

Here, Mendez-Dorantes et al. investigated how far a chromosomal double-strand break (DSB) can be positioned from a repeat sequence to induce repeat-mediated rearrangements in mammalian cells. Using a novel reporter assay in mouse embryonic stem cells, they found that a DSB separated from the 3′ repeat by 28.4 kb can still substantially induce RMDs, indicating that a DSB is sufficient to induce RMDs at a relatively far distance.
Keywords: chromosomal rearrangement, homologous recombination, repeat divergence, MSH2, KU70, BRCA1
Abstract
Chromosomal deletion rearrangements mediated by repetitive elements often involve repeats separated by several kilobases and sequences that are divergent. While such rearrangements are likely induced by DNA double-strand breaks (DSBs), it has been unclear how the proximity of DSBs relative to repeat sequences affects the frequency of such events. We generated a reporter assay in mouse cells for a deletion rearrangement involving repeats separated by 0.4 Mb. We induced this repeat-mediated deletion (RMD) rearrangement with two DSBs: the 5′ DSB that is just downstream from the first repeat and the 3′ DSB that is varying distances upstream of the second repeat. Strikingly, we found that increasing the 3′ DSB/repeat distance from 3.3 kb to 28.4 kb causes only a modest decrease in rearrangement frequency. We also found that RMDs are suppressed by KU70 and RAD51 and promoted by RAD52, CtIP, and BRCA1. In addition, we found that 1%–3% sequence divergence substantially suppresses these rearrangements in a manner dependent on the mismatch repair factor MSH2, which is dominant over the suppressive role of KU70. We suggest that a DSB far from a repeat can stimulate repeat-mediated rearrangements, but multiple pathways suppress these events.
Chromosomal structural variations (SVs), both inherited and somatic, are associated with several human diseases, including cancer (Carvalho and Lupski 2016). SVs often involve repetitive elements, which comprise over half of the human genome, including the ∼1 million copies of Alu-type short interspersed elements (Lander et al. 2001; Batzer and Deininger 2002; Belancio et al. 2010). The BRCA1 gene alone contains 129 Alu elements, which represent ∼40% of the gene, and these Alu elements appear at the junctions of several pathogenic SVs in BRCA1 (Pavlicek et al. 2004). Such SVs include repeat-mediated deletions (RMDs), in which the intervening sequence between two repeats, along with one of the repeats, is deleted. The Alu elements associated with RMDs can be separated by very short distances (such as <1 kb) or span a large fraction of a gene (e.g., >20 kb and encompassing several exons) (Kolomietz et al. 2002; Pavlicek et al. 2004; Belancio et al. 2010). Furthermore, sequence divergence between Alu elements can range between 0% and 30%, depending on the family/subfamily of the pair of repeats (Batzer and Deininger 2002; White et al. 2015). Thus, defining the influence of interrepeat distance and repeat divergence on RMDs as well as the DNA repair pathways that influence these variables will provide insight into the etiology of this class of SVs.
Another variable related to interrepeat distance is the location of the inducing DNA lesion relative to each repeat. For example, a likely inducing agent for RMDs is a chromosomal double-strand break (DSB) that is positioned between the repeats. In particular, repair of such DSBs by the single-strand annealing (SSA) pathway causes RMDs (Symington and Gautier 2011; Bhargava et al. 2016). The SSA pathway involves DSB end resection to form 3′ ssDNA that enables annealing of flanking repeats, which is followed by end processing and ligation to generate an RMD (Symington and Gautier 2011; Bhargava et al. 2016). Considering this mechanism, the distance between a DSB and a repeat defines the amount of end resection required for an RMD. In Saccharomyces cerevisiae, DSBs can readily induce an RMD with a repeat that is ≥25 kb away from the edge of the DSB (Vaze et al. 2002). In contrast, DSB-induced RMD events examined in mammalian cells have involved repeats separated from the edge of the DSB by <3 kb (Stark et al. 2004; Weinstock et al. 2006a; Morales et al. 2015). Thus, in mammalian cells, the effect of DSB/repeat distance on RMD formation, which is linked to the extent of DSB end resection, has been unclear.
Another likely determinant of RMD formation is the degree of homology between repeats, in that repeat divergence can significantly limit the frequency of homologous recombination (Waldman and Liskay 1988; Elliott et al. 2005; Weinstock et al. 2006b; Morales et al. 2015; Chakraborty et al. 2016). The likely mechanism by which repeat divergence suppresses RMDs is heteroduplex rejection, in which mismatches in the annealed intermediate are recognized by the components of the mismatch repair pathway to stimulate unwinding of the heteroduplex (Sugawara et al. 2004; George and Alani 2012; Chakraborty et al. 2016). In this study, we sought to examine the interrelationship between DSB/repeat distance and repeat divergence in RMD formation in mammalian cells, which is linked to the regulation of DSB end resection and heteroduplex rejection, respectively.
Results
Influence of DSB/repeat distance on RMD frequency
We sought to understand the influence of the distance between a DSB and a repeat on the formation of RMDs. For this, we developed a green fluorescent protein (GFP)-based reporter assay (RMD-GFP) that uses two 287-base-pair (bp) repeat sequences separated by 0.4 Mb on mouse chromosome 17 (Fig. 1A). The size of the repeat was chosen to approximate the length of Alu elements, which are between 280 and 300 bp (Batzer and Deininger 2002; White et al. 2015). The 5′ repeat is a segment of exon 2 of the Cdkn1A gene. We then introduced this sequence into the Pim1 locus to generate the 3′ repeat, which is also fused to the GFP-coding sequence. Accordingly, an RMD between these repeats would generate a Cdkn1A-GFP fusion gene, which can be measured as GFP+ cells with flow cytometry.
Figure 1.
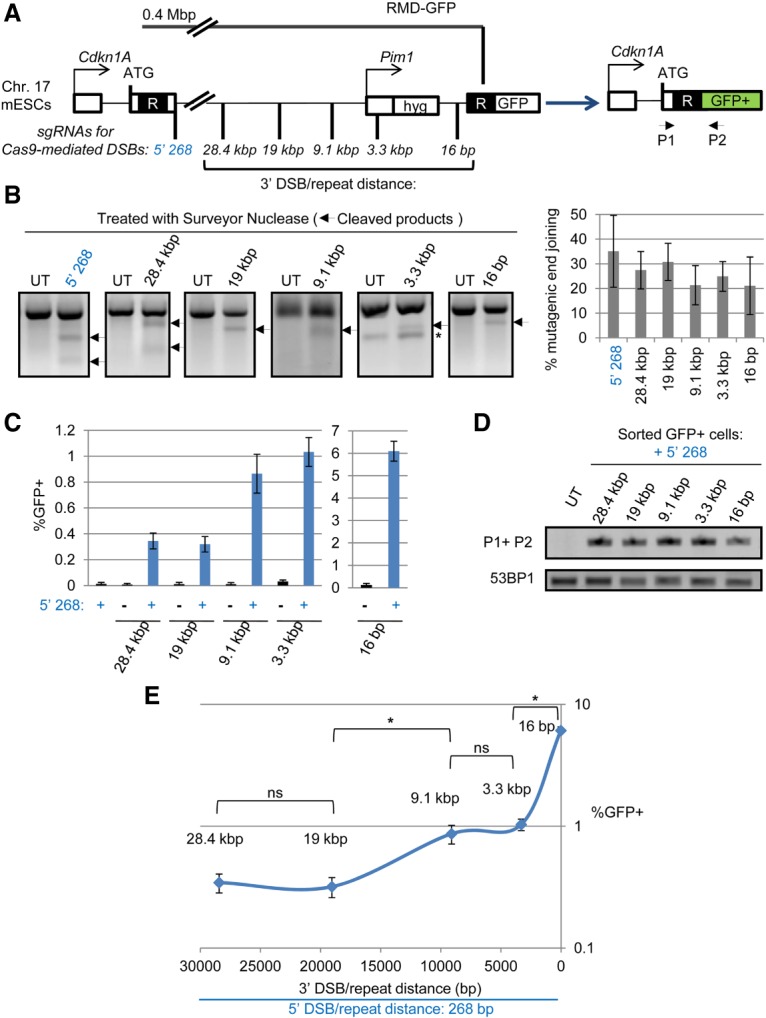
The effect of DSB/repeat distance on the frequency of RMD rearrangements. (A) Diagram of the RMD-GFP reporter. Two tandem repeat (R) sequences are separated by 0.4 Mb and positioned such that an RMD generates a Cdkn1a-GFP fusion gene. The RMD is induced by two DSBs between the repeats: one DSB downstream from the 5′ repeat (5′268) and a second DSB at various distances upstream of the 3′ repeat. Shown are the single-guide RNA (sgRNA)/Cas9 targeting sites for each DSB. (B) Each sgRNA/Cas9-mediated DSB induces mutagenic end joining at a similar frequency. Shown are representative surveyor assays from wild-type RMD-GFP mouse embryonic stem cells (mESCs) that were untransfected (UT) or transfected with expression plasmids for Cas9 and each sgRNA shown in A. (Arrows) Cleaved products; (*) nonspecific band. Also shown is the frequency of mutagenic end joining using the surveyor nuclease assay, normalized to transfection efficiency, for each sgRNA/Cas9-mediated DSB. n = 6. Error bars indicate SD. P > 0.05, comparing all frequencies using one-way ANOVA with Tukey's test. (C) Induction of RMDs using pairs of sgRNAs with Cas9. Shown are the frequencies of GFP+ cells normalized to transfection efficiency for RMD-GFP wild-type mESCs transfected with expression plasmids for Cas9 and the sgRNAs shown in A. (Black) n = 8 for single sgRNAs; (blue) n = 6 for sgRNA pairs. Error bars indicate SD. (D) GFP+ cells harbor the predicted fusion gene. Shown are PCR amplification products from untransfected and sorted GFP+ cells from the sgRNA/Cas9 pairs as in C using primers in Cdkn1A (P1) and GFP (P2). Amplification of a fragment from 53bp1 was used as a control. (E) Chromosomal DSB/repeat distance has a biphasic influence on the frequency of RMDs in wild-type mESCs. Shown are the data from the sgRNA pairs as in C but plotted as the percentage of GFP+ cells versus the distance (in base pairs) between the 3′ chromosomal DSB and the 3′ repeat. n = 6. Error bars indicate SD. (ns) Not significant; (*) P ≤ 0.0018 using one-way ANOVA with Tukey's test.
