

Abstract
The twin-arginine translocation (Tat) system that comprises the TatA, TatB, and TatC components transports folded proteins across energized membranes of prokaryotes and plant plastids. It is not known, however, how the transport of this protein cargo is achieved. Favored models suggest that the TatA component supports transport by weakening the membrane upon full translocon assembly. Using Escherichia coli as a model organism, we now demonstrate in vivo that the N terminus of TatA can indeed destabilize the membrane, resulting in a lowered membrane energization in growing cells. We found that in full-length TatA, this effect is counterbalanced by its amphipathic helix. Consistent with these observations, the TatA N terminus induced proton leakage in vitro, indicating membrane destabilization. Fluorescence quenching data revealed that substrate binding causes the TatA hinge region and the N-terminal part of the TatA amphipathic helix to move toward the membrane surface. In the presence of TatBC, substrate binding also reduced the exposure of a specific region in the amphipathic helix, indicating a participation of TatBC. Of note, the substrate-induced reorientation of the TatA amphipathic helix correlated with detectable membrane weakening. We therefore propose a two-state model in which membrane-destabilizing effects of the short TatA membrane anchor are compensated by the membrane-immersed N-terminal part of the amphipathic helix in a resting state. We conclude that substrate binding to TatABC complexes switches the position of the amphipathic helix, which locally weakens the membrane on demand to allow substrate translocation across the membrane.
Keywords: protein translocation, membrane protein, protein-protein interaction, Escherichia coli (E. coli), conformational change, twin-arginine translocation
Introduction
The Tat3 system serves to transport folded proteins in bacteria, archaea, plant plastids, and possibly plant mitochondria (1–4). In Escherichia coli, the Tat system consists of TatA, TatB, and TatC components. TatA and TatB are similar, and two-component “minimal” Tat systems exist in which TatA exerts functions of TatA and TatB (5). TatA/B components are N-terminally membrane-anchored by a very short hydrophobic transmembrane domain, which is followed by a short hinge region, an amphipathic helix (APH), and a variable C-terminal domain (6). TatC is a polytopic membrane protein with six transmembrane domains (7, 8). TatB tightly interacts with TatC (9, 10). TatA associates with these TatBC complexes and is thought to permeabilize the membrane for protein transport (11–14). TatBC complexes recognize and tightly bind the signal peptides of the cargo proteins throughout the translocation process (15, 16).
The mechanism by which this translocation is achieved must be unusual and is not understood (17). A currently favored model suggests that the N termini of multiple TatA molecules at the translocon site weaken the membrane upon substrate binding, thereby permitting a TatC-mediated pulling of the substrate through the destabilized membrane (“membrane-weakening and pulling mechanism”; see Ref. 18). Molecular dynamics simulations support this view and suggest that multiple TatA N termini can cause a local thinning of the membrane (6). However, direct experimental evidence for a thinning stress imposed by the N terminus of TatA is lacking.
Here we demonstrate that the N terminus of TatA alone can indeed destabilize the membrane. This membrane stress relates to the short length of the TatA membrane anchor. In full-length TatA, the N-terminal half of the APH immerses into the membrane and counterbalances the effects of its own N-terminal membrane anchor. Importantly, substrate association with TatA induces a reorientation of the APH and TatA and thereby destabilizes the membrane to a measurable extent. In the absence of TatBC, the same substrate-induced changes occur in the N-terminal half of the APH, but the C-terminal part of the APH is not influenced. The data suggest that local membrane destabilization is indeed part of TatABC-catalyzed transport, and substrate association triggers this destabilization on demand.
Results
A cell-growth effect of the TatA membrane anchor that is compensated by the TatA amphipathic helix
TatA is a key component of the Tat system and considered to locally weaken the membrane, thereby, in conjunction with the other Tat system components, allowing the passage of folded proteins (18, 19). TatA is anchored by its N terminus in the cytoplasmic membrane (20). The N-terminal 6 residues are in the periplasmic surface of the membrane (6). The transmembrane helix (TMH) starts at Trp7/Gln8 on the periplasmic side and ends at Phe20 on the cytoplasmic side (see Fig. S1). The hydrophobic residues Leu9–Phe20 actually cross the lipid bilayer (21, 6). As the extremely short membrane-spanning region of TatA has been suggested to have the membrane-weakening effect, we tested whether production of the TatA membrane anchor (TatA-NT, residues 1–21) affects membrane stability. In earlier studies, we had already fused TatA-NT to a small, extremely stable soluble protein (the mature domain of the Tat substrate HiPIP from Allochromatium vinosum) (22), which stabilizes the peptide in vivo and renders it readily detectable by SDS-PAGE/Western blotting (23). In that earlier study, we had recognized that the TatA-NT-Hip construct induces the phage shock protein membrane stress response, indirectly indicating a membrane destabilization (23).
In continuation of that study, we systematically analyzed possible membrane-destabilizing effects of the TatA membrane anchor and included TatA-NT extensions to Gly33 (TatA-33), Ala42 (TatA-42), Lys49 (TatA-49), and full-length TatA to assess a possible influence of the adjacent amphipathic helix and further C-terminal regions (Fig. 1A). TatA-33 possesses the hinge region and the residues of the amphipathic helix up to the conserved flexible position Gly33. TatA-42 extends the construct to Ala42 near the end of the amphipathic helix, and TatA-49 further includes a highly charged stretch of residues (Asp-Asp-Glu-Pro-Lys) that is proposed to be important for function (24, 25). As TatA-NT was stabilized by a fusion to HiPIP, we also fused the other constructs C-terminally to HiPIP. Their production had differential effects on growth (Fig. 1B). Growth readily ceased upon induction of TatA-NT-Hip production. This effect was gradually reduced in longer constructs, in the order TatA-NT-Hip > TatA-33-Hip > TatA-42-Hip > TatA-49-Hip > full-length TatA-Hip. The differential effects of the various constructs were not caused by variation of protein abundances, as the two constructs with the strongest effects were even less abundant than the others (Fig. 1C). The two longest constructs showed heterogeneous migration behavior that might relate to distinct stable conformations. All constructs were stably integrated into membranes, as evidenced by carbonate washes (Fig. 1D).
Figure 1.

Inhibition of growth after production of TatA-NT-Hip. A, scheme of TatA, indicating the positions of the TMH, the hinge, the APH, the highly charged patch behind the APH, and the unstructured C-terminal domain. Numbered are positions to which the C-terminally Strep-tagged mature domain of HiPIP (experiments in Fig. 1) or only the Strep-tag (experiments in Fig. 2) have been fused to create truncated TatA variants (see “Experimental procedures”). Residues in capital letters have been structurally solved. B, growth curves of the tatAE-deletion strain JARV16 with recombinant rhamnose-induced production of either TatA-Hip, TatA-49-Hip, TatA-42-Hip, TatA-33-Hip, or TatA-NT-Hip. Induction of the pBW expression vectors was done with 0.1% rhamnose at the indicated time point. S.D. values (error bars) were calculated from three independent cultures. C, SDS-PAGE/Western blot analysis of subcellular fractions of the cultures analyzed in B. S, soluble fraction; M, membrane fraction. unspecific, a cross-reaction of the antibody. D, carbonate washes of the membrane-targeted constructs described above. M, membranes; S, supernatant after carbonate wash; P, pellet fraction after carbonate wash. Western blots were developed using specific HiPIP antibodies and ECL reaction.
To control whether the HiPIP fusion might have had effects that influenced the outcome, we also generated all constructs without fused HiPIP, although we knew that TatA-NT alone was unstable in vivo (Fig. 2A). TatA-NT still had some inhibitory effect. The extended TatA-NT constructs showed the same order with respect to their growth-inhibitory effect as the HiPIP-fused constructs, with TatA-33 > TatA-42 > TatA-49 > TatA (Fig. 2A). Again, the abundance and membrane-targeting of the proteins was assessed by SDS-PAGE/Western blotting (Fig. 2B). As expected, TatA-NT was extremely low abundant. The three extended constructs TatA-33, TatA-42, and TatA-49 were more stable than TatA-NT. Full-length TatA was more abundant than any other construct and had to be 4-fold diluted to achieve a comparable concentration that permitted a good ECL detection of the less abundant constructs. All constructs were membrane-targeted. Membrane integration was verified by carbonate washes (Fig. 2C). It is known that a portion of TatA can be extracted from membranes (26), and we observed this also for TatA-49. Together, the in vivo data indicated that the membrane anchor of TatA can strongly affect growth, and such effects are counterbalanced in full-length TatA and TatA-49. All characteristics that counterbalance the toxic effect of the TatA membrane anchor in these longer TatA constructs can be attributed to the region from Thr22 to Lys49, which is the amphipathic helix and the adjacent charged patch.
Figure 2.

