

ABSTRACT
Macroautophagy/autophagy influences onset and progression of several human neurodegenerative diseases, because of its critical role as a regulator of neuronal proteostasis and organelle quality control. In many neurodegenerative diseases, impairment in autophagy is thought to play a fundamental part in the terminal phases of cellular degeneration and death. However, the ultimate mechanism of neuronal cell death remains elusive. In a recent study we have identified a new form of regulated cell death, which arises upon autophagy inhibition.
KEYWORDS: Atrophin, autophagy, cell death, EPG5, neurodegeneration, nucleophagy, polyglutamine
A number of alternative and noncanonical autophagy pathways have been identified but their impact during neurodegenerative processes has not been established. In a recent study we show that LMNB1 (lamin B1)-associated nucleophagy and Golgi-mediated degradation play a fundamental role in the terminal phases of neurodegeneration in a mouse model for dentatorubral-pallidoluysian atrophy (DRPLA), a degenerative polyglutamine ataxia, caused by the expansion of a CAG stretch in the ATN1 (atrophin 1) gene.
We demonstrate that, in disease-relevant areas, canonical autophagy is initially stimulated in DRPLA mice; however, this initial upregulation fails to progress towards clearance and is followed by a second phase in which canonical signaling is downregulated, possibly as a result of a feedback loop involving downregulation of the TFEB transcription factor, a master controller of the autophagy-lysosome system. Therefore, at disease symptomatic stage and at endstage, affected neurons in DRPLA mouse models display a complete stall in canonical autophagy pathways.
Our study also provides new in vivo evidence for the insurgence of selective and alternative autophagy pathways. Aggresome-like perinuclear structures, which form only upon autophagy induction, are evident in DRPLA neurons. Nucleophagy-linked LMNB1 increases its association with LC3 and its localization to the cytoplasm from early stages and throughout the terminal stages in affected neurons. The Golgi complex, although initially showing fragmentation in correlation with the initial stimulation of autophagy, displays terminally a tendency towards enlargement and formation of large double-membrane vesicular structures. The activation of these pathways correlates with striking alterations of nuclear shape morphology, with heterochromatin appearing to bud off the nucleus, increase in DNA damage, decrease in DAPI intensity, and dramatic cell atrophy.
Following in vivo analysis, we proceeded to mechanistic analysis in cell culture. In vitro data clarify that any sustained genetic or pharmacological inhibition of autophagy leads to dramatic redistribution of LMNB1 to the cytoplasm, alteration of nuclear shape and cell loss. In addition, the article reports that inhibition of autophagy results in striking excretion of LMNB1 (and SQSTM1/p62) in large microvesicles budding off from challenged cells, leading to loss of cell material into the medium (Figure 1). Most interestingly, microvesicles can be taken up by neighboring cells and can be seen localizing in the perinuclear space. Finally, the study clarifies that loss of Golgi integrity increases the amount of LMNB1 found in the cell cytoplasm, but does not significantly reduce the excretion of microvesicles containing LMNB1 and SQSTM1.
Figure 1.
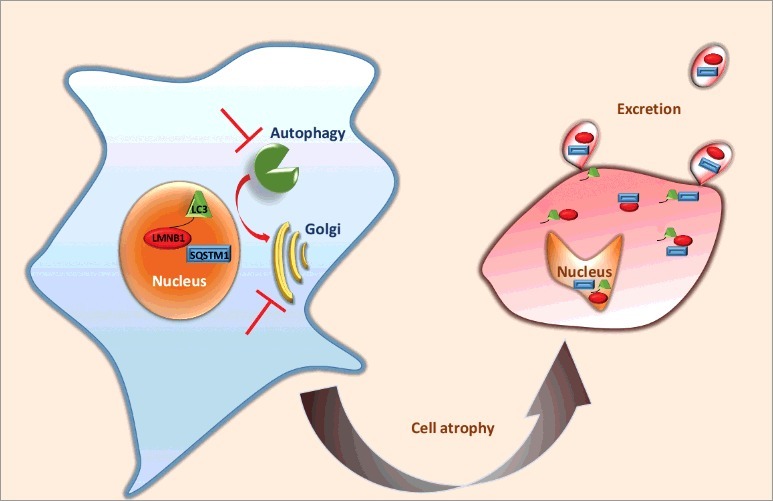
Schematic representation of karyoptosis. Upon blockage of autophagy and Golgi-mediated degradation, LMNB1, LC3 and SQSTM1 are excreted in microvesicles, the cell atrophies, cytoplasm shrinks and the nucleus degenerates, losing a regular shape and accumulating DNA damage.
These findings suggest a model for a newly discovered type of cell death brought about by autophagy inhibition and that is based on nucleophagy and excretion of nuclear components. LMNB1 association with LC3 for nucleophagy and its excretion in moderate quantities appears to happen also in healthy cells. The significance of this process is unknown but it may either underlie normal senescence or, rather, constitute a mechanism for rejuvenating the nuclear matrix and perhaps clearing the nucleus of toxic proteins. However, under conditions in which canonical autophagy is chronically inhibited, LMNB1 is increasingly accumulated in the cytoplasm as a fragment and is excreted, together with other cell components, in the extracellular space. This leads, perhaps by maladaptive excessive excretion of irreplaceable nuclear components, to a falling apart of the nuclear shape, substantial DNA damage and atrophy of the cytoplasm. In our opinion these events define a terminal degeneration event that constitutes a novel type of regulated cell death. We propose to call this process karyoptosis, from the Greek words karyon (nucleus) and apoptosis (falling apart).
Karyoptosis appears to be a combination of the already described nucleophagy and exopher expulsion. It is distinguished from apoptosis for the lack of formation of apoptotic bodies, and from necroptosis by the cell shrinkage, opposite to necroptotic cell swelling. However, karyoptosis’ precise molecular mechanism(s), as well as the pathway(s) and event(s) triggering it remain to be defined. How the Golgi contributes to karyoptosis and LMNB1 degradation also remains to be established.
In a cell nonautonomous perspective, a most interesting question relates to the fate of the excreted microvesicles and their influence on neighboring cells. Do these vesicles signal any information to the surroundings? Are they pro-inflammatory? Because other cells take them up and place them in the perinuclear space, are they forming the aggresome-like structures visualized in vivo?
Thus, our study newly defines a basic biological process and opens up a whole new field of investigation that promises to be of utmost relevance for human health. Because of the great importance of autophagy in long-lived cells (e.g., neurons), and because of the reported impairment of autophagy in many chronic diseases (e.g., as all major neurodegenerative disorders and dementias such as Alzheimer disease, Parkinsonism, Huntington disease and amyotrophic lateral sclerosis/fronto-temporal dementia), we think karyoptosis may represent the so-far vague process underlying cell loss and atrophy in chronic, slowly progressing diseases, in which autophagy is impaired. This is likely not limited to the nervous system. Because autophagy slows down during normal ageing, karyoptosis may be involved in aging processes affecting all organisms. Other long-lived cells such as cardiomyocytes are also heavily dependent on autophagy and may be affected by karyoptosis.
Finally, it appears pivotal to define in the future whether any environmental factor (pollution or other external stressors) known to affect the autophagy-lysosome system may also trigger karyoptosis. If so, this may identify a contributing cause for nongenetic forms of chronic disease, such as sporadic dementia.
Funding Statement
Ataxia UK [grant number A43]; Henry Smith Charity [grant number 20121109].
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.