To induce this RMD, we targeted Cas9-mediated DSBs between the repeats using combinations of single-guide RNAs (sgRNAs). The design of the reporter requires use of two DSBs because the repeats are separated by 0.4 Mb. Specifically, we generated one DSB 268 bp downstream from the 5′ repeat (5′268) and a second DSB upstream of the 3′ repeat. We varied the distance of the DSB upstream of the 3′ repeat using five distances: 16 bp, 3.3 kb, 9.1 kb, 19 kb, and 28.4 kb (Fig. 1A).
Several controls were performed to validate the reporter, which was integrated into wild-type mouse embryonic stem cells (mESCs). For one, we examined the efficiency of each sgRNA to target Cas9-induced DSBs by quantifying mutagenic end joining at the predicted target site using the surveyor nuclease assay. We found that each sgRNA induced a similar frequency of mutagenesis (Fig. 1B). We also confirmed the requirement for both the 5′268 DSB and a second 3′ DSB to induce GFP+ cells (Fig. 1C). In addition, for each pair of sgRNAs, we sorted GFP+ cells to examine the predicted Cdkn1A-GFP fusion gene by PCR analysis and found that only sorted GFP+, but not untransfected, cells contained the fusion gene (Fig. 1D). Finally, we performed an independent test for deletion of the chromosomal segment between the repeats; namely, the RMD-GFP reporter also contains a hygromycin (hyg) resistance gene between the repeats such that an RMD would cause the loss of the intervening sequence containing the hyg gene. Thus, we predicted that GFP+ cells should be sensitive to hygromycin. To test this hypothesis, we transfected cells with the relevant sets of sgRNAs and Cas9. Three days after transfection, we cultured cells with and without hygromycin for an additional 3 d before flow cytometry analysis (Supplemental Fig. S1A). We found that treatment with hygromycin caused a marked reduction in GFP+ cells (Supplemental Fig. S1B). Thus, repair events that generate GFP+ cells also cause loss of the hyg gene, which is consistent with the structure of the predicted RMD.
We then used this assay to evaluate the influence of DSB/repeat distance on the frequency of RMDs in wild-type mESCs (Fig. 1E; Supplemental Fig. S1C). As described above, we induced two DSBs: the first DSB at a constant distance downstream from the 5′ repeat (5′268) and the second DSB at varying distances upstream of the 3′ repeat. From this analysis, we found that 3′ DSB/repeat distance had a biphasic effect on the frequency of RMDs in wild-type mESCs (Fig. 1E); namely, we found that increasing the 3′ DSB/repeat distance from 16 bp to 3.3 kb caused a sixfold reduction in RMD frequency, reflecting an approximately twofold reduction per kilobase. In contrast, we found that increasing the 3′ DSB/repeat distance from 3.3 to 28.4 kb caused a threefold reduction in the frequency of RMDs, reflecting a 0.12-fold reduction per kilobase. Moreover, we found that RMD frequencies were similar with 3′ DSB/repeat distances of 3.3 versus 9.1 kb as well as 19 versus 28.4 kb. These findings indicate that DSB/repeat distance is most inhibitory for RMDs within the first few kilobases. Furthermore, RMD events can be induced in mammalian cells by a DSB that is 28.4 kb away from a repeat, which is within the range of many disease-associated RMDs (Kolomietz et al. 2002; Pavlicek et al. 2004; Belancio et al. 2010).
RMDs at DSB/repeat distances ≥3.3 kb are inhibited by RAD51 but promoted by RAD52
To begin to examine the regulation of these RMD events, we tested the influence of the strand exchange factor RAD51 (Sung et al. 2003). This factor is critical for homology-directed repair (HDR), which involves homologous pairing to initiate nascent DNA synthesis to bridge the DSB (Kass and Jasin 2010; Ceccaldi et al. 2016). In contrast, RAD51 has been demonstrated to inhibit DSB-induced RMDs for repeats separated by <3 kb (Stark et al. 2004). Thus, we hypothesized that RAD51 might also inhibit RMD events at much longer DSB/repeat distances. To disrupt RAD51, we used two dominant-negative proteins: a short peptide fragment of BRCA2 (BRC3), which we fused to Flag tag and nuclear-localizing sequences (3xf-NLS-BRC3), and a mutant of full-length RAD51 (RAD51-K133R) (Stark et al. 2004). The BRC3 peptide has been demonstrated to inhibit RAD51 filament formation, whereas RAD51-K133R is deficient in ATP hydrolysis and shows disrupted filament dynamics (Davies et al. 2001; Robertson et al. 2009).
To validate this dominant-negative approach, we examined the influence of expression vectors for 3xf-NLS-BRC3 and RAD51-K133R on the frequency of HDR. For this, we used the DR-GFP reporter for HDR, targeted to the Pim1 locus in wild-type mESCs. This reporter assay typically involves using I-SceI to induce the DSB, but, to remain consistent across assays, we used an sgRNA to target Cas9 to the I-SceI site in DR-GFP (Supplemental Fig. S2), as described previously (Munoz et al. 2014). We found that expression vectors for 3xf-NLS-BRC3 and RAD51-K133R, compared with control empty vector (EV), each caused a substantial decrease in HDR (Fig. 2A). Next, since BRC3 has been shown to disrupt RAD51 filament formation (Davies et al. 2001), we posited that this peptide might inhibit RAD51 recruitment to DNA damage, measured as RAD51 ionizing radiation (IR)-induced foci (IRIFs) using immunofluorescence analysis. Thus, we transfected wild-type mESCs with the 3xf-NLS-BRC3 expression vector, compared RAD51 IRIF formation in cells with or without Flag costaining, and found the cells expressing 3xf-NLS-BRC3 showed a marked reduction of RAD51 IRIF (Fig. 2B).
Figure 2.
