

Abstract
Cancers are not merely composed of cancer cells alone and, instead, are complex ‘ecosystems’ comprising many different cell types and noncellular factors. The tumour stroma is a critical component of the tumour microenvironment, where it has crucial roles in tumour initiation, progression, and metastasis. Most anticancer therapies target cancer cells specifically, but the tumour stroma can promote resistance of cancer cells to such therapies, eventually resulting in fatal disease. Therefore, novel treatment strategies should combine anticancer and antistroma agents. Herein, we provide an overview of the advances in understanding the complex cancer cell–tumour stroma interactions, and discuss how this knowledge can result in more effective therapeutic strategies, which might ultimately improve patient outcomes.
Introduction
Despite the increasing availability of therapeutic options, cancer remains the second leading cause of death in the USA (>600,000 estimated deaths in 2017)1. However, the rationale for most anticancer therapies is to target malignant cancer cells while largely ignoring the surrounding noncancer cells components of the tumour, or tumour microenvironment (TME). The TME is comprised of all the nonmalignant host cellular and noncellular components of the tumor niche, including, but not limited to, the immune system, blood cells, endothelial cells, fat cells, and the stroma. Over the past decades, the role of the TME in determining disease progression and treatment outcomes has become increasingly evident. Models that describe the effect of the TME on cancer behaviour have been inspired in a number of ecological paradigms, including Paget’s ‘seed and soil’ hypothesis, ecosystems networks, and the optimal foraging theory2–6. These models highlight the complexity of cellular and noncellular interactions within a tumour, many of which support tumour growth and confer resistance to therapies targeting cancer cells. Studies in experimental cancer models have provided ample evidence to support these theories and emphasize the need for therapeutic agents that target the TME.
As a critical component of the TME, the tumour stroma has a profound effect on many hallmarks of cancer7. The stroma is comprised of acellular and noncellular connective tissue that supports functional tissue. Though this paradigm took decades to gain acceptance, the stroma has been demonstrated to have crucial roles in tumorigenesis, cancer progression, metastasis, and therapy resistance. These effects are achieved through the intrinsic properties of the stroma and through additional tumour-promoting properties gained as part of an adaptive response to therapeutic intervention. The combination of cancer cell-autonomous mutations (and other alterations) coupled with changes to the tumour stroma drives tumorigenesis and, ultimately, results in fatal disease. As such, cancer therapeutic strategies that do not take the stroma into account are inadequate. The curative effects of such therapies would be greatly enhanced by combining them with strategies to inhibit the tumour-promoting properties of the stroma. Extensive work has been done to explore the interactions between cancer cells and the stroma, but these advancements remain to be translated into anticancer therapy design. Herein, we address the current state of tumour stroma research and efforts to target the tumour stroma.
Components of the stroma
In any tissue, the main function of stromal factors is to structure and remodel functional tissue. These actions require a variety of macromolecules and cells, each contributing in different ways; understanding the physiological roles of each component is critical to understanding how they affect tumour behaviour. The stroma is composed of specialized connective-tissue cells, including fibroblasts, mesenchymal stromal cells, osteoblasts, and chondrocytes, and the extracellular matrix (ECM) (FIG. 1). Other researchers in the TME field occasionally include other specialized cell types, such as endothelial cells, pericytes, adipocytes, and immune cells, as members of the stromal compartment, but we posit that these cells are more accurately defined as nonstromal cells within the TME; although we define these cells as nonstromal, they substantially influence tumour growth, metastasis, and therapeutic resistance. For example, endothelial cells provide nutrients for tumour growth, constitute routes for metastatic dissemination through angiogenesis, and contribute to resistance to chemotherapies and radiation8–10. Pericytes also contribute to angiogenesis and confer resistance to antiangiogenic therapy11,12. Adipocytes support cancers mainly through the secretion of growth factors and cytokines, and have also been shown to have roles in resistance to chemotherapies, radiotherapy, hormone therapy, and targeted therapies13. Immune cells influence protumorigenic phenotypes (epithelial-to-mesenchymal transition, angiogenesis, and therapy resistance) and antitumour phenotypes (immune surveillance) through diverse and complex mechanisms11,14–16. We recognize the importance of these and other cells in cancer progression and therapy; however, their function is beyond the scope of this Review, in which we focus on the ECM, fibroblasts, mesenchymal stromal cells, osteoblasts, and chondrocytes.
Figure 1. Comparison of nonmalignant stroma and tumour strom.
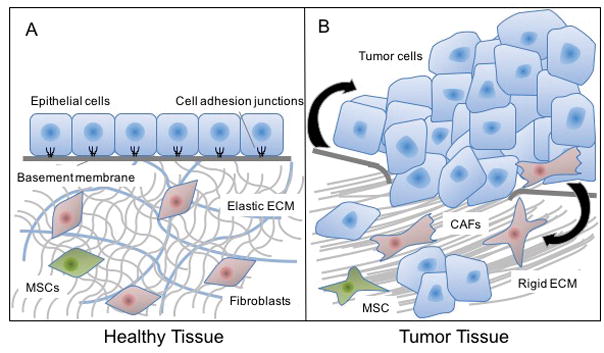
a. Nonmalignant epithelial tissue is supported by a stroma composed of extracellular matrix (ECM), fibroblasts, mesenchymal stromal cells (MSCs), osteoblasts (in bone), and/or chondrocytes (in joints). Cells in the nonmalignant stroma are usually in a quiescent state and maintain homeostasis in the ECM and epithelial compartment, in part by negatively regulating the proliferation, motility, and invasion of cells in the epithelial layer. When cancer develops, the stroma undergoes vast changes to become fibrotic and activated. The ECM becomes denser and more rigid, and is composed of alternative forms of connective fibres, such as tenascin and fibronectin, which cancer cells can invade through. Fibroblasts and MSCs change shape and expression profiles and become more proliferative and secrete higher levels of growth factors, cytokines, and chemokines (black arrows). Stromal fibroblasts in the tumour microenvironment are referred to as cancer-associated fibroblasts (CAFs) or myofibroblasts. The tumour stroma promotes cancer progression and metastasis, and leads to resistance to therapy and disease recurrence.
The extracellular matrix
The ECM provides structure and support for the cellular components in the extracellular space of tissues and organs, and contributes to paracrine cellular signalling17. The ECM is comprised of highly organized interactions of fibrous molecules, proteoglycans, glycoproteins, glycosaminoglycans, and other macromolecules. Approximately 300 different proteins have been catalogued as ECM components18. During embryonic development, the composition of the ECM becomes specialized in each individual organ to suit its unique needs and functions19.
The ECM is slowly and constantly degraded by enzymes, such as collagenases and matrix metalloproteases (MMPs)19,20, and replaced by fibroblast secretions21. This constant remodelling process provides the tissue with a continuous supply of growth factors, which supports tissue homeostasis and wound repair.
The ECM has two main compartments: the basement membrane and the interstitial ECM19. The basement membrane separates epithelial cells from interstitial ECM and other tissues. Epithelial cell attachment to the basement membrane involves integrin binding25. Stromal fibroblasts are usually unable to penetrate the epithelial compartment though the basement membrane, but other cell types (such as immune cells or nerve-cell protrusions) can penetrate this barrier. The interstitial ECM occupies the extracellular space between the epithelium and other tissues. Collagen is the most abundant protein in the interstitial ECM, although many noncollagenous macromolecules, including elastin, fibronectin, tenascin, or laminin, have critical roles within this compartment. All components of the interstitial ECM contain binding domains for other macromolecules and cell-membrane receptors, enabling them to fulfill their structural and signalling roles.
Fibroblasts
Fibroblasts have multiple functions that shape the ECM in which they reside. For example, they build the ECM by secreting collagens and other fibrous macromolecules, but also degrade this network by releasing proteolytic enzymes, such as the MMPs, which enables increased cell mobility throughout the ECM (insert reference from list: Alexander et al 2016). The interactions between fibroblasts and the ECM or surrounding cells, mediated by integrin signalling, affects collagen-fibre alignment and tension (insert reference from list: Alexander et al 2016). Likewise, collagen fibres have an effect on the distribution of fibroblasts throughout the ECM (insert reference from list: Laurent et al. 2007).
Fibroblasts also have an important role in tissue maintenance and homeostasis by expressing enzymes from the cytochrome P450 (CYP) family, which degrade foreign and potentially toxic molecules. These cells also recruit and regulate leukocyte infiltration and inflammation via secretion of cytokines, chemokines, and growth factors26,27. Apart from these canonical functions, fibroblasts might have unique functions depending on the tissue in which they reside28–30.
Fibroblasts exert many of their functions in a nonproliferative state, in which they are able to migrate and secrete ECM components and signalling protein. Tissue wounds and certain pathological conditions can activate fibroblasts in such a way that they proliferate and secrete higher levels of proteins in an aberrant way. As such, fibroblasts act as sentinel cells in these conditions, in which the accumulation of fibroblasts and stroma can be one of the first symptoms. Myofibroblasts are further differentiated fibroblasts that are present in the reactive stroma during wound healing or in other processes34–36. Reactive stroma refers to the process of stromal cell infiltration into a wound site or tumour and the resulting deposition of proteins like collagens, fibrin, and fibronectin (insert reference from list: Dvorak 1986). In cancer, and owing to the presence of an activated stroma, fibroblasts typically resemble myofibroblasts37,38.
Mesenchymal stromal cells
The definition of the characteristics of mesenchymal stromal cells have evolved over time, and their characterization remains an active area of research39. This cell population was first isolated from bone marrow and, owing to being distinct from the haematopoietic lineage, referred to as colony-forming unit fibroblasts40–43. Further preclinical studies showed that these bone marrow stromal cells could differentiate into osteoblasts, chondrocytes, adipocytes, or myocytes depending on the culture conditions44–46. Currently, multipotent stromal cells are commonly referred to as mesenchymal stem cells (MSCs), although this moniker is often replaced with the broader term mesenchymal stromal cells (also abbreviated MSCs) because of the uncertainties regarding their stem-cell nature47,48 — the defining characteristics of MSCs are not consistent in the literature. A further source of uncertainty comes from the fact that fibroblasts and MSCs express similar cell-surface markers and have a similar phenotype48,49. Moreover, both fibroblasts and MSCs have been shown to have multipotent and immunomodulatory functions50–52.
To avoid confusion, in this Review we define MSCs as cells that are able to adhere to plastic surfaces, have the capacity to differentiate into osteoblasts, chondrocytes or adipocytes in culture, and express the cell surface markers CD73, CD90, and CD105, but not any leukocyte markers53,54. These criteria distinguish MSCs from fibroblasts, although some similarities between both cell types remain49,54. While both MSCs and fibroblasts occupy the stroma of many tissues, MSCs are capable of migrating through the body via blood vessels, whereas less evidence exists that fibroblasts migrate via circulation55–57. Relative to fibroblasts and other stromal cell types discussed herein, MSCs are rare58. Their primary functions are to regulate the immune response and to promote tissue regeneration44,59–62. They are also the source of osteoblasts and chondrocytes in bones and joints, respectively.
Osteoblasts
Osteoblasts are unique stromal cells responsible for building bone, a highly specialized ECM63 consisting of a mixture of collagen and noncollagenous proteins that is subsequently calcified with hydroxyapatite64,65. Osteoblasts become embedded within this specialized matrix, eventually becoming osteocytes, which form a network of cells within bone66. During bone formation, osteoblasts secrete soluble molecules that activate osteoclasts (bone-resorbing cells). Bone formation then takes place in pockets of osteoclast-mediated resorption67. Thus, bone is continually formed and resorbed in a tightly controlled process68.
Chondrocytes
Chondrocytes, also differentiated from MSCs, are a major stromal component. Chondrocytes produce cartilage, the specialized ECM present in joints and other cartilaginous tissues69. Cartilage also provides a structure for bone formation during embryonic development70. Chondrocyte are thought to have a lower proliferation rate than other stromal cell types71.
Stroma–cancer interactions
Tumours are composed of cancer cells and surrounding stromal cells (FIG. 1). Tumorigenesis, cancer progression and metastasis are strongly dictated by cell-autonomous genetic and epigenetic changes, but tumour–stroma interactions have also been demonstrated to be critical in these processes7,72. Paget’s description of the ability of cancer cells (‘seeds’) to grow only in certain secondary sites depending on their microenvironment (‘soil’)85 remains accurate. Herein we will focus on carcinomas, which arise from the epithelium and represent 90% of all cancers, as well as on haematological malignancies, in which most of the research into the roles of the tumour stroma on cancer therapy has been conducted, rather than on sarcomas, which are cancers that originate directly from stromal cells.
Stating that the stroma is always tumour-promoting would be inexact; indeed, the stroma is tumour-suppressing in some situations. Tumours behave as an ecosystem within which stromal cells in the TME dynamically interact with cancer cells, and can either compete or cooperate with them — resulting in suppression or promotion of further tumour progression. These interactions have been shown to alter the genotype and phenotype of cancer cells86. Herein, we will discuss the main advances in this area of intense investigation, mostly concerning fibroblasts.
Tumour-suppressing actions of the stroma
Stromal cells usually exist in a nonproliferative state, and therefore have historically been referred to as ‘bystanders’ during cancer development87. In most nonmalignant tissues, the stroma is tumour-suppressing by nature. Indeed, the stroma has crucial roles in tightly regulating the ability of differentiated epithelial cells to proliferate, move, and invade the ECM, in order to maintain organ size and structure88,89. Because of this regulatory action, the nonmalignant stroma also suppresses tumorigenic hallmarks, such as cell proliferation and invasion. The pancreatic stroma, in particular, is recognized as having tumour suppressive effects in the premalignant context90.
