

Abstract
Ras/MAPK pathway signaling is a major participant in neurodevelopment, and evidence suggests that BRAF, a key Ras signal mediator, influences human behavior. We studied the role of the mutation BRAFQ257R, the most common cause of cardiofaciocutaneous syndrome (CFC), in an induced pluripotent stem cell (iPSC)-derived model of human neurodevelopment. In iPSC-derived neuronal cultures from CFC subjects, we observed decreased p-AKT and p-ERK1/2 compared to controls, as well as a depleted neural progenitor pool and rapid neuronal maturation. Pharmacological PI3K/AKT pathway manipulation recapitulated cellular phenotypes in control cells and attenuated them in CFC cells. CFC cultures displayed altered cellular subtype ratios and increased intrinsic excitability. Moreover, in CFC cells, Ras/MAPK pathway activation and morphological abnormalities exhibited cell subtype-specific differences. Our results highlight the importance of exploring specific cellular subtypes and of using iPSC models to reveal relevant human-specific neurodevelopmental events.
Introduction
Mental health disorders account for 13% of the global burden of disease1. Five major psychiatric disorders—autism spectrum disorder (ASD), attention deficit-hyperactivity disorder, bipolar disorder, major depressive disorder, and schizophrenia—have substantial heritability and shared genetic etiology, indicating common pathophysiologies2. Genome-wide association studies found that the most significant shared signaling pathway is the Ras/MAPK pathway3, which regulates cellular proliferation, differentiation, and survival across many cell types and is critical in neurodevelopment4, 5.
Germline mutations in the Ras/MAPK pathway cause a class of Mendelian genetic syndromes termed RASopathies5, which include cardiofaciocutaneous syndrome (CFC; OMIM #115150). CFC individuals have increased risk for behavioral manifestations affecting cognitive systems, social processes, and arousal/regulatory systems6, 7, all relevant to the symptomology of major psychiatric disorders. CFC is an autosomal dominant disorder primarily caused by gain-of-function mutations in the BRAF gene (73% of CFC subjects5), which is ubiquitously expressed in the developing human brain with peak expression between postconceptional weeks 8 and 128. Individuals with CFC commonly present developmental delay, intellectual disability (more than 80%), increased risk for ASD (54–64%), seizures (45%) and a higher incidence of structural brain abnormalities6, 7, 9–14. A systematic study of 66 CFC subjects observed increased ventricular size and cortical atrophy14. Other common neurologic manifestations of CFC are abnormal EEG findings (including hypsarrhythmia), agenesis of the corpus callosum, abnormal myelination, and Chiari malformation7, 9, 14. There are many structural abnormalities in common between CFC and major psychiatric disorders: cortical atrophy is observed in schizophrenia15, 16, thinner corpus callosum in ASDs17, 18, and enlarged ventricles in both15, 16, 18, 19.
Attempts to mechanistically model CFC in mice have led to mixed results. The ablation of Braf in the developing mouse brain led to increased frequency of repetitive movements, seizures, hyperactivity, and impairments in sociability and learning20–22. However, mouse models with gain-of-function mutations in Braf failed to recapitulate the central nervous system (CNS) structural defects found in CFC subjects23, 24 and led to embryonic lethality or overall reduced viability. Human induced pluripotent stem cells (iPSCs) have proven to comprise a successful platform that allows for the direct examination of human neuronal development25–31. Our recent studies on another RASopathy, Costello syndrome32, 33, indicated that an activating mutation in HRAS led to an extended progenitor phase and subsequent increase in the number of cortical neurons33 as well as excessive astrocyte-to-neuron signaling32, all of which correlated with the progressive postnatal brain overgrowth in Costello syndrome34.
Because Ras/MAPK signaling controls differentiation in most tissues, we hypothesized that the BRAF gain-of-function p.Q257R mutation would affect neuronal maturation. We generated iPSC lines from four CFC subjects carrying BRAFQ257R (the most common CFC mutation5) and differentiated them towards several neuronal lineages. We characterized the biochemical consequences of aberrant Ras/MAPK signaling in CFC-derived neural cultures and revealed cellular properties associated with altered maturation, including morphological and physiological features.
Material and Methods
Subjects
All human research was consistent with IRB approved protocols. All participants or their parents provided written consent as approved by the UCSF Human Research Protection Program (CHR #10-02794). We selected subjects based on the presence of BRAF mutation p.Q257R. Recruitment was described previously 6: all CFC subjects were recruited at national RASopathy family meetings and two control participants at a UCSF Neurofibromatosis Symposium. The third control line used in our study, HS1-11, was kindly provided by Dr. Arnold Kriegstein’s lab (UCSF). Control lines were sex- and age-matched to the subject lines (Supplementary Table 1; clinical features in Supplementary Table 2).
