

Abstract
Recognition of dsRNA molecules activates the MDA5–MAVS pathway and plays a critical role in stimulating type-I interferon responses in psoriasis. However, the source of the dsRNA accumulation in psoriatic keratinocytes remains largely unknown. A-to-I RNA editing is a common co- or post-transcriptional modification that diversifies adenosine in dsRNA, and leads to unwinding of dsRNA structures. Thus, impaired RNA editing activity can result in an increased load of endogenous dsRNAs. Here we provide a transcriptome-wide analysis of RNA editing across dozens of psoriasis patients, and we demonstrate a global editing reduction in psoriatic lesions. In addition to the global alteration, we also detect editing changes in functional recoding sites located in the IGFBP7, COPA, and FLNA genes. Accretion of dsRNA activates autoimmune responses, and therefore the results presented here, linking for the first time an autoimmune disease to reduction in global editing level, are relevant to a wide range of autoimmune diseases.
Keywords: RNA editing, A-to-I, psoriasis, interferon
INTRODUCTION
Psoriasis is a chronic autoimmune disease affecting 2%–4% of the population worldwide and characterized clinically by well-demarcated, erythematous, scaly plaques. The main histological features are epidermal hyperplasia, aberrant keratinocyte proliferation, and differentiation and infiltration of T lymphocytes, dendritic cells, and neutrophils. The pathogenesis of psoriasis involves both innate and adaptive immune regulation (Krueger 2002; Lowes et al. 2014).
Interferon-α (IFNα) and IFNβ, collectively known as type I IFNs, are the major effector cytokines of the host immune response, closely linking the innate and the adaptive immune responses (González-Navajas et al. 2012). Type I IFNs can be induced in response to bacterial and viral pathogens through Toll-like receptor (TLR)-dependent pathways (Monroe et al. 2010; Trinchieri 2010). In addition, TLR-independent pathways, including the cytoplasmic sensors of the dsRNA-retinoic-acid-inducible gene I (RIG-I) and the melanoma differentiation-associated gene 5 (MDA5), may induce IFNs through their common adaptor, mitochondrial antiviral signaling protein (MAVS) (Reikine et al. 2014). Accumulating evidence suggests that type I IFNs play a role in the initiation of psoriasis in conjunction with the TNFα and IL-23/IL-17 pathways. Clinically, systemic treatment with IFN-α and β (La Mantia and Capsoni 2010; Afshar et al. 2013), as well as external application of imiquimod cream (which induces the release of IFN from plasmacytoid dendritic cells), lead to induction and exacerbation of psoriasis (Fanti et al. 2006; Rajan and Langtry 2006). Psoriatic skin has an increased activation of downstream targets of the type I interferon signaling pathways, including signal transducer and activator of transcription 1 (STAT1), interferon regulatory factor 9 (IRF-9) and IRF-7 (van der Fits et al. 2004). More direct evidence comes from the observation that mice lacking IRF-2, a negative regulator of the IFN signaling pathway, develop psoriasis-like skin lesions. This phenotype can be partially rescued by knocking out positive regulators of the IFN-α/β signaling pathway (Hida et al. 2000). IFN-α/β are thought to be produced by pathologic dendritic cells, macrophages and keratinocytes, although the pathological mechanism leading to overproduction of type I IFNs is still not fully understood (Perera et al. 2012).
A recent study reported that keratinocytes produce IFN-β in response to dsRNA (Zhang et al. 2016), a finding that implicates the increased levels of dsRNA seen in psoriasis as a cause of the increased IFN levels. Further support for the role of the dsRNA sensing pathway in psoriasis comes from Li et al. (2010) who identified two rare SNPs in MDA5 as associated with a decreased risk of psoriasis.
A process that directly influences dsRNA levels is A-to-I RNA editing, a post-transcriptional modification that diversifies the sequence of RNA from that encoded in the DNA (Bass 2002). A-to-I editing is catalyzed by members of the ADAR (adenosine deaminase acting on RNA) family of dsRNA-binding enzymes. There are two active ADAR proteins: ADAR2 (ADARB1) and ADAR1 (ADAR), which have alternative promoters that generate 2 isoforms: ADARp110 and ADARp150. While ADARp110 is expressed constitutively, ADARp150 is induced by interferon (Patterson and Samuel 1995; George and Samuel 1999; Savva et al. 2012). Editing of dsRNA by ADAR enzymes introduces mismatches resulting in unwinding of the double-stranded base-pairing and consequent reduction of dsRNA levels (Bass and Weintraub 1987; Nishikura 2010).
A number of recent studies point out that A-to-I RNA editing plays a critical role in regulating the innate immune response (Hartner et al. 2009; Toth et al. 2009; Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). The main function of ADAR1 is not to create proteomic diversity (Heraud-Farlow et al. 2017), but rather to suppress the antiviral interferon response by preventing MDA5 from sensing endogenous dsRNA as nonself (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). This newly established intricate interplay between RNA editing and the immune response suggests that editing may play a role in a variety of autoimmune diseases. Indeed, ADAR1 mutations have been linked to Aicardi–Goutieres syndrome (AGS), a severe auto-inflammatory disease (Rice et al. 2012), and hypoediting was observed following up-regulation of PDE8A1 in T cells in systemic lupus erythematosus (SLE) (Orlowski et al. 2008). Consistently, ADAR1 knockout mice die embryonically, showing an elevated expression of type I IFNs (Hartner et al. 2009).
In this study, we demonstrate a global reduction of A-to-I RNA editing in psoriasis that may account for the accumulation of dsRNA (Bass 2002; Nishikura 2010), which in turn stimulates the production of IFNs and is instrumental in triggering the disease. We verified that both IFN pattern recognition receptors and IFN stimulated genes are up-regulated in psoriatic lesions (Yao et al. 2008; Rasheed et al. 2016; Ruano et al. 2016). Editing of coding mRNA may lead to recoding (nonsynonymous changes in the mRNA) (Bahn et al. 2012; Ramaswami et al. 2013; Pinto et al. 2014; Ramaswami and Li 2014). Thus, we also investigated the editing levels in specific recoding sites and identified reduced levels of editing in three evolutionarily conserved recoding sites located in the COPA, FLNA, and IGFBP7 genes. Our findings suggest that reduced editing might play a role in the pathogenesis of psoriasis and other autoimmune diseases.