Effect on growth after production of TatA and truncated TatA variants without HiPIP fusion. A, growth curves of the tatAE-deletion strain JARV16 with recombinant rhamnose-induced production of either TatA, TatA-49, TatA-42, TatA-33, or TatA-NT. All constructs were Strep-tagged. Induction of the pBW expression vectors was done with 0.1% rhamnose at the indicated time point. S.D. values (error bars) were calculated from three independent cultures. B, Tricine-SDS-PAGE/Western blot analysis of fractioned cells of JARV16 producing the indicated TatA-NT variants or TatA. S, soluble fraction; M, membrane fraction. C, carbonate washes of TatA and the truncated TatA variants. M, membranes; S, supernatant after carbonate wash; P, pellet fraction after carbonate wash. Western blots were developed using Strep-Tactin-AP conjugate.
The short length, not the sequence, of the hydrophobic TatA anchor region contributes to the growth phenotype
The above-described effect of the TatA membrane anchor raised the question whether the short membrane-spanning region in that anchor (Leu9–Phe20) was causing membrane damage. We assessed this aspect with further constructs. A TatA-NT(STGG)3-Hip construct was generated in which the 12 hydrophobic residues were substituted by a (STGG)3 linker. This substitution removes the hydrophobicity, which should result in a soluble protein. In addition, a TatA-NT(LA)6-Hip construct was generated in which Leu9–Phe20 were substituted by the artificial transmembrane domain LALALALALALA, which has been used in the past for analyses of artificial transmembrane helices (27). This construct should address questions of potential sequence specificity of observed effects. In construct TatA-NT(+6L)-Hip, we extended the hydrophobic transmembrane region by six leucine residues. This insertion was done behind the WQ motif that defines the beginning of the TMH (6). This third construct addressed the question of whether the short length of the TatA transmembrane region is responsible for the growth inhibition.
The growth was not influenced by TatA-NT(STGG)3-Hip, indicating that neither the unaltered 8 residues at the extreme N terminus nor the fused HiPIP domain had any negative effect (Fig. 3A). The construct was stable and fully soluble and tended to dimerize (Fig. 3B). Apparently, the growth defects depended on membrane targeting or the sequence of the transmembrane helix. In addition, this result confirmed that the fused HiPIP domain did not cause any toxic effect. The TatA-NT(LA)6-Hip construct demonstrated that the specific sequence of the membrane-spanning region was not relevant for the effect, as this construct showed the same strong effect on growth as TatA-NT-Hip (Fig. 3A). The TatA-NT(LA)6-Hip construct exerted this strong inhibition, albeit it was much less abundant than TatA-NT-Hip (Fig. 3B). The membrane-targeted TatA-NT(LA)6-Hip was stably membrane-integrated (Fig. 3C). Finally, the analysis of the TatA-NT(+6L)-Hip construct showed that the extension of the membrane-spanning hydrophobic stretch by 1½ helical turns already reduced the inhibitory effect (Fig. 3A), albeit the construct was more abundant than all the others (Fig. 3B). The membrane-targeted TatA-NT(+6L)-Hip was likewise stably membrane-integrated (Fig. 3C).
Figure 3.

The short membrane anchor of TatA is responsible for the growth defect. A, effect on growth of recombinant TatA-NT(STGG)3-Hip, TatA-NT(LA)6-Hip, and TatA-NT(+6L)-Hip. All constructs were Strep-tagged. Growth curves of the tatAE-deletion strain JARV16 with recombinant rhamnose-induced production of indicated TatA-NT-Hip variants. pBW expression vectors were induced by the addition of 0.1% rhamnose at the indicated time point. S.D. values (error bars) were calculated from three independent cultures. B, SDS-PAGE/Western blot analysis of membrane (M) and soluble (S) fractions of JARV16 producing the indicated TatA-NT-Hip variants, using HiPIP-specific antibodies and ECL detection. *, TatA-NT(STGG)3-Hip dimer. C, carbonate washes of membrane-targeted TatA-NT-Hip variants. Western blots were developed as described in B. M, membranes; S, supernatant after carbonate wash; P, membranes after carbonate wash.
The energization of the cytoplasmic membrane can be compromised by the membrane anchor of TatA
The growth defect was a first experimental hint of the previously postulated membrane-destabilizing effect of the TatA membrane anchor, as membrane destabilization can diminish transmembrane ion gradients and consequently ATP synthesis, transport processes, and metabolism, ultimately resulting in growth inhibition. To address potential effects on membrane energization more directly in vivo, we stained cells with JC-1, a green fluorescent dye that forms red aggregates in cells with energized membranes (28). Cells containing TatA-Hip, TatA-NT-Hip, TatA-NT(LA)6-Hip, and TatA-NT(STGG)3-Hip were examined JC1 fluorescence microscopy. Compared with TatA-Hip and TatA-NT(STGG)3-Hip, the TatA-NT-Hip and TatA-NT(LA)6-Hip constructs led to a clear reduction in red JC-1 fluorescence (Fig. 4A). The strains containing the TatA-NT-Hip, TatA-NT(LA)6-Hip, and TatA-NT(STGG)3-Hip constructs all formed cell chains, which is due to the nonfunctional Tat system in these strains (29). Due to the chain formation, counting of red fluorescent cells was not feasible. Nevertheless, we achieved a quantitative measure for cell energization by determining the green/red JC1-fluorescence ratio using fluorescence spectroscopy (Fig. 4B). Although this method gave quite large error bars, the results demonstrated a significant de-energization by TatA-NT-Hip, and the data also agree with the microscopically observed effect of TatA-NT(LA)6-Hip. As the (STGG)3 substitution abolished the effects on membrane energization seen by TatA-NT-Hip, any observed reductions in membrane energization were unrelated to Tat system nonfunctionality (Fig. 4B).
Figure 4.

TatA-NT-Hip reduces the membrane potential. A, fluorescence microscopy of JC1 staining of JARV16 producing either TatA-Hip, TatA-NT-Hip, TatA-NT(LA)6-Hip, or TatA-NT(STGG)3-Hip. All constructs were Strep-tagged. Formation of red J-aggregates was strongly reduced in TatA-NT-Hip– and TatA-NT(LA)6-Hip–producing cells. B, ratio of J-aggregates to JC1 monomers (590 nm/530 nm) of JARV16 producing the indicated TatA-NT-variant or TatA-Hip. Emission spectra were recorded with an excitation of 485 nm and emission from 500 to 650 nm. Ratios of peak maxima at 590 and 530 nm were calculated. Error bars, S.D.
Demonstration of proton leakage in vitro
The in vivo data indicated a negative influence of the TatA membrane anchor on cytoplasmic membrane energization. To overcome protein stability and growth effect issues that impeded experimental approaches in vivo, we decided to switch to a more defined experimental in vitro setup to study effects of the different TatA-NT constructs. In a first approach, we analyzed the membrane destabilization directly by acridine orange fluorescence quenching (Fig. 5). This approach monitors proton leakage as a measure for membrane destabilization. In this method, inverted cytoplasmic membrane vesicles (IMVs) are prepared and energized by the addition of ATP, which triggers the proton-pumping activity of ATP synthase (30). In the case of intact membranes, acridine orange that is present in the assay becomes protonated inside the acidified vesicles, which causes a fluorescence quenching. When the membranes show proton leakage, the proton gradient is less pronounced, resulting in a reduced acridine orange fluorescence quenching (23).
Figure 5.

The short membrane anchor of TatA can cause membrane defects. A and C, acridine orange quenching of TatBC IMVs (A) or DADE IMVs (C) with the indicated reconstituted in vitro translated TatA variants. B and D, comparison of relative abundance of the membrane-inserted TatA constructs in the vesicles used for the quenching experiments in A and C via Strep-Tactin Chromeo 488 labeling and fluorescence spectroscopy. E, quantification of the data shown in the traces below A and C, as derived from the recovery of fluorescence after CCCP addition. F, Tricine-SDS-PAGE/Western blot analysis of carbonate washes of membrane inserted TatA variants in TatBC IMVs (top) or DADE IMVs (bottom). All constructs were Strep-tagged. Western blots were developed using Strep-Tactin-HRP and ECL detection. M, vesicle membranes; P, pellet after carbonate wash; S, supernatant after carbonate wash. AU, arbitrary units.
