

Abstract
A new self-assembly method is used to rapidly functionalize the surface of liposomes without perturbing the membrane integrity or causing leakage of the aqueous contents. The key molecule is a cholesterol-squaraine-PEG conjugate with three important structural elements: a cholesterol membrane anchor, a fluorescent squaraine docking station that allows rapid and high-affinity macrocycle threading, and a long PEG-2000 chain to provide steric shielding of the decorated liposome. The two-step method involves spontaneous insertion of the conjugate into the outer leaflet of pre-formed liposomes followed by squaraine threading with a tetralactam macrocycle that has appended targeting ligands. A macrocycle with six carboxylates permitted immobilization of intact fluorescent liposomes on the surface of cationic polymer beads, whereas a macrocycle with six zinc(II)-dipicolylamine units enabled selective targeting of anionic membranes, including agglutination of bacteria in the presence of human cells.
Keywords: liposomes, membranes, supramolecular chemistry, fluorescent probes, cell recognition
Graphical Abstract
Insertion of a cholesterol-squaraine-PEG conjugate into the outer leaflet of a pre-formed liposome followed by macrocycle threading of the squaraine component produces a functionalized deep-red fluorescent liposome.
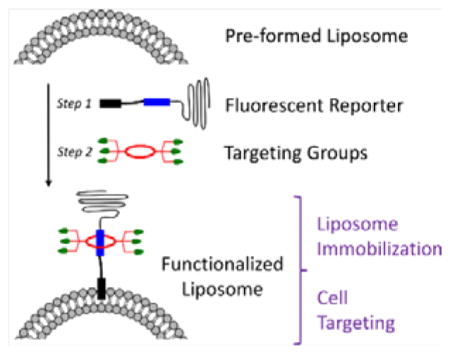
Introduction
Liposomes have been investigated extensively for use as nanocapsules for nanotechnology, drug delivery and molecular imaging.[1,2] The first generation of therapeutic liposomes to receive clinical approval were non-targeted carriers of a pharmaceutical payload,[3] and building on this success is an ongoing effort to develop next generation surface-functionalized nanoparticles for cell or tissue selective targeting.[4–6] The design of targeted liposomes must satisfy several requirements for successful operation in living subjects.[7] The liposome exterior must contain multiple copies of an effective targeting agent, but it also must be protected by long hydrophilic polymers such as polyethylene glycol (PEG) that stabilize the colloidal particles and block undesired uptake by the reticuloendothelial system (RES).[8,9] In addition, there is great value gained by incorporating a reporter group into the liposome structure to permit imaging experiments that can assess the success of the targeting strategy.[10] The simplest way to fabricate targeted liposomes is to disperse a pre-mixed film containing the various membrane components. However this method results in unwanted decoration of the liposome inner monolayer, which wastes valuable targeting components and also may complicate the subsequent loading of liposome payload.[11] Ideally, surface functionalization should be done after liposome fabrication and payload incorporation. Most studies that decorate the surface of liposomes use covalent conjugation chemistry, and while useful targeted systems have been developed, the bimolecular reactions are relatively slow and it is hard to ascertain if and when they have reached completion.[12–20] This is potentially problematic because it leaves reactive groups on the liposome surface which could abrogate selective targeting, thus requiring an additional chemical capping step.[21] Rapid surface functionalization is highly desirable with pre-formed liposomes that are filled with payload since it minimizes the time period for unwanted background leakage of the payload. In principle, the liposome exterior could be rapidly decorated using non-covalent assembly methods that have inherently low activation barriers, but progress has been limited, in large part because there are very few association systems that have the desired combination of supramolecular properties. Two early approaches, using biotin/(strept)avidin[22] or polyhistidine/Ni(II) chelate[23] as the affinity partners to attach proteins to liposomes, were shown to have various performance drawbacks. More recent designs, using liposomes with embedded container molecules such amphiphilic cyclodextrin[24] or calixarene[25] derivatives as docking sites for targeting ligands, have produced interesting in vitro results but are likely to have limited in vivo utility due to the modest association constants.
In recent years we have developed a new non-covalent, self-assembly process that we call Synthavidin (Synthetic Avidin).[26] In short, a deep-red fluorescent squaraine dye is encapsulated by a water-soluble tetralactam macrocycle to give an extremely stable, threaded complex that is well suited for fluorescence imaging. To date, we have pre-assembled fluorescent molecular probes that selectively target bone, dead/dying mammalian tissue, and various microbial species.[27,28] One of the major advantages of Synthavidin self-assembly is the capability to form threaded complexes with long PEG chains appended to the ends of the squaraine dye.[29,30] This means the length of the PEG chains is a structural parameter that can be systematically varied in order to optimize the pharmacokinetic properties of the pre-assembled probes. In this report we describe for the first time how Synthavidin self-assembly can be exploited as a new and generalizable non-covalent method to functionalize the surface of pre-formed liposomes. A summary of the two-step liposome surface functionalization method is shown in Scheme 1. The central molecule is the newly designed cholesterol-squaraine-PEG conjugate, CSP, which has three important structural elements: a cholesterol membrane anchor, a fluorescent squaraine docking station that allows rapid and high-affinity macrocycle threading, and a long PEG-2000 chain to provide steric shielding of the decorated liposome. The first step in the method is spontaneous insertion of the CSP conjugate into the outer leaflet of pre-formed liposomes without disturbing the bilayer. In step two the squaraine component of the anchored CSP is threaded with a tetralactam macrocycle that has appended targeting ligands. The study employed two different tetralactam macrocycles, 6C with six carboxylates which enhance liposome affinity for cationic surfaces, and 6Z with six zinc(II)-dipicolylamine (ZnDPA) units which enable selective targeting of anionic membranes and cell surfaces.[31] A major attribute of this surface functionalization method is its efficiency, as it simultaneously endows the liposomes with targeting ligand, protective PEG chains, and deep-red fluorescence reporter group. A potential concern at the start of the study was whether the surrounding corona of PEG chains would attenuate the targeting capabilities of the sheltered ligands. The limited literature on this broadly important question suggested that the outcome was context dependent.[21,32–34]
Scheme 1.