RESULTS
Global editing is decreased and dsRNA structures are increased in psoriatic lesions
To characterize RNA editing in psoriasis, we analyzed RNA-seq data from 78 psoriatic lesions, 27 psoriatic uninvolved skin (normal looking skin from psoriasis patients), and 86 healthy control samples (data taken from the studies of Li et al. 2014 and Tsoi et al. 2015; see Materials and Methods).
ADAR enzymes edit adenosines that reside in dsRNA. In humans, almost all of the editing activity occurs in Alu repetitive elements because of their tendency to form double-stranded structures (Bazak et al. 2014a). As a result of the mismatches introduced during the editing, the double-stranded structures unwind, decreasing the amount of the total dsRNA. Therefore, quantification of the relative edited Alu elements can serve as a proxy for the amount of dsRNA structures formed by the available expressed Alu repeats. Using an Alu-specific RNA editing detection algorithm (Bazak et al. 2014a), we estimated the amount of edited Alu elements in the psoriatic lesions, uninvolved skin and healthy control samples. The relative amount of edited Alu (the number of edited Alu elements per reads that were aligned to Alu) was lower in samples from lesions than in healthy controls (Fig. 1A; P-value = 9.2 × 10−6, two tailed t-test). Interestingly, there was an intermediate amount of relative edited Alu elements in the uninvolved skin. The Alu index (Bazak et al. 2014b) measure (indicating the average number of A-to-G mismatches across all Alu adenosines weighted by the total coverage at Alu adenosines positions) showed the same trend: a significant reduction in editing in psoriatic lesion compared to healthy control samples (Fig. 1B; P-value = 0.001, two-tailed t-test). Importantly, we have verified that differences in target expression level do not account for the observed differential editing.
FIGURE 1.

A decrease in global editing in psoriatic lesions. (A) The relative number of edited Alu in healthy controls (blue) is higher than in psoriatic lesion (red) and uninvolved skin (pink) samples. The bar-plot represents the average ±SEM of the number of edited Alu per thousand reads that were aligned to Alu. P-value (healthy versus psoriasis) = 9.2 × 10−6. (B) Measuring the Alu editing index in psoriatic lesions, uninvolved skin and healthy control tissues emphasizes the decreased editing activity in psoriasis. P-value = 0.001 (C) Complementary hyperediting screening reveals a decrease in the number of editing sites in psoriatic lesions. The distribution of the normalized number of hyperediting sites is shown. P-value = 2.6 × 10−17. (D) The relative expression of Alu repeats is lower in psoriatic tissues compared to normal controls, suggesting that the source for an elevated level of dsRNA is not an increased expression of dsRNA-forming transcripts. P-value = 7×10−11; all analyses are two-tailed t-tests.
Taking into account events of excessive editing in clusters of adenosines is crucial in order to accurately estimate the scope of RNA editing. Therefore, global editing activity based on clusters of editing (hyperediting) sites was also evaluated. These clusters can be recognized in reads that are usually ignored by the standard editing detection methodologies, since they differ too much from the corresponding DNA sequence and cannot be aligned to the reference sequence using the conservative aligners (Carmi et al. 2011). To overcome these difficulties, we used a designated hyperediting detection scheme (see Materials and Methods) (Porath et al. 2014). Here too, psoriatic lesions showed a significant reduction in editing compared to healthy controls (Fig. 1C; P-value = 2.6 × 10−17, two-tailed t-test). Interestingly, hyperediting in uninvolved skin tissues did not differ from the healthy control samples.
The reduction in editing is expected to increase the amount of endogenous dsRNA structures. However, this could be a secondary effect to an elevated expression of dsRNA-forming transcripts, putting an increased load on the ADAR enzymes. In order to test this hypothesis, we compared the fraction of reads that map to Alu repeats, the major source for endogenous dsRNA structures in human, between healthy and psoriatic tissues. Interestingly, we found that this fraction, measuring the relative expression of Alu repeats and estimating the potential for dsRNA formation, is 14% lower in psoriatic tissues, compared to normal controls (Fig. 1D; P-value = 7 × 10−11, two-tailed t-test). Therefore, the source for an elevated level of dsRNA is not an increased expression of dsRNA-forming transcripts, and may be attributed to lowered editing, increasing the potential of the expressed repeats to form dsRNA structures.
In order to confirm our findings, we have analyzed additional independent RNA-seq data (Ahn et al. 2016; Gupta et al. 2016) containing 18 psoriatic lesion samples and 16 healthy skin controls. Applying the same designated hyperediting detection scheme, psoriatic lesions showed a significant reduction of ∼30% in editing compared to the healthy controls (Fig. 2A; P-value = 0.008, two-tailed t-test).
FIGURE 2.

A second, independent set of psoriatic RNA-seq data show a decrease of editing in psoriatic lesions. (A) Total RNA-seq data set analysis repeats the trend of a decrease in the number of hyperediting sites in psoriatic lesions. Interestingly, after 1 mo of treatment with adalimumab, the editing levels restored back to a similar level of healthy control samples. The figure shows the distribution of the normalized number of hyperediting sites. P-value = 0.008, two-tailed t-test. (B) Healthy control and treated with adalimumab after 1-mo groups were joined together in order to achieve a better statistical power. Complementary hyperediting screening reveals a decrease in the number of editing sites in psoriatic lesions. P-value = 0.0004, two-tailed t-test. (C) Matched comparison of hyperediting in a sample between before and after treatment. P-value = 0.004, two-tailed t-test.