First, we used TatBC-containing vesicles in which TatA was functionally inserted by in vitro translation. The identical in vitro translation conditions were then used for all TatA-NT constructs. Note that HiPIP was not fused in these experiments, as in vitro translated HiPIP does not assemble its iron-sulfur cofactor and thus cannot fold. This was irrelevant, as HiPIP was not required for TatA-NT stabilization under the in vitro conditions (Fig. 5, B and D). After optimization of the in vitro translations for the individual constructs, we could obtain quantitatively comparable amounts of all constructs inserted with surface-exposed C termini in the IMVs (Fig. 5, B and D). The constructs were quantified by the fluorescence of the Strep-Tactin Chromeo 488-conjugate. All constructs were stably integrated into the membrane (Fig. 5F). In the acridine orange assay, full-length TatA had only a slight effect on membrane stability (Fig. 5, A and E), which has been reported before (23). Quenching efficiency was determined as the percentage of quenching relative to the fluorescence level after carbonyl cyanide m-chlorophenyl hydrazone (CCCP)-induced fluorescence recovery. The effect of TatA-49 was comparable with TatA. Strikingly, TatA-NT strongly reduced the proton gradient that could be generated. TatA-33 and TatA-42 also showed a strong decrease in quenching efficiency that was slightly reduced but in the same range as TatA-NT. To assess whether the short length of the hydrophobic region in the TatA membrane anchor was responsible for this phenotype as it was in the in vivo assays, we tested also the TatA-NT(+6L) construct and found that the elongation of the hydrophobic helix by about 1.5 turns already resulted in clearly reduced membrane damage. We also checked whether the effect depended on the sequence of the TatA TMH using our TatA-NT(LA)6 construct. Again in agreement with the in vivo data, we could observe a strong effect of the TatA-NT(LA)6 on the acridine orange fluorescence quenching that was comparable with TatA-NT. We than asked the question of whether TatBC somehow contribute to the above described effects and performed the acridine orange quenching in IMVs derived from the tat-deficient E. coli strain DADE (31). As expected from the in vivo data, we found that TatBC did not play a role for the above described effects because the quenching efficiencies were comparable with the TatBC IMV data (Fig. 5, C and E). Again, the stable membrane insertion was confirmed (Fig. 5F). Together, these data demonstrate that the membrane anchor of TatA indeed can cause a TatBC-independent membrane defect that most likely relates to its very short length and not to its sequence, but in the longer TatA constructs, this defect is largely counterbalanced by the presence of the amphipathic helix and its neighboring charged region up to Lys49.
A conformational switch of the APH in response to substrate binding
As full-length TatA as well as TatA-NT-49 clearly masked the membrane defects generated by its own membrane anchor, we considered a direct role of the APH in membrane stabilization during a resting state. NMR studies (32) and biochemical accessibility studies (33) already indicated that the APH of TatA can adopt a tilted orientation at the membrane surface. The APH could directly contribute to membrane thickness at the membrane-anchor sites. To address this aspect, we introduced rhodamine modifications into single-cysteine variants of TatA and checked their position relative to the membrane. Depending on the degree of exposure, the fluorescence of rhodamine can be quenched by added potassium iodide (KI). Titration of KI into the sample can be used to calculate the Stern–Vollmer constant (KSV) for the individual rhodamine positions (see “Experimental procedures”). The higher this constant, the more solvent-exposed is the respective position. This method is a valuable and well-established tool to analyze protein accessibility in soluble and membrane-integral proteins (34, 35).
The positions for the rhodamine dye were chosen to address various aspects (Fig. 6A); position 2 faces the lumen of the IMVs and thus served as negative control. Within the TMH, we labeled positions 13, 16, 17, 18, and 19, the latter four covering a whole helical turn at the end of the hydrophobic transmembrane segment. Accessibility of this segment in any orientation to the aqueous surface in the course of substrate-induced conformational changes should thus become detectable. Dyes at positions 22, 23, 26, 27, 28, 32, 33, 34, 35, 42, and 45 cover the region from the hinge to the APH and an adjacent conserved negatively charged stretch. Position 51 served as positive control for quenching, as it is already positioned in an exposed region that is not required for functionality (25). Before carrying out the quenching experiments, we analyzed the functionality and membrane insertion of the selected single-cysteine variants in our in vitro translocation system with the model Tat substrate HiPIP (Fig. 6B). HiPIP is Tat exclusively transported due to the presence of a [4Fe-4S]2+/3+ cluster (22). In vitro translated TatA completes the active translocon in TatB/TatC-containing membrane vesicles. Notably, although transport with reconstituted TatA was generally less efficient than transport with TatABC-containing membranes, most TatA single cysteine variants were active to a detectable extent and transported the model Tat substrate HiPIP. Only TatA-K23C, TatA-L32C, and TatA-G33C constructs were inactive. In agreement with this, these variants were already described to inactivate TatA function in vivo (37). The transport mediated by TatA-T22C was strongly affected, which similarly agrees with this in vivo study. For TatA-A42C, we noted transport of HiPIP in vitro, although we could confirm the reported block of TorA transport for that variant (37). The inability to support TorA transport may thus relate to the size or other specific properties of TorA. All cysteine variants of TatA were integrated into the membrane and thus resistant to carbonate wash treatment (Fig. 6C).
Figure 6.

Quencher accessibility assays indicate a conformational switch of the APH upon substrate binding. A, TatA structural model (based on Protein Data Bank entry 2MN7 (21)), indicating positions of analyzed labels. Position Asp51 was not in the structure. B, in vitro HiPIP transport assays with in vitro translated WT TatA and TatA variants mutated at the indicated positions. All constructs were Strep-tagged. Transport assays with IMVs generated from MC4100 pABS-tatABC (pABS-tatABC) and with IMVs lacking TatA are shown as controls. T, lanes with thermolysine-digested IMVs after transport. Fluorescein-labeled HiPIP was used, and fluorescence of the precursor (pre, at ∼14 kDa) and mature (mat, at ∼10 kDa) forms was detected after SDS-PAGE (see “Experimental procedures”). Functional (+) and nonfunctional (−) transport are indicated. C and E, analysis of carbonate washes of TatBC IMVs (C) or DADE IMVs (E) with the reconstituted rhodamine-labeled TatA constructs. Rhodamine fluorescence was detected at ∼17 kDa after SDS-PAGE. M, membrane fraction before carbonate wash; P and S, pellet and supernatant fractions after carbonate wash. D and F, KSV values at the indicated positions for TatABC IMVs, in which TatBC-containing membranes were used for TatA insertion by in vitro translation (D), or for IMVs containing no TatBC and only in vitro translated TatA (F). Quenching results from experiments without substrate are shown as black columns, and results after the addition of 2 μm HiPIP precursor are shown in gray columns. Error bars, S.D.
The KI-quenching data for these TatABC membrane vesicles with rhodamine-labeled TatA variants generated by in vitro translation supported the reported tilted position of the APH relative to the membrane surface (32) and indicated clear differences in solvent exposure along the APH (Fig. 6D, dark columns). In the resting state (i.e. in the absence of Tat substrates), the APH intrudes into the membrane with its N-terminal half, as reflected by the low KSV values. This N-terminal region of the APH therefore appears to contribute to the thickness of the membrane at the position of the membrane anchor, thereby counterbalancing the membrane stress of the short membrane anchor. As expected for a transmembrane domain, the positions in the TMH of the membrane anchor were highly protected, indicating little or no access of the quencher KI. Beginning with the flexible and highly conserved Gly33 position (1), the APH was more surface-exposed with the exception of Ala42.
We then assessed potential effects of substrate binding by adding fully folded HiPIP precursor (Fig. 6D, gray columns). The most striking effect was a switch of the N-terminal half of the APH to a highly accessible, surface-exposed orientation. Also, the hinge region that connects the membrane-anchor with the APH moved to a surface-exposed position. In contrast, the rhodamine at positions 33, 34, and 35 became protected from quencher, and at positions 42, 45, and 51 the substrate addition had no effect. Together, the data thus indicate that the environment of the APH switches upon substrate interaction with the TatABC system. It is interesting that we did not detect any quencher accessibility to the transmembrane domain after substrate binding, although we have placed rhodamine dyes at four succeeding positions (positions 16–19) that cover a complete turn and thus all orientations of the membrane-spanning α-helix. Substrate binding thus does not seem to influence solvent accessibility of the TMH of TatA. It is also interesting that the region close to the very C terminus of the APH was not influenced by substrate binding.
Because it is known that TatA per se can interact with Tat substrates (26), we considered that TatA alone might already be able to make this switch. We thus carried out the same assays in the absence of other Tat components (IMVs prepared from the tatABCD-tatE deletion strain DADE). Again, all constructs were carbonate-resistantly membrane-integrated (Fig. 6E). The result was partially very similar (Fig. 6F); as in the case of membranes with complete TatABC systems, TatA alone showed a clear protection of the N-terminal half of the APH, suggesting a contribution to membrane thickness at the site of the TatA membrane anchor. Also as in the case of complete Tat systems, substrate binding resulted in a reorientation of the hinge and N-terminal half of the APH to a more surface-exposed position. However, in contrast to the observations in the complete TatABC system, positions 33–35 of the APH did not change their accessibility significantly when TatB and TatC were absent. It thus appears that from Gly33 on, there are no TatBC-independent changes in response to substrate binding, and therefore (i) there is probably flexibility at or near Gly-33, and (ii) the protection of Gly33–Ser35 upon substrate binding to TatABC systems depends on TatBC (see “Discussion”). Interestingly, in the absence of substrate, position 34 is somewhat less exposed with TatA alone than in the presence of TatBC, which might relate to a pre-existing TatBC interaction around Ala34, which is the region where we see the only TatBC effects. With respect to the method, it can be concluded that the use of single rhodamine dyes for fluorescence labeling did not interfere with a substrate-induced switch in the region from position 22 to 28, and it also permitted TatBC-dependent changes at positions 33–35.