One of the ways stromal cells regulate epithelial cell function is through the secretion of growth factors. TGFβ, commonly secreted by fibroblasts, can either suppress or promote tumorigenesis depending on the context (insert references from list: Bhowmick et al 2004; Keight et al 2012; and Calon et al 2012). In a mouse model, inactivation of one of the TGFβ receptors in fibroblasts resulted in intraepithelial neoplasia of the prostate, a lesion that can precede cancer formation, and in invasive squamous cell carcinoma of the stomach92.
During tumour development, an inactive stroma can also have suppressive properties. In certain cancer types (such as pancreatic ductal adenocarcinoma, or PDAC), stromal cells can prevent existing tumours from progressing to advanced-disease stages. In mice genetically engineered to develop PDAC via conditional deletion of p53 and Sonic hedgehog (Shh) and simultaneous expression of oncogenic Kras, it was observed that more aggressive tumours, measured by tumour size, metastasis, and survival, had less stromal content, measured by alpha-smooth muscle actin (ACTA2) expression93. Depletion of cancer-associated fibroblasts (CAFs) in PDAC has also been shown to cause immunosuppression90.
The expression of proteins from the metalloproteinase inhibitor (TIMP) family in fibroblasts controls the structural organization of the ECM and stromal cells94. These proteins are endogenous negative regulators of MMP activity; many cancers have aberrant expression of TIMPs and/or MMPs95. Loss or reduction of expression TIMPs causes an increase in MMP function and enables activation of the stroma and subsequent tumour progression96,97. Overexpression of TIMPs, on the other hand, reduces tumorigenesis, growth, angiogenesis, and metastasis in cancer models, such as those of the pancreas98.
In metastatic disease, tissue-specific stromal phenotypes might suppress metastasis. As Paget described85, cancer cells only grow in secondary organs with which they are compatible, implying that certain TMEs do not support the outgrowth of disseminated tumour cells while others are permissive99,100. In general, the stroma can be considered tumour-suppressive in the early stages of the natural history of cancer or metastasis. When tumours within an epithelium or secondary cancer site get an advanced-stage phenotype, forming an acidic and hypoxic TME2, the signalling context leads to activation of the stromal cells.
Tumour-promoting actions of the stroma
A publication from 1986 described the physiological similarity between the stroma of a wound and that of a tumour101. This activated stroma is called desmoplasia or fibrosis, and is frequently observed by pathologists in sections of tumour tissue. In advanced-stage tumours, the activated stroma promotes the further acquisition of genetic and epigenetic changes in cancer cells102–104, and supports cancer progression84. Other researchers propose that cell-autonomous changes in the stroma can induce tumorigenesis, posing a ‘chicken or egg’ quandary, although most of the evidence points to cancer cells activating the stroma, and not the other way around. Herein we present specific examples of the tumour-promoting effects of the stroma with the caveat that it is impossible to discuss every facet of this complex field.
CAFs are perhaps the best studied cell type in the TME105. Unlike fibroblasts in nonmalignant tissue, CAFs are not quiescent, inert supportive cells in the ECM but, instead, are proliferative, migratory, and highly secretory cells87,106. Because they have an altered shape (multispindled rather than single-spindled105) and express ACTA2 and prolyl endopeptidase (FAP), CAFs are often referred to as myofibroblasts106. MSCs are also sometimes included in the CAF population49. CAFs secrete ECM factors that are different to those secreted by nontransformed fibroblasts, including tenascin, periostin, SPARC, and collagens105,107–113. The tumour ECM has an altered organization, is more rigid and contractile than that in the nonmalignant stroma and, among other effects, can downregulate the expression of the tumour suppressor PTEN in cancer cells114. This altered ECM promotes the growth, survival, and migration of cancer cells, and drives angiogenesis115–119. Kaukonen et al. showed that breast cancer cells cultured with ECM derived from nonactivated fibroblasts proliferated significantly less than those cultured with an ECM derived from CAFs120. Similarly, in comparison with nontransformed fibroblasts, CAFs were able to promote their proliferation of co–cultured SV40 T-immortalized prostate epithelial cells in vitro and to induce tumour growth in vivo when transplanted together in the renal capsule of mice121.
The expression of MMPs and other enzymes that degrade and metabolize the ECM is increased in CAFs, enabling cell penetration through the ECM122–124. In comparison with non-activated fibroblasts, CAFs also secrete higher amounts of growth factors, cytokines, and chemokines, which promote cancer cell-intrinsic hallmarks of cancer in autocrine and paracrine fashions. Tumour-promoting factors secreted by CAFs include, among others, TGFβ, FGFs, HGF, PDGF, VEGF, transcription factor p65 (commonly known as NF-κB), TNFα, IFNγ, SDF-1α, IL-6, CTGF, EGF, growth arrest-specific protein 6 (GAS6), galectin-1, secreted frizzled-related protein 1, sonic hedgehog (SHH), and BMPs92,116,125–146. For example, when media from TGFβ2 receptor-null fibroblasts was added to breast cancer cells in culture, the cancer cells migrated and scattered147. This migratory phenotype was largely induced by secretion of HGF by fibroblasts, which signalled through hepatocyte growth factor receptor (MET) and macrophage-stimulating protein receptor (Ron) on breast cancer cells. These findings indicate a complex signalling regulatory mechanism whereby inhibition of one signalling pathway in the cancer cells lead to secretion of a different growth factor by fibroblasts and subsequent activation of the cognate receptor on the cancer cells. A breast cancer xenograft study showed that the invasive tumour stroma contains chemokines differentially throughout tumour progression. Early in tumour development, cancer cells were positive for SDF1 (also known as CXCL12) and its receptor CXC-chemokine receptor 4 (CXCR4), while the stroma was negative for these markers. When the tumour became invasive, however, the stromal cells expressed SDF1 and CXCR4, suggesting that cancer–stroma interaction became more permissive to invasion148. A systems biology approach to determining the tumorigenic properties of secreted fibroblast factors has been undertaken in a breast cancer setting, in which three out of five selected secreted factors promoted tumorigenicity, while the remaining two had little impact on tumour growth149. Moving forwards, this type of systemic analysis will be important to elucidate targetable stromal factors, as well as to determine which combinations of factors has synergistic effect on tumour growth.
With increasing human age, CAFs can convert into a senescent state. Senescent fibroblasts acquire alternative phenotypes that promote cancer progression, a phenomenon known as senescence-associated secretory phenotype (SASP), which is characterized by the secretion of molecular factors such as cytokines, chemokines, and metabolites that drive malignant transformation150,151. For example, senescent osteoblasts drive increased osteoclastogenesis and tumour cell seeding in the bone by secreting high levels of IL-6; thus, senescence-induced changes of the bone stroma can change the ability of tumour cells to seed and/or grow in this milieu152. Osteopontin, secretion of which undetectable in proliferating fibroblasts, is upregulated in senescent fibroblasts and necessary and sufficient to drive tumorigenesis in preneoplastic cells in in vitro and murine models153.
Similarly to circulating tumour cells (CTCs), CAFs seem to circulate in the blood154. Circulating CAFs (based on FAP and ACTA2 expression) were found in 88% of patients with metastatic breast cancer and in 23% of patients with nonmetastatic disease154. Similarly, circulating fibroblast-like cells (positive for vimentin expression and negative for cytokeratin expression) were found in 58% of patients with metastatic prostate cancer, but were absent in patients with nonmetastatic disease155. These vimentin-positive/cytokeratin-negative cells might be cancer cells that have undergone epithelial-to-mesenchymal transition, although the absence of cytokeratin expression suggests that they are likely to be fibroblast-like cells (CTCs are typically defined as cytokeratin-positive). Interestingly, stromal annexin A2 expression was higher in the tumours of patients who had a majority of CTCs with a mesenchymal phenotype than in those with a majority of epithelial-like CTCs156. These data suggest that circulating CAFs might have a role in the metastatic process, as they are found in patients with metastatic disease at a higher rate than those with localized disease. Some have suggested that CAFs play a role in preparing a premetastatic niche for cancer cells, although this has yet to be firmly established157.
The bone marrow is an important source of stromal cells that can promote primary tumour growth and progression and, ultimately, metastasis to bone. The results of several studies138, 158–160 have shown that bone marrow-derived stromal cells circulate to primary tumour sites where they promote tumorigenic activities and properties. Other researchers argue that the tumour stroma is primarily derived from precursor cells present within the local primary tumour, while bone marrow-derived circulating precursor cells are rare161. In a metastatic setting, MSCs and osteoblasts attract tumour cells to the bone marrow by secreting a local gradient of chemoattractants, such as SDF1 or GAS6143,162–164. Once in the bone marrow, breast cancer cells can sequester SDF1 and affect the response of haematopoietic cells to this signalling factor in in vitro co-culture assays165. Similarly, prostate cancer cells compete with haematopoietic stem cells (HSCs) for space and nutrients within the osteoblastic niche of the bone marrow164,166. Cancer cells of bone-invasive tumours also secrete ligands, such as SHH, which up-regulate RANKL expression in osteoblasts, leading to osteoclast activation and bone resorption167,168.
Tumour-induced bone resorption leads to expression and release of factors such as TGFβ, which can induce tumour growth and proliferation and further bone resorption in mouse models169. This signalling interaction is an example of the so-called ‘vicious cycle’ whereby tumour cells, osteoblasts, and osteoclasts secrete stimulatory factors that promote the activation of each cell type, typically leading to net osteolysis and tumour growth and invasion170,171. Osteoblasts themselves might be able to initiate this cycle172. In osteoclasts, expression of RUNX2 has been linked in experimental models and patient samples to the upregulation of factors that promote cancer metastasis to the bone, such as the BMPs173. Tumour-induced pressure causes increased osteocyte secretions (for example, of CCL5 and MMPs), which promote the growth of prostate cancer-derived bone metastasis in mouse models174.
Some of the earliest studies of the profound effect of the stroma on the epithelium were conducted by Cunha and collaborators in the context of prostate development and tumorigenesis175–179. These investigators first found that, depending on the type of urogenital mesenchyme used in in vivo epithelial-mesenchymal tissue recombination experiments, epithelial cells would differentiate and grow in vastly dissimilar ways176–179. For example, they showed that when the testicular feminization syndrome (Tfm) mesenchyme was recombined with wild-type mouse epithelium and grown in the mouse renal capsule, female sex organs developed; alternatively, when wild-type mesenchyme was recombined with Tfm epithelium, male sex organs developed 175. These results indicate that the stroma can drive the phenotypic determination of the epithelium, even when the latter is genetically predisposed to display a different phenotype. Similarly, when fully differentiated bladder cells were combined with a neonatal rat seminal vesicle mesenchyme and placed into mice, it resulted in prostate glandular and acinar differentiation180. Similarly, nonmalignant prostate cells can be transformed into permanently malignant cancer cells by mixing them with CAFs in both in vitro and in vivo experiments121,181. Moreover, the irradiated mammary gland stroma promoted tumour growth of non-irradiated epithelial cells182.
The stroma has also been shown to promote the acquisition of stem-like properties by cancer cells, providing a fertile niche in which primary cancer cells can grow, and metastatic cancer cells can colonize183,184. The early stages of colon cancer are thought to be driven by the stem cell niche: stromal fibroblasts secrete Wnt ligands that promote β-catenin-dependent signalling in colon epithelial cells, driving a cancer stem cell-like phenotype185. In this setting, colon cancer cells located at longer distances from the stroma had lower β-catenin activity and a more differentiated phenotype than those in close vicinity of the stroma185. In a metastatic setting, the lung stroma has also been shown to provide a fertile niche for colonization by cancer cells186.
In addition to their reactive nature, and similarly to cancer cells, stromal cells can undergo genetic changes187. Mutations in the tumour suppressor genes TP53 and PTEN have been found in the stromal compartment of human cancers (separated from epithelial cells using laser microdissection)188,189. Depletion of APC in the stromal compartment can lead to endometrial cancer tumorigenesis in a mouse model190. An activating β-catenin mutation in osteoblasts precedes acute myelogenous leukaemia (AML) in approximately 38% of patients191. These data indicate that mutations might occur in stromal cells before they affect the cancer cells, subsequently driving tumorigenesis. Further efforts to determine the molecular underpinnings on the conversion of non-activated fibroblasts to CAFs in both patient-derived tumour samples and mouse models have revealed several signalling mechanisms, including activation of the Hippo pathway (reference is Calvo 2013), loss of p53 (reference is Procopio 2015), or activation of heat shock factor 1 (HSF1) (reference is Scherz-Shouval 2014), that enhance tumour-promoting properties of CAFs in several tumour types (Tyekucheva 2017 is a general reference to the molecular undrpinnings of stromal-epitehlial interactions)192–195.
Epigenetic mechanisms can also lead to phenotypic changes in CAFs; the differential gene expression observed in CAFs relative to non-activated fibroblasts196–199 is largely the result of changes in DNA methylation and other epigenetic alterations187,200–204. A specific example has been shown in the conversion of non-activated lung fibroblasts into CAFs via p300-histone acetyltransferase-mediated acetylation of STAT3, which causes an invasive phenotype in CAFs after exposure to the cytokine leukaemia inhibitory factor (LIF), derived from either tumour cells or other stromal cells204,205. Global hypomethylation, commonly observed in epithelial cells in carcinoma, has also been observed in CAFs from gastric cancer206.
