.intimp.2015.08.023. [DOI] [PubMed] [Google Scholar]
- Harrington AM, Peck CJ, Liu L, Burcher E, Hutson JM, Southwell BR. Localization of muscarinic receptors M1R, M2R and M3R in the human colon. Neurogastroenterol Motil Off J Eur Gastrointest Motil Soc. 2010;22:999–1008. e262–3. doi: 10.1111/j.1365-2982.2009.01456.x. [DOI] [PubMed] [Google Scholar]
- Head B, Aballay A. Recovery from an Acute Infection in C. elegans Requires the GATA Transcription Factor ELT-2. PLoS Genet. 2014;10:e1004609. doi: 10.1371/journal.pgen.1004609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota CL, McKay DM. Cholinergic regulation of epithelial ion transport in the mammalian intestine. Br J Pharmacol. 2006;149:463–479. doi: 10.1038/sj.bjp.0706889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. The neuronal genome of Caenorhabditis elegans. WormBook. 2013:1–106. doi: 10.1895/wormbook.1.161.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV. Chapter 3 - Epithelial Cell Contributions to Intestinal Immunity. In: Alt FW, editor. Advances in Immunology. Academic Press; 2015. pp. 129–172. [DOI] [PubMed] [Google Scholar]
- Hwang JM, Chang DJ, Kim US, Lee YS, Park YS, Kaang BK, Cho NJ. Cloning and functional characterization of a Caenorhabditis elegans muscarinic acetylcholine receptor. Receptors Channels. 1999;6:415–424. [PubMed] [Google Scholar]
- Irazoqui JE, Ng A, Xavier RJ, Ausubel FM. Role for beta-catenin and HOX transcription factors in Caenorhabditis elegans and mammalian host epithelial-pathogen interactions. Proc Natl Acad Sci U S A. 2008;105:17469–17474. doi: 10.1073/pnas.0809527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 2010;6:e1000982. doi: 10.1371/journal.ppat.1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremias IC, Victorino VJ, Barbeiro HV, Kubo SA, Prado CM, Lima TM, Soriano FG. The Role of Acetylcholine in the Inflammatory Response in Animals Surviving Sepsis Induced by Cecal Ligation and Puncture. Mol Neurobiol. 2016;53:6635–6643. doi: 10.1007/s12035-015-9538-y. [DOI] [PubMed] [Google Scholar]
- Jönsson M, Norrgård O, Forsgren S. Presence of a marked nonneuronal cholinergic system in human colon: study of normal colon and colon in ulcerative colitis. Inflamm Bowel Dis. 2007;13:1347–1356. doi: 10.1002/ibd.20224. [DOI] [PubMed] [Google Scholar]
- Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. 2016;529:307–315. doi: 10.1038/nature17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MRI, Anisuzzaman ASM, Semba S, Ma Y, Uwada J, Hayashi H, Suzuki Y, Takano T, Ikeuchi H, Uchino M, et al. M1 is a major subtype of muscarinic acetylcholine receptors on mouse colonic epithelial cells. J Gastroenterol. 2013;48:885–896. doi: 10.1007/s00535-012-0718-5. [DOI] [PubMed] [Google Scholar]
- Khan RI, Yazawa T, Anisuzzaman ASM, Semba S, Ma Y, Uwada J, Hayashi H, Suzuki Y, Ikeuchi H, Uchino M, et al. Activation of focal adhesion kinase via M1 muscarinic acetylcholine receptor is required in restitution of intestinal barrier function after epithelial injury. Biochim Biophys Acta. 2014;1842:635–645. doi: 10.1016/j.bbadis.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Kini AT, Thangaraj KR, Simon E, Shivappagowdar A, Thiagarajan D, Abbas S, Ramachandran A, Venkatraman A. Aberrant Niche Signaling in the Etiopathogenesis of Ulcerative Colitis: Inflamm. Bowel Dis. 2015:1. doi: 10.1097/MIB.0000000000000523. [DOI] [PubMed] [Google Scholar]
- Koslowski MJ, Kübler I, Chamaillard M, Schaeffeler E, Reinisch W, Wang G, Beisner J, Teml A, Peyrin-Biroulet L, Winter S, et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS ONE. 2009;4:e4496. doi: 10.1371/journal.pone.0004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowski MJ, Teltschik Z, Beisner J, Schaeffeler E, Wang G, Kübler I, Gersemann M, Cooney R, Jewell D, Reinisch W, et al. Association of a functional variant in the Wnt co-receptor LRP6 with early onset ileal Crohn’s disease. PLoS Genet. 2012;8:e1002523. doi: 10.1371/journal.pgen.1002523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Kim SK. Genetic Analysis of the Caenorhabditis elegans MAP Kinase Gene mpk-1. Genetics. 1998;150:103–117. doi: 10.1093/genetics/150.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of Synaptic Transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG Binding to UNC-13 Is Required to Stimulate Acetylcholine Release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Lehner B, Tischler J, Fraser AG. RNAi screens in Caenorhabditis elegans in a 96-well liquid format and their application to the systematic identification of genetic interactions. Nat Protoc. 2006;1:1617–1620. doi: 10.1038/nprot.2006.245. [DOI] [PubMed] [Google Scholar]