Summary of two-step method for surface functionalization of POPC liposomes.
Results and Discussion
The CSP conjugate was synthesized in straightforward fashion by conducting two sequential azide/alkyne cycloaddition reactions that attached azido-cholesterol and azido-PEG-2000 components to each end of a central squaraine bis(alkyne) dye (Scheme S2). The squaraine dye has two important structural features: (a) its C2O4 core is flanked by two 2-aminothiophene rings which donate electron density to the core and provide high chemical stability in aqueous solution, and (b) an N-methyl group at the end of the dye ensures rapid and high-affinity macrocycle threading.[30] As a free compound, the CSP conjugate is soluble in water but self-aggregated, as indicated by a broadened squaraine absorption band, quenched fluorescence and observation of ~7 nm micelles using dynamic light scattering (Figures 1A and S1). Mixing an aqueous solution of self-aggregated CSP with an aliquot of pre-formed, unilamellar 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes (200 nm diameter) resulted in fluorescence turn-on as the CSP inserted into the membrane outer leaflet to create POPC@CSP liposomes. This membrane insertion step was complete within 60 s (Figure 1B) and the squaraine fluorescence remained with the liposomes after they were filtered through a size exclusion column, indicating that the CSP was embedded in the liposomes. This conclusion was confirmed by separates experiments that inserted CSP into POPC liposomes containing the lipophilic fluorescence resonance energy transfer (FRET) partner DiI and observing FRET from the DiI donor to CSP acceptor (Figure S2). The demonstration that CSP inserts strongly into the liposomes is important since recent literature indicates that not all cholesterol-based amphiphiles are well anchored in bilayer membranes.[35,36,37]
Figure 1.
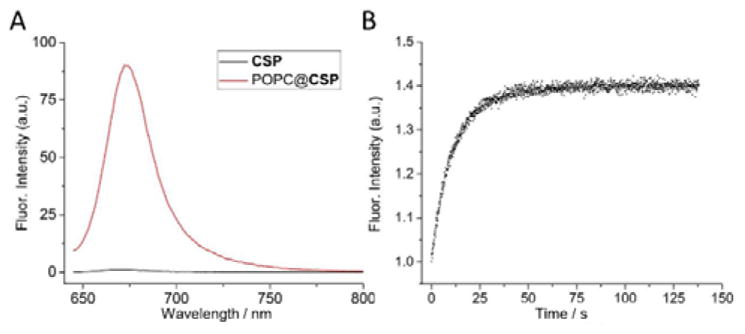
(A) Fluorescence spectra (ex: 630 nm) of CSP (1 μM) in water or after insertion into POPC liposomes. (B) Change in CSP fluorescence (ex: 630 nm, em: 680 nm) over time after addition of POPC liposomes and waiting ~10 s. In both panels, CSP was 0.3 mol% respective to POPC.
Step two of the liposome surface functionalization method is macrocycle threading of the squaraine dye that projects from the exterior of POPC@CSP liposomes. The feasibility of this threading step was initially tested using the multianionic macrocycle, 6C, which exhibits “ideal” supramolecular behaviour (i.e., the multianionic macrocycle and its threaded complexes have no tendency to self-aggregate) while conferring the liposome with the ability to target cationic surfaces. Shown in Figure 2A–B are the spectral changes observed when POPC@CSP liposomes were treated with tetralactam macrocycle 6C to produce POPC@CSP⊃6C liposomes. The threading process was indicated by diagnostic ~20–40 nm red-shifts in the deep-red squaraine absorption/fluorescence maxima (Table S2), and formation of the threaded complex was confirmed by observing efficient fluorescence energy transfer from the anthracene sidewalls of the surrounding macrocycle (ex: 390 nm) to the encapsulated squaraine. The threading-induced red-shift in squaraine emission maxima made it easy to conduct fluorescence titration experiments that added aliquots of 6C to POPC@CSP liposomes where the loading of CSP was 0.3 mol% respective to POPC. Macrocycle threading was complete within seconds after each aliquot addition, and as shown in Figure 2C the titration isotherms fitted well to a 1:1 association model. A remarkably high Ka of (2.8 ± 0.2) × 108 M−1 was determined which is quite close to the Ka previously determined for threading of 6C by a water-soluble analogue of CSP (Scheme S3 and Table S1) and shows that the surface of the POPC@CSP liposomes does not inhibit squaraine threading of 6C.[38] Passage of the surface functionalized POPC@CSP⊃6C liposomes through a size exclusion column did not lower the fluorescence signal, indicating that the threaded complex remained embedded in the liposomes. Furthermore, a series of FRET experiments using liposomes with embedded DiI as a FRET partner confirmed that there was no transfer of the threaded complex out of the POPC@CSP⊃6C liposomes into a large excess of neighbouring liposomes (Figures S2–S4). A series of comparative fluorescence experiments also proved that there was no translocation of liposome-embedded CSP or the CSP⊃6C complex across the bilayer membrane. This was achieved by comparing the absorption and fluorescence spectra for a series of liposome dispersions with the following sets of labelling conditions: (a) CSP or CSP⊃6C complex embedded only in the membrane outer leaflet (Figure S5), (b) CSP or CSP⊃6C complex embedded equally in both membrane leaflets, and (c) CSP⊃6C complex only in the membrane outer leaflet and CSP only in the membrane outer leaflet (Figure S6). The spectra showed that the fraction of CSP or CSP⊃6C complex in each membrane leaflet was unchanged over time. Analyses using dynamic light scattering (DLS) indicated that the average liposome size did not change during the two-step surface functionalization process to make the POPC@CSP⊃6C liposomes (Figure 2D). Additionally, liposome leakage experiments revealed that the surface functionalization procedure induced no significant release of liposome aqueous contents (chloride anions) (Figure S7).