Generation of pluripotent stem cell lines
Dermal fibroblasts were isolated from skin biopsies as previously described35. When the dermal fibroblast culture reached passage 5, they were detached using 0.25% trypsin/EDTA, spun at 500xg for 5 minutes, resuspended in DMEM high-glucose media. We used 360,000 cells per individual per reprogramming. Before electroporation, one μg of each of the plasmids pCXLE-hOCT3/4-shp53-F, pCXLE-hSK, pCXLE-hUL, and pCXLE-EGFP (Addgene)36 was mixed with 100ul Resuspension Buffer R (Neon™ Transfection System, Life Technologies), and added to the cells to be transformed. We pipetted 100μl of the cell suspension into the Neon Transfection System chamber (Life Technologies) and electroporated the cell suspension with three 1,650V, 10ms pulses. Immediately after electroporation, the cell suspension was pipetted directly into a gelatin-coated well containing a warmed fibroblast medium (DMEM High Glucose supplemented with 10% fetal bovine serum) and incubated in a 37ºC, 5% CO2 incubator. GFP-positive cells were examined during the first five days post-electroporation to monitor transfection efficiency. After day 5, transfected fibroblasts were detached and plated onto Mitomycin C treated Mouse Embryonic Fibroblasts (Millipore) at 10,000 cells/cm2. Until iPS-like colonies were ready to be picked and passaged, we fed the cells with hES-KSR Basal Medium - Knockout DMEM (Life Technologies), 20% Knockout Serum Replacement (Life Technologies), 5% L-glutamine (Life Technologies), 10% Non-essential amino acids (Life Technologies), and beta-mercaptoethanol and 10ng/ml bFGF (GlobalStem). Clonal colonies displaying iPSC morphology were manually selected and subsequently cultured on a Matrigel substrate (BD Biosciences) with mTeSR1 media (Stem Cell Technologies). All lines were shown to be pluripotent using a random differentiation protocol37. The presence of the BRAF Q257R point mutation in the CFC subjects was confirmed by Sanger sequencing of exon 6 of the BRAF gene for all the cell lines before and after reprogramming (forward primer: 5′-GGGAGAGAAATACTGTCCATTCCA-3′; reverse primer: 5′-GCTTGAAATCAGTTGCCAGCC -3′; annealing temperature= 58°C). Both DNA fingerprinting analysis and genotyping using the Affymetrix Axiom EUR array38 were performed by the Genomics Core Facility at UCSF using standard protocols before reprogramming, after reprogramming and in one-week old neural differentiation cultures. Subclones utilized for each cell line and independent differentiations performed are listed in Supplementary Table 3.
Immunofluorescence
At the previously described time points, cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature and permeabilized with 90% ice cold methanol in PBS for 10 min. Nonspecific binding was blocked with IF blocking buffer [2% BSA (Sigma), 1% Fish Skin Gelatin (Sigma) and 0.2% Saponin (Sigma) in PBS] for 1h at room temperature. Cells were incubated with primary antibody (Supplementary Table 3) overnight at 4°C, followed by three 5 minute PBS washes. Cells were incubated with appropriate secondary antibodies (Supplementary Table 4) for 1h at room temperature. Slides were mounted with Prolong Gold with DAPI mounting solution (Invitrogen). All images in the same set were obtained using the same exposure and speed parameters. Images were captured using the EVOS® FL imaging system (Life Technologies) and analyzed using the “Cell Counter” plugin in ImageJ software.
Protein expression
We prepared total protein extracts using Phosphosafe extraction reagent (EMD Biosciences) supplemented with complete protease inhibitors (Roche), according to manufacturer’s protocol. 5μg of protein per well was loaded into a gradient 4–12% Bis-Tris SDS-PAGE Bolt gel (Invitrogen) under denaturing conditions, along with 4ul of PageRuler prestained protein ladder (Thermo Fisher). Proteins were transferred to PVDF membranes (BioRad), blocked in 5% BSA in TBS-T (0.1% Tween-20 in TBS), and blots were incubated overnight at 4°C using the antibodies specific to proteins of the Ras/MAPK pathway and PI3K pathways in Supplementary Table 5. All antibodies targeting phosphorylated proteins recognize the activated form of the proteins. The next day, blots were exposed to anti-rabbit IgG–peroxidase conjugated secondary antibody (1:10,000; Sigma-Aldrich) for 1h at room temperature. Following TBST washes, blots were exposed to chemiluminescent detection substrate ECL prime (GE Healthcare) for 1 minute at room temperature. Next, they were exposed to x-ray film Amersham Hyperfilm ECL (GE Healthcare) for up to 1h. Films were scanned with a Canon LiDE210 Flatbed Scanner (Canon) and analyzed using the gel analysis method, as outlined in the ImageJ documentation: https://imagej.nih.gov/ij/docs/menus/analyze.html#gels.
Gene expression
Total RNA was isolated using the RNeasy Mini kit (Qiagen), following manufacturer’s instructions. Complementary DNA (cDNA) was produced from 1μg of total RNA using High Capacity RNA-to-cDNA Kit (Life Technologies). The qRT-PCR assay was performed using 20ng of cDNA and TaqMan gene expression master mix in a QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems). We determined expression levels by relative quantification in comparison to the endogenous control gene GUSB. Expression of each target gene was assessed relative to a control sample using the 2−ΔΔCT method39. Samples were run in technical triplicates, and the threshold suggested by the instrument’s software was used (after visual confirmation) to calculate the Ct. Outlier replicate samples (any data point more than 1.5 interquartile ranges below the first quartile or above the third quartile) were excluded from analysis. The TaqMan probes used in this study are summarized in Supplementary Table 6.
Neural progenitor cell culture
To induce neural differentiation, free floating iPSC aggregates were formed for 24h in mTeSR1 culture medium (Stemcell Technologies). After 24h, the iPSC aggregates were switched into a neural induction medium [DMEM/F12 (Invitrogen), N2 supplement (Invitrogen), MEM-NEAA (Gibco) and 2μg/ml heparin (Sigma-Aldrich)]40 with media exchange every other day. To promote neural induction, we added small molecule TGF-βR inhibitor, SB431542 (5μM, Stemgent), and BMP signaling inhibitor, LDN-193189 (0.25μM, Stemgent), for 48h. On day 3, aggregates were attached to 6-well plates and cultured in neural media for an additional week during which rosettes appeared in the colonies. On day 11, neuroepithelial cells in the center of the colonies were mechanically removed and kept as free-floating aggregates. At day 25 of neural differentiation, neurospheres were dissociated into single cells using Accutase (Stem Cell Technologies) and cultured as monolayer neural progenitor cells (NPCs). NPCs were maintained at high density, grown on polyornithine/laminin-coated plates in NPC media (STEMdiff™ Neural Progenitor Medium, Stem Cell Technologies) and split ~1:5 every week with Accutase (Millipore, Billerica, MA, USA). All subsequent experiments utilized NPCs between passages 4 to 6, when cultures were 80% confluent.