Interestingly, this set includes also 18 samples of the same psoriatic patients after 1 mo of treatment with adalimumab. Adalimumab is a recombinant, fully human Immunoglobulin G1 monoclonal antibody that binds with high affinity and specificity to TNF-α and have high efficacy against psoriasis (Alwawi et al. 2008). The treated samples showed a significant increase in editing compared to psoriatic lesions samples, back to a level similar to that of the healthy control samples (Fig. 2A; P-value = 0.004, two-tailed t-test). The normalization of editing levels by adalimumab is in-line with the modulation of other biological markers such as inflammatory cytokines by this drug (Langkilde et al. 2016) as well as other effective anti-psoriatic treatments (Johnson-Huang et al. 2010; Russell et al. 2014)
Improved statistical power is gained when adding the healthy control samples and the treated samples groups together, as more significant differences in hyperediting are achieved (Fig. 2B, P-value = 0.0004, two-tailed t-test).
Overexpression of MDA5 pathway and IFN stimulated gene signature in psoriatic lesions
The observed decrease in global editing activity and consequent putative increase in dsRNA might be expected to activate the innate immune response due to recognition of the dsRNA as “foreign” by MDA5 (Pestal et al. 2015). Indeed, increased MDA5 expression was previously demonstrated in psoriatic lesions (Prens et al. 2008). To confirm these results, we quantified the mRNA expression levels of several genes related to the TLR-independent IFN activation pathway (MDA5, IRF7, NFKB1, STAT1, and STAT3), as well as the Th1 and Th17 cytokine signature, known to be up-regulated in psoriasis, as a positive control. As expected, psoriatic lesions displayed a significant up-regulation of these cytokines as well as the TLR independent IFN activity pathway genes compared to healthy controls and uninvolved skin (Fig. 3A,B). Interestingly, the MAVS and IRF3 genes, which are also involved in the MDA5 pathway, showed only a very slight insignificant up-regulation. It is possible that additional regulatory mechanisms, such as protein phosphorylation, are involved in the regulation of these genes (Liu et al. 2015).
FIGURE 3.

Overexpression of MDA5 pathway and IFN stimulated gene signature in psoriatic lesions. (A) Up-regulation of known Th1 and Th17 cytokines signature in psoriasis. Distribution of IL12B, IL17A, IL17F, IL22, and IL23A gene expression in psoriatic lesions (red), uninvolved skin (pink), and healthy control (blue) samples. Expression levels are represented by the normalized values from DESeq analysis. (B) Elevated mRNA expression of genes in the MDA5 pathway: MDA5 (P-value = 7.7 × 10−20 and 4.0 × 10−57, respectively, two tailed t-test); IRF7 (P-value = 1.5 × 10−17 and 2.6 × 10−44, respectively, two tailed t-test); NFKB1 (P-value = 5.8 × 10−6 and 5.4 × 10−16, respectively, two tailed t-test); STAT1 (P-value = 9.9 × 10−50 and 5.0 × 10−136, respectively, two tailed t-test); and STAT3 (P-value = 5.2 × 10−24 and 5 × 10−56, respectively, two tailed t-test).
Editing in conserved sites is reduced in psoriatic lesions
While the vast majority of A-to-I editing activity in human RNA occurs in noncoding primate-specific Alu elements, a small fraction of editing sites are conserved within mammalian evolution (Pinto et al. 2014) and are presumed to have an important biological function. The conserved sites tend to reside in coding regions and have higher editing and expression levels than the Alu sites.
To investigate editing that may derive direct functional changes in psoriasis, we tested the editing levels for 58 conserved mammalian sites (Pinto et al. 2014). The global conserved-editing index, reflecting the overall editing levels at these sites (see Materials and Methods), shows a significant reduction in psoriasis (9.5% in psoriatic lesion samples compared with 15.5% in healthy controls and 16.2% in uninvolved skin samples) (Fig. 4A; P-values = 3.2 × 10−21 and 8.4 × 10−17, respectively, two-tailed t-test). Notably, editing levels of the recoding sites of the healthy controls was comparable to that for normal skin tissues in a recent report (see Supplemental Table 1; Tan et al. 2017). These results support the global editing trend already identified.
FIGURE 4.

Editing of conserved sites is reduced in psoriatic lesions. (A) The editing index of the conserved sites displays a lower signal in psoriatic lesions compared to uninvolved skin and healthy control samples. (B) Significantly decreased editing in conserved sites in psoriatic lesions. The editing activity in the sites that exhibit higher editing leads to nonsynonymous mutations in the COPA, FLNA, and IGFBP7 proteins. (C) IGFBP7 expression distribution in psoriatic lesions, uninvolved skin and healthy control samples. Down-regulation of IGFBP7 in psoriatic lesions as previously reported in the literature emphasizes the significant involvement of the gene in the pathology of psoriasis.
Focusing on specific editing sites, we studied eleven conserved sites that passed the expression cutoffs (see Materials and Methods) and found a significant decrease of editing in six of them (Fig. 4B). The three sites exhibiting the highest editing levels (more than 10% editing in at least one group) are recoding sites located in the Coatomer Protein Complex Subunit Alpha (COPA), Filamin A (FLNA) and the Insulin-like growth factor-binding protein 7 (IGFBP7) genes (Fig. 4B). Notably, editing levels of these three targets is altered in various types of tumors (Paz-Yaacov et al. 2015). Interestingly, IGFBP7 has been previously linked to psoriasis, as its expression is down-regulated in psoriatic lesions and increases following phototherapy (Hochberg et al. 2007; Nousbeck et al. 2011). Indeed, our RNA-seq data results confirmed a down-regulation of IGFBP7 in psoriatic lesions compared to uninvolved skin and healthy control samples (Fig. 4C; P-values = 0.009 and 2.4 × 10−5, respectively).
Expression of ADARs and possible regulation
Altered editing levels in psoriasis could result from a reduced expression of ADAR enzymes. In accordance with this possibility, we found a significant reduction of ADAR2 in psoriatic lesions (compared with uninvolved skin and healthy control samples), consistent with the perception that ADAR2 mediates editing in most of the conserved editing sites (Fig. 5A; P-values = 0.03 and 0.005, respectively). In contrast, there was an increase in ADAR1 mRNA (Fig. 5B; P-values = 4.3 × 10−8 and 3.5 × 10−15, respectively). In addition, based on immunohistochemical staining of psoriatic skin lesions from our pathological archive, ADAR1 protein is up-regulated in psoriatic lesions compared to uninvolved skin and normal skin samples (Fig. 5C; see Materials and Methods).