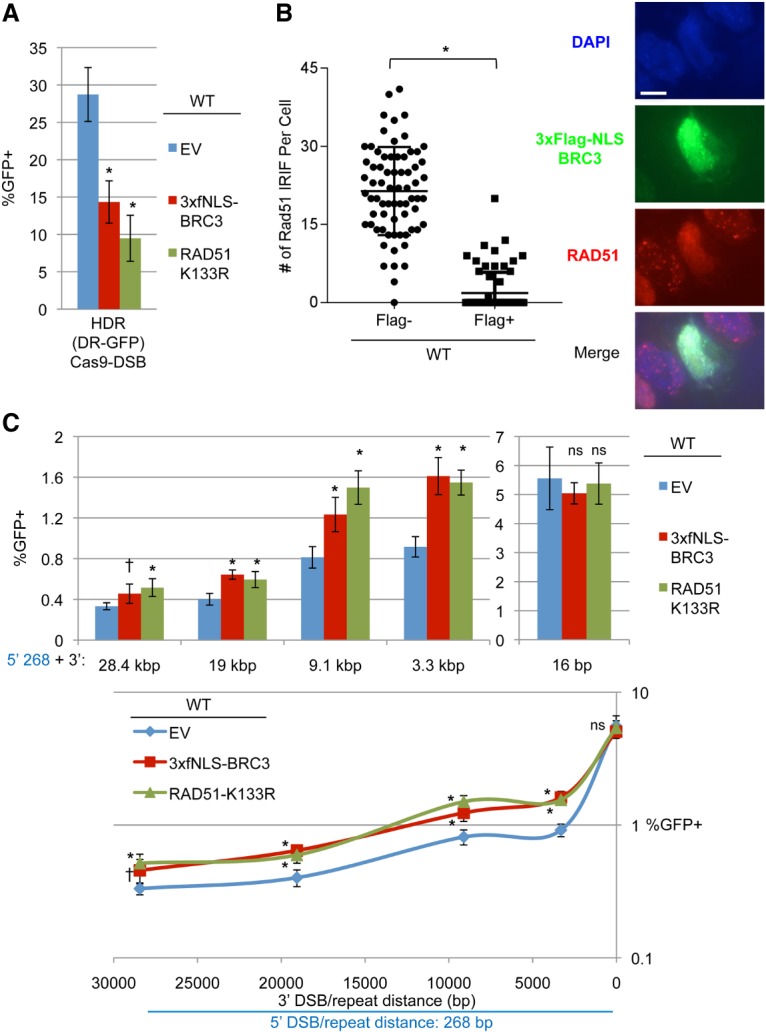
RMDs are suppressed by RAD51 when the 3′ DSB is ≥3.3 kb. (A) Two dominant-negative inhibitors of RAD51—3xFlag-NLS-BRC3 (3xfNLS-BRC3) and RAD51-K133R—disrupt HDR. Wild-type mESCs with DR-GFP were transfected with expression plasmids for an sgRNA targeting this reporter (gDR) and Cas9 along with expression plasmids for 3xfNLS-BRC3, RAD51-K133R, or a control EV. Shown are GFP+ frequencies normalized to transfection efficiency. n = 6. Error bars indicate SD. (*) P = 0.0001 compared with EV using one-way ANOVA with Dunnett's test. (B) Expression of 3xfNLS-BRC3 impairs RAD51 IRIF formation. Shown are representative images of DAPI, Flag, and RAD51 staining in wild-type mESCs transfected with 3xfNLS-BRC3 and exposed to 10 Gy IR followed by 6 h of recovery. Bar, 10 µm. Also shown is the number of RAD51 IRIFs for individual Flag− and Flag+ cells. n = 70. Lines represent the mean with SD. (*) P < 0.0001 using an unpaired two-tailed t-test. (C) The effect of 3xfNLS-BRC3 and RAD51-K133R on RMD frequency. Shown are the frequencies of GFP+ cells normalized to transfection efficiency for RMD-GFP in wild-type mESCs transfected with expression plasmids for a series of sgRNA pairs and Cas9 along with expression plasmids for 3xfNLS-BRC3, RAD51-K133R, or control EV. Also shown are the data plotted as percentage of GFP+ versus 3′ DSB/repeat distance as in Figure 1E. n = 6. Error bars indicate SD. (*) P ≤ 0.0017; (†) P = 0.0260, each compared with EV using one-way ANOVA with Dunnett's test.
We then examined the influence of these two dominant-negative proteins on the formation of RMDs at multiple DSB/repeat distances. For this, we performed the RMD-GFP reporter assay in wild-type mESCs, including the expression vectors for 3xf-NLS-BRC3, RAD51-K133R, or a control EV, using the same concentrations of these vectors as for the DR-GFP experiment above. We found that 3xf-NLS-BRC3 and RAD51-K133R failed to inhibit RMDs at each 3′ DSB/repeat distance and indeed caused a significant increase in the frequency of RMDs at all 3′ DSB/repeat distances except 16 bp (Fig. 2C). These results indicate that RAD51 does not mediate the formation of RMDs but rather has an inhibitory role in such events.
We next examined the role of RAD52, which can mediate annealing of homologous sequences (Hanamshet et al. 2016; Brouwer et al. 2017). For this, we performed the RMD assay in Rad52−/− mESCs (Stark et al. 2004), which we confirmed lack RAD52 mRNA by RT–PCR analysis (Fig. 3A). We also included either an expression vector for V5-immunotagged RAD52 or EV and confirmed expression of V5-RAD52 (Fig. 3A; Supplemental Fig. S3A). From these experiments, at the 16-bp 3′ DSB/repeat distance, we found that RAD52 was dispensable for RMDs. In contrast, at the 3.3-kb distance, RAD52 appears to promote RMDs, but the influence of RAD52 is significant only when comparing Rad52−/− versus the complemented condition. Furthermore, at distances ≥9.1 kb, RAD52 had a profound effect on RMD frequency: Rad52−/− cells showed a marked reduction compared with wild-type, and the complementation vector caused a substantial increase in RMD frequencies. Indeed, RMD frequencies for the complemented condition at ≥3.3 kb were significantly higher than wild type. This increase is likely due to overexpression of RAD52 in Rad52−/− mESCs, although we cannot formally test this possibility due to the lack of a functional anti-mouse RAD52 antibody. Nevertheless, these findings indicate that RAD52 promotes RMDs at 3′ DSB/repeat distances of ≥3.3 kb.
Figure 3.
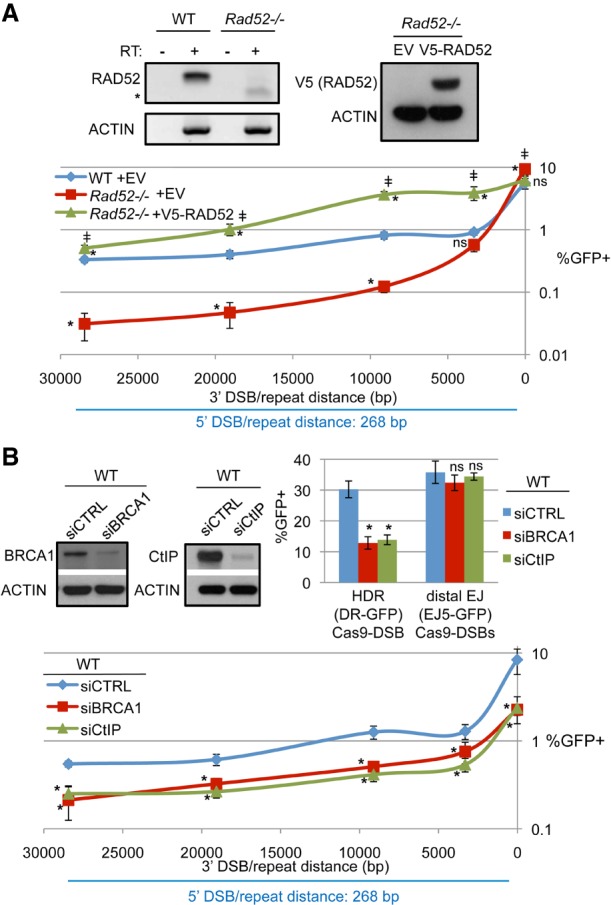
RAD52, CtIP, and BRCA1 promote RMDs. (A) RAD52 promotes RMDs for 3′ DSB/repeat distances of ≥3.3 kb. Wild-type or Rad52−/− mESCs with RMD-GFP were transfected with the respective sgRNA/Cas9 expression vectors along with a control EV or V5-RAD52 complementation vector. Shown is the percentage of GFP+ from these experiments, normalized to transfection efficiency. n = 6. Error bars indicate SD. (*) P ≤ 0.0009 distinct from wild-type EV; (‡) P ≤ 0.0025 for Rad52−/− mESCs EV versus V5-RAD52, both using one-way ANOVA with Tukey's test. Wild-type frequencies are the same as in Figure 2C. Also shown is RT–PCR analysis of the RAD52 transcript in wild-type and Rad52−/− mESCs using RNA either treated with reverse transcriptase (RT) or mock-treated as well as immunoblotting analysis confirming RAD52 expression using the V5 immunotag. (*) Nonspecific band. (B) Depletion of BRCA1 or CtIP causes a reduction in HDR and RMDs but not end joining (distal EJ). Wild-type mESCs with various reporters were transfected with the respective sgRNA/Cas9 expression plasmids along with a nontargeting siRNA (siCTRL), a pool of four BRCA1 siRNAs (siBRCA1), or a pool of four CtIP siRNAs (siCtIP). Shown is the percentage of GFP+ from these experiments, normalized to transfection efficiency. n = 6 for DR-GFP and EJ5-GFP; n = 9 for RMD-GFP. Error bars indicate SD. (*) P = 0.0001 versus siCTRL using one-way ANOVA with Dunnett's test. Also shown is immunoblotting analysis confirming depletion of BRCA1 and CtIP by the respective siRNAs.
End resection factors are critical for regulation of RMDs
The regulatory step of RMDs that is most likely affected by DSB/repeat distance is end resection, since the distance between the DSB and the repeat defines the amount of end resection required to uncover the repeat as ssDNA. Thus, we hypothesized that end resection is a key regulatory step for RMDs. We first examined the end resection mediators BRCA1 and CtIP (Schlegel et al. 2006; Sartori et al. 2007) using RNAi depletion with a pool of four siRNAs targeting each factor (siBRCA1 and siCtIP, respectively), which we confirmed deplete the expected target protein (Fig. 3B). We first tested these siRNAs for effects on HDR, which requires end resection, using the DR-GFP assay described above. As an additional control, we examined an assay for end joining between distal ends of two Cas9-induced DSBs (distal EJ [EJ5-GFP]) (Supplemental Fig. S2; Munoz et al. 2014). We found that depletion of BRCA1 or CtIP caused a reduction in HDR but not distal EJ (Fig. 3B). We then examined the influence of siBRCA1 and siCtIP on the RMD-GFP reporter assay and found that depletion of BRCA1 or CtIP caused a significant reduction in the frequency of RMDs at all 3′ DSB/repeat distances (Fig. 3B; Supplemental Fig. S3B). Together, these results indicate that the end resection mediators BRCA1 and CtIP are important for RMD formation at multiple DSB/repeat distances (i.e., 16 bp to 28.4 kb).