Neural differentiation
In this protocol, we obtain not only diverse types of neurons, but glial cells as well40, therefore we refer to it as neural differentiation (protocols for differentiation directed towards specific neuronal cell types is described below). NPCs were plated into poly-ornithine/laminin-coated plates at 50,000 cells/cm2 and fed with neural medium [Neurobasal medium (Invitrogen), supplemented with N2 supplement (Invitrogen), and B27 supplement (Invitrogen)]40. Cells were fed twice a week and analyzed at one-week and five-weeks after plating. The neural differentiation protocol was performed in three independent batches for each cell line.
Cell cycle exit analysis
A single dose of BrdU (Sigma, 15ng/ml) was added to NPCs for 4h before differentiation into forebrain neurons and fixed after 48h of culture in the neural medium. Neural cultures were processed for immunostaining with mouse anti-BrdU (1:100, BD Biosciences) and rabbit anti-Ki67 antibodies (1:400, Abcam). Cell cycle exit was established by counting all BrdU+ but Ki67− cells over the total number of BrdU+ cells per coverslip. The percentage of (BrdU+/Ki67−)/BrdU+ cells is reported as the percentage of cells that exited cell cycle.
For analysis of the role of ERK1/2 and AKT phosphorylation in early maturation, DMSO (control), MEK inhibitor U0126 (10μM, Cell Signaling Technologies), PI3K inhibitor Wortmannin (1μM, Cell Signaling Technologies), a combination of both U0126 (10μM) and Wortmannin (1μM), or AKT activator SC79 (10μg/ml, Millipore Sigma) were added to the single dose of BrdU for 4h prior to changing the NPC medium to neural medium. The following steps were performed as described above.
Electrophysiology
To label neurons for visual targeting, cultures were infected with a lentivirus that expresses Discosoma Red Fluorescent Protein (DsRFP) under the synapsin promoter (Addgene). Coverslips containing cultures differentiated for 5 weeks in neural medium were transferred to a recording chamber in an upright light microscope (Nikon Eclipse, Japan). The coverslips were immersed in recording solution (in mM: 140 NaCl, 5 KCl, 10 HEPES, 10 D-glucose, 2 MgCl2, 2 CaCl2, pH 7.4) and maintained at room temperature. Whole-cell patch clamp recordings were obtained from DsRFP+ neurons (total neurons analyzed for control lines: n=15; CFC: n=18; Wortmannin pre-treated controls: n=7) under visual guidance using borosilicate glass pipette electrodes. Electrodes were filled with a K-gluconate based internal solution (in mM: 123 K-gluconate, 12 KCl, 10 HEPES-KOH pH 7.2, 1 EGTA, 0.1 CaCl2, 1 Mg-ATP, 0.2 Na4GTP and 4 D-glucose; adjusted to 320 mOsm with added sucrose). Electrophysiological signals were acquired using a patch clamp amplifier (Multiclamp 700B, Axon Instruments/ Molecular Devices) and digitizer (Digidata 1440A, Axon Instruments/ Molecular Devices), and recorded using pCLAMP 10 software (Axon Instruments/ Molecular Devices). For current-clamp recordings, signals were filtered at 10kHz and sampled at 25kHz. In the voltage-clamp recording mode, signals were filtered at 4 kHz and sampled at 10kHz. Whole cell capacitance and series resistance compensation were applied.
Differentiation into glutamatergic cortical neurons
Differentiation of NPCs to an enriched glutamatergic population was adapted from previously described protocols 28,40. NPCs between passages 4 and 6 were plated onto poly-ornithine/laminin-coated wells at a density of 10–20,000 cells/ well on a 24-well plate in glutamatergic neuron medium [Neurobasal medium (Invitrogen), supplemented with N2 supplement (Invitrogen), B27 supplement (Invitrogen) and laminin (Sigma, 1μg/ml), with the addition of fresh BDNF and GDNF (PeproTech, 10 ng/mL), cAMP (0.1 μM) (Sigma) and ascorbic acid (Sigma, 200μg/ml)]. In order to promote differentiation, γ-secretase inhibitor XXI (Millipore, 0.2μM) was added to the culture for the first 24h following plating. Cells were fed every other day and were analyzed at one-week and five-weeks after plating.
Differentiation into GABAergic interneurons
To obtain cell populations enriched for GABAergic interneurons from NPCs, we used a previously developed protocol41. NPCs between passages 4 and 6 were plated onto polyornithine/laminin-coated wells at a density of 10–20,000 cells/well on a 24-well plate in GABAergic neuron medium [Neurobasal medium (Invitrogen), supplemented with N2 supplement (Invitrogen), B27 supplement without vitamin A(Invitrogen) and laminin (Sigma, 1μg/ml), with the addition of fresh BDNF, GDNF and IGF1 (all PeproTech, 10ng/ml)]. SHH (necessary to induce the expression of NKX2.1; Peprotech 500ng/ml) and γ-secretase inhibitor XXI (to promote differentiation; Millipore, 0.2μM) were added to the culture for the first 24h following plating. Cells were fed every other day and were analyzed one-week and five-weeks after plating.
Morphological analysis
Two days before cell fixing, five-week old glutamatergic cortical neurons and GABAergic interneuron cultures were transfected with 500ng per well of pCXLE-eGFP plasmid (Addgene plasmid 27082), as previously described42. Cells were then fixed in 4% PFA, and we proceeded with the immunofluorescence staining protocol described above.