Many stromal cell types have been clearly shown to have a role in supporting, and potentially initiating, tumour growth, with most of the evidence coming from studies of CAFs, but also of MSCs and osteoblasts. Of note, little to no work has been done on the roles of chondrocytes in tumorigenesis158.
Effects of tumour stroma on therapy
After receiving any modality of anticancer therapy, cancers tend to recur, even in patients who had a favourable response. The degree to which a patient with cancer will respond to a therapy depends strongly on the extent to which the stroma has become activated207. For example, the presence of myofibroblasts can enable prediction of biochemical recurrence in patients with prostate cancer208, and stromal expression of FAPα is prognostic of resistance to chemotherapy and recurrence in patients with ovarian cancer209. Herein, we discuss examples of how specific therapies are hampered by the tumour stroma, with the caveat that this area of study needs additional exploration.
Limiting drug access
The tumour stroma can limit access of therapeutic agents to their target tissues in three ways: fibrosis, high interstitial pressure, and degradation of drugs by stromal enzymes (FIG. 2a). The buildup of a rigid ECM (fibrosis) around and throughout a tumour creates a physical barrier that reduces diffusion of therapeutic agents to cancer cells. A dense ECM can reduce blood vessel density and lead to vessels embedding within the matrix, creating a tough barrier that drugs cannot perfuse, as has been observed in PDAC211. In addition, cancer cells can strongly adhere to ECM proteins in order to evade chemotherapy in a process known as cell adhesion-mediated drug resistance (CAM-DR)212,213. CAM-DR operates chiefly through direct binding of cancer cells to fibronectin, which is associated with changes in other signalling proteins214–217. Interstitial pressure is higher in the TME than in nonmalignant tissue 218–221. This difference can affect drug diffusion and delivery, as demonstrated in PDAC222,223, melanoma224 or glioma225.
Figure 2. Tumour stroma-mediated chemoresistance.
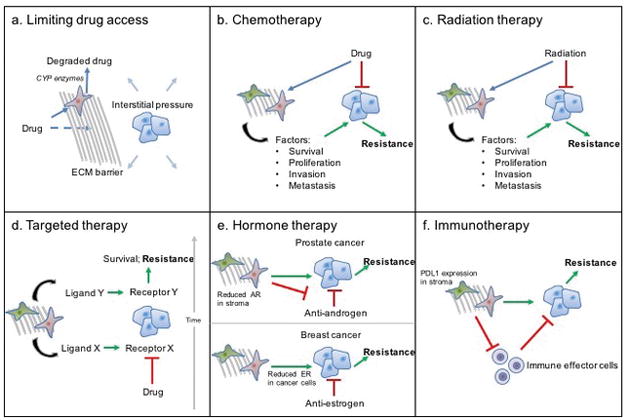
In response to anticancer therapy, the tumour stroma mediates resistance to therapy and disease recurrence. a | Dense fibrosis causes limited access of cancer cells to therapeutic agents in three ways: creating an extracellular matrix (ECM) barrier that such agents cannot diffuse through; promoting stromal cytochrome P450 (CYP)-mediated degradation of drugs; and increasing interstitial pressure that prevents therapeutic agents from entering the tumour. b | In response to chemotherapy or c | radiation therapy, cancer-associated fibroblasts (CAFs) and mesenchymal stromal cells (MSCs) secrete different growth factors, cytokines, and chemokines that promote cancer-cell survival, proliferation, invasion, and metastasis, leading to resistance. d | Targeted inhibition of a specific pathway (ligand–receptor X) results in the stromal secretion of new ligands (ligand–receptor Y), resulting in survival and resistance. e | In prostate cancer, decreased androgen receptor (AR) expression in the stroma leads to resistance to androgen-deprivation therapies. In breast cancer; the stroma promotes decreased oestrogen receptor (ER) expression in cancer cells, leading to resistance to antihormonal therapies. f | CAFs, MSCs, and ECM suppress effector immune cell activation and tumour infiltration.
Fibroblasts, particularly in the bone marrow stroma, express CYPs that metabolize a variety of potentially toxic molecules, including chemotherapeutic drugs such as docetaxel226. CYP3A4 is expressed by stromal cells in the bone marrow niche, commonly seen as a chemoprotective microenvironment. Upon docetaxel treatment, tumours co-cultured with primary human bone marrow fibroblasts negative for CYP3A4 expression (via shRNA knockdown) reached smaller sizes than those co-cultured with fibroblasts from the same source expressing CYP3A4227. In a mouse model of multiple myeloma, bone marrow fibroblast expression of CYP26 resulted in a microenvironment with low expression of retinoic acid, which promoted a bortezomib-resistant phenotype228.
Resistance to cytotoxic chemotherapy
Classic cytotoxic chemotherapy targets rapidly proliferating cells. Stromal secretion of ligands promoting proliferation is one of the reasons why cancer cells proliferate rapidly. Chemotherapy cannot eliminate 100% of cancer cells partially due to intrinsic resistant phenotypes within the cancer cell population. Moreover, the tumour stroma can promote cancer cell survival and proliferation in a treatment-naïve setting and in response to treatment, which does not eliminate nonproliferating stromal cells105; this intrinsic resistance might be at least partially the result of induction of autophagy rather than apoptosis in response to external agents229 and enables stromal cells to divert their response to a nonlethal stress response.
Chemotherapy-induced DNA damage in the TME causes a stress response in stromal cells, which then secrete many factors that promote cancer cell survival, proliferation, invasion, and metastasis (FIG. 2b). For example, in a prostate cancer model, DNA damage induced by docetaxel or mitoxantrone caused increase expression of GDNF in stromal cells, which promoted tumour cell proliferation, invasion, and chemotherapy resistance in a paracrine fashion230. Docetaxel and mitoxantrone also caused increased stromal secretion of WNT16B, which promotes prostate cancer cell survival in the presence of chemotherapeutic agents231. Similarly, in a lymphoma model, doxorubicin led stromal cells to secrete IL-6 and TIMP-1, both of which supported cancer cell survival in the thymus232. In other in vitro studies, the addition of conditioned media from an immortalized fibroblast cell line to head and neck squamous cell carcinoma (HNSCC)-derived cells increased resistance to cisplatin two-fold relative to cells not treated with conditioned medium233. Along the same lines, chemotherapy can cause non-activated fibroblasts to develop a CAF-like phenotype, secreting factors that promote a stem-cell-like phenotype in breast cancer cells234. In a colon cancer model, a similar secretory phenotype was observed in fibroblasts treated with 5-fluorouracil or oxaliplatin, which induced a stem-cell-like, chemotherapy-resistant phenotype in cancer cells via a mechanism involving exosomes235. Finally, the release of cysteine and glutathione from primary CAFs isolated from ovarian cancer patients was described to lead to ovarian cancer cell resistance to platinum-based therapy by preventing accumulation of platinum in the cell nuclei236.
Resistance to radiation therapy
Approximately 50% of patients with cancer receive radiation therapy237, which, like chemotherapy, leads to DNA damage. Similarly to tissue wounding or injury, radiation therapy also results in fibrosis. This response leads to survival and expansion of the number of stromal cells and ECM, which in turn promotes survival and radiation resistance of cancer cells, in addition to providing signals that stimulate their proliferation and invasion (FIG. 2c). Integrin expression has been shown to be consistently upregulated in fibroblasts after exposure to ionizing radiation in in vitro, in vivo, and ex vivo (from cancer patients) studies238–243. For example, pancreatic stellate cells (a specialized stromal cell type) upregulate β1 integrin in response to radiation exposure in a FAK-dependent manner, protecting PDAC cells from radiation241. β1 integrin expression, along with AKT signaling, is also associated with protection of breast cancer cells from ionizing radiation242. Finally, integrin αvβ6 expression increases in lung cancer cells following radiation exposure and precedes the secretion of TGFβ and the development of lung fibrosis243.
Irradiated fibroblasts also have an increased ability to secrete factors that induce chemoresistance244. In in vitro models, squamous cancer cell lines co-cultured with irradiated fibroblast cell lines were more proliferative than those co–cultured with non-irradiated fibroblasts 245. Irradiated fibroblasts also increased the activation of proliferation signalling mechanisms (RAS and the mitogen-activated protein kinase cascade), invasion pathways (MMPs, laminin 5, and filamin A), and TGFβ signalling in cancer cells245. In a similar model of PDAC, irradiated fibroblasts increased HGF/MET signalling in neighbouring cancer cells, which enhanced their mobility 246.
Resistance to targeted therapies
Therapies based on agents that inhibit specific molecules or pathways are associated with two major issues: cancers are heterogeneous and thus, only certain cancer cells might be addicted to a particular oncogene; and targeting one protein or pathway typically results in the upregulation of a another pathway, resulting in adaptation, resistance, and recurrence. The stroma assists cancer cells in this adaptation (FIG. 2d).
Bevacizumab is an antibody that targets VEGFA. In a mouse model of lung cancer, treatment with bevacizumab led to acquired resistance, at least partially via upregulation of VEGFA, FGF2, FGFR2, and PDGFRA in stromal cells248. In addition, in patients with lung adenocarcinoma, the number of FSP-1-positive fibroblasts in tumours was higher in those who received bevacizumab than in those who did not248. Treatment of mice with subcutaneous syngeneic myeloma tumours with anti-VEGF antibody resulted in tumours with therapeutic resistance with CAFs present in the resistant tumours reactivating angiogenesis via PDGFC signalling133.
Receptor tyrosine kinases (RTKs) are common targets of inhibitory therapies because they are frequently upregulated in cancer cells and promote several hallmarks of cancer. In lung cancer, EGFR is one such receptor, and many patients respond to EGFR inhibitors. However, as is the case of most tyrosine kinase inhibitors (TKIs) of RTKs, resistance develops. In a study of co-cultured lung cancer cells, resistance to EGFR-TKIs was driven by CAFs expressing podoplanin, although the molecular mechanism underlying resistance were unclear250. In a breast cancer model, resistance to TKIs was the result of HGF secretion by fibroblasts251. HGF also caused resistance to BRAF TKIs in melanoma, colorectal, and glioblastoma-derived cell lines, indicating consistency across cancer types252. A seminal study conducted with a melanoma mouse model showed that BRAF-driven tumours develop rapid resistance to anti-BRAF therapies primarily owing to the paradoxical activation of CAFs by such therapies. CAF activation, in turn, signalling downstream of BRAF in resistant cancer cells and promoted their survival253. Interestingly, a therapeutic response was observed when CAFs were targeted with an inhibitor of FAK, indicating that cancer cell resistance to targeted therapies can be overcome using fibroblast-targeting strategies.
Resistance to hormone antagonists
Therapies that target hormone signalling are used to treat patients with prostate or breast cancers, which are usually dependent on androgens or oestrogen, respectively. In prostate epithelium, androgen receptor (AR) activity promotes proliferation254, but in the stroma, AR expression and activation by androgen binding inhibits prostate epithelial proliferation, whereas loss of AR in the stroma correlates with prostate cancer progression, although it is unknown how this observed phenomenon is manifested (please insert reference: Singh et al. 2014). This phenomenon suggests that bipolar androgen therapy (introduction of supraphysiological levels of androgen after androgen deprivation therapy) could be an effective therapeutic strategy255. Notably, stromal AR expression decreases with prostate cancer progression, and is associated with increased epithelial cell proliferation and poor outcomes in patients (FIG. 2e)256,257.
Co-culturing with CAFs has been shown to reduce oestrogen receptor (ER) expression in breast cancer cells, which precedes a hormone-independent state and, therefore, resistance to hormone antagonists (FIG. 2e)258. Analysis of conditioned media from co-cultured cancer cells showed a >5-fold upregulation of 46 proteins (including MMPs and TGFβ) compared with media from non co-cultured breast cancer cells258. Other studies have shown that the loss of caveolin 1 expression in breast cancer stromal tissue enables prediction of a lack of response to tamoxifen, an ER antagonist259,260. Most mechanisms driving resistance to hormone therapy are thought to be cancer cell-autonomous, but these data indicate that, in prostate or breast tumours, the stroma could have important, clinically relevant roles; further studies need to be conducted.
Resistance to immunotherapy
The goal of cancer immunotherapy is to induce a TH1 immune response, primarily driven by cytotoxic T cells and macrophages (referred to herein as effector immune cells), to eliminate tumour cells that present mutation-associated neoantigens261; an effective response to immunotherapy relies on effector immune cells. Tumour-associated ECM and fibroblasts have immune modulatory effects 262,263 (FIG. 2f). One example is the fact that ECM-sequestered tenascin can prevent the infiltration of effector immune cells, which has been evidenced in mice null for tenascin expression, which have higher effector immune cell infiltration in tumours than wild-type mice264. The organization of the molecular components of the tumour ECM also has a key role in regulating the localization and migration of effector immune cells throughout the tumour stroma (insert reference from list: Gajewski et al. 2013). For instance, activated T cells were observed in regions of loose fibronectin and collagen, but were not abundant in denser regions of tissue sections from human non-small cell lung cancer tumours265. In addition, T and B cell abundance was directly correlated with vessel density in tumour stroma in tissue sections from cancer patients with solid tumours266.
Inoculation of human tumours into mice has long been known to be more effective in the presence of human stromal factors, an interaction that was thought to be caused by the immunosuppressive functions of the tumour stroma267. This effect was confirmed in a lung cancer mouse model, in which FAP-positive fibroblasts suppressed effector immune cell infiltration into tumours268. Stromal cells express the immune-checkpoint protein programmed cell death 1 ligand 1 (PD-L1). The effect of stromal PDL1 expression on immunotherapy outcomes is currently unclear, with some studies showing an association with a better prognosis for patients with breast cancer269, but also a poor prognosis for patients with adult T-cell leukaemia270.