Substrate-induced membrane destabilization by TatA
As TatA alone already showed the switch with respect to the N-terminal half of the APH, it was possible that some substrate-induced membrane weakening could be already detectable in the absence of TatBC. We thus carried out acridine orange fluorescence quenching with TatA IMVs that were either preincubated with Tat substrate precursor or mock-treated (Fig. 7). The results showed a reduced fluorescence quenching when Tat substrate was present, suggesting that indeed the above described reorientation of the APH in response to substrate interactions results in a measurable destabilization of IMVs. As it was in principle possible that Tat substrates alone already destabilized the membranes due to their reported lipid bilayer interaction (38–40), we carried out the same experiments with IMVs prepared from the Tat-deficient strain DADE. Notably, the preincubation of DADE IMVs with HiPIP precursor had no effect on the degree of fluorescence quenching in the assay, demonstrating that the effects were only due to substrate-TatA interactions and not due to a Tat-independent membrane interaction of Tat substrate.
Figure 7.
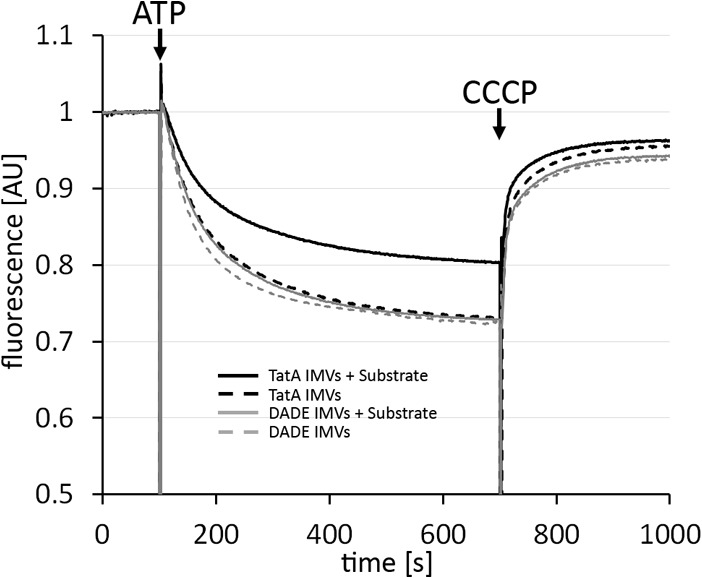
Substrate association with TatA alone can already affect membrane stability. Acridine orange fluorescence quenching with TatA IMVs without or with preincubation with 2 μm HiPIP precursor was performed. As control, the same experiment has been also carried out with IMVs from strain DADE (no Tat component), which demonstrates that HiPIP precursor has only a very weak effect in the absence of TatA. Time points of ATP and CCCP additions are indicated. All constructs were Strep-tagged. AU, arbitrary units.
Discussion
Substrate-induced reorientation of TatA amphipathic helices coincides with the induction of membrane defects at Tat translocation sites
We herein report a possible mechanism to induce local membrane stress on demand for Tat-dependent protein translocation. The N-terminal transmembrane domain of TatA can in principle generate membrane defects, but full-length TatA counterbalances these deleterious effects in the absence of Tat substrates. These conclusions are based on in vivo and in vitro approaches that demonstrate energization deficiencies of cells and membrane vesicles in response to the TatA constructs that are shorter than TatA-49. It thus is clear that the APH (∼Lys24–Met43) and most likely also the highly charged adjacent region (Asp45–Lys49) are counterbalancing detrimental effects of the TMH. This fully agrees with the minimally required length for TatA functionality as determined by in vivo studies (25).
The short length of the TMH is probably the reason for the destabilization, as this domain alone can reduce membrane energization, and this effect depends on the short length (Figs. 1, 2, 4, and 5). An elongation by 1½ helical turns markedly reduces the inhibitory effect (Figs. 3 and 5). The exact sequence of the TMH is not relevant, as an artificial TMH in TatA-NT-(LA)6 had the same effect (Figs. 3–5). This fits to the recent observation that the exact sequence of that region is not a determinant for function, as it can be exchanged between E. coli and thylakoid systems (41). Our data now experimentally show the postulated membrane weakening by the unusual TMH of TatA (18), which was so far supported by molecular dynamics simulations (6). In this TMH, only 12 hydrophobic residues cross the lipid bilayer. Notably, a stretch of 12 hydrophobic residues, although very short, is sufficiently long for a complete membrane integration of a peptide by the Sec61 translocon in eukaryotes (42).
A membrane-weakening as the basis for Tat transport would need to be transient and local. The Tat system needs to minimize constitutive membrane stress under “resting” conditions (i.e. when no substrate is present to be transported). According to our data, this can be in principle achieved by the influence of the substrate-dependent orientational switch of the APH. TatA alone is able to achieve the counterbalancing of membrane weakening (Fig. 7). The N-terminal part of its APH can immerge into the lipid bilayer in the absence of substrate, and it becomes surface-exposed in response to added substrate. TatA can thus sense and respond to substrate already in the absence of TatBC.
What drives the conformational switch?
One important question is the understanding of the driving force for the reorientation of the APH in response to substrate. We have recently demonstrated that TatA can interact with Tat substrates in a signal peptide–dependent, RR-independent manner (26). In that study, also molecular dynamics simulations were carried out that indicated intensive contacts between the APH of TatA and mainly the C-domain of the signal peptide. This fully agrees with the fact that the RR motif is not recognized by TatA (26).
Molecular dynamics simulations of TatA in membranes suggest that only oligomeric TatA assemblies can destabilize the membrane (6). As we see a membrane destabilization, substrate binding therefore probably induces the conformational switch of TatA that is organized in clusters. These clusters may transiently form or pre-exist at TatBC. TatA alone can also destabilize membranes, and it is therefore likely that the known TatBC-independent TatA clusters (43) can already associate with substrate. The group of Ken Cline (12, 44, 45) demonstrated in seminal studies substrate-induced TatA-TatBC and TatA-TatA cross-links that can be explained by TatA recruitments, rearrangements, or conformational transitions. Later, a substrate-induced conformational transition of the APH that is similar to the one that we now show for the E. coli system had been clearly demonstrated for the thylakoid system by Aldridge et al. (33). The data of that study are largely consistent with our data, and in this study, it was already hypothesized that a moved APH could induce a membrane weakening. We now provide experimental evidence for such a membrane destabilization and relate this to a substrate-induced conformational change that restricts the membrane thickness to the short length of the TMH, which is probably the physiological function. As the switch of the APH conformation is a consequence of a substrate interaction, the APH itself is the most likely interaction site. An APH interaction with mature domains of Tat substrates had been for the first time experimentally demonstrated by the group of Carole Dabney-Smith (46) for the thylakoidal system. Also, in the E. coli system, mature domains of Tat substrates have been shown to interact with TatA (16, 47–49), and it has been demonstrated that TatA has the capacity to interact with Tat substrates in a TatBC-independent manner (26). For the thylakoid system, it has been suggested that the APH interactions relate to nonspecific passive contacts before or during the membrane passage of mature domains, as a cross-link to a position at the end of the APH (Phe48) was TatB- and proton-motive force-dependent (46). In agreement with the thylakoidal system data, we also found no substrate effects at the C-terminal end of the APH in the absence of TatBC (Fig. 6F). The APH thus most likely binds Tat substrates around the N terminus of the APH, as evidenced by large changes in response to substrate addition in that region (Thr22–Ile28) in the presence as well as in the absence of TatBC (Fig. 6, D and F). Three of all analyzed positions were inactivated by their exchange to cysteine. The accessibility assays with these positions were nevertheless similar to neighboring positions, and effects were detected in response to substrate (K23C) or TatBC (G33C). Most likely, the absence of unexpected effects for these positions can be explained by the fact that the accessibility assays are end point measurements that do not monitor transport kinetics or overall functionality. If, for example, an exchange slows an important movement by 3 orders of magnitude from a millisecond to a second range, which would be a virtual block of transport, the assay would not differentiate this and recognize the movement. It also could be that the reversibility of a movement can be affected by inactivating mutations. We consider both options but favor the kinetic argumentation. The data thus can be carefully taken, and they agree with the overall conclusions.
In conclusion, we propose that the interaction of Tat substrates with the APH induces the conformational switch most likely by rearranging the APH-APH interactions in TatA clusters. Future studies will hopefully clarify this aspect.