Morphological analysis of glutamatergic neurons was performed in TUJ1+GFP+ neurons (20 neurons per line, a total of 60 control neurons and 80 CFC neurons analyzed), while analysis of GABAergic interneurons was conducted in GABA+GFP+ neurons (20 neurons per line, a total of 60 control neurons and 80 CFC neurons analyzed). To measure soma size and count the number of neurites and branches, we used the default tools of Image J software. Neurite length, branch length, and branching analyses were evaluated using the Sholl analysis plugin and Simple Neurite Tracer plugin for ImageJ. Morphological analysis was performed in three independent differentiations for all cell lines and each differentiation protocol.
Statistics
Continuous variables are expressed as mean±SD. The different categories were compared by repeated measures ANOVA, which accounts for biological and technical replicate differences. A p-value ≤ 0.05 was considered statistically significant. In all figures, p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
Results
BRAFQ257R does not affect reprogramming efficiency or competency to generate neural progenitor cells
To evaluate the effect of the BRAFQ257R mutation in neurodevelopment, we generated iPSCs from dermal fibroblasts of 4 unrelated CFC individuals and 3 sex- and age-matched controls (Supplementary Table 1 and Supplementary Figure 1a) using electroporation of nonintegrating episomal plasmids to express reprogramming factors acutely and avoid viral-mediated mutagenesis36. For the number of subclones and independent experiments performed, as well as individual values for each cell line, refer to Supplementary Table 3 and Supplementary Table 7. We tested the pluripotency of the iPSC lines generated (Supplementary Table 3 and Supplementary Figure 1b) and observed no differences in their ability to proliferate, differentiate into the three germ layers (Supplementary Figure 1c), or activate the MAPK and PI3K signaling pathways (Supplementary Figure 1d).
Subsequently, we differentiated each iPSC line towards a neural fate and generated neural progenitor cells (NPCs), which can differentiate into all three CNS cell types40 (Supplementary Figure 1e). All NPC cultures were positive for βIII-tubulin (TUJ1), a marker of early postmitotic and differentiated neurons and some mitotically active neuronal precursors43, with no significant difference in the proportion of cells expressing this marker between CFC and control lines (Supplementary Figure 1f and 1g). Across CFC and control lines, most cells were positive for NPC markers: NESTIN, SOX2, and PAX6 (Supplementary Figure 1h, 1i, and 1j). We detected no difference in the proliferation of CFC and control cultures via Ki67 staining (Supplementary Figure 1f and 1k) or population doubling time (Supplementary Figure 1l). Likewise, the cell cycle profile of CFC and control NPCs was similar (Supplementary Figure 1m). Therefore, CFC and control NPCs showed no apparent proliferation or differentiation differences. Although we observed a high inter-individual variation in protein expression, CFC NPCs exhibited a significant decrease in the phosphorylation of ERK1 (60.7% decrease, p=0.006, Figure 1a and 1b), ERK2 (26.9% decrease, p=0.0008, Figure 1a and 1b), and AKT (71.7% decrease, p=0.002, Figure 1a and 1b). This difference remained significant when analyzing the ratio of activated protein over total protein for of p-ERK1/ERK1 and p-AKT/AKT, but not p-ERK2/ERK2 (Figure 1b). This observation suggests that activation of ERK1 and AKT may play an important role in later CFC phenotypes.
Figure 1. CFC neural cultures show progenitor pool depletion, early maturation, and imbalance of neural cell types.

(A) Western blot analysis of the components of the Ras/MAPK pathway and its phosphorylation status, and quantification of the blots (B) from protein lysates extracted from CFC and control NPC cultures. The results were normalized with their corresponding α-tubulin values (assigned a value of 1) and are mean ±SD of two independent experiments (C) Double immunostaining for BrdU (green) and Ki67 (red) of early neuronal progenitors within 48h of differentiation; progenitors that exited the cell cycle, %(BrdU+Ki67−)/BrDU+ cells (orange arrows) were quantified. (D) Cleaved caspase 3 staining for cell death of the same NPCs and subsequent quantification. (E) Immunostaining of TUJ1, MAP2, NEUN, GABA, and TBR1 of CFC lines and control lines at week 1 of neural differentiation. Quantification of these stainings are shown as percentage of TUJ1+ cells (F), MAP2+ cells (G), NEUN+ cells (H), average neurite length of TUJ1+ cells (I), average number of neurites per TUJ1+ cell (J), percentage of GABA+ cells (K), and TBR1+ cells (L). (M) mRNA levels of MAP2, deep layer markers TBR1, CTIP2, and FEZF2, upper layer markers CUX1 and SATB2, and GABA precursors GAD1 and GAD2 in one-week old neural cultures. ΔCt values were calculated by normalizing the average Ct value of each cell line by the average Ct value of GUSB of the same cell line; ΔΔCt was calculated by normalizing the ΔCt value of each line to the average ΔCt value of the control lines. (N) Immunostaining of TUJ1, GFAP, CD44, S100, MAP2, FOXP2, and CUX1 at week 5 of neural differentiation. Quantification of these stainings is shown as a percentage of cells expressing GFAP (O), MAP2 (P), FOXP2 (Q) and CUX1 (R). (S) mRNA level analysis of astroglial cell markers GFAP, S100B, CD44, neuronal marker MAP2, deep layer markers TBR1, CTIP2, and FEZF2, upper layer markers CUX1 and SATB2 in five-week old neural cultures. ΔCt values were calculated by normalizing the average Ct value of each cell lines by the average Ct value of GUSB of the same cell line; ΔΔCt was calculated by normalizing the ΔCt value of each line to the average ΔCt value of the control lines. For all immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. For all scatter plots illustrated, values represent mean±SD per condition, calculated with repeated measures ANOVA, accounting for biological and technical replicate values. Number of subclones and independent experiments performed are detailed in Supplementary Table 3. Values for mean and standard deviation of each control and CFC subject for each experiment are detailed in Supplementary Table 7. p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
CFC neural cultures display early maturation and imbalance of neural cell types
We first investigated the cell cycle exit of differentiating NPCs to determine if the BRAF mutation affects the neural progenitor cell population. We added BrdU to the culture medium of NPCs for 4h before the neural differentiation protocol, changed the NPC media to neural media and assayed for the original pool of progenitor cells (BrdU+) and progenitor cell pool depletion [(%BrdU+Ki67−)/%BrdU+] after 48h. Control and CFC lines exhibit the same percentage of progenitor cells prior to the neuronal differentiation protocol (%BRDU+; Figure 1c). Yet, we observed an increase in post-mitotic cells, indicating a depletion of the progenitor cell pool in CFC cultures (BRAFWT= 48.44±16.71%, vs. BRAFQ257R= 76.23±10.46%, p=0.002; Figure 1c). By probing these cells for the apoptosis marker cleaved caspase 3, we confirmed that the loss of progenitor cells is unlikely to be due to cell death (Figure 1d). Altogether, our results suggested that cells in the CFC cultures were leaving the mitotic progenitor state more rapidly compared to controls.