Tumour-stroma targeting strategies
Cancer therapies should include strategies to target and constrain the tumour stroma, which can both suppress and promote tumorigenesis. In situations in which the tumour stroma promotes cancer hallmarks and induces resistance to anticancer therapy, the use of therapies targeting the stroma could have curative outcomes. However, such a strategy would be counter-productive in a context in which the stroma is tumour-suppressive and thus, caution must be taken90. Reliable biomarkers of stromal activity and tumour-promoting properties will guide these critical therapeutic decisions.
When considering therapeutic strategies to eliminate cancer, stroma-targeting agents must be considered (TABLE 1), although targeting the stroma alone will likely not eliminate the entire tumour. Cancer cells would retain their genetic and epigenetic alterations and, therefore, would likely revert the phenotype of a reactive stroma that might have been rendered completely inert. Agents targeting the tumour stroma should therefore be administered as combination therapies with cytotoxic agents (FIG. 3).
Table 1.
Targeting tumor stroma for cancer therapy
| Example stromal targets (Preclinical study references) | Example agents involved in cancer clinical trials (ClinicalTrials.gov identifier for representative clinical trial) |
|---|---|
| CXC-chemokine receptor 4 (Shiozawa, Y. et al. 2011; Domanska, U. M. et al. 2014; Domanska, U. M. et al. 2012) | Plerixafor (NCT01610999) |
| Cytochrome P450 3A4 (Alonso, S. et al. 2015) | Clarithromycin (NCT03043989) Itraconazole (NCT02157883) |
| FAK (Mantoni, T. S. et al. 2011) | Defactinib (NCT03287271) |
| Prolyl endopeptidase FAP (Hofheinz, R. D. et al. 2003; Scott, A. M. et al. 2003; Mersmann, M. et al. 2001; Welt, S. et al. 1994) | Sibrotuzumab (NCT02198274) RO6874813 (NCT02558140) |
| FGF pathway (Mitsuhashi, A. et al. 2015; Bai, A. et al. 2010; Chae, Y. K. et al. 2017; Katoh, M. et al. 2014; Bello, E. et al. 2011; Gozgit, J. M. et al. 2012) | Dovitinib (NCT01548924) AZD4547 (NCT01791985) |
| Hyaluronic acid (Provenzano, P. P. et al. 2012; Kultti, A. et al. 2009; Hajime, M. et al. 2007) | PEGPH20 (NCT01453153) |
| TGFβ pathway (Biswas, S. et al. 2007) | Galunisertib (NCT02304419) Fresolimumab (NCT02581787) |
Figure 3. Targeting tumour stromal cells in addition to cancer cells.
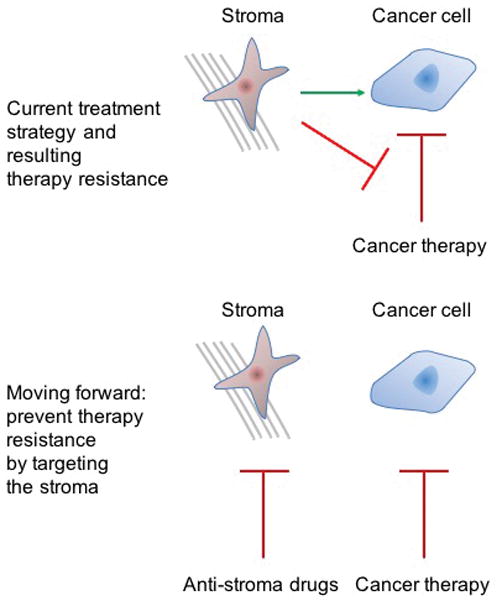
a | Currently, most antitumour therapies target and eliminate cancer cells, and are not design to directly affect the tumour stroma. b | However, tumour recurrence can result from the interactions of the tumour stroma with both cancer cells and anticancer therapies. Through its interaction with cancer cells, the stroma promotes the hallmarks of cancer and can induce a therapy-resistant phenotype. Through its direct interaction with anticancer therapy, the stroma can prevent the action of such therapies on cancer cells. (FIG. 2). c | We posit that, in addition to targeting cancer cells, anticancer therapeutic strategies should include methods to target and constrain the stroma, or to revert it to a tumour-suppressive state.
Targeting the ECM
Several strategies exist to prevent the ECM from acting as a barrier by reducing the density of its components and enable diffusion of therapeutic agents. Halofuginone inhibits the synthesis of type I collagen, which has been shown to reduce desmoplasia271–273. In a melanoma xenograft model, halofuginone reduced osteolysis and bone metastasis273. Likewise, PEGylated hyaluronidase and 4-methylumbelliferone inhibited the secretion of hyaluronic acid in the stroma, thereby decreasing interstitial pressure and enabling the uptake of therapeutic agents223,274–276. Lysyl oxidase (LOX) catalyzes collagen crosslinking, and has high levels of activity in the tumour stroma277,278. Agents targeting LOX activity could decrease collagen crosslinking, thereby reducing the density of the tumour stroma and increase the effectiveness of anticancer therapies279–282. The vitamin D receptor ligand calcipotriol 283 and all-trans retinoic acid (ATRA)284 are other agents that reduced fibrosis in experiments with PDAC models.
Some therapeutic strategies are focused on penetrating the ECM rather than degrading it. Nanoparticle albumin-bound paclitaxel (nab-paclitaxel) is a taxane conjugated to albumin, which increases the solubility of paclitaxel in the tumour stroma285. When combined with gemcitabine, nab-paclitaxel has substantial antitumour activity in patients with PDAC286. A novel strategy is the use of amniotic MSCs to deliver cytotoxic drugs to the tumour site287. MSCs from other origins can also be used; for example, a prodrug in which the active toxin Leu12ADT is released upon cleavage by prostate-specific antigen (PSA) was bound to microparticles that were coated onto bone marrow-derived MSCs and injected into mice, resulting in tumour shrinkage288. These strategies might be effective in clinical settings because MSCs tend to migrate towards areas of wound healing or tumour growth289,290.
ECM-based approaches to target the TME
Instead of targeting either cancer cells or the stroma directly, some strategies exploit stromal components for the therapeutic agent to be delivered at the tumour site. One of the oldest examples of this strategy is the use of bisphosphonates to treat bone metastases. Bisphosphonates do not target cancer cells directly (although evidence of their antitumour effects has become available over time291) but, instead, uncouple bone turnover, thereby halting the vicious cycle in the bone metastases and depriving cancer cells of bone marrow-derived growth factors291. Bisphosphonates are taken up by osteoclasts and inhibit their ability to resorb bone, which also reduces the activity of osteoblasts in the bone stroma292. While bisphosphonates decrease the overall number of skeletal-related events in patients with bone metastasis, their administration is rarely associated with improvements of overall survival. Some investigators hypothesize that, by the time bone metastases are clinically relevant and detectable, it is too late for bisphosphonates to be effective. Rather, if these agents were administered to patients before tumour cells colonize the bone pre-metastatic niche, overt metastatic lesions might be preventable (insert new reference: Dhesy-Thind, S, et al. Use of adjuvant bisphosphonates and other bone-modifying agents in breast cancer: a cancer care Ontario and American Society of Clinical Oncology Practice Guideline. J Clin Oncol 2017).
223Ra is a radioisotope used to treat patients with bone metastases because it mimics calcium, and therefore substitutes for calcium in the bone mineral matrix during bone formation295. Once enmeshed in the bone, 223Ra releases alpha particles that eliminate surrounding cells, including cancer and stromal cells, in the bone metastatic microenvironment. Thus, this strategy exploits the unique properties of the bone ECM and the high bone turnover of bone metastases to exert cytotoxic effects with a lower risk of adverse effects than with other therapies. This FDA-approved agent increases overall survival in patients with metastatic castration-resistant prostate cancer296.
Targeting proteins expressed by stromal cells
Similarly to targeted therapy of cancer cells, the tumour stroma can be modulated by targeting specific proteins expressed by stromal cells that are involved in their proliferation, protein secretion, and ECM formation. FAP is a major focus for targeted therapy of stromal cells297. In the late 1990s and early 2000s, the anti-FAP monoclonal antibody F19 was tested in clinical trials for the treatment and imaging (via 131I–mAbF19) of colorectal tumours but failed to impart a clinical benefit, likely because the antibodies used did not mediate antibody-dependent cellular toxicity of FAP-expressing stromal cells298–301. Subsequently, this strategy evolved and anti-FAP antibodies were conjugated to drugs. FAP5–DM1 is one of such conjugates using the cytotoxic drug maytansine (DM1). The anti-FAP antibody portion delivers the conjugate to FAP-positive fibroblasts, which internalize the compound, cleave the chemical linker, and free the drug portion that induces cell death302. FAP5–DM1 induced long-lasting tumour regression with substantial reduction and reorganization of the stromal compartment in xenograft models of pancreatic, lung, colorectal, and head and neck cancers when compared to mice treated with vehicle or the antibody alone303. Anti-FAP antibodies labelled with radioactive tumoricidal 177Lu had a similar action in the stroma of melanoma xenograft mice304.
Another strategy exploits the endogenous protease activity of FAP to cleave and subsequently activate the prodrug promelittin to release the cytolytic toxin melittin, that eliminates neighbouring cells, including CAFs305. This strategy is less specific to CAFs, but the broader effects of this compound might have greater tumoricidal properties; however, the possibility of widespread toxicity is also a concern. ATRA is currently used to treat multiple cancers and, in addition to other activities, it inhibits FAP, αSMA, and TGFβR expression in CAFs, thereby reducing ECM and cytokine secretion306,307, and subsequently, cancer cell proliferation and invasion. ATRA reverses chemoresistance through multiple mechanisms, but is metabolized by CYP26, which reduces its effects in multiple myeloma228,309; inhibition of CYP26 with R115866 reversed the bortezomib-resistant phenotype of multiple myeloma-derived cells in pre-clinical co-culture and xenograft models228. Similarly, CYP3A4 metabolizes docetaxel, thereby reducing its cytotoxic activity, and inhibition of stromal CYP3A4 with clarithromycin reversed the chemoprotective effect of this enzyme on multiple myeloma cells co-cultured with primary human bone marrow cells expressing CYP3A4227. This strategy could be beneficial to treat patients with prostate cancer bone metastasis, because docetaxel is one of the treatments approved for this population, but cancer recurrence after treatment with this agent is virtually universal310.
Targeting cancer cell–stroma signalling interactions
The targeted inhibition of cell surface receptors for stromal-secreted factors in cancer cells can be considered an indirect mechanism of targeting the stroma; although, owing to the expression of these receptors on cancer cells, such agents are in fact targeting cancer cells. Moreover, mutation or overexpression of many RTKs leads to tumour cell growth independent of stromal cell-secreted growth factors. Nonetheless, this strategy can be considered to be inhibitory of the effects of the tumour stroma.
In many cancers, CAFs secrete FGFs. Many therapeutic strategies target FGF receptors, from RTK inhibitors (such as dovitinib, which targets FGFR3) to monoclonal antibodies (such as GP369, which targets FGFR2-IIIb) 311–315. Targeting the FGF2/FGF2R axis in xenograft models has been shown to prevent resistance to bevacizumab248. Another novel strategy is to prevent stromal cell-secreted FGFs from binding to their receptors on cancer cells; for example, with a ligand trap for FGF using a soluble fusion protein (FGFR1-IIIc fused to the Fc domain of IgG1)312.
Combination therapies can generate considerably greater tumour responses than monotherapies. For example, in a mouse model of breast cancer316, high levels of TGFβ were found in the blood after radiation or chemotherapy. In these mice, treatment with a TGFβ-neutralizing antibody led to a reduction in the numbers of CTCs and lung metastases316. In another example, the chemokine SDF1, abundantly secreted by osteoblasts, was shown to act as a chemoattractant for prostate cancer cells in the bone marrow HSC niche140,164. Treatment with AMD3100, an inhibitor of the SDF1 receptor CXCR4, resulted in efflux of prostate cancer cells from the bone marrow niche and into the blood circulation, where they are more sensitive to therapy164,317,318. In addition, targeting of CXCR4 in PDAC stroma caused increased immune cell infiltration into the tumour and enhanced responses to immunotherapy in syngeneic mouse models319. Thus, the combination of AMD3100 and cytotoxic therapy or immunotherapy holds great promise.
Targeting stromal antigens with immune cells
Chimeric antigen receptor-T (CAR-T) cells are T cells that have been genetically engineered to express antigen-recognizing ectodomains fused to signalling domains from T cell receptors and co–stimulatory molecules320. CAR-T cells have the ability to gain access to the epithelial cell compartment by invading through the ECM and evading immunosuppressive signals321. In preclinical studies CAR-T cells have been reprogrammed to recognize FAP-positive cells and target stromal cells specifically322. Such FAP-specific T cells had single-agent antitumour activity in a mouse lung cancer model, but were associated with sustained antitumour effects and prolonged survival when combined with CAR-T cells specific for the cancer cell antigen erythropoietin-producing hepatocellular carcinoma A2 (EphA2)323. Cancer vaccines against the FAP antigen are another option for CAF targeting324–327; T cell-mediated elimination FAP-positive CAFs (comparison of treatment with the vaccine or an empty vector control in tumor-bearing mice) caused a reduction in collagen density in the ECM, enabling greater uptake of several chemotherapeutic drugs326. FAP-specific vaccines cause tumour regression without the addition of any cytotoxic drugs by stimulation of antitumour immunity, but in combination with other agents, these vaccines might make this strategy more effective.