The involvement of TatBC-independent steps in Tat transport is not unexpected
It has been believed for a long time that all Tat transport is initiated by an interaction of the signal peptide twin-arginine motif with TatBC and that TatA is recruited thereafter. This view is currently challenged by the observation that TatA has binding sites at “resting” TatBC complexes (50, 51) and that TatA can be co-purified with TatBC equally well when the substrate-binding site of TatC is inactivated (52). The initial experimental evidence for a transient recruitment of TatA to TatBC upon substrate binding to TatBC is based on cross-link data that may also have resulted from rearrangements of TatA at TatBC upon substrate binding (12, 44, 45). Other lines of evidence come from TatA that has been labeled with fluorescent protein (XFP) tags (53, 54). The problem of these approaches is that the dissociation of such TatA-XFP fusions from TatBC could be triggered by the ∼26-kDa XFP tag attached to the 9.6-kDa TatA. In agreement with this, it has been observed that TatA-YFP was functionally inactive, and activity could be only re-established by co-production of nontagged TatA or TatE (54). Substrate binding might influence the affinity of TatBC for TatA, as mutations that enhance the affinity of TatBC to Tat substrates show a constitutive TatA-YFP/TatBC interaction (55). An increased affinity of TatBC for TatA could therefore compensate for XFP-tag effects. The transient recruitment of nontagged TatA to TatBC thus remains to be shown. Based on the substrate-independent TatA/TatBC interactions, we currently favor the view that TatA and TatBC always function hand in hand and remain in close proximity.
The RR motif of Tat signal peptides can remain associated with TatC during translocation (15), and Tat substrates are contacting TatA during transport with their mature domains (26, 46, 49). TatA recognizes the transported domains to initiate the conformational switch, but TatBC recognizes the RR motif, and transport is only achieved when both activities cooperate. As TatA and TatBC act synergistically on distinct regions of the transported substrate, it is not surprising that the individual TatA/Tat substrate interaction does not require an RR recognition. It would be delusive to neglect the TatBC-independent TatA interactions; the membrane destabilization appears to rely on them.
TatBC alters the accessibility of a specific region of the APH in the presence of Tat substrate
As TatA and TatBC cooperate for Tat transport, it is important to recognize that we could detect an influence of TatBC on the accessibility of TatA regions. The main difference between the observations with TatA vesicles versus TatABC vesicles is related to the Gly33/Ala34/Ser35 region of the APH. Whereas the accessibility of this region is essentially unaltered upon substrate interaction in the absence of TatBC, this region becomes protected upon substrate interaction in the presence of TatBC, suggestive of a TatBC “footprint” on the TatA APH that indicates a steric hindrance of solvent accessibility by a TatBC-dependent environment. This may be (i) TatBC itself, which changes conformation upon substrate binding, or it may be (ii) the substrate, which might tightly associate with this region of the TatA-APH. Both scenarios are conceivable, and a direct substrate interaction with the APH has been already demonstrated (46). Also, the substrate-induced accessibility differences that have been detected in the thylakoid system were considered to potentially indicate a steric hindrance by substrate (33). In this context, one significant difference of our data from the data reported by Aldridge et al. (33) may be highly important; the movement of the hinge region preceding the APH was not detected in that study. We think that this difference is caused by the much larger size of the modifying agent (methyl-PEG12)3-PEG4-maleimide (2360 Da) that had to be used to detect modifications by shifts in SDS-PAGE analyses. We used iodide ions (127 Da), which can access positions that are sterically inaccessible for larger molecules. It is tempting to speculate that the substrate is positioned on top of the hinge region, ready to be transported, and thereby reduces the accessibility for larger agents.
In summary, we propose the following mechanism of membrane destabilization and counterbalancing (Fig. 8). The hydrophobic region of the TatA membrane-anchor is very short and destabilizes the membrane if the N-terminal region of the APH does not immerse into the membrane and thereby elongates the membrane-spanning section. Full-length TatA controls this membrane destabilization with its APH, whose N-terminal region is able to switch its orientation in response to substrate association. This switch restricts membrane thickness to the length of the TMH, which results in a membrane destabilization on demand that can be demonstrated by substrate-induced proton leakage in vitro. The essential role of TatBC as translocon core and motor is not called into question by the membrane-weakening role of TatA. Future experiments will surely clarify these aspects, leading to a deeper understanding of the translocation mechanism.
Figure 8.
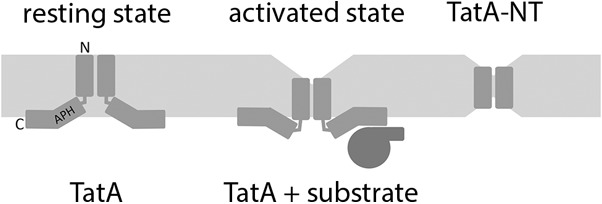
A model for the generation of substrate-induced membrane stress at Tat translocons. Whereas the APH of TatA contributes to membrane thickness in a resting state (left), substrate binding causes a conformational switch that reduces membrane thickness (middle). The N-terminal transmembrane domain of TatA alone (TatA-NT) destabilizes membranes by membrane thinning (right). See “Discussion” for details.
Experimental procedures
Strains and growth conditions
E. coli strain MC4100 and indicated derivatives thereof were used for growth experiments and in vitro studies, and E. coli XL1-blue was used for cloning. Strains were grown aerobically at 37 °C in LB medium (1% tryptone, 1% NaCl, 0.5% yeast extract) in the presence of 100 μg/ml ampicillin. For growth curves, 25-ml cultures were inoculated with OD600 of 0.1 and grown aerobically. The OD600 was determined in 30-min intervals. For induction of gene expression, 0.1% rhamnose was added after 90 min.
Genetic methods and plasmids
All constructs used in this study were Strep-tagged for their detection. Construction of pBW-tatA-strep and pBW-tatA-NT-mhip-strep were described elsewhere (23). pBW-tatA-33-strep, pBW-tatA-42-strep, and pBW-tatA-49-strep were cloned via standard methods with forward primer tatA-NdeI-F (5′-TCT TCT CAT ATG GGT GGT ATC AGT ATT TGG C-3′) and the corresponding reverse primer tatA(G33)-BamHI-R (5′-TTA AAG GAT CCA CCA AGA TCG GAA CCG ATG G-3′), tatA(A42)-BamHI-R (5′-TTA AAG GAT CCT GCT TTT TTA AAG CCT TTG ATC G-3′), or BamHI-tatA(K49)-R (5′-GAC GGA TCC CTT TGG TTC ATC ATC GCT C-3′), respectively. mhip fusions were cloned via BglII/BamHI, and pBW-tatA-NT-strep were cloned using pBW-tatA-strep with an additional BamHI site after G21 (23), cut with BamHI, and religated. pBW-tatA-NT(STGG)3-mhip-strep, pBW-tatA-NT(LA)6-mhip-strep, and pBW-tatA(9 + 6L)-mhip-strep were constructed using pBW-tatA-NT-mhip-strep as template and forward primers NdeI-tatA-NT-(LA)6-mhip-F (5′-TAG CCA TAT GGG TGG TAT CAG TAT TTG GCA GTT AGC GCT TGC ATT GGC ACT GGC TCT CGC GCT TGC AGG CAC CAA AAA GCT CGG CTC CAT CGG-3′), NdeI-tatA-NT(STGG)3-mhip-F (5′-ATA TAC ATA TGG GTG GTA TCA GTA TTT CGA CGG GTG GGG GCA GTA CGG GTG GCG GGA GTA CGG GTG GGG GCT CCG CTC CCG CCA ATG CCG TGG CC-3′), and NdeI-tatA-NT(+6L)-F (5′-AAG TAC ATA TGG GTG GTA TCA GTA TTT GGC AGC TGC TCT TAC TGT TGC TGT TAT TGA TTA TTG CCG TCA TC-3′) together with reverse primer hip-BamHI-R (5′-ATA TAT AGG ATC CGC CGG CCT TCA GGG TC-3′) for amplification. PCR products were cut with NdeI/BamHI and cloned in the corresponding sites of pBW-tatA-NT-mhip-strep, thereby exchanging the fragment. All constructs were verified via restriction analysis and sequencing.
For in vitro transcription and translation, the tatA gene or the Strep-tagged N-terminal membrane anchor–encoding sequence of TatA were cloned into pGEM-5Zf (Promega) using the forward primer tatA-NdeI-F (5′-TCT TCT CAT ATG GGT GGT ATC AGT ATT TGG C-3′) in combination with tatA-PstI-R (5′- GGA CTG CAG TTA CAC CTG CTC TTT ATC GTG-3′) and tatA-NTstrep-PstI-R (5′-GAC TGC AGT TAT TTT TCG AAC TGC GGG TGG CTC CAG GTG CCA AAA AGC AGT ACA AC-3′), resulting in pGEM-tatA or pGEM-tatA-NTstrep. For the construction of the pGEM-5Zf–based in vitro transcription system for tatA-NT(+6L), primers NdeI-tatA-NT(+6L)-F and tatA-NTstrep-PstI-R were combined for PCR, and the product was cloned into the corresponding sites of pGEM-5Zf. For the construction of tatA-33-strep, tatA-42-strep and tatA-49-strep in vitro transcription vectors, the corresponding pBW-tatA-33-strep, pBW-tatA-42-strep and pBW-tatA-49-strep vectors were digested with NdeI/SalI, and the fragments were cloned into the corresponding sites of pGEM-5Zf. The indicated codon mutations in tatA were introduced into pGEM-tatA by QuikChange mutagenesis (Stratagene). All constructs were confirmed by sequencing.