The increased percentage of progenitor cells exiting the cell cycle is suggestive of premature maturation of the CFC neural cultures. We sought to determine whether other evidence supported the rapid cellular maturation phenotype observed in CFC. Thus, we matured the NPCs into neural cultures for up to 5 weeks40. This protocol allows for the generation of a mixed neural culture containing diverse types of neurons, as well as glial cell types40. We confirmed the onset of increasing expression of BRAF, MAPK3 (which encodes ERK1), and MAPK1 (which encodes ERK2) genes in both CFC and control lines, as well as the onset of increase in the protein levels of BRAF and ERK1/2 (Supplementary Figure 2).
At week 1 of differentiation, both CFC and control cells were positive for TUJ1, and there was no significant difference in the expression of TUJ1 (Figure 1e and 1f). At this early time point, the expression of mature neuronal markers, such as MAP244 and NEUN45, is not expected. Yet, one-week old CFC neural cultures were significantly enriched for both MAP2 (BRAFWT= 3.57±3.06%, vs. BRAFQ257R= 17.91±7.99%, p=0.0005; Figure 1e and 1g) and NEUN (BRAFWT=6.76±6.7%, vs. BRAFQ257R= 22.778±11.23%, p=0.0005, Figure 1e and 1h). Moreover, we observed that CFC TUJ1+ cells displayed morphology associated with more mature neurons46: more neurites (neurites/cell: BRAFWT=2.87±0.92, vs. BRAFQ257R=3.87±1.17; p=0.003; Figure 1e and 1j) and longer neurites (BRAFWT=79.30±57.13 μm, vs. BRAFQ257R=170.74±139.72 μm; p=0.0009; Figure 1e and 1i),
Primate corticogenesis involves a variety of different lineage precursors in the subventricular zone47. This niche contains distinct glutamatergic neuronal precursors, GABAergic neuronal precursors, and glial precursors47. To confirm the observation of premature differentiation of CFC NPCs, we expected an increase in those cell types formed earlier in development, such as deep layer glutamatergic neurons, while the depletion of progenitors would cause a decrease in late-born cells, such as upper layer cortical neurons and glia, including astroglial cells. Therefore, we further characterized the different neural cellular subtypes in CFC and control cultures. We generated a consistent population of glutamatergic cortical neurons, confirmed by the expression of the subtype-specific cortical marker, TBR1 (Figure 1e). One-week old CFC cultures were significantly enriched for early-born deep layer cortical cell marker TBR1 (cortical layers 5 and 6 marker; BRAFWT=13.68±9.52%, vs. BRAFQ257R=52.91±24.00%, p=0.0034; Figure 1e and 1l). We also detected an increase in the proportion of cells expressing the inhibitory neurotransmitter GABA in CFC cultures (BRAFWT=0.16±0.34%, vs. BRAFQ257R=4.87±4.60%, p=0.029; Figure 1e and 1k). We additionally confirmed the imbalance in CFC neural cell types through mRNA levels of MAP2, deep layer markers TBR1, CTIP2 and FEZF2, upper layer markers CUX1and SATB2, and GABA precursors GAD1 and GAD2 (Figure 1m).
After five weeks of differentiation, we measured the expression of the mature neuron marker, MAP2, and observed that its expression was equivalent between control and CFC neural cultures (Figure 1n and 1p). At this time point, we assessed the proportions of late-born neural cells, such as upper layer glutamatergic neurons (CUX1+, cortical layer 2/3 and 4 marker) and astroglial cells (GFAP+, CD44+, and S100+), compared to early born deep layer cortical neurons (FOXP2+). Overall, for both control and mutant lines, most of the cells produced were neurons (%MAP2+ cells: 59.5±7.5%). Among these, the cells were mostly glutamatergic from layer VI and layer II/III (%(FOXP2+/MAP2+) +%(CUX1+/MAP2+)= 19.97±4.3%), while very few were GABAergic (%GABA+/MAP2+ = 1.53%±0.6) (data not shown). As we predicted, there was an increase in deep layer cortical neurons (FOXP2+ cells; BRAFWT=4.44±0.78%, vs. BRAFQ257R=10.83±1.92%; p<0.0001, Figure 1n and 1q), depletion of upper layer cortical neurons (CUX1+ cells; BRAFWT=3.56±3.35%, vs. BRAFQ257R=1.39±0.91%; p=0.039; Figure 1n and 1r), and depletion of GFAP+, CD44+ or S100+ astroglial cells in the CFC-derived cultures (GFAP+ cells; BRAFWT=9.83±6.14%, vs. BRAFQ257R=1.93±1.74, p=0.0006 Figure 1n and 1o). We further confirmed the imbalance of cell types in five-week old CFC neural culture through mRNA level analysis of astroglial cell markers GFAP, S100B, CD44, deep layer markers TBR1, CTIP2 and FEZF2, and upper layer markers CUX1and SATB2, (Figure 1s). Overall, our results indicate that under neural differentiation conditions, CFC neural cultures undergo a depletion of progenitor cells and early maturation.