Figure 2.
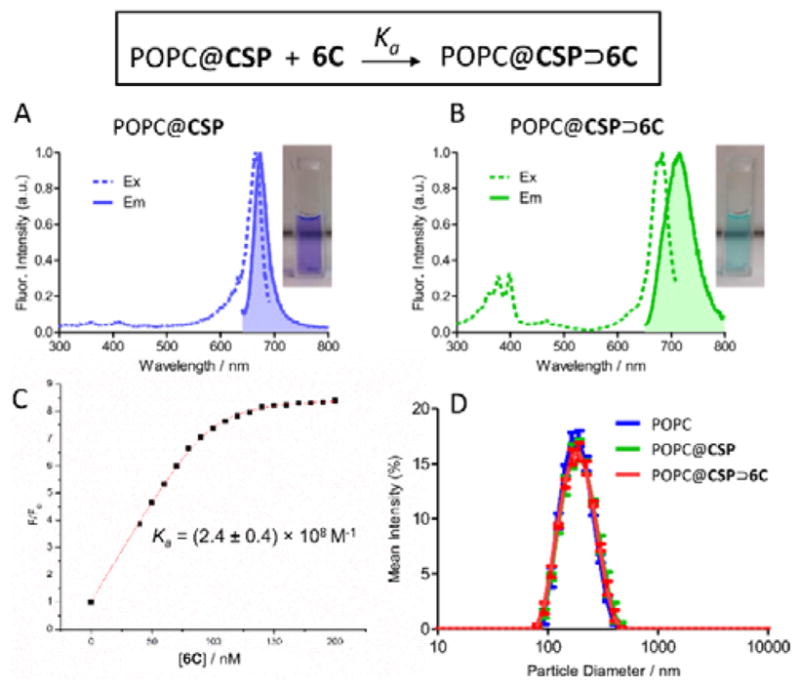
Fluorescence excitation/emission spectra of; (A) POPC@CSP liposomes and (B) POPC@CSP⊃6C liposomes. (C) Fluorescence titration that added 6C to POPC@CSP liposomes and monitored increase in fluorescence intensity (F/Fo, ex: 690 nm, em: 700 nm) due to formation of POPC@CSP⊃6C. (D) Dynamic light scattering showed that liposome surface functionalization produced no change in liposome size. In all panels, the CSP was 0.3 mol% respective to POPC.
In order to prepare a cell targeting system, we chose to decorate the liposome surface with ZnDPA units. Fluorescence titration experiments mixed POPC@CSP liposomes with the macrocycle 6Z (appended with six ZnDPA units) to produce targeted multivalent POPC@CSP⊃6Z liposomes. Consistent with the preparation of POPC@CSP⊃6C liposome described above, the threading of 6Z was indicated by a red-shift in squaraine absorption/emission bands and the Ka was determined to be (2.4 ± 0.4) × 108 M−1 (Figure S8C). Additional experiments showed that the two-step surface functionalization process to produce POPC@CSP⊃6Z liposomes caused no change in liposome size (Figure S8D) and no significant leakage of liposome aqueous contents (sulforhodamine dye) (Figure S13). Furthermore, the embedded CSP⊃6Z complex remained in the liposome outer leaflet and did not transfer over time to other liposomes in the sample (Figure S9) or translocate to the inner leaflet of the liposome membrane (Figure S10–S11). However, the threading to produce POPC@CSP⊃6C and POPC@CSP⊃6Z liposomes differed in two major ways. One was the relatively slow rate of 6Z threading on the surface of the POPC@CSP liposomes. As shown in Figure 3A the threading of 6C was complete within a few seconds, whereas the threading of 6Z required ~20 minutes (Figure 3A). Previous studies have shown that the threading of 6Z by a dispersed, water soluble squaraine is fast[28] so the slow formation of POPC@CSP⊃6Z must be a liposome surface effect. The most likely explanation is surface promoted association of the ZnDPA units in the newly docked 6Z with neighbouring CSP conjugates, driven by Lewis acid/base interactions between the Zn(II) cations and the electron rich squaraine oxygen atoms, as reported for related metal cation/dye association systems.[39] While the threading rate to form POPC@CSP⊃6Z liposomes was slower than POPC@CSP⊃6C liposomes, it is important to emphasize that both processes reached stoichiometric completion in a practically useful time period. The second major difference between POPC@CSP⊃6C and POPC@CSP⊃6Z liposomes was the fluorescence brightness. As shown in Figure 3B the fluorescence intensity of POPC@CSP⊃6Z liposomes was about four times lower, which matches previous observations that fluorescent ZnDPA complexes have a tendency to become self- aggregated and self-quenched on a membrane surface.[28] As shown below, relief of this self-quenching effect can be exploited to achieve target-activated turn-on fluorescence.
Figure 3.
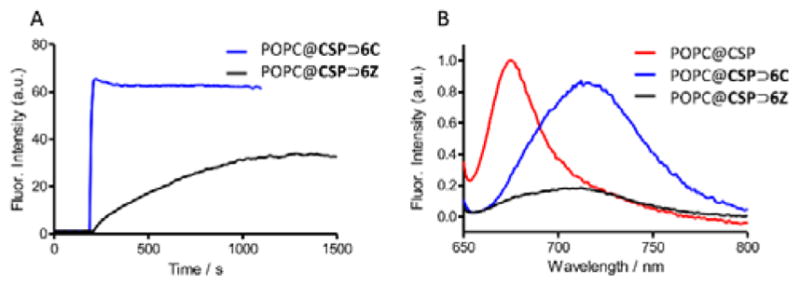
(A) Kinetics of association as measured by fluorescence (ex: 685 nm em: 710 nm). Threading by 6C occurred in seconds while complete threading by 6Z required ~20 min. (B) Emission spectra (ex: 640 nm) demonstrating POPC@CSP⊃6Z was partially quenched compared with POPC@CSP⊃6C.