Next, we tested the converse of this hypothesis: that end resection inhibitors are important to suppress RMDs. Specifically, we examined the influence of KU70 and 53BP1 on RMD formation, since these factors inhibit end resection (Zhou et al. 2014). We also examined the DNA ligase 4 cofactor XRCC4, which is in the same DSB repair pathway as KU70 (i.e., nonhomologous end joining) and has been shown to inhibit homologous recombination (Bennardo et al. 2009; Chang et al. 2017).
Beginning with KU70 and XRCC4, we examined Ku70−/− and Xrcc4−/− cells and compared both with wild type as well as the complemented condition (i.e., cells transfected with an expression vector for KU70 or XRCC4, respectively) (Fig. 4A,B). From these experiments, at the 16-bp 3′ DSB/repeat distance, we found that loss of KU70 or XRCC4 caused a marked increase in RMD frequency (Fig. 4A,B; Supplemental Fig. S4A,B). At the 3.3-kb distance, loss of KU70 caused a significant increase, and XRCC4 had no effect on RMD frequency. At ≥9.1 kb, loss of KU70 or XRCC4 caused a significant increase in RMD frequency, although, for ≥19 kb, the effect of XRCC4 loss was evident only relative to the complemented condition. These findings indicate that XRCC4 and KU70 suppress RMDs at multiple DSB/repeat distances, although the greatest effect is at 16 bp.
Figure 4.
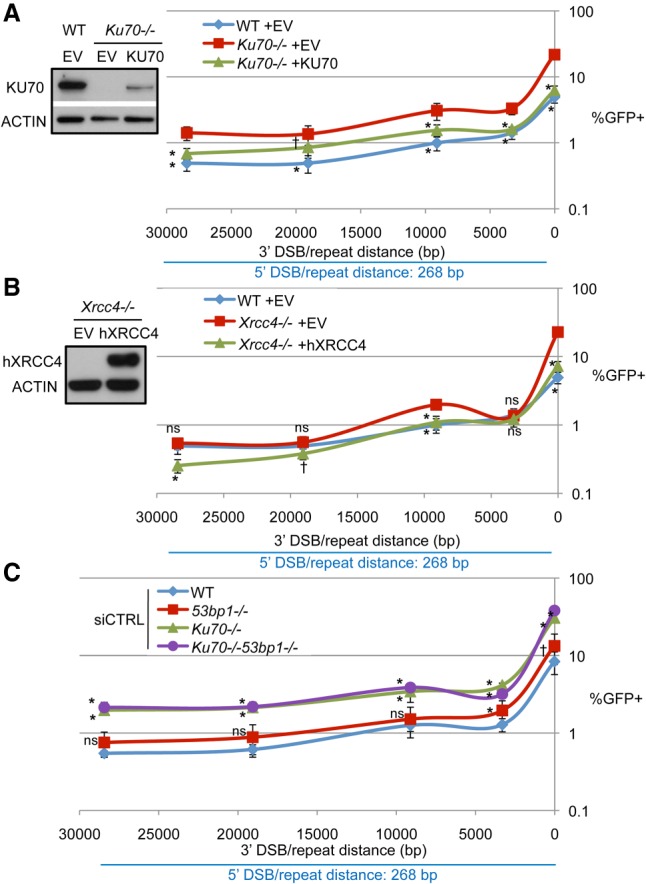
KU70 and XRCC4 suppress RMDs, whereas 53BP1 does not have a substantial influence. (A) KU70 suppresses RMDs. Wild-type or Ku70−/− mESCs with RMD-GFP were transfected with the respective sgRNA/Cas9 expression vectors along with control EV or KU70 complementation vector. Shown are the percentages of GFP+ frequencies from these experiments, normalized to transfection efficiency. Also shown are immunoblots confirming KU70 expression. n = 6 for wild-type EV; n = 9 for Ku70−/− EV and KU70 complemented. Error bars indicate SD. (*) P = 0.0001; (†) P = 0.0077, for Ku70−/− EV versus wild-type EV or for Ku70−/− EV versus KU70 complemented using one-way ANOVA with Dunnett's test. (B) XRCC4 suppresses RMDs. Xrcc4−/− mESCs with RMD-GFP were transfected with the respective sgRNA/Cas9 expression vectors along with a control EV or an expression vector for human XRCC4 (hXRCC4). Shown are the frequencies of the percentages of GFP+ cells from these experiments, normalized to transfection efficiency. The wild-type frequencies from A are included for comparison. n = 6. Error bars indicate SD. (*) P ≤ 0.0003; (†) P ≤ 0.0313, for Xrcc4−/− EV versus wild type or for Xrcc4−/− EV versus hXRCC4 using one-way ANOVA with Dunnett's test. Also shown is an immunoblot confirming expression of hXRCC4. Wild-type mESC immunoblotting signals are not shown, since this antibody does not recognize mouse XRCC4. (C) 53BP1 does not have an obvious effect on the frequency of RMDs. The mESC lines wild type, 53bp1−/−, Ku70−/−, and Ku70−/−53bp1−/−, each with RMD-GFP, were transfected with the respective sgRNAs/Cas9 plasmids, and nontargeting siCTRL was also included. Shown are the percentages of GFP+ cells from these experiments, normalized to transfection efficiency. Wild-type frequencies are the same as in Figure 3B. n = 9. Error bars indicate SD. (*) P ≤ 0.0021; (†) P = 0.0389, comparisons versus wild-type using one-way ANOVA with Dunnett's test.
We also performed the RMD-GFP assay in 53bp1−/− mESCs as well as a 53bp1−/−Ku70−/− mESC line that we generated (Fig. 4C; Supplemental Fig. S4C,D). From these experiments, we found that loss of 53BP1 did not have an obvious effect on RMD frequency either on its own or in combination with loss of KU70 (Fig. 4C; Supplemental Fig. S4C), although 53BP1 loss alone caused a minor increase in RMD frequency at short 3′ DSB/repeat distances (i.e., 3.3 kb and 16 bp) (Fig. 4C; Supplemental Fig. S4C).
The influence of 53BP1 on inhibiting homologous recombination is magnified in BRCA1-deficient cells (Bunting et al. 2010; Onyango et al. 2016). Thus, we considered the possibility that loss of 53BP1—and perhaps KU70—might suppress the requirement of BRCA1 and/or CtIP for RMD formation. For this, we performed the RMD-GFP reporter assay in wild-type, 53bp1−/−, Ku70−/−, and Ku70−/−53bp1−/− mESCs that were transfected with siBRCA1, siCtIP, or siCTRL. We confirmed depletion of BRCA1 and CtIP in each of these mESC lines (Fig. 5A,B).
Figure 5.

Loss of KU70 and 53BP1 affects the requirement for BRCA1 and CtIP for RMDs. (A) Depletion of BRCA1 fails to reduce RMD frequency in cells deficient in KU70 and/or 53BP1 and indeed causes an increase in KU70-deficient cells. Cell lines shown in Figure 4C were transfected with the respective sgRNA/Cas9 expression plasmids along with either siCTRL or siBRCA1. Shown are the percentages of GFP+ cells from these experiments, normalized to transfection efficiency. The frequencies for wild type and each cell line with siCTRL are the same as Figure 4C. n = 9. Error bars indicate SD. (*) P ≤ 0.0003 for siCTRL versus siBRCA1 for all cell lines at each sgRNA pair using an unpaired t-test with the Holm-Sidak correction. Also shown is immunoblotting to confirm depletion of BRCA1 in each cell line. The wild-type immunoblot is the same as Figure 3B and is shown for comparison. (B) Depletion of CtIP shows diminished effects on RMDs in cells deficient in KU70 and/or 53BP1 compared with wild type. Cell lines were transfected as in A but including siCtIP. Shown are the frequencies of GFP+ cells normalized to transfection efficiency for these experiments. As in A, the frequencies for wild type and each cell line with siCTRL are the same as Figure 4C; namely, the data shown are from experiments with siCTRL, siBRCA1, and siCtIP tested in parallel for each cell line. n = 9. Error bars indicate SD. (*) P ≤ 0.003; (†) P < 0.03 for siCTRL versus siCtIP for all cell lines at each sgRNA pair using an unpaired t-test with the Holm-Sidak correction. Also shown is immunoblotting to confirm depletion of CtIP in each cell line. The wild-type immunoblot is the same as Figure 3B and is shown for comparison.