CFC-derived neurons have increased intrinsic excitability
The bona fide marker of cortical neuronal functional maturation is the ability to maintain moderate frequency action potential (AP) firing48. We hypothesized that if CFC-derived neural cultures do mature faster, they would display an increase in the frequency of AP firing. To this end, we utilized whole-cell patch-clamp electrophysiology to examine various measures of intrinsic excitability in CFC and control neurons. To specifically target neurons within the mixed neural cultures, we transduced five-week old neural cultures with SYN1-DsRFP lentivirus to label mature neurons at four days before recording (Figure 2a).
Figure 2. Increased intrinsic excitability in CFC-derived neurons.
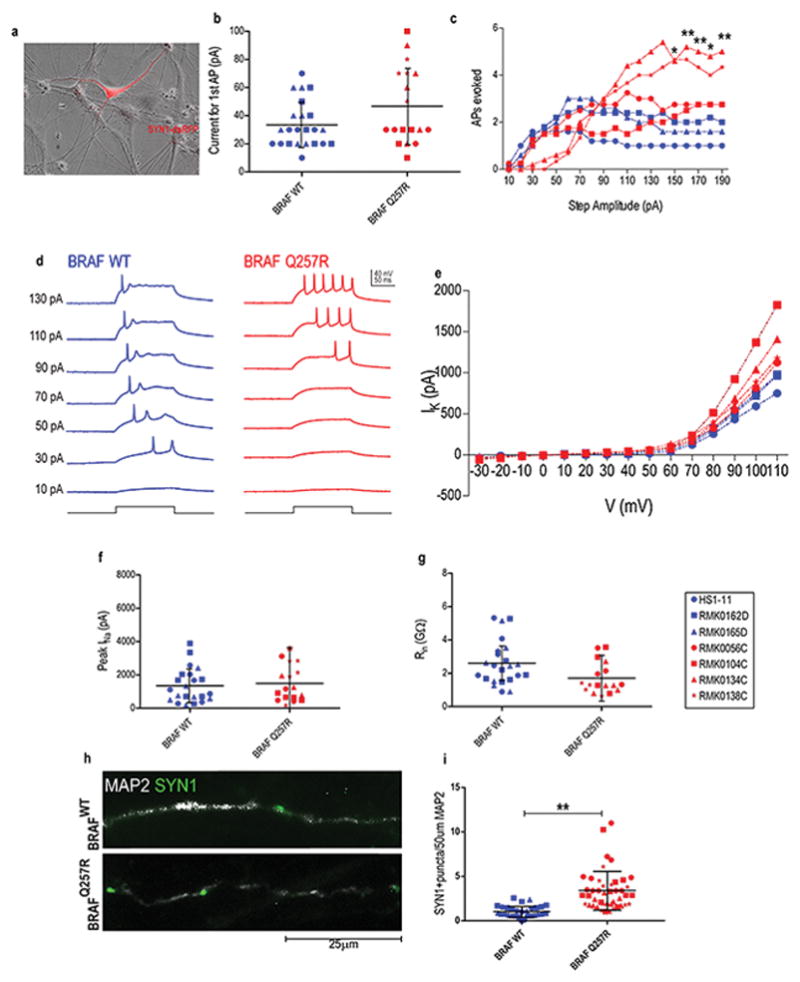
(A) Overlaid dsRFP fluorescence and phase-contrast images depicting a SYN1−DsRFP+ neuron. For the patch clamp experiments (B–G), we performed two independent blind experiments, with one subclone per cell line. (B) The average current per individual required to elicit a single AP in neurons from control-derived neurons (blue), CFC-derived neurons (red). (C) The quantified AP response evoked by step currents of increasing amplitude. (D) Example current-clamp recordings from control-derived and CFC-derived neurons. Additional measures of intrinsic excitability: IK (E), Peak INa (F), Rin (G). (H) Immunostaining of Synapsin1+ puncta on MAP2+ control- and CFC-derived neurons at week 5 of differentiation. (I) Quantification of Synapsin1+ puncta per 50 μm neurite length in control- and CFC-derived neurons. For all immunostainings depicted, at least ten MAP2+ neurons per coverslip from three independent experiments were counted. For all scatter plots illustrated, values represent mean±SD per condition, with significance calculated using repeated measures ANOVA, accounting for biological and technical replicates. Number of subclones and independent experiments performed are detailed in Supplementary Table 3. Values for mean and standard deviation of each control and CFC subject for each experiment are detailed in Supplementary Table 7. p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
In order to elicit AP firing, in current-clamp configuration, a series of positive current steps of successively increasing amplitude (200ms, 10–190pA) were delivered to neurons maintained at a holding potential of −70mV. We found that the overwhelming majority of DsRFP+ neurons fired APs (49/52 cells), illustrating the usefulness of SYN1-DsRFP lentiviral transduction to label neurons. The amount of current to elicit a single AP was similar in control and CFC (Figure 2b). CFC-derived neurons had markedly distinct AP firing behavior at higher amplitude stimuli (Figure 2c and 2d). Control neurons fired a maximum of 2 AP in response to a 70-pA step current, on average, with a decrease in the average number of APs fired using higher amplitude stimuli. In contrast, in CFC-derived neurons, although higher amplitude current was required to reach maximal AP firing rates (~110–160pA), the maximal rate was superior to that of controls (BRAFWT: 2.87±1.51 APs vs. BRAFQ257R: 4.12±2.34 APs). Moreover, CFC-derived neurons could maintain this maximal firing rate even at the highest amplitude stimuli. When we assayed other metrics of intrinsic excitability, including potassium/sodium currents and input resistance (Figure 2e, 2f, and 2g), we did not observe further significant differences except for an increased potassium current at the highest tested voltage step (Figure 2e), suggestive of neurons with a larger area. Our results showed that CFC neural cultures exhibit accelerated functional neuronal maturation, which was associated with 2.5-fold increase in the number of SYN1+ puncta in dendrites of CFC neurons (p= 0.0016; Figure 2h and 2i).