The next stage of the research was to assess the targeting capabilities of the functionalized POPC@CSP⊃6C and POPC@CSP⊃6Z liposomes. We expected that the anionic POPC@CSP⊃6C liposomes would associate with cationic surfaces, and this was confirmed by conducting a series of simple experiments using rigid cationic polymeric beads (0.7 mm diameter). As shown in Figure 4A–B, beads treated for 30 minutes with POPC@CSP⊃6C liposomes, followed by extensive washing, acquired a characteristic squaraine color and fluorescence, while beads treated with untargeted POPC@CSP liposomes did not. Evidence that the adhered POPC@CSP⊃6C liposomes were structurally intact was gained by repeating the experiments using liposomes containing rhodamine 123 (Rh) as a fluorescent marker of aqueous contents. There was no escape of Rh from the adhered liposomes until the sample was treated with detergent as a membrane lysis agent (Figure 4C). These results demonstrate how Synthavidin self-assembly can be used to immobilize intact fluorescent liposomes on synthetic surfaces for possible use in nanotechnology applications such as sensing arrays.[40]
Figure 4.
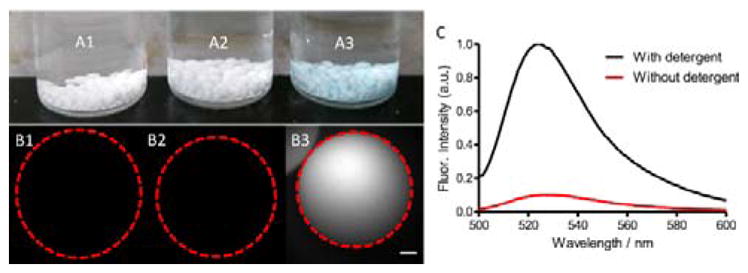
(A) Photograph of three vials containing cationic beads treated with (A1) buffer, (A2) POPC@CSP, and (A3) POPC@CSP⊃6C. (B) Fluorescent micrographs (Ex: 635–675 nm, Em: 696–736 nm) showing an individual bead from each of the corresponding samples in A. Red dashed line denotes bead perimeter. Scale bar is 100 μm. (C) Fluorescence spectra (ex: 490 nm) of supernatant from a sample of beads coated with POPC@CSP⊃6C liposomes containing fluorescent rhodamine 123, before (red) and after (black) lysis of the adhered liposomes with detergent.
A more biomedically relevant set of targeting studies examined cationic POPC@CSP⊃6Z liposomes with the expectation they would selectively associate with anionic membranes and cell surfaces. A series of model target liposomes were prepared with membranes comprised of zwitterionic POPC mixed with 0–20 mol% of anionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS). Also incorporated into each of the target membranes was 1 mol% of DiI which enabled membrane targeting to be measured by FRET experiments (Figure 5A).[41] Separate samples of functionalized and target liposomes were mixed and monitored for FRET from the DiI donor in the target liposomes to the squaraine acceptor in the functionalized liposomes. The results with POPC@CSP⊃6Z liposomes are in Figure 5B and show that the extent of FRET increased with the fraction of POPS in the target liposomes, and that the targeting process reached completion after a few minutes. As expected, there was no similar FRET increase when control experiments were conducted using POPC@CSP⊃6C liposomes (Figure S12). Each liposome mixture was also evaluated by DLS and only the system that mixed POPC@CSP⊃6Z liposomes and POPS (20%) liposomes produced a large increase in particle size (Figure 5C). It is worth noting that the aggregated liposomes did not precipitate, most likely because the surrounding corona of PEG chains provided the aggregates with colloidal stability. The liposome aggregation process was further characterized by first measuring the amount of lipid mixing. Recalling that the embedded CSP⊃6Z complex was self-quenched in the POPC@CSP⊃6Z liposomes, a convenient lipid mixing assay was devised to measure the rate that self-quenching was relieved. Thus, POPC@CSP⊃6Z liposomes were prepared with ten times higher loading (3.0 mol%) of the embedded CSP⊃6Z complex. Mixing these self-quenched liposomes with target anionic POPS (20%) liposomes resulted in a large increase in CSP⊃6Z fluorescence over a period of ~1 hour (Figure 6), indicating relief of self-quenching due to relatively slow dilution of the embedded CSP⊃6Z into the associated POPS (20%) liposomes. Complementary information concerning leakage of aqueous contents was gained by mixing POPC@CSP⊃6Z liposomes with POPS (20%) liposomes that contained a high concentration of the fluorescent dye, sulforhodamine B (the chloride leakage assay used above is not compatible with ZnDPA compounds). Self-quenching of the sulforhodamine dye is relieved when it escapes the liposomes. As shown in Figure S14 there was very little enhanced leakage from the POPS (20%) liposomes after they were targeted by the POPC@CSP⊃6Z liposomes. Furthermore, the same small level of leakage was observed when the sulforhodamine dye was inside both the POPC@CSP⊃6Z liposomes and POPS (20%) liposomes (Figure S14), indicating negligible mixing of liposome aqueous contents. Taken together, the data suggests that the aggregated POPC@CSP⊃6Z and POPS (20%) liposomes are not fully fused. That is, there is slow transfer of lipids between the apposed bilayers but the aqueous interiors are not connected. This conclusion is not surprising for a couple of reasons; (a) the liposomes are primarily comprised of POPC, a cylindrical polar lipid that is known to energetically favour a planar lamellar structure and disfavour the curved surfaces that are formed during the process of full membrane fusion,[42] and (b) the long PEG chains that project from the membrane exterior hinder close contact of the aggregated bilayers.[43,44] While full membrane fusion and aqueous contents mixing is needed for effective cytosol delivery of polar payload such as large macromolecules, selective targeting of stable liposomes to cell surfaces is also a desirable process for controlled delivery of lipophilic drugs[45] and also for many imaging applications.[7]
Figure 5.