FIGURE 5.

Expression of ADARs and possible regulation. Based on the RNA-seq data, ADAR2 is decreased (A) while ADAR1 is increased (B) in psoriatic lesions. (C) Up-regulation of ADAR1 was also observed at the protein level based on histochemical staining. (D) Global editing levels based on a set of 6583 known editing sites, and subgroups of this set that were reported to be specifically edited by ADAR1 (n = 4458) and by ADAR2 (n = 502). There is a common trend of editing decrease in targets of both ADAR enzymes. (E) Up-regulation of ADAR1-binding proteins’ expression in psoriatic lesions may provide a possible explanation for the decrease in editing in psoriatic lesions, and the observation emphasizes the complexity of A-to-I RNA editing regulation. DICER1: P-value = 1.4 × 10−7 and 1.8 × 10−17, respectively. ELAVL1: P-value = 0.025 and 0.005, respectively. ILF2: P-value = 7.6 × 10−7 and 8.0 × 10−11, respectively. ILF3: P-value = 0.0007 and 9.6 × 10−6, respectively. All analyses are two tailed t-tests.
In order to test whether the increase in ADAR1 expression is accompanied by an increase in its editing activity, we analyzed a set of sites known to be edited exclusively by ADAR1 (4458 sites) or ADAR2 (502 sites) (Wang et al. 2013). The results demonstrated that the editing activity of both enzymes is actually decreased in psoriatic lesions compared to uninvolved skin and healthy control samples (Fig. 5D. ADAR1 targets: P-values = 0.0004 and 0.0003, respectively. ADAR2 targets: P-values = 0.012 and 1.4 × 10−5, respectively), raising a question about the regulation mechanisms affecting ADARs' editing activity in psoriasis.
ADAR1 forms homodimer or heterodimer complexes with ADAR1-binding proteins. The homodimerzation is essential for the A-to-I RNA editing activity (Cho et al. 2003; Valente and Nishikura 2007). Therefore, a possible explanation for the discrepancy between the global editing decrease and ADAR1 elevation in the disease may be provided by an observed up-regulation of the ADAR1-binding proteins. In accordance with this proposition, we detected an up-regulation of ADAR1-binding protein genes such as DICER1 (Ota et al. 2013), ELAVL1 (Wang et al. 2013), ILF2 (Nie et al. 2005), and ILF3 (Nie et al. 2005) in psoriatic lesions (Fig. 5E). This complex scenario emphasizes the intricacy of the A-to-I RNA editing regulation and its close relation to the immune process.
DISCUSSION
Psoriasis is a chronic autoimmune disease mediated by inflammatory cytokines, including type I IFNs (Lowes et al. 2014). Recent reports have suggested a role for A-to-I RNA editing in the suppression of the interferon response. Here we suggest two different routes by which decreased A-to-I editing in keratinocytes could be involved in the exacerbation of psoriasis (Fig. 6): First, decreased editing in three highly conserved recoding sites in the genes COPA, FLNA, and IGFBP7, might alter their function, leading to keratinocyte proliferation, and impaired differentiation. Second, a global decrease in editing activity can lead to an accumulation of dsRNA with consequent activation of the MDA5/MAVS pathway which in turn triggers the type I IFN response as well as NFkB associated cytokine production.
FIGURE 6.
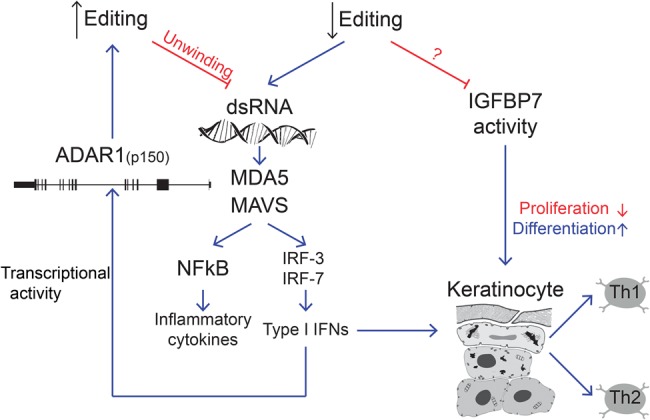
Proposed model for the A-to-I RNA editing involvement in psoriasis. Decreased editing in coding regions modulates genes such as IGFBP7 and COPA. In addition, the reduction in global editing triggers the accumulation of dsRNA molecules that activate the MDA5/MAVS pathway and stimulate type I IFN responses as well as NFkB associated cytokine production. A feedback mechanism up-regulates ADAR1.
Of the three recoding targets identified, two are known to be associated with autoimmune diseases. COPA encodes the COPA subunit of the seven member coat protein complex I (COPI), a key component of the vesicular trafficking machinery in eukaryotic cells mediating bidirectional membrane traffic between the endoplasmic reticulum (ER) and the Golgi (Quek and Chow 1997). Recently, mutations in COPA have been linked to autoimmune diseases characterized by high-titer autoantibodies, inflammatory arthritis, and interstitial lung disease (Watkin et al. 2015). Expression of mutant COPA leads to endoplasmic reticulum stress and the up-regulation of Th17 cytokines, a pathway known to be implicated in psoriasis. As editing of COPA is expected to lead to changes in the protein function (Solomon et al. 2014), decreased editing might have a role in the pathogenesis of psoriasis. IGFBP7 is a secreted protein with various functions within the cell, including mitogenic effects and tumor suppressor functions, as well as inhibition or stimulation of angiogenesis. Editing of IGFBP7 results in a reduction of proteolytic cleavage (Godfried Sie et al. 2012), that in turn modulates its biological activity. In psoriasis, IGFBP7 expression has been shown to be down-regulated whereas phototherapy treatment increases its levels (Hochberg et al. 2007; Nousbeck et al. 2011). In this way, since the edited form of IGFBP7 has been shown to inhibit proliferation and induce senescence in cultured keratinocytes (Hochberg et al. 2013), hypoediting of IGFBP7 might have a pathogenic role in the epidermal proliferation seen in psoriasis.