Beginning with the effects of BRCA1 on RMDs, we found that siBRCA1 treatment failed to reduce the RMD frequency at all 3′ DSB/repeat distances in 53bp1−/−, Ku70−/−, and Ku70−/−53bp1−/− mESCs (Fig. 5A). Indeed, for the KU70-deficient cells, we found that depletion of BRCA1 causes a significant increase in the frequency of RMDs at the 3′ DSB/repeat distances of 9.1 and 3.3 kb (Fig. 5A). These results indicate that loss of 53BP1 and/or KU70 suppresses the requirement of BRCA1 for RMD formation and that 53BP1 and KU70 are epistatic for this effect, since Ku70−/−53bp1−/− mESCs were not distinct from Ku70−/− (Fig. 5A). Furthermore, these findings indicate that in KU70-deficient cells, BRCA1 appears to inhibit RMDs.
We also found that loss of 53BP1 and KU70 affected the influence of CtIP on RMDs; namely, in 53bp1−/− mESCs, while siCtIP treatment caused a significant reduction in RMD frequency at most DSB/repeat distances, the fold effects were often diminished compared with wild-type mESCs (Fig. 5B). Furthermore, in Ku70−/− and Ku70−/− 53bp1−/− mESCs, siCtIP treatment failed to cause a reduction in RMD frequency for DSB/repeat distances ≥9.1 kb and caused only a minor effect at shorter DSB/repeat distances (i.e., ≤1.2-fold decrease vs. ≥2.4-fold in wild type) (Fig. 5B). Notably, CtIP levels were effectively depleted by siCtIP treatment in each cell line, based on immunoblotting analysis (Fig. 5B). These results indicate that loss of 53BP1 and/or KU70 diminishes the requirement of CtIP for RMDs.
Repeat sequence divergence limits the formation of RMDs at multiple DSB/repeat distances in a manner dependent on MSH2
Previous studies using assays with repeats separated from a DSB by <3 kb have shown that sequence divergence between repeats limits the formation of RMDs (Elliott et al. 2005; Weinstock et al. 2006b; Morales et al. 2015). We sought to examine the influence of sequence divergence on RMDs that are induced at multiple DSB/repeat distances. We generated variants of the RMD-GFP reporter by introducing sequence divergence in the 3′ repeat fused to GFP using mutations that are equally dispersed across the repeat and do not affect the respective codon (i.e., are silent mutations). Specifically, we introduced three and eight mutations to generate 1% RMD-GFP and 3% RMD-GFP, respectively (Fig. 6A; Supplemental Fig. S5A–C).
Figure 6.
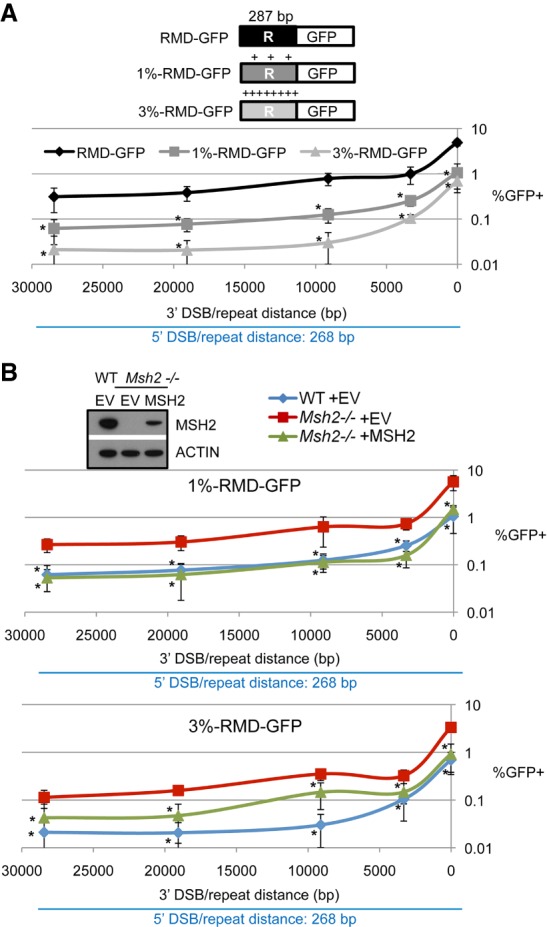
Introducing sequence divergence between the repeats substantially suppresses RMD in a manner dependent on the mismatch repair factor MSH2. (A) Introducing sequence divergence between the repeats markedly suppresses RMD frequency at multiple DSB/repeat distances. Shown are variants of the RMD-GFP reporter: 1% RMD-GFP and 3% RMD-GFP, with three and eight mutations, respectively, which are equally dispersed in the 3′ repeat (R) fused to GFP and do not affect the respective codon in the repeat (i.e., are silent mutations). Each reporter was integrated into wild-type mESCs and transfected with the respective sgRNA/Cas9 pairs shown in Figure 1A. Shown is the frequency of GFP+ cells normalized to transfection efficiency for these experiments. n = 15 for RMD-GFP. n = 6 from Figure 4A, with additional replicates performed later with the divergent reporter cell lines; n = 9 for 1% RMD-GFP and 3% RMD-GFP. Error bars indicate SD. (*) P = 0.0001 comparisons versus RMD-GFP using one-way ANOVA with Dunnett's test. (B) Loss of MSH2 increases the frequency of RMDs between divergent repeats. Msh2−/− mESCs with the 1% and 3% reporters shown in A were transfected with the respective sgRNA/Cas9 expression vectors along with a control EV or MSH2 complementation vector. Shown are the percentages of GFP+ frequencies normalized to transfection efficiency along with the wild-type frequencies from A. n = 9. Error bars indicate SD. (*) P ≤ 0.001 comparisons versus Msh2−/− EV using one-way ANOVA with Dunnett's test. Also shown is immunoblot analysis confirming MSH2 expression.
With these reporters, we examined the influence of sequence divergence between repeats on the frequency of RMDs at distinct DSB/repeat distances using the same sgRNAs described for the parental reporter (Fig. 1A). From these experiments, we found that both 1% and 3% sequence divergence significantly reduced the frequency of RMDs at all DSB/repeat distances compared with the parental reporter (Fig. 6A; Supplemental Fig. S6A). Furthermore, 3% sequence divergence caused a lower RMD frequency compared with 1% divergence (Fig. 6A; Supplemental Fig. S6A). We also performed additional control experiments. For one, we sorted GFP+ cells to confirm the predicted Cdkn1A-GFP fusion using PCR analysis for the divergent reporters using a representative pair of DSBs (5′268 combined with the 3′ DSB at 16 bp) (Supplemental Fig. S5B). In addition, we tested for loss of the hyg selectable marker in GFP+ cells for the divergent reporters using two representative pairs of DSBs (5′268 combined with the 3′ DSB at 3.3 kb and 16 bp) (Supplemental Fig. S5C). We found that treatment of hygromycin caused a significant reduction in GFP+ cells, which is consistent with the predicted RMD (Supplemental Fig. S5C). Together, these results indicate that sequence divergence between repeats significantly limits the formation of RMDs at multiple DSB/repeat distances.
We then posited that the reduction in RMD frequency between divergent repeats is caused by heteroduplex rejection, which requires the mismatch repair factor MSH2 (Sugawara et al. 2004; George and Alani 2012; Morales et al. 2015; Chakraborty et al. 2016). To test this hypothesis, we performed the RMD reporter assays in Msh2−/− mESCs, which we compared with wild-type as well as Msh2−/− cells transfected with an MSH2 complementation vector (Fig. 6B). With the parental reporter with identical repeats (RMD-GFP), we found that Msh2−/− mESCs showed an increase in the frequency of RMDs at most 3′ DSB/repeat distances, except at 28.4 kb, compared with wild-type mESCs (Supplemental Fig. S6B). However, we found that adding the MSH2 complementation vector had no effect on RMD frequency in Msh2−/− mESCs (Supplemental Fig. S6B). Because MSH2 complementation had no effect, we suggest that the increase of RMD frequency between identical repeats in Msh2−/− mESCs cannot necessarily be attributed to loss of MSH2. Importantly, however, in contrast to RMDs between identical repeats, with 1% and 3% divergent repeats, we found that Msh2−/− mESCs showed a marked increase in the frequency of RMDs at all 3′ DSB/repeat distances compared with wild-type mESCs, which are reduced with the MSH2 complementation vector (Fig. 6B; Supplemental Fig. S6B), although, in the case of the 3% RMD-GFP reporter, we found that MSH2 complementation did not entirely restore RMD frequencies to wild type at all 3′ DSB/repeat distances, which may reflect the limitations of transient complementation to precisely mimic wild-type protein levels. In any case, these results indicate that MSH2 suppresses the formation of RMDs between divergent repeats in a manner that is independent of DSB/repeat distance.