(A) Association of POPC@CSP⊃6Z liposomes with anionic target liposomes containing POPS (20%) is indicated by FRET. (B) Change in FRET with different target liposomes containing POPS (0–20%). (C) Dynamic light scattering showing size distributions at ten minutes after liposome mixing.
Figure 6.

Association of POPC@CSP⊃6Z liposomes (CSP was 3 mol% respective to POPC) with anionic target liposomes containing POPS (20%) produces an increase in squaraine fluorescence (ex. 377 nm).
From a molecular recognition perspective, it is important to appreciate that the ZnDPA targeting units in POPC@CSP⊃6Z liposomes are located near the liposome surface, at the base of lengthy PEG-2000 chains. But the long PEG chains apparently do not block the ZnDPA units from contacting the exposed surface of target POPS (20%) liposomes. This finding encouraged us to determine if POPC@CSP⊃6Z liposomes could selectively target a structurally more complicated biological cell surface and we chose to test the clinically important bacterium Staphylococcus aureus. We have previously shown that fluorescent ZnDPA probes can selectively stain the anionic bacterial envelope in preference to the near zwitterionic surfaces of healthy mammalian cells.[46] Furthermore, multivalent ZnDPA probes are capable of strongly agglutinating bacteria in the presence of mammalian cells.[46,47] With this precedence in mind we mixed separate samples of planktonic S. aureus with either targeted POPC@CSP⊃6Z liposomes or control non-targeted POPC@CSP liposomes. As shown by the representative micrographs in Figure 7, the control liposomes had no measurable affinity for the bacteria, whereas the targeted liposomes produced immediate agglutination and co-localization of the bacterial clusters with the liposome’s deep-red fluorescence. Another set of cell microscopy experiments assessed the ability of the POPC@CSP⊃6Z liposomes to target the S. aureus bacteria in the presence of healthy human cells. Separate samples of dispersed human cells (Jurkat) and S. aureus were stained with a non-specific, fluorescent membrane stain (DiI). Then the two cell populations were mixed and treated with POPC@CSP⊃6Z liposomes. As shown by the brightfield image in Figure 8, the bacterial cells were strongly agglutinated and easily observed as clusters mixed with the much larger human calls (additional images in Figures S15 and S16). As expected, the exterior of both the human cells and the bacteria were stained by the green-emitting DiI. In contrast, the deep-red fluorescence of the POPC@CSP⊃6Z co-localized only with the agglutinated bacteria. These microscopy studies clearly demonstrate the selective targeting ability of the functionalized POPC@CSP⊃6Z liposomes, and we infer that the targeting capabilities of the ZnDPA units located near the liposome surface are not abrogated by the corona of long PEG chains that extend above. This favourable outcome suggests that liposome functionalization using Synthavidin self-assembly is a potentially new way to solve the PEG dilemma,[9] which is the contradictory vision of a liposome surface that is coated with targeting units and protective PEG chains.[48,49]
Figure 7.
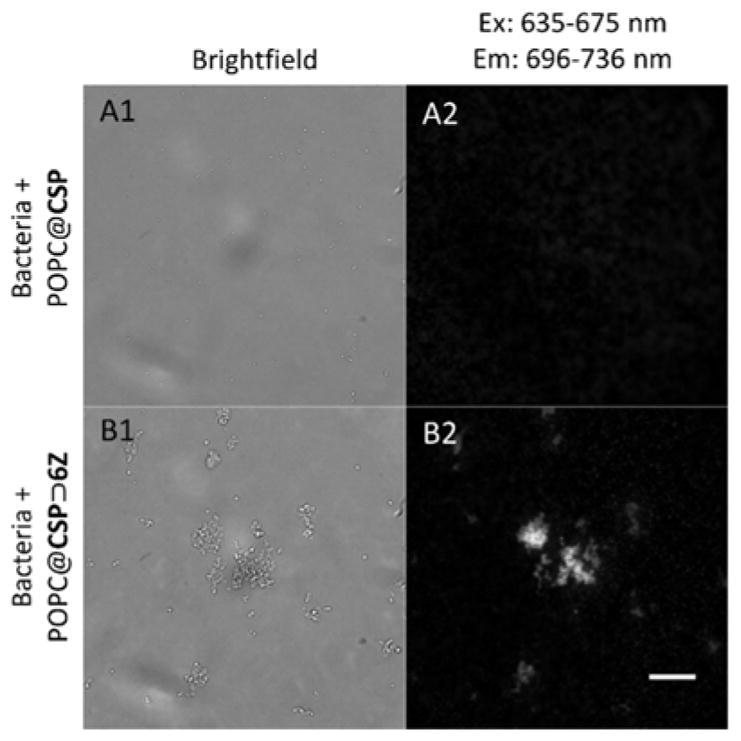
Representative micrographs showing POPC@CSP⊃6Z liposomes targeted and agglutinated S. aureus bacteria (B1,B2) but POPC@CSP liposomes did not (A1,A2). Scale bar is 20 μm.
Figure 8.