The recoding sites may be functionally important, but comprise only a tiny fraction of the total editing activity. The vast majority of editing sites reside in dsRNA structures within noncoding RNA, a known modulator of innate immunity. The IFN pathways are known to be crucial for the initiation phase of psoriasis (Lowes et al. 2014). Furthermore, several transcriptome analysis studies have identified type I IFN-inducible gene signatures as a major pathological pathway in psoriatic skin. Despite these observations, the mechanisms leading to this aberrant IFN production are not well understood. One possible mechanism was suggested to be related to the Koebner reaction, i.e., formation of a new psoriatic plaque following minor tissue injury, observed in some patients. This type of injury is thought to lead to the accumulation of self-DNA and self-RNA molecules that complex with the endogenous antimicrobial peptide LL37 and delivery to the endosomal compartments of dendritic cells (pDC). Subsequently, the complexes are recognized by TLRs, leading to IFN production (Lande et al. 2007; Ganguly et al. 2009). Recently, Zhang and coworkers reported that LL37/dsRNA complexes also activate the MAVS-RIG-I-MDA5 signaling pathway and trigger IFNβ within keratinocytes (Zhang et al. 2016). Here we present an additional source of dsRNA and MAVS pathway activation in psoriatic keratinocytes: Global reduction of A-to-I editing is expected to lead to the accumulation of self dsRNA independently of tissue injury. As the Koebner reaction is responsible for triggering psoriasis in only a minority of cases, the reduction in editing might represent a more general stimulus and could have a role in both the initiation and exacerbation of psoriasis. In addition, since keratinocytes comprise the vast majority of cells analyzed in this study, our results support the role of keratinocytes as a source of IFN (in accordance with Zhang et al. 2016).
The main sources for dsRNA are embedded mobile elements that are localized in close proximity in reverse orientation within transcribed genes (editing is usually taking place in the preRNA, before splicing). These repetitive-elements-derived dsRNAs, and their editing, are seen in all metazoans, including mouse (where B1 and B2 elements are the main editing targets) (Porath et al. 2017). The degree to which a transcriptome of a given species undergoes hyperediting is governed by the repertoire of repeats in the underlying genome and its ability to form dsRNA structures. Editing levels vary considerably across species, and normal mouse exhibit much less editing, globally, compared with the diseased human samples studied here (Porath et al. 2017). However, it is the reduction in editing compared with the homeostatic state that is likely to result in activation of the innate immunity system (Liddicoat et al. 2015). The reduced editing level in psoriasis seems to be at odds with the elevated ADAR expression level. Indeed, complex nonlinear regulation of editing by ADARs was observed in several reports (Jacobs et al. 2009; Wahlstedt et al. 2009; Ring et al. 2010; Garncarz et al. 2013). A possible explanation might be the up-regulation of the ADAR1-binding proteins (Fig. 5E) that compete with ADAR1 for targets. In addition, lower ADAR expression in the nucleus might lead to lower editing and accumulation of dsRNA, which in turn induces cytoplasmic ADAR p150 expression that probably does not contribute much to editing. As we cannot distinguish between the ADAR isoforms, it is possible that the observed increase of expression of the total level of ADAR represents an elevation in a less active version of the genes. Another possible scenario that can explain the results is an altered nucleocytoplasmic partitioning of endogenous dsRNAs in the psoriatic lesions, leading to more dsRNAs in the cytoplasm where IFN signaling is triggered by the dsRNA. Further experimental validation is needed to reveal the actual mechanism.
It should be noted that the data were generated from tissues with a heterogeneous cellular composition, and we appreciate that this could affect the observed editing levels (Gal-Mark et al. 2017). It will be important to repeat the analysis on more homogeneous cell populations once methods of obtaining cell-type–specific data have progressed sufficiently.
Initial evidence for impaired global A-to-I editing has also been demonstrated in two other autoimmune diseases: SLE and the rare hereditary disease AGS. In SLE, Orlowski et al. (2008) reported A-to-I hypoediting followed by up-regulation of the PDE8A1 transcripts in SLE T cells. In AGS, a mutation in ADAR1 activates type I IFN signaling (Rice et al. 2012). Here we demonstrate, for the first time, global editing impairment in psoriasis, a multifactorial autoimmune disease. Taken together, these results highlight the importance of the fine balance between IFN pathways and RNA editing for a precise immune response.
MATERIALS AND METHODS
Preprocessing, quality control, alignment, and editing detection
RNA-seq data of 104 psoriatic lesions, 27 uninvolved skin, and 93 healthy control skin samples were downloaded from the Gene Expression Omnibus (under the GEO accretion number GSE63980) (Li et al. 2014; Tsoi et al. 2015). From this set, we chose only samples labeled as SINGLE (Library Layout = “SINGLE”). In cases where one sample has more than one FASTQ file, we chose the file with the higher number of reads. Then, the command fastq-quality-filter from the fastx-toolkit was applied to remove reads with a low quality score, using parameters that demand at least 20 Phred score (−q 20) through 70% of the reads length (−p 70). Samples with an insufficient number of reads (below 20 M) were then removed, leaving 78 psoriatic lesions, 27 uninvolved, and 86 healthy control samples. Sequence read quality was evaluated using the FastQC quality control tool (Andrews 2010) to ensure that the data are qualitative.
The reads were mapped to the hg19 reference genome using the STAR aligner (Dobin et al. 2013). Only uniquely aligned reads (outFilterMultimapNmax = 1) were used. We then applied two strategies to estimate global editing levels by evaluating the editing in Alu repetitive elements and in hyperediting clusters. In addition, we analyzed the editing in specific genomic positions known to be editing sites.