Macrophages have both profibrotic and antifibrotic properties328, dependent on their status in the spectrum between M1 and M2 polarization329. Agonists of CD40, a co–stimulatory molecule commonly expressed by antigen-presenting cells, can activate macrophages and induce them to degrade the ECM. In several studies, activation of the MMPs in monocytes and macrophages treated with CD40 agonists led to degradation of the ECM and improved tumour infiltration of immune cells as well as response to checkpoint blockade inhibition, warranting further exploration of this strategy in the future328,330–333.
Finally, chondrocytes, cells often excluded in discussions about the tumour stroma, can be engineered to deliver cytotoxic drugs to tumours. Fibroblasts differentiated to chondrocytes ex vivo were engineered to express high levels of IL-12, and irradiated to prevent their proliferation. When these cells were injected into mice bearing colon tumours, they secreted high levels of IL-12, and caused a sizable increase in T cell and natural killer cells infiltration, which eliminated cancer cells and reduced tumour angiogenesis334. This unique strategy might have potential in future combinatorial studies.
Conclusions
Cancer develops as a result of an accumulation of genetic and epigenetic alterations346–348. Increasing evidence suggests that, in addition to cancer cell-autonomous genetic mutations, stromal pathology often contributes to tumorigenesis121,181,189–191. We have presented ample evidence from preclinical and clinical studies that clarify the critical importance of the interactions between cancer cells and the stromal compartment in tumour progression and therapeutic resistance. Upon exposure to therapies that target cancer cells, the tumour stroma supports cancer-cell survival and recurrence, leading to fatal metastatic disease. To combat this resistance, drugs that target stromal components are in various stages of preclinical and clinical development. In many cases, these stroma-targeting drugs reverse or abrogate resistance to cancer cell-targeting therapies, leading to strong anticancer responses. Despite these discoveries, completely curative strategies are not currently available. The substantial gap in knowledge regarding the optimal sequence and composition of combinations comprising stromal-targeting agents and cancer-targeting agents prevents such strategies from being offered to patients349. The order in which these agents are administered is important, because certain sequences can result in adverse effects. For example, when chemotherapy is administered after an immune response has been initiated, it could cause the death of immune cells, thereby reversing their cytotoxic effect. Ongoing clinical trials are assessing the activity and response of the tumour stroma before and after anticancer therapy (NCT03165487). Discovery studies involving laser capture microdissection of stroma350, coupled with analyses of genetic mutations351 and epigenetic changes352 can uncover novel therapeutic targets. These types of studies will provide information about how to better target the stroma in the future.
An important consideration is that many of the experiments addressing cancer cell–stroma interactions are performed using imperfect models, such as co-cultures and xenograft or syngeneic mouse models. These experimental models are fundamental to preclinical cancer research, but substantial gaps exist between the results of these experiments and those obtained in clinical settings. Communication between researchers and medical professionals will be crucial in determining the next steps to bring discoveries to patients. As we continue to develop a better understanding of the complex interactions between a heterogeneous milieu of cellular and noncellular contributors in the TME, we will be able to improve stroma-targeting strategies and design more-effective anticancer therapies.
Box 1. Biomarkers of stromal involvement.
Owing to the evident involvement of the stroma in tumour progression and therapy resistance, ideally, specific cell types and/or targetable molecular lesions should be identifiable within the tumour microenvironment or in liquid biopsies that would enable targeting the stroma with therapeutic interventions or stratification of patients into treatment subgroups335. The stromal compartment, separate from cancer-specific alterations, might have independent prognostic value 335. Cancer-associated fibroblasts (CAFs) typically express prolyl endopeptidase and alpha smooth muscle actin. Detection of these markers using immunostaining can enable the identification of CAFs in tumour biopsy samples to assess the extent of fibroblast involvement336. This approach would provide evidence on the outcomes of tumour-targeting strategies. Other markers, such as podoplanin, CD99, matrix metalloproteinases, and the transcription factor forkhead box F1 have also been associated with the tumour stroma, but none have been successfully used for clinical purposes337–340. An example of a marker with potential clinical utility is the loss of expression of caveolin-1 in the tumour stroma as a predictor of recurrence in patients with breast cancer259. In patients with pancreatic cancer, the presence of intratumoural hyaluronic acid could predict the ability of a drug to penetrate the stroma and access the tumour276. Beyond single protein markers, gene signatures based on the expression of selected genes could represent the most effective method341–345. Ideally, such signatures should be assessed with blood-based assays; similarly to cancer cells, stromal cells are detectable in the blood of patients with cancer154. However, little evidence supports the clinical relevance of such a biomarker at present, arguably, as a result of the scarcity of clinical data. Future studies (likely using a systemic approach) should identify clinically relevant molecules of stromal origin that would predict whether specific therapies are effective in individual patients and/or provide a readout of the outcomes of a therapy after it has been administered.
Key points.
Tumours are comprised of cancer cells as well as a stromal compartment with cellular and noncellular components
The tumour stroma has critical roles in cancer development, progression and metastasis
Typically, anticancer therapies predominantly target cancer cells, and their effect on the tumour stroma is not taken into account
The tumour stroma responds to anticancer therapies by inducing therapeutic resistance, which can ultimately lead to fatal disease
Anticancer therapies should target both cancer cells and the stromal compartment to be effective and result in improved patient outcomes
Acknowledgments
The work of the K.J.P., K.C.V., and A.E.dG. is supported by the National Cancer Institute (NCI) grants U54CA143803, CA163124, CA093900, CA143055 as well as the Prostate Cancer Foundation, and the Patrick C. Walsh Fund. The work of K.C.V. is supported by the NCI grant F32CA206394. The authors are grateful to S. Amend for editing the manuscript.
Footnotes
Author contributions
K.C.V. and A.E.dG. wrote the manuscript and prepared display items. K.J.P. contributed to discussions about the contents of the manuscript. All authors reviewed/edited the manuscript before submission
Competing interests statement
The authors declare no competing interests.
ClinicalTrials.gov https://clinicaltrials.gov/ct2/home
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Amend SR, Pienta KJ. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget. 2015;6:9669–9678. doi: 10.18632/oncotarget.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amend SR, Roy S, Brown JS, Pienta KJ. Ecological paradigms to understand the dynamics of metastasis. Cancer letters. 2016;380:237–242. doi: 10.1016/j.canlet.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camacho DF, Pienta KJ. Disrupting the networks of cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:2801–2808. doi: 10.1158/1078-0432.CCR-12-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Groot AE, Roy S, Brown JS, Pienta KJ, Amend SR. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Molecular cancer research: MCR. 2017;15:361–370. doi: 10.1158/1541-7786.MCR-16-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maley CC, et al. Classifying the evolutionary and ecological features of neoplasms. Nature reviews. Cancer. 2017;17:605–619. doi: 10.1038/nrc.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Brown JM. Vasculogenesis: a crucial player in the resistance of solid tumours to radiotherapy. Br J Radiol. 2014;87:20130686. doi: 10.1259/bjr.20130686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hida K, Akiyama K, Ohga N, Maishi N, Hida Y. Tumour endothelial cells acquire drug resistance in a tumour microenvironment. J Biochem. 2013;153:243–249. doi: 10.1093/jb/mvs152. [DOI] [PubMed] [Google Scholar]
- 10.Kibria G, Hatakeyama H, Harashima H. Cancer multidrug resistance: mechanisms involved and strategies for circumvention using a drug delivery system. Arch Pharm Res. 2014;37:4–15. doi: 10.1007/s12272-013-0276-2. [DOI] [PubMed] [Google Scholar]
- 11.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ, Thijssen VL, Griffioen AW. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev. 2015;67:441–461. doi: 10.1124/pr.114.010215. [DOI] [PubMed] [Google Scholar]
- 13.Choi J, Cha YJ, Koo JS. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog Lipid Res. 2017;69:11–20. doi: 10.1016/j.plipres.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Kozin SV, et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer research. 2010;70:5679–5685. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribas A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015;5:915–919. doi: 10.1158/2159-8290.CD-15-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roca H, et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PloS one. 2013;8:e76773. doi: 10.1371/journal.pone.0076773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development. 2010;137:1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hynes RO, Naba A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4:a004903. doi: 10.1101/cshperspect.a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laurent GJ, Chambers RC, Hill MR, McAnulty RJ. Regulation of matrix turnover: fibroblasts, forces, factors and fibrosis. Biochem Soc Trans. 2007;35:647–651. doi: 10.1042/BST0350647. [DOI] [PubMed] [Google Scholar]
- 22.Alexander J, Cukierman E. Stromal dynamic reciprocity in cancer: intricacies of fibroblastic-ECM interactions. Current opinion in cell biology. 2016;42:80–93. doi: 10.1016/j.ceb.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 24.Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen. 2011;19:134–148. doi: 10.1111/j.1524-475X.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alberts B, et al. Molecular Biology of the Cell. 2. Garland Publishing, Inc; 1989. [Google Scholar]
- 26.Buckley CD, et al. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 27.Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. The American journal of pathology. 1997;151:317–322. [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider EL, Mitsui Y, Au KS, Shorr SS. Tissue-specific differences in cultured human diploid fibroblasts. Exp Cell Res. 1977;108:1–6. doi: 10.1016/s0014-4827(77)80002-5. [DOI] [PubMed] [Google Scholar]
- 29.Zamansky GB, Arundel C, Nagasawa H, Little JB. Adaptation of human diploid fibroblasts in vitro to serum from different sources. J Cell Sci. 1983;61:289–297. doi: 10.1242/jcs.61.1.289. [DOI] [PubMed] [Google Scholar]
- 30.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Driskell RR, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HY, et al. Localized Smooth Muscle Differentiation Is Essential for Epithelial Bifurcation during Branching Morphogenesis of the Mammalian Lung. Dev Cell. 2015;34:719–726. doi: 10.1016/j.devcel.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shyer AE, Huycke TR, Lee C, Mahadevan L, Tabin CJ. Bending gradients: how the intestinal stem cell gets its home. Cell. 2015;161:569–580. doi: 10.1016/j.cell.2015.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011;57:376–379. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res. 2016;5 doi: 10.12688/f1000research.8190.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinz B, et al. The myofibroblast: one function, multiple origins. The American journal of pathology. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiga K, et al. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel) 2015;7:2443–2458. doi: 10.3390/cancers7040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuxhorn JA, et al. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8:2912–2923. [PubMed] [Google Scholar]
- 39.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3:393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 41.Friedenstein AJ, Piatetzky S, II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16:381–390. [PubMed] [Google Scholar]
- 42.Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Experimental hematology. 1976;4:267–274. [PubMed] [Google Scholar]
- 43.Piersma AH, et al. Characterization of fibroblastic stromal cells from murine bone marrow. Experimental hematology. 1985;13:237–243. [PubMed] [Google Scholar]
- 44.Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 45.Rickard DJ, Sullivan TA, Shenker BJ, Leboy PS, Kazhdan I. Induction of rapid osteoblast differentiation in rat bone marrow stromal cell cultures by dexamethasone and BMP-2. Developmental biology. 1994;161:218–228. doi: 10.1006/dbio.1994.1022. [DOI] [PubMed] [Google Scholar]
- 46.Wakitani S, Saito T, Caplan AI. Myogenic cells derived from rat bone marrow mesenchymal stem cells exposed to 5-azacytidine. Muscle Nerve. 1995;18:1417–1426. doi: 10.1002/mus.880181212. [DOI] [PubMed] [Google Scholar]
- 47.Horwitz EM, et al. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7:393–395. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 48.Lindner U, Kramer J, Rohwedel J, Schlenke P. Mesenchymal Stem or Stromal Cells: Toward a Better Understanding of Their Biology? Transfus Med Hemother. 2010;37:75–83. doi: 10.1159/000290897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paunescu V, et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med. 2011;15:635–646. doi: 10.1111/j.1582-4934.2010.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haniffa MA, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. Journal of immunology. 2007;179:1595–1604. doi: 10.4049/jimmunol.179.3.1595. [DOI] [PubMed] [Google Scholar]
- 51.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature biotechnology. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 53.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 54.Alt E, et al. Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-forming potential. Biol Cell. 2011;103:197–208. doi: 10.1042/BC20100117. [DOI] [PubMed] [Google Scholar]
- 55.Battula VL, et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood. 2013;122:357–366. doi: 10.1182/blood-2012-06-437988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. 2000;109:235–242. doi: 10.1046/j.1365-2141.2000.01986.x. [DOI] [PubMed] [Google Scholar]
- 57.Zvaifler NJ, et al. Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res. 2000;2:477–488. doi: 10.1186/ar130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galotto M, et al. Stromal damage as consequence of high-dose chemo/radiotherapy in bone marrow transplant recipients. Experimental hematology. 1999;27:1460–1466. doi: 10.1016/s0301-472x(99)00076-4. [DOI] [PubMed] [Google Scholar]
- 59.Sanchez-Abarca LI, et al. Uptake and delivery of antigens by mesenchymal stromal cells. Cytotherapy. 2013;15:673–678. doi: 10.1016/j.jcyt.2013.01.216. [DOI] [PubMed] [Google Scholar]
- 60.Yagi H, et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–679. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moroni L, Fornasari PM. Human mesenchymal stem cells: a bank perspective on the isolation, characterization and potential of alternative sources for the regeneration of musculoskeletal tissues. Journal of cellular physiology. 2013;228:680–687. doi: 10.1002/jcp.24223. [DOI] [PubMed] [Google Scholar]