Representative micrographs showing a mixture of bacteria (S. aureus) and human (Jurkat) cells treated with DiI (non-selective membrane dye) and bacteria-selective POPC@CSP⊃6C liposomes. (A) Brightfield micrograph. (B) DiI fluorescence. (C) POPC@CSP⊃6C fluorescence. (D) Merge of the three panels, with strong co-localization of DiI and POPC@CSP⊃6C indicated as yellow. Scale bar is 20 μm
Conclusions
A new self-assembly method is used to rapidly functionalize the surface of liposomes without perturbing the membrane integrity or causing leakage of the aqueous contents. The two-step method first inserts the fluorescent conjugate CSP into the outer monolayer of liposomes and then threads a specific tetralactam macrocycle onto the surface anchored CSP. The process simultaneously equips the liposomes with targeting ligands, sterically protective PEG chains, and deep-red fluorescence reporter groups. Studies using a macrocycle bearing multiple carboxylate groups produced anionic liposomes that adhered to the surfaces of rigid cationic polymeric beads. Studies using a macrocycle bearing ZnDPA targeting units produced functionalized liposomes that recognized anionic membranes and selectively agglutinated bacteria in the presence of mammalian cells. It is notable that the corona of PEG chains around the liposomes did not inhibit the targeting capabilities of the sheltered ZnDPA targeting units. Using the same CSP conjugate as a common docking platform, it should be possible to systematically vary the macrocycle structure and create a wide range of targeted liposomes for diagnostic or therapeutic applications.[50] Moreover, Synthavidin self-assembly can be exploited to functionalize the surface of many other types of biomedically important nanoparticles,[51] especially those that are coated with bilayer membranes such as exosomes,[14] microvesicles,[52] and cell-membrane coated nanoparticles.[53]
Experimental Section
Synthesis
The synthetic methods and compound characterization data are described in the Supporting Information.
Liposome Experiments
General procedure
Liposomes were prepared by the following sequence; a film of appropriate phospholipid (1 μmol, purchased from Avanti Polar Lipids) was hydrated with 1 mL of TES (5 mM pH 7.4) NaCl (145 mM), followed by dispersion through brief vortex with a glass ring, five freeze/thaw cycles using liquid N2 and a warm water bath, and extrusion 21 times through a 0.200 μM polycarbonate filter.
Formation of POPC@CSP Liposomes
An appropriate volume of CSP (50 μM) in water was added to a rapidly stirred dispersion of pre-formed liposomes, to produce liposomes with CSP (0.3 mol%, except where noted) only in the outer leaflet. Alternatively, liposomes with CSP in both leaflets were prepared by including the CSP in the lipid film before hydration. Excitation spectra with 720 nm emission and emission spectra with 640 nm excitation were collected using 3 nm slits.
Formation of POPC@CSP⊃6C and POPC@CSP⊃6Z Liposomes
Appropriate quantities of 6C or 6Z (100 μM) in water were added to rapidly stirred dispersions of POPC@CSP liposomes. Excitation spectra with 720 nm emission and emission spectra with 377 nm excitation (anthracene absorption followed by internal energy transfer) were collected using 3 nm slits and a 498 nm long pass filter in the emission pathway to remove double diffracted scattered light.
FRET Experiments
A chloroform solution of phospholipid and DiI (1 mol%) was evaporated to create a film which was hydrated as described above. FRET from DiI to CSP, CSP⊃6C, or CSP⊃6Z was monitored with ex: 545 nm, em: 560–800 nm, with 3 nm slit widths. A decrease in donor DiI emission and increased emission at 690 nm (for the squaraine dye) or 710 nm (for the threaded complex) was indicative of FRET.
Leakage Assays
Chloride Leakage: POPC liposomes containing TES (5 mM, pH 7.4), NaCl (145 mM) were prepared with an external solution of TES (5 mM), NaNO3 (489 mM, pH 7.4). Chloride leakage into the external solution was measured using a chloride selective electrode (Beckman) before and 30 min after addition of 0.3 mol% CSP and again after addition of 1.2 molar equivalents of 6C. Calibration standards were made from isotonic mixtures of the NaCl and NaNO3 buffers.
Sulforhodamine B leakage: POPC liposomes containing sulforhodamine B (20 mM), TES (5 mM, pH 7.4), NaCl (145 mM) were prepared with an external solution of TES (5 mM, pH 7.4), NaNO3 (489 mM). The leakage-induced increase in sulforhodamine B fluorescence (ex: 550 nm, em: 570 nm) was monitored while the liposomes were treated sequentially with 0.3 mol% CSP and then 1.2 molar equivalents of 6Z. The sulforhodamine B assay was also used to measure leakage or mixing of aqueous contents when POPC@CSP⊃6Z liposomes were added to target POPC liposomes (with 0 or 20% POPS).
All leakage assays were normalized to 100% by complete lysis at the end of the experiment using a small volume of Triton X-100. All assays were completed in triplicate.
Cell Culture
Streptococcus aureus (MSSA-476) was cultured on supplemented agar plates for 24 hours at 37°C prior to usage. Jurkat (ATCC® TIB-152™) human leukemia cells were cultured according to the supplier protocols in RPMI media (Life Technologies) supplemented with 10% FBS and 1% streptomycin at 37°C and 5% CO2 over air.
Microscopy
A small colony of S. aureus was diluted in PBS and shaken to disperse the bacteria. Immediately prior to imaging, POPC@CSP or POPC@CSP⊃6Z was added (final [CSP] = 500 nM). For mixed cell experiments, a small population of either Jukat or S. aureus was individually stained with 20 μg/mL DiI for 5 minutes. The cells were then spun down and washed 2x with PBS, resuspended in a small volume of PBS and combined, treated by adding an aliquot of POPC@CSP⊃6Z (final [CSP] = 500 nM) and imaged immediately. Fluorescence microscopy was conducted on a Zeiss Axiovert 100 TV epifluoresence microscope equipped with an X-cite 120 fluorescence illumination system. Images were collected in NIS-elements using an Andor iXon EMCCD camera operating in CCD mode with 2 s acquisition times and 3 MHz readout speed. Filter sets from Semrock were: DAPI-1160B (ex: 387/11 nm, em: 447/60 nm); FITC 2024B (ex: 485/20 nm, em: 524/24 nm) Cy5.5-C (ex: 655/40 nm, em: 716/40 nm) and a custom filter set with UV excitation and far red emission (ex: 387/11 nm, em: 716/40 nm). Images were processed in ImageJ and scaled to the highest intensity image in the set.