MSH2 has a dominant role vs. KU70 to suppress RMDs with divergent repeats
Finally, we examined the influence of KU70 on RMDs with divergent repeats as well as the genetic relationship between KU70 and MSH2 on such events. Beginning with the 3′ DSB/repeat distance of 16 bp, KU70 deficiency caused a substantial increase in RMD frequency with both the 1% and 3% divergent repeats, which is similar to the effect on the parental reporter (Fig. 7A; Supplemental Fig. S7A). Furthermore, the effect of KU70 loss was not distinct from the effect of MSH2 deficiency at the 16-bp 3′ DSB/repeat distance (Fig. 7B; Supplemental Fig. S7B). For the DSB/repeat distances of ≥3.3 kb, KU70 deficiency caused a modest increase in RMDs between divergent repeats, and, indeed, for 1% divergence, the effect of KU70 was evident only compared with the complemented condition (Fig. 7A; Supplemental Fig. S7A). Notably, for most of the 3′ DSB/repeat distances ≥3.3 kb, the frequency of the RMDs between divergent repeats is significantly higher for Msh2−/− versus Ku70−/−, except for 3% divergence at the 3.3-kb and 28.4-kb distances (Fig. 7B; Supplemental Fig. S7B).
Figure 7.

MSH2 has a dominant role compared with KU70 to suppress RMDs between divergent repeats. (A) KU70 suppresses RMDs between divergent repeats. Ku70−/− mESCs with the 1% RMD-GFP and 3% RMD-GFP reporters were transfected as in Figure 4A. Shown are the percentages of GFP+ cells from these experiments, normalized to transfection efficiency, with the wild-type frequencies from Figure 6A shown for comparison. n = 9. Error bars indicate SD. (*) P ≤ 0.0085; (†) P ≤ 0.0432 for Ku70−/− EV versus wild type or for Ku70−/− EV versus KU70 expression vector using one-way ANOVA with Dunnett's test. (B) Loss of both MSH2 and KU70 causes a marked increase in RMDs between divergent repeats compared with the single mutants, and MSH2 loss has a greater effect than KU70 loss. Msh2−/−Ku70−/− mESCs with the 1% RMD-GFP and 3% RMD-GFP reporters were transfected with the respective sgRNA/Cas9 expression plasmids along with control EV. Shown are the percentages of GFP+ cells from these experiments, normalized to transfection efficiency, with the wild-type, Msh2−/−, and Ku70−/− frequencies from Figure 6 and A shown for comparison. n = 9. Error bars indicate SD. (*) P ≤ 0.0003 for Msh2−/−Ku70−/− versus every other cell line; (‡) P ≤ 0.0009 for Msh2−/− versus Ku70−/− using one-way ANOVA with Tukey's test. (C) Expression of KU70 or MSH2 suppresses RMDs between divergent repeats in Msh2−/−Ku70−/− mESCs, with MSH2 having a greater effect. Msh2−/− Ku70−/− mESCs with the 1% RMD-GFP and 3% RMD-GFP reporters were transfected as in B but including expression vectors for KU70, MSH2, or EV. Shown are the percentages of GFP+ cells from these experiments, normalized to transfection efficiency, with wild-type frequencies from Figure 6A shown for comparison. n = 6. Error bars indicate SD. (*) P ≤ 0.0003 for Msh2−/−Ku70−/− EV versus all other conditions; (‡) P ≤ 0.015 for MSH2 versus KU70 complementation in Msh2−/− Ku70−/− cells using one-way ANOVA with Tukey's test.
To further examine the relative influence of KU70 and MSH2 on these events, we performed the RMD reporter assays with the divergent repeats in a Msh2−/−Ku70−/− mESC line that we generated (Supplemental Fig. S7B). Strikingly, we found that the RMD frequencies for the Msh2−/−Ku70−/− cell line were substantially higher than for either single mutant at nearly all 3′ DSB/repeat distances, with only one exception (1% divergence at 28.4 kb not distinct from Msh2−/−) (Fig. 7B; Supplemental Fig. S7B). We then performed complementation analysis with the Msh2−/−Ku70−/− cell lines and found that expression of either KU70 or MSH2 suppressed RMDs at all DSB/repeat distances (Fig. 7C; Supplemental Fig. S8). Notably, the MSH2 complementation caused a substantially greater reduction in RMD frequency versus KU70 (except 1% divergence at 16 bp and 3% divergence at 28.4 kb). Altogether, these results indicate that suppression of RMDs between divergent repeats by MSH2 is dominant over the suppression of these events by KU70.
Discussion
Defining the barriers to the formation of chromosomal SVs is important to understand the mechanisms of genome maintenance. Thus, we sought to characterize the barriers to DSB-induced RMDs in mammalian cells, focusing on the distance between a DSB and a repeat as well as sequence divergence between repeats (Fig. 8). We found that a DSB adjacent to each repeat was proficient at inducing RMDs, whereas increasing the 3′ DSB/repeat distance to 3.3 kb caused a sixfold reduction in these events. Furthermore, we found that increasing this distance from 3.3 to 28.4 kb caused only a threefold reduction in RMD frequency. Notably, DSB/repeat distance likely defines the amount of end resection required for RMDs. Thus, based on our findings, we suggest that the greatest barrier to end resection is within the first few kilobases, but once end resection extends a few kilobases, there is only a modest barrier to much more extensive resection. These findings are consistent with physical measurements of ssDNA at DSBs in mammalian cells, which indicate that end resection is most frequent at <1 kb (Zhou et al. 2014), although the limit of detection of such measurements in wild-type cells is ∼3–4 kb of end resection (Cruz-Garcia et al. 2014; Zhou et al. 2014; Canela et al. 2016) such that the extent of resection beyond this range has been unclear.
Figure 8.
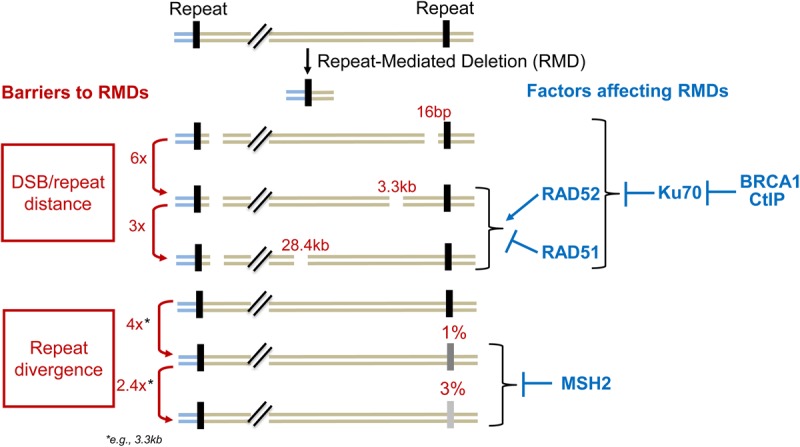
A summary of barriers and mediators of RMD formation.
Consistent with DSB end resection being important for RMDs, we found that several factors that inhibit/mediate end resection (KU70, BRCA1, and CtIP) affect the RMD frequency at multiple DSB/repeat distances (i.e., 16 bp to 28.4 kb). Furthermore, XRCC4, which functions in the same DSB repair pathway as KU70 and inhibits homologous recombination (Bennardo et al. 2009; Chang et al. 2017), also suppresses RMDs. We also found that loss of KU70 diminishes the requirement of CtIP and BRCA1 for RMD formation. These findings indicate that at least part of the function of CtIP and BRCA1 in promoting these events is to inhibit KU70 function. This model is consistent with reports that CtIP in combination with the MRE11 complex can promote removal of the KU70/KU80 heterodimer (KU) from DNA ends, based on single-molecule approaches (Myler et al. 2017) as well as measurements of KU recruitment to replication-associated breaks (Chanut et al. 2016). Similarly, the CtIP homolog/orthologs in Schizosaccharomyces pombe and S. cerevisiae (Ctp1 and SAE2, respectively) appear dispensable for end resection in the absence of KU (Mimitou and Symington 2010; Langerak et al. 2011).
Regarding BRCA1, we found that this factor not only is dispensable for RMDs in KU70-deficient cells but appears to inhibit RMDs in this context. Thus, in KU70-deficient cells, BRCA1 has an effect similar to that of RAD51 in suppressing RMDs. Accordingly, our findings support the notion that BRCA1 mediates at least two distinct steps of homologous recombination: (1) displacement of KU to facilitate end resection and (2) promotion of RAD51 function. Consistent with this notion, while loss of KU was shown to rescue PARP inhibitor sensitivity in BRCA1-deficient cells, indicating at least a partial rescue of homologous recombination, loss of KU caused only a modest increase in RAD51 recruitment to DNA damage (Bunting et al. 2012). Furthermore, this model is supported by a recent report that used several mutants of BRCA1 to separate its functions in HDR versus SSA (i.e., an RMD event) (Anantha et al. 2017). BRCA1 also has been implicated recently in directly promoting RAD51-mediated strand exchange (Zhao et al. 2017). Notably, these findings might explain the variable requirement of BRCA1 for distinct assays for end resection (Schlegel et al. 2006; Cruz-Garcia et al. 2014; Zhou et al. 2014); namely, we speculate that while BRCA1 is a mediator of end resection, its later role in promoting RAD51 function would cause a reduction in the steady-state level of resected ends, which might result in no obvious net change in the frequency of resected ends in some assays.