General biochemical methods
SDS-PAGE and subsequent Western blotting were carried out by standard procedures (56, 57). For small proteins, Schägger gels (16% T, 6 m urea) were performed as described elsewhere (58). Western blots were developed employing specific HiPIP antibodies, Strep-Tactin-AP, or Strep-Tactin-HRP conjugate as indicated according to the manufacturer's instructions (IBA, Göttingen, Germany). Images of Western blots were acquired utilizing the Intas Advanced Imager (INTAS Science Imaging Instruments GmbH, Göttingen, Germany).
For all in vivo experiments, Strep-tagged TatA constructs were produced using pBW22-based plasmids (23, 59). For preparation of proteins, cell fractionation experiments, and carbonate washes, 100-ml cultures of E. coli JARV16 were inoculated with an OD600 of 0.1, and pBW22-based protein production was induced after 90 min when cell densities reached an OD600 of ∼0.6. After 3 h, cells were harvested, and cell densities were normalized to an OD600 of 1 before centrifugation of a 100-ml culture at 4500 × g for 10 min at 4 °C. Cells were resuspended in 2 ml of homogenization buffer (50 mm Tris-HCl, pH 8, 250 mm NaCl). 1 mm PMSF and DNase I were added, and cells were homogenized via French press (2 passages, 800 p.s.i.). After homogenization, cell debris was removed by centrifugation for 10 min at 20,000 × g at 4 °C. For cell fractionation, membranes were sedimented by ultracentrifugation at 130,000 × g for 30 min at 4 °C of one-half of the crude extract. Supernatant (soluble fraction) and membranes (resuspended in 1 volume of homogenization buffer) were analyzed by SDS-PAGE/Western blotting. For carbonate washes, the second half of the crude extract was used and also centrifuged at 4 °C for 30 min at 130,000 × g. The supernatant was carefully discarded, and crude membranes were resuspended with ice-cold 100 mm Na2CO3. After a second centrifugation step at 4 °C for 30 min at 130,000 × g, the supernatant was removed, and washed membranes were resuspended in 1 volume of 100 mm Na2CO3 again. All fractions were analyzed with SDS-PAGE/Western blotting. For JC1 staining, E. coli strain JARV16 was used. 25-ml cultures were inoculated with an OD600 of 0.05, and protein production was induced by adding 0.2% rhamnose directly after inoculation. When the maximum expression level was reached after 3 h, cells were harvested, and the density was normalized to 1 ml of OD600 = 1. After centrifugation at 3000 × g for 10 min, cell pellets were resuspended in 0.5 ml of TEG staining buffer (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 10 mm glucose) complemented with 0.1% (v/v) JC1 (5 mg/ml JC1 stock solution) and incubated for 30 min in the dark on a roller. Cells were sedimented again by centrifugation and washed in 2 volumes of TEG buffer without dye. After centrifugation, cells were resuspended in 1 volume of TEG buffer, and 2 μl of the cell suspension were spotted on an agar slide for fluorescence microscopy. Cells were imaged by fluorescence microscopy, using a Zeiss AxioImager M2 microscope equipped with an AxioCam MRm camera and a Zeiss ×100/numeric aperture 1.3 EC Plan-Neofluar objective. For quantification of JC1 fluorescence, 100 μl of the washed cell suspension were added to 1.4 ml of TEG buffer and transferred into a cuvette. Fluorescence emission spectra were recorded from 500 to 650 nm with an excitation at 485 nm, using the JASCO FP-6500 spectrofluorometer (Jasco International Co., Ltd., Tokyo, Japan). Spectra were analyzed, and 590 nm/530 nm ratios were calculated using JASCO Spectra Manager software.
In vitro translocation assays were carried out as described previously (60). As translocation substrate, we used a T61C variant of the HiPIP precursor, which was fluorescence-labeled by modification of the cysteine with fluorescein-5-maleimide using standard protocols for maleimide coupling (61). HiPIP precursor was refolded from inclusion bodies and purified as described previously (36). For in vitro transcription, the tatA or tatA-NTstrep regions in the corresponding pGEM constructs were amplified using the pGEM-recognizing primers pGEM-5-F (5′-CCC AGT CAC GAC GTT GTA AAA CG-3′) and pGEM-5-R (5′-CTT CCG GCT CGT ATG TTG TG-3′), and the purified PCR product was used as template for SP6 RNA polymerase according to the supplier's protocol (New England Biolabs). Translation was done according to standard protocols (62).
In vitro reconstitution of cysteine-labeled TatA into inverted membrane vesicles
Fluorescence-labeled TatA variants were inserted into IMVs prepared from E. coli strain DADE/pBW-tatBC (52) by employing wheat germ extract–based in vitro translation according to the protocol of Lin et al. (62), but in the presence of IMVs (A280 = 4) and with 600 pmol/ml tetramethylrhodamine (TMR)-cysteine-tRNACys.
For preparation of TMR-cysteine-tRNACys, uncharged tRNACys was partially purified from bakers' yeast tRNA (Sigma) using a Mono Q 5/50 GL (GE Healthcare) strong anion exchanger. 5 mg of bakers' yeast tRNA dissolved in 1 ml of Buffer A (20 mm HEPES, 0.26 m NaCl) was applied on the column equilibrated with the same buffer, and tRNAs were eluted by a linear 0.26–1 m NaCl gradient over 40 ml, and 1-ml fractions were collected. The tRNACys was predominately eluted at 0.5 m NaCl with a yield of ∼250 μg/ml (∼10 μm). Ethanol-precipitated tRNACys was then subjected to aminoacylation with cysteine. A 2-ml aminoacylation reaction contained 250 μg of tRNACys, 1 mm l-cysteine, 100 mm HEPES, pH 8.0, 10 mm MgSO4, 10 mm KCl, 2 mm DTT, 4 mm ATP, 100 mm CTP, and 200 μl of E. coli S-100 fraction (36), and the reaction was incubated at 37 °C for 90 min. After phenolization and ethanol precipitation, the charged Cys-tRNACys was rechromatographed as before. Aminoacylation caused almost no difference on the elution of tRNACys. To reduce the possibly oxidized thiol group of Cys, 10 mm DTT was added to the elution fractions. The fraction containing Cys-tRNACys was dialyzed twice against 1 liter of degassed 50 mm KH2PO4/K2HPO4, pH 7.3, 100 mm NaCl for 1 h in a cold room, and the recovered Cys-tRNACys was labeled with TMR by the addition of 1 mm TMR-5-maleimide (Sigma) for 30 min at 20 °C. Afterward, the unreacted TMR-5-maleimide was quenched by 2 mm DTT and dialyzed against Buffer A at 4 °C. The TMR-Cys-tRNACys was purified by chromatography as before, and the elution fractions containing fluorescent TMR-Cys-tRNACys were combined and ethanol-precipitated. TMR-Cys- tRNACys was stored at −80 °C.
IMVs with fluorescence-labeled TatA were applied onto a 500-μl 0.5 m sucrose cushion and sedimented for 30 min at 130,000 × g and 4 °C to remove soluble compounds. After washing with IMV buffer (0.25 m sucrose, 20 mm HEPES, pH 7.5, 1 mm DTT), the IMVs were suspended in 200 μl of buffer.
Rhodamine collisional fluorescence-quenching assay
To examine collisional quenching of rhodamine fluorescence at specific TatA positions, IMVs containing TMR-labeled TatA were suspended in 1.5 ml of IMV buffer to a final concentration of A280 = 2. TMR emission intensity was measured at λex = 543 nm and λem = 575 nm on a JASCO FP-6500 spectrofluorometer with constant stirring. Fluorescence intensity was averaged from five successive 5-s integrations. The quencher KI was then added from a 4 m stock solution to final concentrations of 10, 20, or 40 mm, and the respective emission intensities were continually recorded. Data were analyzed using the Stern–Volmer equation, (F0/F) − 1 = KSV [Q], where F0 is the net emission intensity in the absence of quencher, [Q] is the concentration of quencher, F is the net emission intensity at [Q], and KSV is the Stern–Volmer constant. A linear dependence of quenching ((F0/F) − 1) on [Q] indicates direct proportionality as expected for collisional quenching. KSV corresponds to the slope of a linear least-squares best fit to the data points in which the line is constrained to go through the origin.
Acridine orange fluorescence-quenching assays
Acridine orange fluorescence-quenching assays were performed on the JASCO spectrofluorometer. A 1.5-ml reaction mixture contained IMVs (A280 = 2) in SHM buffer (250 mm sucrose, 10 mm HEPES, pH 7.5, 5 mm MgSO4), 25 mm creatine phosphate, 50 μg/ml creatine phosphate kinase, and 2 μm acridine orange. The mixture was incubated in a cuvette at 20 °C in the dark with constant stirring, and acridine orange fluorescence was measured in a time-course manner at λex = 494 nm and λem = 540 nm. After 100 s, acridine orange quenching was induced by the addition of 1.25 mm ATP, and the measurement was continued for another 600 s. 3 μl of 5 mm CCCP was then added to the cuvette to abolish the proton gradient, and the change of the acridine orange fluorescence intensity was recorded for another 300 s.