Decreased phosphorylation of AKT leads to progenitor cell pool depletion in CFC
Because the first noticeable differences between CFC and control cultures were the reduction in the levels of phosphorylation of ERK1/2 and AKT (Figure 1a and 1b) and early cell cycle exit during neural differentiation (Figure 1c), we sought to test whether these two observations were related. First, we added either DMSO only, the MEK inhibitor U0126 only (10μM in DMSO), the PI3K inhibitor Wortmannin only (1μM in DMSO), or a combination of both U0126 and Wortmannin to the media of control NPCs for 1h. We then compared the ratio of phosphorylation of ERK1/2 and AKT to DMSO-treated control and CFC NPCs (Supplementary Figure 3a and 3b). Western blot analysis revealed that U0126 inhibited the phosphorylation of ERK1/2 while increasing that of AKT. On the other hand, Wortmannin decreased the phosphorylation of AKT and ERK1/2, similar to our observations in CFC NPCs (Supplementary Figure 3a and 3b). Next, we tested whether these pathways are associated with the accelerated cell cycle exit by pre-treating control NPCs with DMSO, U0126, Wortmannin, or U0126+Wortmannin, to simulate the phosphorylation profile of CFC NPCs. After 48h in neural differentiation medium, double staining for BrdU and Ki67 revealed that exposure to Wortmannin is sufficient to mimic the CFC phenotype in control cells (Supplementary Figure 3c and 3d). Simultaneous pre-exposure to both the U0126 and Wortmannin did not produce effects beyond those of the Wortmannin exposure alone (Supplementary Figure 3c and 3d). Therefore, this observation indicated that the accelerated cell-cycle exit of CFC cultures was associated with the inhibition of the AKT pathway accompanied by decreased activation of the ERK pathway before neural differentiation.
To corroborate the role of decreased AKT activation in the progenitor pool depletion of CFC lines, we tested whether increasing AKT phosphorylation in CFC cultures would rescue this phenotype. We added the AKT activator SC7949 (10μg/ml) to the media of CFC NPCs for 1h and compared the levels of phosphorylation of ERK1/2 and AKT in SC79 pre-treated CFC NPCs to both DMSO-treated control and CFC NPCs (Supplementary figure 3e and 3f). SC79 pre-treated CFC cells showed an increased level of AKT activation compared to the same cells exposed to DMSO only (Supplementary figure 3e and 3f). Following the same cell cycle exit assay described above, we observed that AKT inhibition (Wortmannin treatment) in control cells recapitulated the increase in post-mitotic cells of the CFC lines, while AKT activation (SC79 treatment) in CFC cells prevented the depletion of progenitor cells to levels similar to the observed in control lines (Figure 3b and 3c).
Figure 3. Decreased phosphorylation of AKT is associated with progenitor cell pool depletion in CFC.

(A) Crosstalk between the Ras/Raf/MEK/ERK and Ras/PI3K/AKT pathway, and the targets of Wortmannin and SC79 (adapted from64). (B) Double immunostaining for BrdU (red) and Ki67 (green) of early neuronal progenitors within 48h of differentiation in control NPCs, control NPCs pre-treated with Wortmannin, CFC NPCs, and CFC NPCs pre-treated with SC79; progenitors that exited the cell cycle, %(BrdU+Ki67−)/BrdU+ cells (orange arrows) were quantified and graphed in (C). (D) Immunostaining of MAP2 in control one-week old neural cultures, control one-week old neural cultures pre-treated with Wortmannin, CFC one-week old neural cultures, and CFC one-week old neural cultures pre-treated with SC79. (E) Quantification of the percentage of MAP2 positive DAPI in these stainings. (F) Immunostaining of TUJ1 in control one-week old neural cultures, control one-week old neural cultures pre-treated with Wortmannin, CFC one-week neural cultures, and CFC one-week old neural cultures pre-treated with SC79. Quantification of the average number of neurites per TUJ1+ cell (G). (H) Immunostaining of GFAP and TUJ1 in control five-week old neural cultures, control five-week old neural cultures pre-treated with Wortmannin, CFC five-week old neural cultures, and CFC five-week old neural cultures pre-treated with SC79. (I) Quantification of the percentage of GFAP positive DAPI in these stainings. (J) Immunostaining of MAP2 and FOXP2 in control five-week old neural cultures, control five-week old neural cultures pre-treated with Wortmannin, CFC five-week old neural cultures, and CFC five-week old neural cultures pre-treated with SC79. (K) Quantification of the percentage of FOXP2 positive DAPI in these stainings. (L) Immunostaining of MAP2 and CUX1 in control five-week old neural cultures, control five-week old neural cultures pre-treated with Wortmannin, CFC five-week old neural cultures, and CFC five-week old cultures pre-treated with SC79. (M) Quantification of the percentage of CUX1 positive DAPI in these stainings. For all immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. For all scatter plots illustrated, values represent mean±SD per condition, calculated with repeated measures ANOVA, accounting for biological and technical replicates. Number of subclones and independent experiments performed are detailed in Supplementary Table 3. Values for mean and standard deviation of each control and CFC subject for each experiment are detailed in Supplementary Table 7. p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
Moreover, to assess whether decreased phosphorylation AKT in CFC NPCs and subsequent depletion of progenitor cells was driving the neural maturation phenotype, we compared neural differentiation of control NPCs pre-treated with Wortmannin to CFC NPCs, and we compared CFC NPCs pre-treated with SC79 to control NPCs), all four conditions undergoing the same protocol. We were able to recapitulate early neural maturation of CFC cultures in the control cultures pre-treated with Wortmannin, while mitigating the phenotype in CFC cultures with SC79 pre-treatment (Figure 3d–h), evidenced by MAP2 expression and TUJ1+ cell morphology at one-week old neural differentiation cultures. At week 5, even though control lines pre-treated with Wortmannin showed increased number of deep layer cortical neurons and decreased number of astroglial cells (Figure 3i–n), CFC lines displayed a more accentuated and significant difference compared to untreated control lines (Figure 3i and 3j). On the other hand, pre-treatment of CFC lines with SC79 was able to attenuate the increase in the number of CUX1+ cells and decrease in astroglial cells (Figure 3i–n). These observations could be due to the sustained effect of SC79 in AKT phosphorylation even after its removal from cell culture or in vivo49.