In contrast to KU70, we found that loss of 53BP1 did not have a significant effect on RMD frequency either on its own or in the context of KU70 deficiency. These findings appear inconsistent with reports that 53BP1 disruption can cause an increase in end resection (Bunting et al. 2012; Zhou et al. 2014; Ochs et al. 2016). However, apart from these measurements of end resection per se, 53BP1 deficiency causes a relatively modest effect on homologous recombination unless in the context of BRCA1 deficiency (Bouwman et al. 2010; Bunting et al. 2010, 2012; Onyango et al. 2016). Consistent with these reports, we found that loss of 53BP1 suppressed the effect of BRCA1 depletion on RMD formation and reduced the effect of CtIP depletion on these events. Thus, 53BP1-mediated suppression of end resection does not appear to be critical to limit RMD formation except in the context of depletion of BRCA1 or CtIP. Finally, since end resection has been found to be elevated in S–G2-phase cells (Symington and Gautier 2011), we note that our model system for these experiments (mESCs) is predominantly in S phase, which we confirmed here (Supplemental Fig. S9), including for each of the mutant mESC lines.
Regarding other factors involved in RMDs, we found that RAD52 promotes these events at 3′ DSB/repeat distances ≥3.3 kb. Indeed, at ≥9.1 kb, RAD52 deficiency had a profound effect on RMD frequency. The likely role of RAD52 in RMDs is to stabilize dsDNA between the homologous repeats, which is supported by biochemical studies of RAD52 activity (Hanamshet et al. 2016; Brouwer et al. 2017). Our findings indicate that such RAD52 activity is likely proficient even when homologous sequences are relatively far from DNA ends.
In addition to our analysis of DSB/repeat distance, we found that sequence divergence between the repeats (1%–3%) substantially suppressed RMDs in a manner dependent on MSH2. These findings are consistent with the role of mismatch repair factors in promoting heteroduplex rejection via promoting recruitment of a DNA helicase to displace the mismatched annealing intermediate (Sugawara et al. 2004; George and Alani 2012; Chakraborty et al. 2016). This process in S. cerevisiae has been shown to require at least a short distance between the DSB and the edge of the mismatched heteroduplex (Anand et al. 2017). Consistent with this notion, we found that such heteroduplex rejection is proficient at DSB/repeat distances ranging from 16 bp to 28.4 kb (each paired with a 5′ DSB/repeat distance of 268 bp). Importantly, we also found that combined loss of MSH2 and KU70 caused a marked increase in RMDs between divergent repeats. Furthermore, the effect of MSH2 was significantly greater than KU70. This latter finding indicates that the heteroduplex rejection is a dominant barrier for RMDs between divergent repeats relative to suppression of end resection.
Since RMDs can cause substantial loss of genetic information, these events have the potential to contribute to the increased cancer risk that is caused by inherited mutations of BRCA1 and MSH2 (Lynch et al. 2015; Nielsen et al. 2016). Our findings indicate that disruption of KU70, combined with deficiency in MSH2 or BRCA1, causes a marked increase in the frequency of RMDs. Thus, we speculate that loss of KU function and/or amplification of pathways that inhibit KU has the potential to increase RMDs in cells deficient in MSH2 and/or BRCA1. Such an increase in RMDs could thereby cause genome instability, which has the potential to contribute to cancer development. Similarly, therapeutic disruption of KU function could augment RMD formation during clastogen treatment of such tumors. In conclusion, our study provides evidence that a DSB positioned relatively far from a repeat can induce RMDs, but several pathways and factors are important to suppress these events.
Materials and methods
Plasmids, cell lines, and siRNA
The RMD-GFP reporter plasmid and the variants with different repeat sequences were generated by introducing a repeat sequence (Supplemental Fig. S5A) upstream of the GFP-coding sequence into the pim-EJ6-GFP plasmid (Bhargava et al. 2017). Each sgRNA sequence is shown in Supplemental Table S1 and was expressed from the px330 plasmid, which coexpresses Cas9 (Addgene, 42230; generously deposited by Dr. Feng Zhang) (Ran et al. 2013). The sgRNAs for targeting the DR-GFP and EJ5-GFP reporters were described previously (Munoz et al. 2014). The plasmids pCAGGS-KU70, pCAGGS-MSH2, pCAGGS-RAD51-K133R, pCAGGS-HA-XRCC4, pCAGGS-NZE-GFP, pgk-puro, and pCAGGS-BSKX EV were described previously (Bennardo et al. 2008, 2009; Bhargava et al. 2017). The pCAGGS-V5-RAD52 expression vector was derived from the previously described pCAGGS-RAD52 (mouse) plasmid (Bennardo et al. 2008). The pCAGGS-3xf-NLS-BRC3 expression vector was generated by inserting the 3xFlag sequence into the previously described pCAGGS-NLS-BRC3 vector (Stark et al. 2004). We used the nontargeting siCTRL (GE Dharmacon, D-001810-01) and pools of four siRNAs per gene: siBRCA1 (GE Dharmacon, D-040545-01, D-040545-02, D-040545-03, and D-040545-04) and siCtIP (GE Dharmacon, D-055713-14, D-055713-15, D-055713-16, and D-055713-17).
The wild-type, Ku70−/−, Msh2−/−, Rad52−/−, and 53bp1−/− mESCs were described previously (Gu et al. 1997; Claij and te Riele 2004; Stark et al. 2004; Bhargava et al. 2017). Since the Msh2 and Rad52 genes were disrupted with a hyg cassette, which was also used for Pim1 targeting of the reporter, we first mutated the hyg gene in the Msh2−/− and Rad52−/− mESC lines using an sgRNA with Cas9 followed by screening individual clones for hygromycin sensitivity. Loss of RAD52 RNA in the Rad52−/− mESC line was examined using RT–PCR with primers for RAD52 (5′-TGGGAAGTCTGTGCTCCTCT-3′ and 5′-CCCAGCCATTGTAACCAAAC-3′) and Actin (5′-GGCTGTATTCCCCTCCATCG-3′ and 5′-TCTCCAGGGAGGAAGAGGAT-3′). The Ku70−/−53bp1−/− cell line was derived from the Ku70−/− cell line, and the Msh2−/−Ku70−/− cell line was derived from the Msh2−/− hygromycin-sensitive cell line using previously described sgRNA/Cas9 expression vectors (Howard et al. 2015; Bhargava et al. 2017). The RMD-GFP reporters were integrated into the Pim1 locus as described previously (Bennardo et al. 2008).
DSB reporter assays and surveyor nuclease assay
For the DSB reporter assays, 0.5 × 105 mESCs were plated per well in a 24-well plate. Each well was transfected with 200 ng of each sgRNA/Cas9 plasmid using 1.8 µL of Lipofectamine 2000 in 0.5 mL of antibiotic-free medium. To compare wild type and each mutant cell line, transfections included either 200 ng of control EV or complementation vector (RAD52, KU70, XRCC4, or MSH2). At least two independent Pim1 targeted clones were tested using at least one representative RMD event, whereas one clone was used for the in-depth analysis that is shown. The RAD51 dominant-negative analysis included 200 ng of control EV, 100 ng of 3xf-NLS-BRC3, or 50 ng of RAD51-K133R, and the latter two included additional EV to normalize the total plasmid concentration. For the siRNA analysis, transfections included 5 pmol of siRNA. In addition, 200 ng of EV was included in the DR-GFP experiments to normalize the total concentration of plasmid, since this reporter involves only one sgRNA. Three days after transfection, cells were analyzed by flow cytometry using a CyAn-ADP, as described (Gunn and Stark 2012). For testing the hygromycin sensitivity of the GFP+ cells, 3 d after transfection, cells were replated and cultured with or without 110 µg/mL hygromycin B (Invitrogen) for three additional days prior to flow cytometry analysis. In each experiment, the frequency of GFP+ cells was normalized to transfection efficiency, which was determined using parallel transfections with 200 ng of a GFP expression vector (pCAGGS-NZE-GFP) plus the appropriate concentration of EV, complementation vector, RAD51 dominant-negative plasmid, or siRNA treatment. Statistical analysis of the RMD reporter experiments were performed at each DSB repeat/distance using the methods described in the figure legends. GFP+ cells were isolated using either an Aria III or Aria SORP instrument (Becton Dickinson) and examined by PCR with primers P1 (5′-TCTCATGGTGTGGTGGAAAA-3′) and P2 (5′-AAGTCGTGCTGCTTCATGTG-3′).