- Luallen RJ, Bakowski MA, Troemel ER. Characterization of Microsporidia-Induced Developmental Arrest and a Transmembrane Leucine-Rich Repeat Protein in Caenorhabditis elegans. PLoS ONE. 2015:10. doi: 10.1371/journal.pone.0124065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc. 2006;1:1772–1777. doi: 10.1038/nprot.2006.281. [DOI] [PubMed] [Google Scholar]
- Matera MG, Rogliani P, Cazzola M. Muscarinic receptor antagonists for the treatment of chronic obstructive pulmonary disease. Expert Opin Pharmacother. 2014;15:961–977. doi: 10.1517/14656566.2014.899581. [DOI] [PubMed] [Google Scholar]
- Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. 2013;62:1214–1222. doi: 10.1136/gutjnl-2012-302550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD. The C. elegans intestine. WormBook Online Rev. C Elegans Biol. 2007:1–36. doi: 10.1895/wormbook.1.133.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean LP, Smith A, Cheung L, Sun R, Grinchuk V, Vanuytsel T, Desai N, Urban JF, Zhao A, Raufman J-P, et al. Type 3 Muscarinic Receptors Contribute to Clearance of Citrobacter rodentium. Inflamm Bowel Dis. 2015;21:1860–1871. doi: 10.1097/MIB.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan R, Anderson A, Nurrish S. Behavioral and immune responses to infection require Gαq- RhoA signaling in C. elegans. PLoS Pathog. 2012;8:e1002530. doi: 10.1371/journal.ppat.1002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149:452–466. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB. Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron. 1999;24:323–333. doi: 10.1016/s0896-6273(00)80847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najibi M, Labed SA, Visvikis O, Irazoqui JE. An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense. Cell Rep. 2016;15:1728–1742. doi: 10.1016/j.celrep.2016.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettgen P. Regulation of Vascular Inflammation and Remodeling by ETS Factors. Circ Res. 2006;99:1159–1166. doi: 10.1161/01.RES.0000251056.85990.db. [DOI] [PubMed] [Google Scholar]
- O’rourke D, Baban D, Demidova M, Mott R, Hodgkin J. Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 2006;16:1005–1016. doi: 10.1101/gr.50823006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Heath J, Drachenberg C, Raufman JP, Xie G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer. 2013;13:204. doi: 10.1186/1471-2407-13-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Kratsios P, Serrano-Saiz E, Sheftel H, Mayo AE, Hall DH, White JG, LeBoeuf B, Garcia LR, Alon U, et al. A cellular and regulatory map of the cholinergic nervous system of C. elegans. ELife. 2015;4:e12432. doi: 10.7554/eLife.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochini L, Scalise M, Galluccio M, Pani G, Siminovitch KA, Indiveri C. The human OCTN1 (SLC22A4) reconstituted in liposomes catalyzes acetylcholine transport which is defective in the mutant L503F associated to the Crohn’s disease. Biochim Biophys Acta. 2012;1818:559–565. doi: 10.1016/j.bbamem.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Powell JR, Ausubel FM. Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol Clifton NJ. 2008;415:403–427. doi: 10.1007/978-1-59745-570-1_24. [DOI] [PubMed] [Google Scholar]
- Powell JR, Kim DH, Ausubel FM. The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc Natl Acad Sci U S A. 2009;106:2782–2787. doi: 10.1073/pnas.0813048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP, Gjomarkaj M. Acetylcholine mediates the release of IL-8 in human bronchial epithelial cells by a NFkB/ERK-dependent mechanism. Eur J Pharmacol. 2008;582:145–153. doi: 10.1016/j.ejphar.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Pujol N, Ewbank JJ, Link EM, Liu LX, Kurz CL, Alloing G, Tan MWW, Ray KP, Solari R, Johnson CD. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr Biol CB. 2001;11:809–821. doi: 10.1016/s0960-9822(01)00241-x. [DOI] [PubMed] [Google Scholar]