Additional RNA-seq data of 52 samples (18 psoriatic lesions, 18 after treatment with adalimumab, and 16 healthy control skin samples) were downloaded from the Gene Expression Omnibus (under the GEO accretion number GSE74697) (Ahn et al. 2016; Gupta et al. 2016).
For proper comparison with the first data set (although the use of different library preparation and sequencing protocols prevents a direct numerical comparison), the reads were trimmed from 101 to 79 bp (deleting 11 bp from start and 11 bp from end) using seqtk trimfq tool. Then, the command fastq-quality-filter from the fastx-toolkit was applied to remove reads with a low quality score as before, using the same parameters that demand at least a 20 Phred score (−q 20) through 70% of the reads length (−p 70). Orphan reads were removed by applying fastqCombinePairedEnd.
Sequence read quality and alignment of the second RNA-seq set were done as in the first data set. Hyperediting clusters strategy was applied to determine RNA-editing levels.
Detection of editing in Alu repetitive elements
Alu repetitive elements tend to form double-stranded RNA structures. These structures are the favorite substrates of A-to-I RNA editing by ADAR enzymes. Almost all of the editing activity in humans occurs in Alu elements and millions of sites have been detected to date (Bazak et al. 2014a,b). For this reason, we used the editing signal in Alu elements as an indicator of the global editing activity.
While virtually all adenosines within most of the Alu elements undergo editing, most of them are edited at only a low level (<1%). Thus, because most of the detected sites are a random sample of weakly edited sites, a high depth, much higher than available, is required to quantify editing at all the specific adenosines. We therefore chose to focus on the global editing within Alu elements rather than editing of specific Alu adenosines. The editing level of each sample was evaluated using a previously described algorithm (Bazak et al. 2014a,b), in the same way it was applied recently in a screen of editing in cancer (Paz-Yaacov et al. 2015) (except to one modification described below). The accuracy of the editing detection was extremely high as A-to-G mismatches, which are indicative of A-to-I editing, constituted 94.9% ± 0.1 of the total possible mismatches. The global editing was estimated using the Alu index calculated as the ratio of “G” at all the adenosine positions in Alu, to the A + G at the same genomic locus. This is in contrast to the cancer project where the index calculation was based only on adenosines in Alu with a dominant mismatch of A-to-G. The higher the index, the more editing activity that occurs in a sample. Since the index is based on the combined results of millions of adenosines, we consider it to be a robust measurement of the global editing levels.
Hyperediting
Conventionally, detection of RNA editing is carried out by comparing RNA sequences to their appropriate reference (DNA sequenced or reference genome) and looking for reliable A-to-G mismatches. However, such methodology may fail to recognize heavy editing reads, since they differ from the corresponding DNA sequence and cannot be aligned to the reference sequence using the conservative aligners (Carmi et al. 2011). Using a pipeline developed in our laboratory, we are able to recognize such heavily edited reads (so called hyperediting clusters) (Porath et al. 2014). The idea is to perform transformation of all adenosines and guanosines in both the unmapped RNA sequences and the reference, followed by realignment. Applying the pipeline methodology on reads that could not be mapped using the initial STAR alignment gave a very clean signal with A-to-G mismatches significantly enriched among all possible substitutions (97% ± 0.17). The validity of the method is also supported by the observation that the identified editing sites included the familiar ADAR sequence motif (G depletion upstream and G enrichment downstream from the editing site) and an equivalent signal was observed in both strands without significant strand preference (44.7% ± 0.25 of the A-to-G mismatches were present in the plus strand). To compare groups and minimize the effect of the library size, we normalized the total editing sites in each sample by the number of mapped reads. These normalized values serve as a measure of the levels of hyperediting in each sample.
Editing in known editing sites
In addition to the global measurements of editing, we applied the REDIToolKnown script (Picardi and Pesole 2013) to quantify the editing occurring in a tight set of conserved editing sites (Pinto et al. 2014). As an initial analysis, the intention was to measure the overall global editing level in all the evolutionarily conserved sites. For this purpose, we used permissive parameters that allow minimum one read supporting the variation (−v 1), minimum 0.001 editing frequency (−n 0.001), exploring one base near splice junction (−r 1), minimum one read coverage, and trimming of six bases at both ends of the reads (−T 6-6). The global editing levels in conserved sites were evaluated using the editing index calculated from the ratio of the total number of A-to-G mismatches at the examined genomic locations of the conserved editing sites, to the total number of reads aligned to these positions.
In order to accurately evaluate the editing at individual conserved sites, we focused on the highly expressed sites and filtered sites supported by at least 10 reads in 75% of the samples in at least two of the groups (psoriatic lesion, uninvolved skin, and healthy control). Statistical significance was estimated using a two-tailed t-test followed by the Benjamini–Hochberg false discovery rate (FDR) multiple-testing. In addition, conserved editing sites were annotated using Annovar (Wang et al. 2010).
Gene expression
A gene annotation table was downloaded from the UCSC table browser website (Karolchik et al. 2003). The number of reads aligned to the exons of each gene was assessed using featureCounts (Liao et al. 2013) with the same BAM files used in the editing analysis. The DESeq package (Anders and Huber 2010) in R was used for differential expression evaluation and estimation of the P-values.
Detection of editing by different ADAR enzymes
We built a list of editing sites to be tested based on ADAR targets reported in Wang et al. (2013) and used liftOver tools to convert the hg18 genomic locations in the published list to hg19 genomic positions. We then ran REDIToolKnown script as described above, following annotation by Annovar (Wang et al. 2010) and exclusion of sites located in immunoglobulins (since these are likely to represent mismatches due to somatic hypermutations) and known gSNPs (dbSNP 135). We did include known editing sites reported as SNPs (Eisenberg et al. 2005). Out of 6583 editing sites detected, 4458 sites are edited only by ADAR1 and 502 are reported to be exclusively targets for ADAR2 targets (Wang et al. 2013). The editing index was calculated as described in the specific editing section.