- 62.Wu L, Cai X, Zhang S, Karperien M, Lin Y. Regeneration of articular cartilage by adipose tissue derived mesenchymal stem cells: perspectives from stem cell biology and molecular medicine. Journal of cellular physiology. 2013;228:938–944. doi: 10.1002/jcp.24255. [DOI] [PubMed] [Google Scholar]
- 63.Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000;289:1501–1504. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- 64.Seeman E, Delmas PD. Bone quality--the material and structural basis of bone strength and fragility. N Engl J Med. 2006;354:2250–2261. doi: 10.1056/NEJMra053077. [DOI] [PubMed] [Google Scholar]
- 65.Mackie EJ. Osteoblasts: novel roles in orchestration of skeletal architecture. Int J Biochem Cell Biol. 2003;35:1301–1305. doi: 10.1016/s1357-2725(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 66.Caetano-Lopes J, Canhao H, Fonseca JE. Osteoblasts and bone formation. Acta Reumatol Port. 2007;32:103–110. [PubMed] [Google Scholar]
- 67.Gori F, et al. The expression of osteoprotegerin and RANK ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated. Endocrinology. 2000;141:4768–4776. doi: 10.1210/endo.141.12.7840. [DOI] [PubMed] [Google Scholar]
- 68.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 69.Muir H. The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays. 1995;17:1039–1048. doi: 10.1002/bies.950171208. [DOI] [PubMed] [Google Scholar]
- 70.Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol. 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 71.Urban JP. The chondrocyte: a cell under pressure. Br J Rheumatol. 1994;33:901–908. doi: 10.1093/rheumatology/33.10.901. [DOI] [PubMed] [Google Scholar]
- 72.Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15:1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 75.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 77.Karagiannis GS, et al. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Molecular cancer research: MCR. 2012;10:1403–1418. doi: 10.1158/1541-7786.MCR-12-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle. 2009;8:589–595. doi: 10.4161/cc.8.4.7669. [DOI] [PubMed] [Google Scholar]
- 79.Bremnes RM, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 80.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nature reviews. Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 81.Zigrino P, Loffek S, Mauch C. Tumor-stroma interactions: their role in the control of tumor cell invasion. Biochimie. 2005;87:321–328. doi: 10.1016/j.biochi.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 82.McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 83.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Current opinion in cell biology. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 85.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 86.Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008;1:158–164. doi: 10.1593/tlo.08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev. 2009;19:67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 88.Cunha GR, Bigsby RM, Cooke PS, Sugimura Y. Stromal-epithelial interactions in adult organs. Cell Differ. 1985;17:137–148. doi: 10.1016/0045-6039(85)90481-6. [DOI] [PubMed] [Google Scholar]
- 89.Cunha GR, Donjacour AA, Sugimura Y. Stromal-epithelial interactions and heterogeneity of proliferative activity within the prostate. Biochem Cell Biol. 1986;64:608–614. doi: 10.1139/o86-084. [DOI] [PubMed] [Google Scholar]
- 90.Ozdemir BC, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menen R, et al. Inhibition of metastasis of circulating human prostate cancer cells in the chick embryo by an extracellular matrix produced by foreskin fibroblasts in culture. Anticancer research. 2012;32:1573–1577. [PubMed] [Google Scholar]
- 92.Bhowmick NA, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 93.Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shimoda M, Jackson HW, Khokha R. Tumor suppression by stromal TIMPs. Mol Cell Oncol. 2016;3:e975082. doi: 10.4161/23723556.2014.975082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cruz-Munoz W, Khokha R. The role of tissue inhibitors of metalloproteinases in tumorigenesis and metastasis. Critical reviews in clinical laboratory sciences. 2008;45:291–338. doi: 10.1080/10408360801973244. [DOI] [PubMed] [Google Scholar]
- 96.Shimoda M, et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol. 2014;16:889–901. doi: 10.1038/ncb3021. [DOI] [PubMed] [Google Scholar]
- 97.Tjomsland V, et al. Profile of MMP and TIMP Expression in Human Pancreatic Stellate Cells: Regulation by IL-1alpha and TGFbeta and Implications for Migration of Pancreatic Cancer Cells. Neoplasia. 2016;18:447–456. doi: 10.1016/j.neo.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bloomston M, Shafii A, Zervos EE, Rosemurgy AS. TIMP-1 overexpression in pancreatic cancer attenuates tumor growth, decreases implantation and metastasis, and inhibits angiogenesis. J Surg Res. 2002;102:39–44. doi: 10.1006/jsre.2001.6318. [DOI] [PubMed] [Google Scholar]
- 99.Langley RR, Fidler IJ. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. International journal of cancer. Journal international du cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fidler IJ, et al. Modulation of tumor cell response to chemotherapy by the organ environment. Cancer metastasis reviews. 1994;13:209–222. doi: 10.1007/BF00689637. [DOI] [PubMed] [Google Scholar]
- 101.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 102.Couillard J, Demers M, Lavoie G, St-Pierre Y. The role of DNA hypomethylation in the control of stromelysin gene expression. Biochemical and biophysical research communications. 2006;342:1233–1239. doi: 10.1016/j.bbrc.2006.02.068. [DOI] [PubMed] [Google Scholar]
- 103.Hanson JA, et al. Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst. 2006;98:255–261. doi: 10.1093/jnci/djj051. [DOI] [PubMed] [Google Scholar]
- 104.Lin HJ, et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer research. 2008;68:10257–10266. doi: 10.1158/0008-5472.CAN-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kalluri R. The biology and function of fibroblasts in cancer. Nature reviews. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 106.Micallef L, et al. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair. 2012;5:S5. doi: 10.1186/1755-1536-5-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neuzillet C, et al. Stromal expression of SPARC in pancreatic adenocarcinoma. Cancer metastasis reviews. 2013;32:585–602. doi: 10.1007/s10555-013-9439-3. [DOI] [PubMed] [Google Scholar]
- 108.Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 109.Chiquet-Ehrismann R, Mackie EJ, Pearson CA, Sakakura T. Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell. 1986;47:131–139. doi: 10.1016/0092-8674(86)90374-0. [DOI] [PubMed] [Google Scholar]
- 110.Kyutoku M, et al. Role of periostin in cancer progression and metastasis: inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int J Mol Med. 2011;28:181–186. doi: 10.3892/ijmm.2011.712. [DOI] [PubMed] [Google Scholar]
- 111.Mackie EJ, et al. Tenascin is a stromal marker for epithelial malignancy in the mammary gland. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:4621–4625. doi: 10.1073/pnas.84.13.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ouyang G, et al. Upregulated expression of periostin by hypoxia in non-small-cell lung cancer cells promotes cell survival via the Akt/PKB pathway. Cancer letters. 2009;281:213–219. doi: 10.1016/j.canlet.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 113.Ruan K, Bao S, Ouyang G. The multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci. 2009;66:2219–2230. doi: 10.1007/s00018-009-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mouw JK, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nature medicine. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim W, et al. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-beta signaling. Mol Biol Cell. 2008;19:2135–2146. doi: 10.1091/mbc.E07-10-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:781–791. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu J, Agarwal S. Mechanical signals activate vascular endothelial growth factor receptor-2 to upregulate endothelial cell proliferation during inflammation. Journal of immunology. 2010;185:1215–1221. doi: 10.4049/jimmunol.0903660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pylayeva Y, et al. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. The Journal of clinical investigation. 2009;119:252–266. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tomakidi P, et al. Defects of basement membrane and hemidesmosome structure correlate with malignant phenotype and stromal interactions in HaCaT-Ras xenografts. Differentiation; research in biological diversity. 1999;64:263–275. doi: 10.1046/j.1432-0436.1999.6450263.x. [DOI] [PubMed] [Google Scholar]
- 120.Kaukonen R, et al. Normal stroma suppresses cancer cell proliferation via mechanosensitive regulation of JMJD1a-mediated transcription. Nature communications. 2016;7:12237. doi: 10.1038/ncomms12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer research. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boire A, et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 123.Sternlicht MD, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hotary KB, et al. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 125.Tang D, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. International journal of cancer. Journal international du cancer. 2012;130:2337–2348. doi: 10.1002/ijc.26290. [DOI] [PubMed] [Google Scholar]
- 126.Luttenberger T, et al. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Laboratory investigation; a journal of technical methods and pathology. 2000;80:47–55. doi: 10.1038/labinvest.3780007. [DOI] [PubMed] [Google Scholar]
- 127.Joesting MS, et al. Identification of SFRP1 as a candidate mediator of stromal-to-epithelial signaling in prostate cancer. Cancer research. 2005;65:10423–10430. doi: 10.1158/0008-5472.CAN-05-0824. [DOI] [PubMed] [Google Scholar]
- 128.Bragado P, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013;15:1351–1361. doi: 10.1038/ncb2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Calon A, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carstens JL, et al. FGFR1-WNT-TGF-beta signaling in prostate cancer mouse models recapitulates human reactive stroma. Cancer research. 2014;74:609–620. doi: 10.1158/0008-5472.CAN-13-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Claffey KP, et al. Fibroblast growth factor 2 activation of stromal cell vascular endothelial growth factor expression and angiogenesis. Laboratory investigation; a journal of technical methods and pathology. 2001;81:61–75. doi: 10.1038/labinvest.3780212. [DOI] [PubMed] [Google Scholar]
- 132.Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–651. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Crawford Y, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer cell. 2009;15:21–34. doi: 10.1016/j.ccr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 134.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS medicine. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fukumura D, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 136.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 137.Prabhu VV, Warfel NA, El-Deiry WS. CTGF-mediated autophagy-senescence transition in tumor stroma promotes anabolic tumor growth and metastasis. Cell Cycle. 2012;11:2592–2593. doi: 10.4161/cc.21240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 139.Sun YX, et al. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. The Prostate. 2007;67:61–73. doi: 10.1002/pros.20500. [DOI] [PubMed] [Google Scholar]
- 140.Sun YX, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. Journal of cellular biochemistry. 2003;89:462–473. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 141.LeBedis C, Chen K, Fallavollita L, Boutros T, Brodt P. Peripheral lymph node stromal cells can promote growth and tumorigenicity of breast carcinoma cells through the release of IGF-I and EGF. International journal of cancer. Journal international du cancer. 2002;100:2–8. doi: 10.1002/ijc.10481. [DOI] [PubMed] [Google Scholar]
- 142.Sainaghi PP, et al. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. Journal of cellular physiology. 2005;204:36–44. doi: 10.1002/jcp.20265. [DOI] [PubMed] [Google Scholar]
- 143.Shiozawa Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12:116–127. doi: 10.1593/neo.91384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Taichman RS, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PloS one. 2013;8:e61873. doi: 10.1371/journal.pone.0061873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hugo HJ, et al. Contribution of Fibroblast and Mast Cell (Afferent) and Tumor (Efferent) IL-6 Effects within the Tumor Microenvironment. Cancer Microenviron. 2012;5:83–93. doi: 10.1007/s12307-012-0098-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Molecular cancer research: MCR. 2008;6:1521–1533. doi: 10.1158/1541-7786.MCR-07-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tait LR, et al. Dynamic stromal-epithelial interactions during progression of MCF10DCIS.com xenografts. International journal of cancer. Journal international du cancer. 2007;120:2127–2134. doi: 10.1002/ijc.22572. [DOI] [PubMed] [Google Scholar]
- 149.Rajaram M, Li J, Egeblad M, Powers RS. System-wide analysis reveals a complex network of tumor-fibroblast interactions involved in tumorigenicity. PLoS genetics. 2013;9:e1003789. doi: 10.1371/journal.pgen.1003789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elkhattouti A, Hassan M, Gomez CR. Stromal Fibroblast in Age-Related Cancer: Role in Tumorigenesis and Potential as Novel Therapeutic Target. Front Oncol. 2015;5:158. doi: 10.3389/fonc.2015.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pazolli E, et al. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer research. 2012;72:2251–2261. doi: 10.1158/0008-5472.CAN-11-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Luo X, et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016;14:82–92. doi: 10.1016/j.celrep.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pazolli E, et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer research. 2009;69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ao Z, et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer research. 2015;75:4681–4687. doi: 10.1158/0008-5472.CAN-15-1633. [DOI] [PubMed] [Google Scholar]
- 155.Jones ML, Siddiqui J, Pienta KJ, Getzenberg RH. Circulating fibroblast-like cells in men with metastatic prostate cancer. The Prostate. 2013;73:176–181. doi: 10.1002/pros.22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bystricky B, et al. Relationship Between Circulating Tumor Cells and Annexin A2 in Early Breast Cancer Patients. Anticancer research. 2017;37:2727–2734. doi: 10.21873/anticanres.11624. [DOI] [PubMed] [Google Scholar]
- 157.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature reviews. Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bergfeld SA, Blavier L, DeClerck YA. Bone marrow-derived mesenchymal stromal cells promote survival and drug resistance in tumor cells. Molecular cancer therapeutics. 2014;13:962–975. doi: 10.1158/1535-7163.MCT-13-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Worthley DL, et al. Bone marrow cells as precursors of the tumor stroma. Exp Cell Res. 2013;319:1650–1656. doi: 10.1016/j.yexcr.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brennen WN, Chen S, Denmeade SR, Isaacs JT. Quantification of Mesenchymal Stem Cells (MSCs) at sites of human prostate cancer. Oncotarget. 2013;4:106–117. doi: 10.18632/oncotarget.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Arina A, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:7551–7556. doi: 10.1073/pnas.1600363113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jung Y, et al. Annexin 2-CXCL12 interactions regulate metastatic cell targeting and growth in the bone marrow. Molecular cancer research: MCR. 2015;13:197–207. doi: 10.1158/1541-7786.MCR-14-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mishra A, Shiozawa Y, Pienta KJ, Taichman RS. Homing of cancer cells to the bone. Cancer Microenviron. 2011;4:221–235. doi: 10.1007/s12307-011-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shiozawa Y, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. The Journal of clinical investigation. 2011;121:1298–1312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wobus M, et al. Breast carcinoma cells modulate the chemoattractive activity of human bone marrow-derived mesenchymal stromal cells by interfering with CXCL12. International journal of cancer. Journal international du cancer. 2015;136:44–54. doi: 10.1002/ijc.28960. [DOI] [PubMed] [Google Scholar]