- Qadota H, Inoue M, Hikita T, Köppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173. doi: 10.1016/j.gene.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. Acetylcholine. WormBook Online Rev. C Elegans Biol. 2007:1–21. doi: 10.1895/wormbook.1.131.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raufman J-P, Samimi R, Shah N, Khurana S, Shant J, Drachenberg C, Xie G, Wess J, Cheng K. Genetic Ablation of M3 Muscarinic Receptors Attenuates Murine Colon Epithelial Cell Proliferation and Neoplasia. Cancer Res. 2008;68:3573–3578. doi: 10.1158/0008-5472.CAN-07-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, Function, and Control of Phosphoinositide-Specific Phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Rössler OG, Henß I, Thiel G. Transcriptional response to muscarinic acetylcholine receptor stimulation: Regulation of Egr-1 biosynthesis by ERK, Elk-1, MKP-1, and calcineurin in carbachol-stimulated human neuroblastoma cells. Arch Biochem Biophys. 2008;470:93–102. doi: 10.1016/j.abb.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Gabor GL, Miklos Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, et al. Comparative Genomics of the Eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales ME. Muscarinic receptors as targets for anti-inflammatory therapy. Curr Opin Investig Drugs Lond Engl 2000. 2010;11:1239–1245. [PubMed] [Google Scholar]
- Sarkar A, Lehto SM, Harty S, Dinan TG, Cryan JF, Burnet PWJ. Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. Trends Neurosci. 2016;39:763–781. doi: 10.1016/j.tins.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa H, Korswagen HC. T.C. elegans R. Community, editor. WormBook. 2013. Wnt signaling in C. elegans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-García O, Valdez-Alarcón JJ, Baizabal-Aguirre VM. The Wnt/β-catenin signaling pathway controls the inflammatory response in infections caused by pathogenic bacteria. Mediators Inflamm. 2014;2014:310183. doi: 10.1155/2014/310183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Rafter JD, Melowic HR, Cho W. Mechanism of Diacylglycerol-induced Membrane Targeting and Activation of Protein Kinase Cδ. J Biol Chem. 2004;279:29501–29512. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- Steger KA, Avery L. The GAR-3 muscarinic receptor cooperates with calcium signals to regulate muscle contraction in the Caenorhabditis elegans pharynx. Genetics. 2004;167:633–643. doi: 10.1534/genetics.103.020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. Approaching the Next Revolution? Evolutionary Integration of Neural and Immune Pathogen Sensing and Response. Cold Spring Harb Perspect Biol. 2014 doi: 10.1101/cshperspect.a016360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoyi K, Geldart AM, Christou H, Liu X, Chung SW, Perrella MA. Elk-3 is a KLF4-regulated gene that modulates the phagocytosis of bacteria by macrophages. J Leukoc Biol. 2015;97:171–180. doi: 10.1189/jlb.4A0214-087R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Sci N Y NY. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbout NG, Jacoby DB. Muscarinic Receptor Agonists and Antagonists: Effects on Inflammation and Immunity. In: Fryer AD, Christopoulos A, Nathanson NM, editors. Muscarinic Receptors. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. pp. 403–427. [DOI] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 2007;8:R194. doi: 10.1186/gb-2007-8-9-r194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Dziarski R, Wang Q, Swartz K, Sakamoto KM, Gupta D. Bacterial peptidoglycan-induced tnf-alpha transcription is mediated through the transcription factors Egr-1, Elk-1, and NF-kappaB. J Immunol Baltim Md 1950. 2001;167:6975–6982. doi: 10.4049/jimmunol.167.12.6975. [DOI] [PubMed] [Google Scholar]
- Yoo BB, Mazmanian SK. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity. 2017;46:910–926. doi: 10.1016/j.immuni.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C-M, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, Flatberg A, Johannessen H, Friedman RA, Renz BW, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6:250ra115-250ra115. doi: 10.1126/scitranslmed.3009569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.