Statistical calculations and figure drawing
The statistical calculations and figure drawing were done using R programming (R Core Team 2014)
Immunohistochemistry of human skin tissues
Biopsy specimens from lesions (n = 6), uninvolved skin (n = 5), and normal skin controls (n = 5) were obtained from the Sheba Medical Center Institute of Pathology. Patients were of Caucasian origin aged 18 to 85. All patients were clinically diagnosed with psoriasis vulgaris, and had not received systemic immunosuppressive treatment, phototherapy (Psoralen and UVA [PUVA]/solarium/UVB), or topical therapy for at least 3 wk prior to skin biopsy. The uninvolved skin samples were taken from the same area, ∼5 cm away from the lesion biopsy. All the biopsies were evaluated by a dermatopathologist to confirm the histological diagnosis. The study was approved by the IRB Committee of the Sheba Medical Center (confirmation number: 9776-12-SMC).
For immunostaining of paraffin–embedded slides, 5-µm slides were incubated at 60°C for 1 h, de-waxed in xylene, and rehydrated. Antigen retrieval was done using the Tris Buffer (pH 9.0) antigen retrieval protocol. Sections were blocked by casein and incubated with Anti-ADAR1 Antibody (ATLAS antibodies HPA003890, 1:800). The slides were then incubated with a secondary horseradish peroxidase (HRP) conjugate at 1:300 (Vector Labs). Antibody binding was visualized with the substrate-chromogen AEC and the stained sections were viewed under a light microscope and analyzed by personnel blinded to the origin of the material.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Hagit Porath, Lily Bazak, and Yishay Pinto for assisting with the computational analysis and Orshay Gabay for the graphical help. This work was supported by the Ministry of Science and Technology, Israel, the Japan Science and Technology Agency (JST) (10765-3 to E.Y.L), the European Research Council (311257 to E.Y.L.), the Israel Science Foundation (1380/14 to E.Y.L., 379/12 to E.E.), and the Talpiot Medical leadership program (to S.G.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064659.117.
REFERENCES
- Afshar M, Martinez AD, Gallo RL, Hata TR. 2013. Induction and exacerbation of psoriasis with Interferon-α therapy for hepatitis C: a review and analysis of 36 cases. J Eur Acad Dermatol Venereol 27: 771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn R, Gupta R, Lai K, Chopra N, Arron ST, Liao W. 2016. Network analysis of psoriasis reveals biological pathways and roles for coding and long non-coding RNAs. BMC Genomics 17: 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwawi EA, Mehlis SL, Gordon KB. 2008. Treating psoriasis with adalimumab. Ther Clin Risk Manag 4: 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data. Vol. 2011. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X. 2012. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 22: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1987. A developmentally regulated activity that unwinds RNA duplexes. Cell 48: 607–613. [DOI] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014a. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L, Levanon EY, Eisenberg E. 2014b. Genome-wide analysis of Alu editability. Nucleic Acids Res 42: 6876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi S, Borukhov I, Levanon EY. 2011. Identification of widespread ultra-edited human RNAs. PLoS Genet 7: e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. 2003. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem 278: 17093–17102. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Adamsky K, Cohen L, Amariglio N, Hirshberg A, Rechavi G, Levanon EY. 2005. Identification of RNA editing sites in the SNP database. Nucleic Acids Res 33: 4612–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanti PA, Dika E, Vaccari S, Miscial C, Varotti C. 2006. Generalized psoriasis induced by topical treatment of actinic keratosis with imiquimod. Int J Dermatol 45: 1464–1465. [DOI] [PubMed] [Google Scholar]
- Gal-Mark N, Shallev L, Sweetat S, Barak M, Billy Li J, Levanon EY, Eisenberg E, Behar O. 2017. Abnormalities in A-to-I RNA editing patterns in CNS injuries correlate with dynamic changes in cell type composition. Sci Rep 7: 43421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M. 2009. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 206: 1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garncarz W, Tariq A, Handl C, Pusch O, Jantsch MF. 2013. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol 10: 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. 1999. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci 96: 4621–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfried Sie C, Hesler S, Maas S, Kuchka M. 2012. IGFBP7’s susceptibility to proteolysis is altered by A-to-I RNA editing of its transcript. FEBS Lett 586: 2313–2317. [DOI] [PubMed] [Google Scholar]
- González-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Ahn R, Lai K, Mullins E, Debbaneh M, Dimon M, Arron S, Liao W. 2016. Landscape of long noncoding RNAs in psoriatic and healthy skin. J Invest Dermatol 136: 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM, Pang L, Liddicoat BJ, Gupte A, Li JB, et al. 2017. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol 18: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, Sato T, Hirose S, Shirai T, Taki S, et al. 2000. CD8+ T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-α/β signaling. Immunity 13: 643–655. [DOI] [PubMed] [Google Scholar]
- Hochberg M, Zeligson S, Amariglio N, Rechavi G, Ingber A, Enk CD. 2007. Genomic-scale analysis of psoriatic skin reveals differentially expressed insulin-like growth factor-binding protein-7 after phototherapy. Br J Dermatol 156: 289–300. [DOI] [PubMed] [Google Scholar]
- Hochberg M, Gilead L, Markel G, Nemlich Y, Feiler Y, Enk CD, Denichenko P, Karni R, Ingber A. 2013. Insulin-like growth factor-binding protein-7 (IGFBP7) transcript: A-to-I editing events in normal and cancerous human keratinocytes. Arch Dermatol Res 305: 519–528. [DOI] [PubMed] [Google Scholar]
- Jacobs MM, Fogg RL, Emeson RB, Stanwood GD. 2009. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev Neurosci 31: 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Huang LM, Suárez-Fariñas M, Sullivan-Whalen M, Gilleaudeau P, Krueger JG, Lowes MA. 2010. Effective narrow-band UVB radiation therapy suppresses the IL-23/IL-17 axis in normalized psoriasis plaques. J Invest Dermatol 130: 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res 31: 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JG. 2002. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol 46: 1–23; quiz 23–26. [DOI] [PubMed] [Google Scholar]
- La Mantia L, Capsoni F. 2010. Psoriasis during interferon β treatment for multiple sclerosis. Neurol Sci 31: 337–339. [DOI] [PubMed] [Google Scholar]