Fluorospectrometric estimation of relative recombinant protein concentration in membranes
TatA and its variants used in acridine orange assays possess a C-terminal Strep-tag that is exposed to the outside of the IMVs. Relative amounts of membrane-targeted Strep-tagged protein could therefore be estimated by use of fluorescence-labeled Strep-Tactin. 3 μl of 0.5 mg/ml Strep-Tactin Chromeo 488 (IBA Lifesciences) was added to 50 μl of IMVs (A280 = 20) harboring in vitro–translated TatA-strep and its variants. After incubation at room temperature for 30 min with shaking, the IMVs were purified by sedimentation at 130,000 × g for 30 min through a 0.5 m sucrose cushion. IMVs were washed once with SHM buffer and resuspended in 1.5 ml of SHM buffer. Using an excitation wavelength at 488 nm, the relative fluorescence of IMV-bound Strep-Tactin Chromeo 488 was determined at 520 nm.
Author contributions
B. H. performed the in vitro assays, E. S. H. performed the in vivo experiments, and D. M. performed experiments in early stages of the project. All authors analyzed the data. E. S. H. and T. B. prepared figures, and T. B. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Sybille Traupe and Inge Reupke for technical support.
This work was supported by DFG Grant BR2285/4-2. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
- Tat
- twin-arginine translocation
- APH
- amphipathic helix
- HiPIP
- high-potential iron sulfur protein
- IMV
- inner membrane vesicle
- NT
- N-terminal domain
- CCCP
- carbonyl cyanide m-chlorophenyl hydrazone
- TMR
- tetramethylrhodamine
- TMH
- transmembrane helix
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- YFP
- yellow fluorescent protein.
References
- 1. Hou B., and Brüser T. (2011) The Tat-dependent protein translocation pathway. Biomol. Concepts 2, 507–523 [DOI] [PubMed] [Google Scholar]
- 2. Natale P., Brüser T., and Driessen A. J. (2008) Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane–distinct translocases and mechanisms. Biochim. Biophys. Acta 1778, 1735–1756 10.1016/j.bbamem.2007.07.015 [DOI] [PubMed] [Google Scholar]
- 3. Palmer T., and Berks B. C. (2012) The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10, 483–496 10.1038/nrmicro2814 [DOI] [PubMed] [Google Scholar]
- 4. Carrie C., Weissenberger S., and Soll J. (2016) Plant mitochondria contain the protein translocase subunits TatB and TatC. J. Cell Sci. 129, 3935–3947 10.1242/jcs.190975 [DOI] [PubMed] [Google Scholar]
- 5. Goosens V. J., Monteferrante C. G., and van Dijl J. M. (2014) The Tat system of Gram-positive bacteria. Biochim. Biophys. Acta 1843, 1698–1706 10.1016/j.bbamcr.2013.10.008 [DOI] [PubMed] [Google Scholar]
- 6. Rodriguez F., Rouse S. L., Tait C. E., Harmer J., De Riso A., Timmel C. R., Sansom M. S., Berks B. C., and Schnell J. R. (2013) Structural model for the protein-translocating element of the twin-arginine transport system. Proc. Natl. Acad. Sci. U.S.A. 110, E1092–E1101 10.1073/pnas.1219486110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Behrendt J., Standar K., Lindenstrauss U., and Brüser T. (2004) Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol. Lett. 234, 303–308 10.1111/j.1574-6968.2004.tb09548.x [DOI] [PubMed] [Google Scholar]
- 8. Rollauer S. E., Tarry M. J., Graham J. E., Jääskeläinen M., Jäger F., Johnson S., Krehenbrink M., Liu S.-M., Lukey M. J., Marcoux J., McDowell M. A., Rodriguez F., Roversi P., Stansfeld P. J., Robinson C. V., et al. (2012) Structure of the TatC core of the twin-arginine protein transport system. Nature 492, 210–214 10.1038/nature11683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolhuis A., Mathers J. E., Thomas J. D., Barrett C. M., and Robinson C. (2001) TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J. Biol. Chem. 276, 20213–20219 10.1074/jbc.M100682200 [DOI] [PubMed] [Google Scholar]
- 10. Richter S., and Brüser T. (2005) Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J. Biol. Chem. 280, 42723–42730 10.1074/jbc.M509570200 [DOI] [PubMed] [Google Scholar]
- 11. Cline K., and Mori H. (2001) Thylakoid ΔpH-dependent precursor proteins bind to a cpTatC-Hcf106 complex before Tha4-dependent transport. J. Cell Biol. 154, 719–729 10.1083/jcb.200105149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dabney-Smith C., Mori H., and Cline K. (2006) Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J. Biol. Chem. 281, 5476–5483 10.1074/jbc.M512453200 [DOI] [PubMed] [Google Scholar]
- 13. Blümmel A.-S., Haag L. A., Eimer E., Müller M., and Fröbel J. (2015) Initial assembly steps of a translocase for folded proteins. Nat. Commun 6, 7234 10.1038/ncomms8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hauer R. S., Schlesier R., Heilmann K., Dittmar J., Jakob M., and Klösgen R. B. (2013) Enough is enough: TatA demand during Tat-dependent protein transport. Biochim. Biophys. Acta 1833, 957–965 10.1016/j.bbamcr.2013.01.030 [DOI] [PubMed] [Google Scholar]
- 15. Gérard F., and Cline K. (2006) Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J. Biol. Chem. 281, 6130–6135 10.1074/jbc.M512733200 [DOI] [PubMed] [Google Scholar]
- 16. Alami M., Lüke I., Deitermann S., Eisner G., Koch H. G., Brunner J., and Müller M. (2003) Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell 12, 937–946 10.1016/S1097-2765(03)00398-8 [DOI] [PubMed] [Google Scholar]
- 17. Keegstra K., Werner-Washburne M., Cline K., and Andrews J. (1984) The chloroplast envelope: is it homologous with the double membranes of mitochondria and Gram-negative bacteria? J. Cell. Biochem. 24, 55–68 10.1002/jcb.240240105 [DOI] [PubMed] [Google Scholar]
- 18. Brüser T., and Sanders C. (2003) An alternative model of the twin arginine translocation system. Microbiol. Res. 158, 7–17 10.1078/0944-5013-00176 [DOI] [PubMed] [Google Scholar]
- 19. Cline K. (2015) Mechanistic aspects of folded protein transport by the twin arginine translocase (Tat). J. Biol. Chem. 290, 16530–16538 10.1074/jbc.R114.626820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Porcelli I., de Leeuw E., Wallis R., van den Brink-van der Laan E., de Kruijff B., Wallace B. A., Palmer T., and Berks B. C. (2002) Characterization and membrane assembly of the TatA component of the Escherichia coli twin-arginine protein transport system. Biochemistry 41, 13690–13697 10.1021/bi026142i [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y., Hu Y., Li H., and Jin C. (2014) Structural basis for TatA oligomerization: an NMR study of Escherichia coli TatA dimeric structure. PLoS One 9, e103157 10.1371/journal.pone.0103157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brüser T., Yano T., Brune D. C., and Daldal F. (2003) Membrane targeting of a folded and cofactor-containing protein. Eur. J. Biochem. 270, 1211–1221 10.1046/j.1432-1033.2003.03481.x [DOI] [PubMed] [Google Scholar]
- 23. Mehner D., Osadnik H., Lünsdorf H., and Brüser T. (2012) The Tat system for membrane translocation of folded proteins recruits the membrane-stabilizing Psp machinery in Escherichia coli. J. Biol. Chem. 287, 27834–27842 10.1074/jbc.M112.374983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Warren G., Oates J., Robinson C., and Dixon A. M. (2009) Contributions of the transmembrane domain and a key acidic motif to assembly and function of the TatA complex. J. Mol. Biol. 388, 122–132 10.1016/j.jmb.2009.02.060 [DOI] [PubMed] [Google Scholar]
- 25. Lee P. A., Buchanan G., Stanley N. R., Berks B. C., and Palmer T. (2002) Truncation analysis of TatA and TatB defines the minimal functional units required for protein translocation. J. Bacteriol. 184, 5871–5879 10.1128/JB.184.21.5871-5879.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taubert J., Hou B., Risselada H. J., Mehner D., Lünsdorf H., Grubmüller H., and Brüser T. (2015) TatBC-independent TatA/Tat substrate interactions contribute to transport efficiency. PLoS One 10, e0119761 10.1371/journal.pone.0119761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krishnakumar S. S., and London E. (2007) Effect of sequence hydrophobicity and bilayer width upon the minimum length required for the formation of transmembrane helices in membranes. J. Mol. Biol. 374, 671–687 10.1016/j.jmb.2007.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Becker L. A., Bang I.-S., Crouch M.-L., and Fang F. C. (2005) Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol. Microbiol. 56, 1004–1016 10.1111/j.1365-2958.2005.04604.x [DOI] [PubMed] [Google Scholar]