- 166.Wang N, et al. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis - evidence from in vivo models. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2014 doi: 10.1002/jbmr.2300. [DOI] [PubMed] [Google Scholar]
- 167.Shimo T, et al. The Role of Sonic Hedgehog Signaling in Osteoclastogenesis and Jaw Bone Destruction. PloS one. 2016;11:e0151731. doi: 10.1371/journal.pone.0151731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Heller E, et al. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment. Cancer research. 2012;72:897–907. doi: 10.1158/0008-5472.CAN-11-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Johnson RW, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer research. 2011;71:822–831. doi: 10.1158/0008-5472.CAN-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature reviews. Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 171.Guise TA. The vicious cycle of bone metastases. J Musculoskelet Neuronal Interact. 2002;2:570–572. [PubMed] [Google Scholar]
- 172.Jeong HM, Cho SW, Park SI. Osteoblasts Are the Centerpiece of the Metastatic Bone Microenvironment. Endocrinol Metab (Seoul) 2016;31:485–492. doi: 10.3803/EnM.2016.31.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li XQ, et al. ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGFbeta Signaling Pathway. Cancer research. 2015;75:3302–3313. doi: 10.1158/0008-5472.CAN-15-0240. [DOI] [PubMed] [Google Scholar]
- 174.Sottnik JL, Dai J, Zhang H, Campbell B, Keller ET. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer research. 2015;75:2151–2158. doi: 10.1158/0008-5472.CAN-14-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cunha GR, et al. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J Steroid Biochem Mol Biol. 2004;92:221–236. doi: 10.1016/j.jsbmb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 176.Cunha GR. Epithelio-mesenchymal interactions in primordial gland structures which become responsive to androgenic stimulation. Anat Rec. 1972;172:179–195. doi: 10.1002/ar.1091720206. [DOI] [PubMed] [Google Scholar]
- 177.Cunha GR. Tissue interactions between epithelium and mesenchyme of urogenital and integumental origin. Anat Rec. 1972;172:529–541. doi: 10.1002/ar.1091720307. [DOI] [PubMed] [Google Scholar]
- 178.Cunha GR. The role of androgens in the epithelio-mesenchymal interactions involved in prostatic morphogenesis in embryonic mice. Anat Rec. 1973;175:87–96. doi: 10.1002/ar.1091750108. [DOI] [PubMed] [Google Scholar]
- 179.Cunha GR. Epithelial-stromal interactions in development of the urogenital tract. Int Rev Cytol. 1976;47:137–194. doi: 10.1016/s0074-7696(08)60088-1. [DOI] [PubMed] [Google Scholar]
- 180.Aboseif S, El-Sakka A, Young P, Cunha G. Mesenchymal reprogramming of adult human epithelial differentiation. Differentiation; research in biological diversity. 1999;65:113–118. doi: 10.1046/j.1432-0436.1999.6520113.x. [DOI] [PubMed] [Google Scholar]
- 181.Hayward SW, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer research. 2001;61:8135–8142. [PubMed] [Google Scholar]
- 182.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer research. 2000;60:1254–1260. [PubMed] [Google Scholar]
- 183.Yasuda K, et al. Fibroblasts induce expression of FGF4 in ovarian cancer stem-like cells/cancer-initiating cells and upregulate their tumor initiation capacity. Laboratory investigation; a journal of technical methods and pathology. 2014;94:1355–1369. doi: 10.1038/labinvest.2014.122. [DOI] [PubMed] [Google Scholar]
- 184.Zhao XL, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. The Journal of pathology. 2017;243:376–389. doi: 10.1002/path.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vermeulen L, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 186.Del Pozo Martin Y, et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015;13:2456–2469. doi: 10.1016/j.celrep.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Du H, Che G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol Lett. 2017;13:3–12. doi: 10.3892/ol.2016.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kurose K, et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nature genetics. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 189.Kurose K, et al. Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Human molecular genetics. 2001;10:1907–1913. doi: 10.1093/hmg/10.18.1907. [DOI] [PubMed] [Google Scholar]
- 190.Tanwar PS, Zhang L, Roberts DJ, Teixeira JM. Stromal deletion of the APC tumor suppressor in mice triggers development of endometrial cancer. Cancer research. 2011;71:1584–1596. doi: 10.1158/0008-5472.CAN-10-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kode A, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Calvo F, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Procopio MG, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17:1193–1204. doi: 10.1038/ncb3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Scherz-Shouval R, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tyekucheva S, et al. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nature communications. 2017;8:420. doi: 10.1038/s41467-017-00460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bauer M, et al. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene. 2010;29:1732–1740. doi: 10.1038/onc.2009.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nakagawa H, et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23:7366–7377. doi: 10.1038/sj.onc.1208013. [DOI] [PubMed] [Google Scholar]
- 198.Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer research. 2004;64:6950–6956. doi: 10.1158/0008-5472.CAN-04-0677. [DOI] [PubMed] [Google Scholar]
- 199.Singer CF, et al. Differential gene expression profile in breast cancer-derived stromal fibroblasts. Breast cancer research and treatment. 2008;110:273–281. doi: 10.1007/s10549-007-9725-2. [DOI] [PubMed] [Google Scholar]
- 200.Horie M, et al. TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2017 doi: 10.1152/ajplung.00193.2017. ajplung 00193 02017. [DOI] [PubMed] [Google Scholar]
- 201.Marks DL, Olson RL, Fernandez-Zapico ME. Epigenetic control of the tumor microenvironment. Epigenomics. 2016;8:1671–1687. doi: 10.2217/epi-2016-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mathot P, et al. DNA methylation signal has a major role in the response of human breast cancer cells to the microenvironment. Oncogenesis. 2017;6:e390. doi: 10.1038/oncsis.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rodriguez-Canales J, et al. Identification of a unique epigenetic sub-microenvironment in prostate cancer. The Journal of pathology. 2007;211:410–419. doi: 10.1002/path.2133. [DOI] [PubMed] [Google Scholar]
- 204.Albrengues J, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nature communications. 2015;6:10204. doi: 10.1038/ncomms10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Albrengues J, et al. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014;7:1664–1678. doi: 10.1016/j.celrep.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 206.Jiang L, et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer research. 2008;68:9900–9908. doi: 10.1158/0008-5472.CAN-08-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Velaei K, Samadi N, Barazvan B, Soleimani Rad J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast. 2016;30:92–100. doi: 10.1016/j.breast.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 208.Ayala G, et al. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:4792–4801. [PubMed] [Google Scholar]
- 209.Mhawech-Fauceglia P, et al. Stromal Expression of Fibroblast Activation Protein Alpha (FAP) Predicts Platinum Resistance and Shorter Recurrence in patients with Epithelial Ovarian Cancer. Cancer Microenviron. 2015;8:23–31. doi: 10.1007/s12307-014-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kelly T, Huang Y, Simms AE, Mazur A. Fibroblast activation protein-alpha: a key modulator of the microenvironment in multiple pathologies. Int Rev Cell Mol Biol. 2012;297:83–116. doi: 10.1016/B978-0-12-394308-8.00003-0. [DOI] [PubMed] [Google Scholar]
- 211.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Damiano JS, Hazlehurst LA, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia. 2001;15:1232–1239. doi: 10.1038/sj.leu.2402179. [DOI] [PubMed] [Google Scholar]
- 213.Hazlehurst LA, Dalton WS. Mechanisms associated with cell adhesion mediated drug resistance (CAM-DR) in hematopoietic malignancies. Cancer metastasis reviews. 2001;20:43–50. doi: 10.1023/a:1013156407224. [DOI] [PubMed] [Google Scholar]
- 214.Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR) Oncogene. 2000;19:4319–4327. doi: 10.1038/sj.onc.1203782. [DOI] [PubMed] [Google Scholar]
- 215.Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–2421. doi: 10.1038/sj.onc.1206315. [DOI] [PubMed] [Google Scholar]
- 216.Hazlehurst LA, Argilagos RF, Dalton WS. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br J Haematol. 2007;136:269–275. doi: 10.1111/j.1365-2141.2006.06435.x. [DOI] [PubMed] [Google Scholar]
- 217.Lwin T, et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood. 2007;110:1631–1638. doi: 10.1182/blood-2006-11-060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer research. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 219.Young JS, Lumsden CE, Stalker AL. The significance of the tissue pressure of normal testicular and of neoplastic (Brown-Pearce carcinoma) tissue in the rabbit. J Pathol Bacteriol. 1950;62:313–333. doi: 10.1002/path.1700620303. [DOI] [PubMed] [Google Scholar]
- 220.DuFort CC, DelGiorno KE, Hingorani SR. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2016;150:1545–1557 e1542. doi: 10.1053/j.gastro.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. British journal of cancer. 2013;108:1–8. doi: 10.1038/bjc.2012.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wegner CS, et al. Dynamic contrast-enhanced MRI of the microenvironment of pancreatic adenocarcinoma xenografts. Acta Oncol. 2017;56:1754–1762. doi: 10.1080/0284186X.2017.1343494. [DOI] [PubMed] [Google Scholar]
- 223.Provenzano PP, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ozerdem U, Hargens AR. A simple method for measuring interstitial fluid pressure in cancer tissues. Microvasc Res. 2005;70:116–120. doi: 10.1016/j.mvr.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Munson JM, Bellamkonda RV, Swartz MA. Interstitial flow in a 3D microenvironment increases glioma invasion by a CXCR4-dependent mechanism. Cancer research. 2013;73:1536–1546. doi: 10.1158/0008-5472.CAN-12-2838. [DOI] [PubMed] [Google Scholar]
- 226.Hirth J, et al. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:1255–1258. [PubMed] [Google Scholar]
- 227.Alonso S, et al. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget. 2015;6:14905–14912. doi: 10.18632/oncotarget.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Alonso S, et al. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. The Journal of clinical investigation. 2016;126:4460–4468. doi: 10.1172/JCI88152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Xu K, et al. Autophagy induction contributes to the resistance to methotrexate treatment in rheumatoid arthritis fibroblast-like synovial cells through high mobility group box chromosomal protein 1. Arthritis Res Ther. 2015;17:374. doi: 10.1186/s13075-015-0892-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Huber RM, et al. DNA damage induces GDNF secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget. 2015;6:2134–2147. doi: 10.18632/oncotarget.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sun Y, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nature medicine. 2012;18:1359–1368. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Steinbichler TB, Metzler V, Pritz C, Riechelmann H, Dudas J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget. 2016;7:2508–2518. doi: 10.18632/oncotarget.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Peiris-Pages M, Sotgia F, Lisanti MP. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget. 2015;6:10728–10745. doi: 10.18632/oncotarget.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hu Y, et al. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PloS one. 2015;10:e0125625. doi: 10.1371/journal.pone.0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wang W, et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell. 2016;165:1092–1105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9:193–199. doi: 10.7150/ijms.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cordes N. Integrin-mediated cell-matrix interactions for prosurvival and antiapoptotic signaling after genotoxic injury. Cancer letters. 2006;242:11–19. doi: 10.1016/j.canlet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 239.Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. beta1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene. 2006;25:1378–1390. doi: 10.1038/sj.onc.1209164. [DOI] [PubMed] [Google Scholar]
- 240.Hellevik T, et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat Oncol. 2012;7:59. doi: 10.1186/1748-717X-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer research. 2011;71:3453–3458. doi: 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Park CC, Zhang HJ, Yao ES, Park CJ, Bissell MJ. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer research. 2008;68:4398–4405. doi: 10.1158/0008-5472.CAN-07-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Puthawala K, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82–90. doi: 10.1164/rccm.200706-806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chargari C, Clemenson C, Martins I, Perfettini JL, Deutsch E. Understanding the functions of tumor stroma in resistance to ionizing radiation: emerging targets for pharmacological modulation. Drug Resist Updat. 2013;16:10–21. doi: 10.1016/j.drup.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 245.Kamochi N, et al. Irradiated fibroblast-induced bystander effects on invasive growth of squamous cell carcinoma under cancer-stromal cell interaction. Cancer science. 2008;99:2417–2427. doi: 10.1111/j.1349-7006.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ohuchida K, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer research. 2004;64:3215–3222. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- 247.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med. 2011;3:623–636. doi: 10.1002/emmm.201100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mitsuhashi A, et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nature communications. 2015;6:8792. doi: 10.1038/ncomms9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kinugasa Y, Matsui T, Takakura N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem cells. 2014;32:145–156. doi: 10.1002/stem.1556. [DOI] [PubMed] [Google Scholar]
- 250.Yoshida T, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:642–651. doi: 10.1158/1078-0432.CCR-14-0846. [DOI] [PubMed] [Google Scholar]
- 251.Mueller KL, et al. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012;14:R104. doi: 10.1186/bcr3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hirata E, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Singh M, et al. Stromal androgen receptor in prostate development and cancer. The American journal of pathology. 2014;184:2598–2607. doi: 10.1016/j.ajpath.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Schweizer MT, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Science translational medicine. 2015;7:269ra262. doi: 10.1126/scitranslmed.3010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wikstrom P, Marusic J, Stattin P, Bergh A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. The Prostate. 2009;69:799–809. doi: 10.1002/pros.20927. [DOI] [PubMed] [Google Scholar]