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, et al. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449: 564–569. [DOI] [PubMed] [Google Scholar]
- Langkilde A, Olsen LC, Saetrom P, Drabløs F, Besenbacher S, Raaby L, Johansen C, Iversen L. 2016. Pathway analysis of skin from psoriasis patients after adalimumab treatment reveals new early events in the anti-inflammatory mechanism of anti-TNF-α. PLoS One 11: e0167437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liao W, Cargill M, Chang M, Matsunami N, Feng B-J, Poon A, Callis-Duffin KP, Catanese JJ, Bowcock AM, et al. 2010. Carriers of rare missense variants in IFIH1 are protected from psoriasis. J Invest Dermatol 130: 2768–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, Ding J, Stuart PE, Xing X, Kochkodan JJ, et al. 2014. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol 134: 1828–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2013. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, et al. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Suárez-Fariñas M, Krueger JG. 2014. Immunology of psoriasis. Annu Rev Immunol 32: 227–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, Vance RE. 2010. Induction of type I interferons by bacteria. Cell Microbiol 12: 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Ding L, Kao PN, Braun R, Yang JH. 2005. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol 25: 6956–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousbeck J, Ishida-Yamamoto A, Bidder M, Fuchs D, Eckl K, Hennies HC, Sagiv N, Gat A, Gini M, Filip I, et al. 2011. IGFBP7 as a potential therapeutic target in psoriasis. J Invest Dermatol 131: 1767–1770. [DOI] [PubMed] [Google Scholar]
- Orlowski RJ, O'Rourke KS, Olorenshaw I, Hawkins GA, Maas S, Laxminarayana D. 2008. Altered editing in cyclic nucleotide phosphodiesterase 8A1 gene transcripts of systemic lupus erythematosus T lymphocytes. Immunology 125: 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. 2013. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153: 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, Samuel CE. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15: 5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. 2015. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep 13: 267–276. [DOI] [PubMed] [Google Scholar]
- Perera GK, Di Meglio P, Nestle FO. 2012. Psoriasis. Annu Rev Pathol 7: 385–422. [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Pesole G. 2013. REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29: 1813–1814. [DOI] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Carmi S, Levanon EY. 2014. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5: 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Knisbacher BA, Eisenberg E, Levanon EY. 2017. Massive A-to-I RNA editing is common across the Metazoa and correlates with dsRNA abundance. Genome Biol 18: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prens EP, Kant M, van Dijk G, van der Wel LI, Mourits S, van der Fits L. 2008. IFN-α enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: relevance for psoriasis. J Invest Dermatol 128: 932–938. [DOI] [PubMed] [Google Scholar]
- Quek HH, Chow VT. 1997. Molecular and cellular studies of the human homolog of the 160-kD α-subunit of the coatomer protein complex. DNA Cell Biol 16: 275–280. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rajan N, Langtry JA. 2006. Generalized exacerbation of psoriasis associated with imiquimod cream treatment of superficial basal cell carcinomas. Clin Exp Dermatol 31: 140–141. [DOI] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed H, El-Komy M, Hegazy RA, Gawdat HI, AlOrbani AM, Shaker OG. 2016. Expression of sirtuins 1, 6, tumor necrosis factor, and interferon-γ in psoriatic patients. Int J Immunopathol Pharmacol 29: 764–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikine S, Nguyen JB, Modis Y. 2014. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol 5: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring H, Boije H, Daniel C, Ohlson J, Öhman M, Hallböök F. 2010. Increased A-to-I RNA editing of the transcript for GABAA receptor subunit α3 during chick retinal development. Vis Neurosci 27: 149–157. [DOI] [PubMed] [Google Scholar]
- Ruano J, Suárez-Fariñas M, Shemer A, Oliva M, Guttman-Yassky E, Krueger JG. 2016. Molecular and cellular profiling of scalp psoriasis reveals differences and similarities compared to skin psoriasis. PLoS One 11: e0148450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CB, Rand H, Bigler J, Kerkof K, Timour M, Bautista E, Krueger JG, Salinger DH, Welcher AA, Martin DA. 2014. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol 192: 3828–3836. [DOI] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA, Basillo C, Wahba A, Lengyel P, Speyer J, Ochoa S, Bass B, Weintraub H, et al. 2012. The ADAR protein family. Genome Biol 13: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon O, Bazak L, Levanon EY, Amariglio N, Unger R, Rechavi G, Eyal E. 2014. Characterizing of functional human coding RNA editing from evolutionary, structural, and dynamic perspectives. Proteins 82: 3117–3131. [DOI] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AM, Li Z, Cattaneo R, Samuel CE. 2009. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem 284: 29350–29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. 2010. Type I interferon: friend or foe? J Exp Med 207: 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi LC, Iyer MK, Stuart PE, Swindell WR, Gudjonsson JE, Tejasvi T, Sarkar MK, Li B, Ding J, Voorhees JJ, et al. 2015. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol 16: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente L, Nishikura K. 2007. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J Biol Chem 282: 16054–16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L, van der Wel LI, Laman JD, Prens EP, Verschuren MC. 2004. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-α sensitivity is unaltered. J Invest Dermatol 122: 51–60. [DOI] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Enstero M, Ohman M. 2009. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 19: 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. 2013. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep 5: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, Thamsen M, Santos-Cortez RL, Lee K, Gambin T, et al. 2015. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 47: 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Richman L, Morehouse C, de los Reyes M, Higgs BW, Boutrin A, White B, Coyle A, Krueger J, Kiener PA, et al. 2008. Type I interferon: potential therapeutic target for psoriasis? PLoS One 3: e2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, Adase C, Sanford JA, Gao N, Chensee M, et al. 2016. Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity 45: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.