Cationic Surface Targeting Studies
Amberlite IRA-958(Cl) ion-exchange beads (50 mg) were rinsed with (5 mM TES, pH 7.4), NaCl (145 mM) and treated with POPC@CSP or POPC@CSP⊃6C (100 μL of at 100 μM CSP) for 30 minutes. The beads were washed 5 times with an aqueous solution of TES (5 mM, pH 7.4), NaCl (145 mM). Fluorescence microscopy was conducted as before using a Cy5.5-C filter set. Liposome leakage studies were conducted by filling the liposomes with 30 μg/mL rhodamine 123 (Rh) and removing the unincorporated Rh by size exclusion. The beads were treated with the filled liposomes as before, washed 5 times, then added to the bottom of a fluorescence cuvette with 1 mL of TES (5 mM pH 7.4), NaCl (145 mM) supernatant, and a fluorescence scan of the supernatant was acquired (ex. 490 nM). A small volume of Triton X-100 was then added to release Rh into the supernatant, and another fluorescence scan was acquired.
Supplementary Material
Acknowledgments
This work was supported by grants from the NSF (CHE1401783 to B.D.S.), the NIH (R01GM059078 to B.D.S. and T32GM075762 to S.K.S) and the CONACyT (Mexico).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Abu Lila AS, Ishida T. Biol Pharm Bull. 2017;40:1–10. doi: 10.1248/bpb.b16-00624. [DOI] [PubMed] [Google Scholar]
- 2.Pattni BS, Chupin VV, Torchilin VP. Chem Rev. 2015;115:10938–10966. doi: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- 3.Duggan ST, Keating GM. Drugs. 2011;71:2531–58. doi: 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Marqués-Gallego P, De Kroon AIPM. Biomed Res Int. 2014;2014:129458. doi: 10.1155/2014/129458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang H. Open Access Sci Reports. 2012;1:1–8. [Google Scholar]
- 6.Richards DA, Maruani A, Chudasama V. Chem Sci. 2017;8:63–77. doi: 10.1039/c6sc02403c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hennig R, Pollinger K, Veser A, Breunig M, Goepferich A. J Control Release. 2014;194:20–27. doi: 10.1016/j.jconrel.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 9.Hatakeyama H, Akita H, Harashima H. Adv Drug Deliv Rev. 2011;63:152–160. doi: 10.1016/j.addr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Gray BP, McGuire MJ, Brown KC. PLoS One. 2013;8:e72938. doi: 10.1371/journal.pone.0072938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rillahan CD, Macauley MS, Schwartz E, He Y, McBride R, Arlian BM, Rangarajan J, Fokin VV, Paulson JC. Chem Sci. 2014;5:2398–2406. doi: 10.1039/C4SC00451E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu L, Kate P, Torchilin VP. ACS Nano. 2012;6:3491–3498. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmetiere F, Irwin C, Viola-Villegas NT, Longo V, Cheal SM, Zanzonico P, Pillarsetty N, Weber WA, Lewis JS, Reiner T. Bioconjug Chem. 2013;24:1784–1789. doi: 10.1021/bc400322h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyth T, Petrova K, Payton NM, Persaud I, Redzic JS, Graner MW, Smith-Jones P, Anchordoquy TJ. Bioconjug Chem. 2014;25:1777–1784. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alshaer W, Hillaireau H, Vergnaud J, Ismail S, Fattal E. Bioconjug Chem. 2015;26:1307–1313. doi: 10.1021/bc5004313. [DOI] [PubMed] [Google Scholar]
- 16.Luo W, Westcott N, Dutta D, Pulsipher A, Rogozhnikov D, Chen J, Yousaf MN. ACS Chem Biol. 2015;10:2219–2226. doi: 10.1021/acschembio.5b00137. [DOI] [PubMed] [Google Scholar]
- 17.Vabbilisetty P, Sun XL. Org Biomol Chem. 2014;12:1237. doi: 10.1039/c3ob41721b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oude Blenke E, Klaasse G, Merten H, Plückthun A, Mastrobattista E, Martin NI. J Control Release. 2015;202:14–20. doi: 10.1016/j.jconrel.2015.01.027. [DOI] [PubMed] [Google Scholar]
- 19.Bak M, Jølck RI, Eliasen R, Andresen TL. Bioconjug Chem. 2016;27:1673–1680. doi: 10.1021/acs.bioconjchem.6b00221. [DOI] [PubMed] [Google Scholar]
- 20.Jølck RI, Feldborg LN, Andersen S, Moghimi SM, Andresen TL. In: Biofunctionalization Polym Their Appl. Nyanhongo G, Steiner W, Gübitz G, editors. 2011. pp. 251–280. [Google Scholar]
- 21.Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P, Benz CC, Papahadjopoulos D. Biochemistry. 1997;36:66–75. doi: 10.1021/bi962148u. [DOI] [PubMed] [Google Scholar]
- 22.Loughrey H, Bally MB, Cullis PR. Biochim Biophys Acta. 1987;901:157–160. doi: 10.1016/0005-2736(87)90267-7. [DOI] [PubMed] [Google Scholar]
- 23.Chikh GG, Wai ML, Schutze-Redelmeier MP, Meunier JC, Bally MB. Biochim Biophys Acta - Biomembr. 2002;1567:204–212. doi: 10.1016/s0005-2736(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 24.Kauscher U, Stuart MCA, Drücker P, Galla HJ, Ravoo BJ. Langmuir. 2013;29:7377–7383. doi: 10.1021/la3045434. [DOI] [PubMed] [Google Scholar]