- 257.Li Y, et al. Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J Cell Mol Med. 2008;12:2790–2798. doi: 10.1111/j.1582-4934.2008.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Holton SE, Bergamaschi A, Katzenellenbogen BS, Bhargava R. Integration of molecular profiling and chemical imaging to elucidate fibroblast-microenvironment impact on cancer cell phenotype and endocrine resistance in breast cancer. PloS one. 2014;9:e96878. doi: 10.1371/journal.pone.0096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Witkiewicz AK, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. The American journal of pathology. 2009;174:2023–2034. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Mercier I, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther. 2008;7:1212–1225. doi: 10.4161/cbt.7.8.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Silzle T, Randolph GJ, Kreutz M, Kunz-Schughart LA. The fibroblast: sentinel cell and local immune modulator in tumor tissue. International journal of cancer. Journal international du cancer. 2004;108:173–180. doi: 10.1002/ijc.11542. [DOI] [PubMed] [Google Scholar]
- 264.Talts JF, Wirl G, Dictor M, Muller WJ, Fassler R. Tenascin-C modulates tumor stroma and monocyte/macrophage recruitment but not tumor growth or metastasis in a mouse strain with spontaneous mammary cancer. J Cell Sci. 1999;112(Pt 12):1855–1864. doi: 10.1242/jcs.112.12.1855. [DOI] [PubMed] [Google Scholar]
- 265.Salmon H, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. The Journal of clinical investigation. 2012;122:899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Martinet L, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer research. 2011;71:5678–5687. doi: 10.1158/0008-5472.CAN-11-0431. [DOI] [PubMed] [Google Scholar]
- 267.Singh S, Ross SR, Acena M, Rowley DA, Schreiber H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med. 1992;175:139–146. doi: 10.1084/jem.175.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kraman M, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 269.Li X, et al. Stromal PD-L1 Expression Is Associated With Better Disease-Free Survival in Triple-Negative Breast Cancer. Am J Clin Pathol. 2016;146:496–502. doi: 10.1093/ajcp/aqw134. [DOI] [PubMed] [Google Scholar]
- 270.Miyoshi H, et al. PD-L1 expression on neoplastic or stromal cells is respectively a poor or good prognostic factor for adult T-cell leukemia/lymphoma. Blood. 2016;128:1374–1381. doi: 10.1182/blood-2016-02-698936. [DOI] [PubMed] [Google Scholar]
- 271.Pines M, Knopov V, Genina O, Lavelin I, Nagler A. Halofuginone, a specific inhibitor of collagen type I synthesis, prevents dimethylnitrosamine-induced liver cirrhosis. J Hepatol. 1997;27:391–398. doi: 10.1016/s0168-8278(97)80186-9. [DOI] [PubMed] [Google Scholar]
- 272.Zion O, et al. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas. 2009;38:427–435. doi: 10.1097/MPA.0b013e3181967670. [DOI] [PubMed] [Google Scholar]
- 273.Juarez P, et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer research. 2012;72:6247–6256. doi: 10.1158/0008-5472.CAN-12-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kultti A, et al. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp Cell Res. 2009;315:1914–1923. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 275.Hajime M, et al. Inhibitory effect of 4-methylesculetin on hyaluronan synthesis slows the development of human pancreatic cancer in vitro and in nude mice. International journal of cancer. Journal international du cancer. 2007;120:2704–2709. doi: 10.1002/ijc.22349. [DOI] [PubMed] [Google Scholar]
- 276.Wong KM, Horton KJ, Coveler AL, Hingorani SR, Harris WP. Targeting the Tumor Stroma: the Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20) Curr Oncol Rep. 2017;19:47. doi: 10.1007/s11912-017-0608-3. [DOI] [PubMed] [Google Scholar]
- 277.Erler JT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 279.Cox TR, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer research. 2013;73:1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Gilkes DM, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer research. 2013;73:3285–3296. doi: 10.1158/0008-5472.CAN-12-3963. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 281.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Miller BW, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sherman MH, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chronopoulos A, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nature communications. 2016;7:12630. doi: 10.1038/ncomms12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Alvarez R, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. British journal of cancer. 2013;109:926–933. doi: 10.1038/bjc.2013.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Von Hoff DD, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bonomi A, et al. Human amniotic mesenchymal stromal cells (hAMSCs) as potential vehicles for drug delivery in cancer therapy: an in vitro study. Stem Cell Res Ther. 2015;6:155. doi: 10.1186/s13287-015-0140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Levy O, et al. A prodrug-doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials. 2016;91:140–150. doi: 10.1016/j.biomaterials.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kidd S, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Brennen WN, Denmeade SR, Isaacs JT. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr Relat Cancer. 2013;20:R269–290. doi: 10.1530/ERC-13-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Clezardin P. Mechanisms of action of bisphosphonates in oncology: a scientific concept evolving from antiresorptive to anticancer activities. Bonekey Rep. 2013;2:267. doi: 10.1038/bonekey.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature reviews. Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Bagnato A, Rosano L. The endothelin axis in cancer. Int J Biochem Cell Biol. 2008;40:1443–1451. doi: 10.1016/j.biocel.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 294.Lalich M, McNeel DG, Wilding G, Liu G. Endothelin receptor antagonists in cancer therapy. Cancer Invest. 2007;25:785–794. doi: 10.1080/07357900701522588. [DOI] [PubMed] [Google Scholar]
- 295.Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85:717–724. doi: 10.1016/j.urology.2014.11.031. [DOI] [PubMed] [Google Scholar]
- 296.Nilsson S, et al. Two-year survival follow-up of the randomized, double-blind, placebo-controlled phase II study of radium-223 chloride in patients with castration-resistant prostate cancer and bone metastases. Clin Genitourin Cancer. 2013;11:20–26. doi: 10.1016/j.clgc.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 297.Liu R, Li H, Liu L, Yu J, Ren X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol Ther. 2012;13:123–129. doi: 10.4161/cbt.13.3.18696. [DOI] [PubMed] [Google Scholar]
- 298.Hofheinz RD, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26:44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 299.Scott AM, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:1639–1647. [PubMed] [Google Scholar]
- 300.Mersmann M, et al. Human antibody derivatives against the fibroblast activation protein for tumor stroma targeting of carcinomas. International journal of cancer. Journal international du cancer. 2001;92:240–248. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1170>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 301.Welt S, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1994;12:1193–1203. doi: 10.1200/JCO.1994.12.6.1193. [DOI] [PubMed] [Google Scholar]
- 302.Erickson HK, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer research. 2006;66:4426–4433. doi: 10.1158/0008-5472.CAN-05-4489. [DOI] [PubMed] [Google Scholar]
- 303.Ostermann E, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4584–4592. doi: 10.1158/1078-0432.CCR-07-5211. [DOI] [PubMed] [Google Scholar]
- 304.Fischer E, et al. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6208–6218. doi: 10.1158/1078-0432.CCR-12-0644. [DOI] [PubMed] [Google Scholar]
- 305.LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Molecular cancer therapeutics. 2009;8:1378–1386. doi: 10.1158/1535-7163.MCT-08-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Froeling FE, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486–1497. 1497 e1481–1414. doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 307.Guan J, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer letters. 2014;345:132–139. doi: 10.1016/j.canlet.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 308.Ghiaur G, Wroblewski M, Loges S. Acute Myelogenous Leukemia and its Microenvironment: A Molecular Conversation. Semin Hematol. 2015;52:200–206. doi: 10.1053/j.seminhematol.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 309.Ghiaur G, et al. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16121–16126. doi: 10.1073/pnas.1305937110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Mehra R, et al. Characterization of bone metastases from rapid autopsies of prostate cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:3924–3932. doi: 10.1158/1078-0432.CCR-10-3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Bai A, et al. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer research. 2010;70:7630–7639. doi: 10.1158/0008-5472.CAN-10-1489. [DOI] [PubMed] [Google Scholar]
- 312.Chae YK, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. 2017;8:16052–16074. doi: 10.18632/oncotarget.14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34:280–300. doi: 10.1002/med.21288. [DOI] [PubMed] [Google Scholar]
- 314.Bello E, et al. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer research. 2011;71:1396–1405. doi: 10.1158/0008-5472.CAN-10-2700. [DOI] [PubMed] [Google Scholar]
- 315.Gozgit JM, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Molecular cancer therapeutics. 2012;11:690–699. doi: 10.1158/1535-7163.MCT-11-0450. [DOI] [PubMed] [Google Scholar]
- 316.Biswas S, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. The Journal of clinical investigation. 2007;117:1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Domanska UM, et al. CXCR4 inhibition enhances radiosensitivity, while inducing cancer cell mobilization in a prostate cancer mouse model. Clinical & experimental metastasis. 2014;31:829–839. doi: 10.1007/s10585-014-9673-2. [DOI] [PubMed] [Google Scholar]
- 318.Domanska UM, et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia. 2012;14:709–718. doi: 10.1593/neo.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Feig C, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR, Greenberg PD. Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev. 2014;257:145–164. doi: 10.1111/imr.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Stromnes IM, et al. T Cells Engineered against a Native Antigen Can Surmount Immunologic and Physical Barriers to Treat Pancreatic Ductal Adenocarcinoma. Cancer cell. 2015;28:638–652. doi: 10.1016/j.ccell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Wang LC, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kakarla S, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Chen M, et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci Rep. 2015;5:14421. doi: 10.1038/srep14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Gottschalk S, Yu F, Ji M, Kakarla S, Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PloS one. 2013;8:e82658. doi: 10.1371/journal.pone.0082658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. The Journal of clinical investigation. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wen Y, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer science. 2010;101:2325–2332. doi: 10.1111/j.1349-7006.2010.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Long KB, et al. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400–413. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Winograd R, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res. 2015;3:399–411. doi: 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res. 2015;3:236–244. doi: 10.1158/2326-6066.CIR-14-0226. [DOI] [PubMed] [Google Scholar]
- 333.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Tada H, et al. Reprogrammed chondrocytes engineered to produce IL-12 provide novel ex vivo immune-gene therapy for cancer. Immunotherapy. 2017;9:239–248. doi: 10.2217/imt-2016-0004. [DOI] [PubMed] [Google Scholar]
- 335.Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–68. doi: 10.1016/j.semcancer.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 336.Liao Y, Ni Y, He R, Liu W, Du J. Clinical implications of fibroblast activation protein-alpha in non-small cell lung cancer after curative resection: a new predictor for prognosis. J Cancer Res Clin Oncol. 2013;139:1523–1528. doi: 10.1007/s00432-013-1471-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Edlund K, et al. CD99 is a novel prognostic stromal marker in non-small cell lung cancer. International journal of cancer. Journal international du cancer. 2012;131:2264–2273. doi: 10.1002/ijc.27518. [DOI] [PubMed] [Google Scholar]
- 338.Ishikawa S, et al. Matrix metalloproteinase-2 status in stromal fibroblasts, not in tumor cells, is a significant prognostic factor in non-small-cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:6579–6585. doi: 10.1158/1078-0432.CCR-04-0272. [DOI] [PubMed] [Google Scholar]
- 339.Ono S, et al. Podoplanin-positive cancer-associated fibroblasts could have prognostic value independent of cancer cell phenotype in stage I lung squamous cell carcinoma: usefulness of combining analysis of both cancer cell phenotype and cancer-associated fibroblast phenotype. Chest. 2013;143:963–970. doi: 10.1378/chest.12-0913. [DOI] [PubMed] [Google Scholar]
- 340.Saito RA, et al. Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer research. 2010;70:2644–2654. doi: 10.1158/0008-5472.CAN-09-3644. [DOI] [PubMed] [Google Scholar]
- 341.Chang HY, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS biology. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Chen JL, et al. Stromal responses among common carcinomas correlated with clinicopathologic features. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:5127–5135. doi: 10.1158/1078-0432.CCR-12-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nature medicine. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 344.Navab R, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7160–7165. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Planche A, et al. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PloS one. 2011;6:e18640. doi: 10.1371/journal.pone.0018640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Danaei G, et al. Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366:1784–1793. doi: 10.1016/S0140-6736(05)67725-2. [DOI] [PubMed] [Google Scholar]
- 347.Mandelker D, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. Jama. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Dry JR, Yang M, Saez-Rodriguez J. Looking beyond the cancer cell for effective drug combinations. Genome Med. 2016;8:125. doi: 10.1186/s13073-016-0379-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Micke P, Ostman A. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer. 2004;45(Suppl 2):S163–175. doi: 10.1016/j.lungcan.2004.07.977. [DOI] [PubMed] [Google Scholar]
- 351.Zhang P, Lehmann BD, Shyr Y, Guo Y. The Utilization of Formalin Fixed-Paraffin-Embedded Specimens in High Throughput Genomic Studies. Int J Genomics. 2017;2017:1926304. doi: 10.1155/2017/1926304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Dietrich D, et al. Analysis of DNA methylation of multiple genes in microdissected cells from formalin-fixed and paraffin-embedded tissues. J Histochem Cytochem. 2009;57:477–489. doi: 10.1369/jhc.2009.953026. [DOI] [PMC free article] [PubMed] [Google Scholar]