For the surveyor nuclease assay, cells were transfected as described above with 200 ng of each sgRNA/Cas9 plasmid, with 400 ng of EV to maintain a constant total plasmid concentration. The assay and analysis were performed with the Surveyor mutation detection kit (IDT) as described previously (Bhargava et al. 2017) using the primers listed in Supplemental Table S1. All target sites were amplified with Platinum HiFi Supermix (Thermo Fisher) except the 3.3-kb site, which was amplified with Phusion high-fidelity PCR master mix with GC buffer (New England Biolabs).
Immunoblotting, immunofluorescence, and cell cycle analysis
For immunoblotting analysis with the complementation vectors, we used the same concentration of complementation vector as the reporter assays (scaled four times), with EV to maintain the same total concentration of plasmid, and protein was extracted 2 d after transfection. For analysis with the siRNAs, we used the same concentration of siRNA as the reporter assays (scaled four times) but including 1.2 µg of pgk-puro plasmid. One day after transfection, cells were treated with 1.5 µg/mL puromycin, and protein was extracted 2 d later. For protein extraction, cells were lysed using NETN buffer (20 mM Tris at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Igepal, 1.25 mM DTT, Roche protease inhibitor) with several freeze/thaw cycles ELB lysis buffer (250 mM NaCI, 5 mM EDTA, 50 mM HEPES, 0.1% NP-40 [Ipegal], Roche protease inhibitor) with sonication (Qsonica, Q800R). Extracts were probed with antibodies for BRCA1 (Barlow et al. 2013), CtIP (Active Motif, 61141), V5 (Thermo Fisher, R96125), XRCC4 (Santa Cruz Biotechnology, sc-271087), 53BP1 (Abcam, ab36823), KU70 (Santa Cruz Biotechnology, sc-1487), MSH2 (Bethyl Laboratories, A300-452A), and Actin (Sigma, A2066). ECL reagent (Thermo Scientific) was used to develop HRP signals.
For immmunofluorescence analysis, cells were transfected with 3xf-NLS-BRC3 using the same plasmid concentration as the reporter assays. Two days after transfection, cells were treated with 10 Gy of IR (Gammacell 3000) followed by a 6-h recovery and affixed to slides using a Cytospin 4 (Thermo Fisher). Cells were fixed with 4% paraformaldehyde, permeabilized, and stained with antibody for the Flag epitope (Sigma, F3165) and RAD51 (Santa Cruz Biotechnology, sc-8349) followed by Alexa fluor 488 goat anti-mouse antibody (Thermo Fisher, A11029), Alexa flour 568 goat anti-rabbit antibody (Thermo Fisher A11036), and DAPI using VectaShield mounting medium (Vector Laboratories, H1500). Images were acquired using the Zeiss Observer Z1 with a 40× objective using ZEN Black software.
For cell cycle analysis, mESCs were treated with 10 µM BrdU (Becton Dickinson, 51-2420KC) for 30 min prior to fixation in 70% ethanol and staining with FITC-BrdU (Becton Dickinson, 51-33284X) and propidium iodide with RNase (Sigma, P4170 and R4642, respectively). Cells were analyzed on a CyAN-ADP.
Supplementary Material
Acknowledgments
We thank Gabriella Lee for technical assistance, and Dr. Andre Nussenzweig for the BRCA1 antibody. This study was funded in part by the National Cancer Institute of the National Institutes of Health (R01CA120954 and R01CA197506 to J.M.S., and P30CA33572 for City of Hope Core Facilities). C.M.-D. is supported by a Ford Foundation Predoctoral Fellowship.
Author contributions: C.M.-D., R.B., and J.M.S. designed research, developed plasmid and cell line reagents, and analyzed data; C.M.-D. performed the experiments; and C.M.-D. and J.M.S. wrote the manuscript with input from R.B.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311084.117.
References
- Anand R, Beach A, Li K, Haber J. 2017. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544: 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, Ganesan S, Xia B. 2017. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife 6: e21350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. 2013. Identification of early replicating fragile sites that contribute to genome instability. Cell 152: 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379. [DOI] [PubMed] [Google Scholar]
- Belancio VP, Roy-Engel AM, Deininger PL. 2010. All y'all need to know ’bout retroelements in cancer. Semin Cancer Biol 20: 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. 2008. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4: e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Gunn A, Cheng A, Hasty P, Stark JM. 2009. Limiting the persistence of a chromosome break diminishes its mutagenic potential. PLoS Genet 5: e1000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, Stark JM. 2016. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava R, Carson CR, Lee G, Stark JM. 2017. Contribution of canonical nonhomologous end joining to chromosomal rearrangements is enhanced by ATM kinase deficiency. Proc Natl Acad Sci 114: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17: 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer I, Zhang H, Candelli A, Normanno D, Peterman EJG, Wuite GJL, Modesti M. 2017. Human RAD52 captures and holds DNA strands, increases DNA flexibility, and prevents melting of duplex DNA: implications for DNA recombination. Cell Rep 18: 2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. 2012. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 46: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, Nussenzweig A. 2016. DNA breaks and end resection measured genome-wide by end sequencing. Mol Cell 63: 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26: 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty U, George CM, Lyndaker AM, Alani E. 2016. A delicate balance between repair and replication factors regulates recombination between divergent DNA sequences in Saccharomyces cerevisiae. Genetics 202: 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut P, Britton S, Coates J, Jackson SP, Calsou P. 2016. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat Commun 7: 12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claij N, te Riele H. 2004. Msh2 deficiency does not contribute to cisplatin resistance in mouse embryonic stem cells. Oncogene 23: 260–266. [DOI] [PubMed] [Google Scholar]
- Cruz-Garcia A, Lopez-Saavedra A, Huertas P. 2014. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep 9: 451–459. [DOI] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. 2001. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7: 273–282. [DOI] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Jasin M. 2005. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell 17: 885–894. [DOI] [PubMed] [Google Scholar]
- George CM, Alani E. 2012. Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA. Crit Rev Biochem Mol Biol 47: 297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Jin S, Gao Y, Weaver DT, Alt FW. 1997. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci 94: 8076–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, Stark JM. 2012. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 920: 379–391. [DOI] [PubMed] [Google Scholar]
- Hanamshet K, Mazina OM, Mazin AV. 2016. Reappearance from obscurity: mammalian Rad52 in homologous recombination. Genes 7: E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SM, Yanez DA, Stark JM. 2015. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet 11: e1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Jasin M. 2010. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 584: 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. 2002. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35: 97–112. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Langerak P, Mejia-Ramirez E, Limbo O, Russell P. 2011. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet 7: e1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. 2015. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer 15: 181–194. [DOI] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2010. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29: 3358–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, White TB, Streva VA, DeFreece CB, Hedges DJ, Deininger PL. 2015. The contribution of alu elements to mutagenic DNA double-strand break repair. PLoS Genet 11: e1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz MC, Yanez DA, Stark JM. 2014. An RNF168 fragment defective for focal accumulation at DNA damage is proficient for inhibition of homologous recombination in BRCA1 deficient cells. Nucleic Acids Res 42: 7720–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler LR, Gallardo IF, Soniat MM, Deshpande RA, Gonzalez XB, Kim Y, Paull TT, Finkelstein IJ. 2017. Single-molecule imaging reveals how Mre11–Rad50–Nbs1 initiates DNA break repair. Mol Cell 67: 891–898 e894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen FC, van Overeem Hansen T, Sorensen CS. 2016. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer 16: 599–612. [DOI] [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, Lukas C. 2016. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol 23: 714–721. [DOI] [PubMed] [Google Scholar]
- Onyango DO, Howard SM, Neherin K, Yanez DA, Stark JM. 2016. Tetratricopeptide repeat factor XAB2 mediates the end resection step of homologous recombination. Nucleic Acids Res 44: 5702–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek A, Noskov VN, Kouprina N, Barrett JC, Jurka J, Larionov V. 2004. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet 13: 2737–2751. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RB, Moses DN, Kwon Y, Chan P, Chi P, Klein H, Sung P, Greene EC. 2009. Structural transitions within human Rad51 nucleoprotein filaments. Proc Natl Acad Sci 106: 12688–12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. 2007. Human CtIP promotes DNA end resection. Nature 450: 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel BP, Jodelka FM, Nunez R. 2006. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res 66: 5181–5189. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. 2004. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. 2004. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci 101: 9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P, Krejci L, Van Komen S, Sehorn MG. 2003. Rad51 recombinase and recombination mediators. J Biol Chem 278: 42729–42732. [DOI] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. [DOI] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10: 373–385. [DOI] [PubMed] [Google Scholar]
- Waldman AS, Liskay RM. 1988. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 8: 5350–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock DM, Elliott B, Jasin M. 2006a. A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood 107: 777–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock DM, Richardson CA, Elliott B, Jasin M. 2006b. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst) 5: 1065–1074. [DOI] [PubMed] [Google Scholar]
- White TB, Morales ME, Deininger PL. 2015. Alu elements and DNA double-strand break repair. Mob Genet Elements 5: 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, Kwon Y, Rao T, Wang W, Sheng C, et al. 2017. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550: 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, Paull TT. 2014. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res 42: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.