- 25.Wang YX, Zhang YM, Wang YL, Liu Y. Chem Mater. 2015;27:2848–2854. [Google Scholar]
- 26.Smith BD. Beilstein J Org Chem. 2015;11:2540–2548. doi: 10.3762/bjoc.11.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peck EM, Battles PM, Rice DR, Roland FM, Norquest KA, Smith BD. Bioconjug Chem. 2016;27:1400–1410. doi: 10.1021/acs.bioconjchem.6b00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roland FM, Peck EM, Rice DR, Smith BD. Bioconjug Chem. 2017;28:1093–1101. doi: 10.1021/acs.bioconjchem.7b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peck EM, Liu W, Spence GT, Shaw SK, Davis AP, Destecroix H, Smith BD. J Am Chem Soc. 2015;137:8668–8671. doi: 10.1021/jacs.5b03573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W, Peck EM, Hendzel KD, Smith BD. Org Lett. 2015;17:5268–5271. doi: 10.1021/acs.orglett.5b02633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice DR, Clear KJ, Smith BD. Chem Commun. 2016;52:8787–8801. doi: 10.1039/c6cc03669d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaasgaard T, Mouritsen OG, Jørgensen K. Int J Pharm. 2001;214:63–65. doi: 10.1016/s0378-5173(00)00633-5. [DOI] [PubMed] [Google Scholar]
- 33.Pantos A, Tsiourvas D, Sideratou Z, Paleos CM, Giatrellis S, Nounesis G. Langmuir. 2004;20:6165–6172. doi: 10.1021/la040026u. [DOI] [PubMed] [Google Scholar]
- 34.Tomatsu I, Marsden HR, Rabe M, Versluis F, Zheng T, Zope H, Kros A. J Mater Chem. 2011;21:18927. [Google Scholar]
- 35.Versluis F, Voskuhl J, van Kolck B, Zope H, Bremmer M, Albregtse T, Kros A. J Am Chem Soc. 2013;135:8057–8062. doi: 10.1021/ja4031227. [DOI] [PubMed] [Google Scholar]
- 36.Tokunaga T, Kuwahata K, Sando S. Chem Lett. 2013;42:127–129. [Google Scholar]
- 37.Thomas F, Voigt M, Worm M, Negwer I, Muller SS, Kettenbach K, Ross TL, Roesch F, Koynov K, Frey H, Helm M. Chem Eur J. 2016;22:11578–11582. doi: 10.1002/chem.201602758. and references therein. [DOI] [PubMed] [Google Scholar]
- 38.Control titration experiments added 6C to a solution of free, but self-aggregated CSP in water (micellar CSP). In this case, threading of 6C took several minutes to complete after each aliquot addition and the value of Ka was 100 times lower than Ka for 6C association with POPC@CSP liposomes (Table S1). The difference suggests that macrocycle access to the squaraine component of CSP is quite hindered when the free CSP is self-aggregated as micelles in water, but apparently not hindered when the CSP conjugate is embedded at low concentration in a POPC liposome membrane.
- 39.Avirah RR, Jyothish K, Ramaiah D. J Org Chem. 2008;73:274–279. doi: 10.1021/jo702209a. [DOI] [PubMed] [Google Scholar]
- 40.Christensen SM, Stamou DG. Sensors. 2010;10:11352–11368. doi: 10.3390/s101211352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plaunt AJ, Harmatys KM, Wolter WR, Suckow MA, Smith BD. Bioconjug Chem. 2014;25:724–737. doi: 10.1021/bc500003x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang ZY, Smith BD. Bioconjug Chem. 2000;11:805–814. doi: 10.1021/bc000018z. [DOI] [PubMed] [Google Scholar]
- 43.Mondal Roy S, Sarkar M. J Lipids. 2011;2011:528784. doi: 10.1155/2011/528784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Käsbauer M, Lasic DD, Winterhalter M. Chem Phys Lipids. 1997;86:153–159. [Google Scholar]
- 45.Fahr A, Van Hoogevest P, May S, Bergstrand N, Leigh MLS. Eur J Pharm Sci. 2005;26:251–265. doi: 10.1016/j.ejps.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 46.Turkyilmaz S, Rice DR, Palumbo R, Smith BD. Org Biomol Chem. 2014:5645–5655. doi: 10.1039/c4ob00924j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao S, Abu-Esba L, Turkyilmaz S, White AG, Smith BD. Theranostics. 2013;3:658–666. doi: 10.7150/thno.6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Trends Biotechnol. 2014;32:32–45. doi: 10.1016/j.tibtech.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 49.Pearson RM, Sen S, Hsu HJ, Pasko M, Gaske M, Král P, Hong S. ACS Nano. 2016;10:6905–6914. doi: 10.1021/acsnano.6b02708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Accardo A, Morelli G. Biopolymers. 2015;104:462–479. doi: 10.1002/bip.22678. [DOI] [PubMed] [Google Scholar]
- 51.Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, Medintz IL. Chem Rev. 2013;113:1904–2074. doi: 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez-Trillo F, Grover LM, Stephenson-Brown A, Harrison P, Mendes PM. Angew Chem Int Ed. 2017;56:3142–3160. doi: 10.1002/anie.201607825. [DOI] [PubMed] [Google Scholar]
- 53.Kroll AV, Fang RH, Zhang L. Bioconjug Chem. 2016;28:23–32. doi: 10.1021/acs.bioconjchem.6b00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.