- 29. Ize B., Stanley N. R., Buchanan G., and Palmer T. (2003) Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol. Microbiol. 48, 1183–1193 10.1046/j.1365-2958.2003.03504.x [DOI] [PubMed] [Google Scholar]
- 30. Warnock D. G., Reenstra W. W., and Yee V. J. (1982) Na+/H+ antiporter of brush border vesicles: studies with acridine orange uptake. Am. J. Physiol. 242, F733–F739 [DOI] [PubMed] [Google Scholar]
- 31. Wexler M., Sargent F., Jack R. L., Stanley N. R., Bogsch E. G., Robinson C., Berks B. C., and Palmer T. (2000) TatD is a cytoplasmic protein with DNase activity: no requirement for TatD family proteins in sec-independent protein export. J. Biol. Chem. 275, 16717–16722 10.1074/jbc.M000800200 [DOI] [PubMed] [Google Scholar]
- 32. Walther T. H., Grage S. L., Roth N., and Ulrich A. S. (2010) Membrane alignment of the pore-forming component TatA(d) of the twin-arginine translocase from Bacillus subtilis resolved by solid-state NMR spectroscopy. J. Am. Chem. Soc. 132, 15945–15956 10.1021/ja106963s [DOI] [PubMed] [Google Scholar]
- 33. Aldridge C., Storm A., Cline K., and Dabney-Smith C. (2012) The chloroplast twin arginine transport (Tat) component, Tha4, undergoes conformational changes leading to Tat protein transport. J. Biol. Chem. 287, 34752–34763 10.1074/jbc.M112.385666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mori H., Tsukazaki T., Masui R., Kuramitsu S., Yokoyama S., Johnson A. E., Kimura Y., Akiyama Y., and Ito K. (2003) Fluorescence resonance energy transfer analysis of protein translocase. SecYE from Thermus thermophilus HB8 forms a constitutive oligomer in membranes. J. Biol. Chem. 278, 14257–14264 10.1074/jbc.M300230200 [DOI] [PubMed] [Google Scholar]
- 35. Hackeng T. M., Yegneswaran S., Johnson A. E., and Griffin J. H. (2000) Conformational changes in activated protein C caused by binding of the first epidermal growth factor-like module of protein S. Biochem. J. 349, 757–764 10.1042/bj3490757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson A. E., Woodward W. R., Herbert E., and Menninger J. R. (1976) Ne-acetyllysine transfer ribonucleic acid: a biologically active analogue of aminoacyl transfer ribonucleic acids. Biochemistry 15, 569–575 10.1021/bi00648a018 [DOI] [PubMed] [Google Scholar]
- 37. Greene N. P., Porcelli I., Buchanan G., Hicks M. G., Schermann S. M., Palmer T., and Berks B. C. (2007) Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the bacterial twin arginine translocase. J. Biol. Chem. 282, 23937–23945 10.1074/jbc.M702972200 [DOI] [PubMed] [Google Scholar]
- 38. Hamsanathan S., Anthonymuthu T. S., Bageshwar U. K., and Musser S. M. (2017) A hinged signal peptide hairpin enables Tat-dependent protein translocation. Biophys. J. 113, 2650–2668 10.1016/j.bpj.2017.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shanmugham A., Wong Fong Sang H. W., Bollen Y. J., and Lill H. (2006) Membrane binding of twin arginine preproteins as an early step in translocation. Biochemistry 45, 2243–2249 10.1021/bi052188a [DOI] [PubMed] [Google Scholar]
- 40. Brehmer T., Kerth A., Graubner W., Malesevic M., Hou B., Brüser T., and Blume A. (2012) Negatively charged phospholipids trigger the interaction of a bacterial Tat substrate precursor protein with lipid monolayers. Langmuir 28, 3534–3541 10.1021/la204473t [DOI] [PubMed] [Google Scholar]
- 41. Hauer R. S., Freudl R., Dittmar J., Jakob M., and Klösgen R. B. (2017) How to achieve Tat transport with alien TatA. Sci. Rep. 7, 8808 10.1038/s41598-017-08818-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaud S., Fernández-Vidal M., Nilsson I., Meindl-Beinker N. M., Hübner N. C., Tobias D. J., von Heijne G., and White S. H. (2009) Insertion of short transmembrane helices by the Sec61 translocon. Proc. Natl. Acad. Sci. U.S.A. 106, 11588–11593 10.1073/pnas.0900638106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Leeuw E., Porcelli I., Sargent F., Palmer T., and Berks B. C. (2001) Membrane interactions and self-association of the TatA and TatB components of the twin-arginine translocation pathway. FEBS Lett. 506, 143–148 10.1016/S0014-5793(01)02904-0 [DOI] [PubMed] [Google Scholar]
- 44. Mori H., and Cline K. (2002) A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid ΔpH/Tat translocase. J. Cell Biol. 157, 205–210 10.1083/jcb.200202048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dabney-Smith C., and Cline K. (2009) Clustering of C-terminal stromal domains of Tha4 homo-oligomers during translocation by the Tat protein transport system. Mol. Biol. Cell 20, 2060–2069 10.1091/mbc.E08-12-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pal D., Fite K., and Dabney-Smith C. (2013) Direct interaction between a precursor mature domain and transport component Tha4 during twin arginine transport of chloroplasts. Plant Physiol. 161, 990–1001 10.1104/pp.112.207522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maurer C., Panahandeh S., Jungkamp A.-C., Moser M., and Müller M. (2010) TatB functions as an oligomeric binding site for folded Tat precursor proteins. Mol. Biol. Cell 21, 4151–4161 10.1091/mbc.E10-07-0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Panahandeh S., Maurer C., Moser M., DeLisa M. P., and Müller M. (2008) Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. J. Biol. Chem. 283, 33267–33275 10.1074/jbc.M804225200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taubert J., and Brüser T. (2014) Twin-arginine translocation-arresting protein regions contact TatA and TatB. Biol. Chem. 395, 827–836 [DOI] [PubMed] [Google Scholar]
- 50. Habersetzer J., Moore K., Cherry J., Buchanan G., Stansfeld P. J., and Palmer T. (2017) Substrate-triggered position switching of TatA and TatB during Tat transport in Escherichia coli. Open Biol. 7, 170091 10.1098/rsob.170091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aldridge C., Ma X., Gerard F., and Cline K. (2014) Substrate-gated docking of pore subunit Tha4 in the TatC cavity initiates Tat translocase assembly. J. Cell Biol. 205, 51–65 10.1083/jcb.201311057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Behrendt J., and Brüser T. (2014) The TatBC complex of the Tat protein translocase in Escherichia coli and its transition to the substrate-bound TatABC complex. Biochemistry 53, 2344–2354 10.1021/bi500169s [DOI] [PubMed] [Google Scholar]
- 53. Rose P., Fröbel J., Graumann P. L., and Müller M. (2013) Substrate-dependent assembly of the Tat translocase as observed in live Escherichia coli cells. PLoS One 8, e69488 10.1371/journal.pone.0069488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alcock F., Baker M. A. B., Greene N. P., Palmer T., Wallace M. I., and Berks B. C. (2013) Live cell imaging shows reversible assembly of the TatA component of the twin-arginine protein transport system. Proc. Natl. Acad. Sci. U.S.A. 110, E3650–E3659 10.1073/pnas.1306738110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang Q., Alcock F., Kneuper H., Deme J. C., Rollauer S. E., Lea S. M., Berks B. C., and Palmer T. (2017) A signal sequence suppressor mutant that stabilizes an assembled state of the twin arginine translocase. Proc. Natl. Acad. Sci. U.S.A. 114, E1958–E1967 10.1073/pnas.1615056114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 58. Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protoc. 1, 16–22 10.1038/nprot.2006.4 [DOI] [PubMed] [Google Scholar]
- 59. Wilms B., Hauck A., Reuss M., Syldatk C., Mattes R., Siemann M., and Altenbuchner J. (2001) High-cell-density fermentation for production of l-N-carbamoylase using an expression system based on the Escherichia coli rhaBAD promoter. Biotechnol. Bioeng. 73, 95–103 10.1002/bit.1041 [DOI] [PubMed] [Google Scholar]
- 60. Stolle P., Hou B., and Brüser T. (2016) The Tat substrate CueO is transported in an incomplete folding state. J. Biol. Chem. 291, 13520–13528 10.1074/jbc.M116.729103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim Y., Ho S. O., Gassman N. R., Korlann Y., Landorf E. V., Collart F. R., and Weiss S. (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 19, 786–791 10.1021/bc7002499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin P.-J., Jongsma C. G., Liao S., and Johnson A. E. (2011) Transmembrane segments of nascent polytopic membrane proteins control cytosol/ER targeting during membrane integration. J. Cell Biol. 195, 41–54 10.1083/jcb.201103117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.