BRAFQ257R causes cellular subtype-specific phenotypes
To explore potential differences between CFC and control specific neuronal subtype populations that are independent of the progenitor pool depletion, we used previously established protocols to differentiate CFC and control NPCs into two distinct neuronal cell lineages: cortical glutamatergic neurons28 and GABAergic interneurons41. In both protocols, NPCs were first treated with Compound E (γ-secretase inhibitor XXI) during the first 24h of the neuronal induction protocol to inhibit Notch signaling. The Notch signaling pathway suppresses differentiation and promotes proliferation: ligand-receptor binding activates the pathway through the cleavage and subsequent nuclear translocation of Notch intracellular domain by γ-secretase50. Consequently, by adding Compound E, we overcame the initial progenitor cell pool depletion observed in the CFC neural cultures (Figure 4a). To confirm that these protocols generated subtype specific enriched populations, these methodologies were directly compared. Five-week old GABAergic cultures on average displayed 5.99±3.85% FOXP2+ cells and no expression of CUX1+ cells, compared to 29.49±12.33 FOXP2+ cells and 25.52±20.28% CUX1+ cells in five-week old glutamatergic cultures (Supplementary Figure 4a–f). Conversely, in five-week old glutamatergic cultures, an average of 0.82±1.07% cells was positive for both GAD65/GAD67, and showed no expression of GABA, compared to 70.37±14.86% GAD65+GAD67+ and 81.28±15.78% GABA+ cells in five-week old GABAergic cultures (Supplementary Figure 4g–l).
Figure 4. CFC cortical glutamatergic neuronal cultures show increased number of neurites per cell and decreased phosphorylation of BRAF.

(A) Double immunostaining for BrdU and Ki67 in early neuronal progenitors within 48h of differentiation with and without the addition of Compound E; progenitors that exited the cell cycle, %(BrdU+Ki67−)/BrDU+ cells were quantified. (B) Immunostaining of TUJ1 and TBR1 at week 1 of glutamatergic cortical neuron differentiation, and (E) of MAP2, TBR1, CUX1, and FOXP2 at week 5 of glutamatergic cortical neuron differentiation. Quantification of these staining is shown in (C–I). For the above immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. (J) Fluorescent microscopy analysis of GFP-labelled five-week old control-derived and CFC-derived neurons. For the morphometric analysis, 20 GFP+TUJ1+ neurons per coverslip from three independent experiments were counted. Image J analysis of soma size (K), neurites per cell (L), neurite length (M), branches per neurite (N), branch length (O), and Sholl analysis of the number of intersecting neurites (P). Western blot analysis of the components of the Ras/MAPK pathway and its phosphorylation status (Q), and quantification of the blots (R) from protein lysates extracted from five-week old CFC and control glutamatergic cortical neuron cultures. The results were normalized with their corresponding α-tubulin values (assigned a value of 1) and are mean±SD of two independent experiments. For all immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. For all scatter plots illustrated, values represent mean±SD per condition, calculated with repeated measures ANOVA, accounting for biological and technical replicates. Number of subclones and independent experiments performed are detailed in Supplementary Table 3. Values for mean and standard deviation of each control and CFC subject for each experiment are detailed in Supplementary Table 7. p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
First, we differentiated the NPCs towards enriched cortical glutamatergic cultures28, confirmed by expression of TBR1, FOXP2, and CUX1 (Figure 4b and 4e). One-week old CFC cultures had no significant difference in the expression of neuronal marker TUJ1 (Figure 4b and 4c) or the layer 5/6 marker TBR1 (Figure 4b and 4d). Five-week old CFC cultures also showed no significant difference in expression of the mature neuronal marker MAP2 (Figure 4e and 4f) or of deep layer glutamatergic neuronal markers TBR1 (Figure 4e and 4g), FOXP2 (Figure 4e and 4h), and upper layer CUX1 (Figure 4e and 4i). These results suggested that the imbalance in different glutamatergic neurons subtypes in the CFC mixed neural cultures is associated with the depletion of the progenitor cell pool (Figure 1c and 4a). On the other hand, the morphometric analysis showed that CFC glutamatergic neurons exhibited more neurites per cell (BRAFWT=1.93±1.28, vs. BRAFQ257R=2.67±1.43, p=0.0078; Figure 4j and 4l), as well as shorter branches (BRAFWT=100.42±226.4 μm, vs. BRAFQ257R=50.17±96.47 μm, p=0.0089; Figure 4j and 4o). Analysis by western blot revealed that CFC cortical glutamatergic neuronal cultures have a decrease in the phosphorylation of serine 445 of BRAF (56.2% decrease, p=0.02), but no difference in the phosphorylation levels at site T202/Y204 of ERK1/2 or T308 of AKT (Figure 4q and 4r).
Next, using a previously published protocol41, we performed GABAergic inhibitory neuronal differentiation of CFC and control NPCs, confirmed by positive GABA, NKX2.1, and GAD65/GAD67 (the two isoforms of the enzymes that catalyze the decarboxylation of glutamate to GABA) staining (Figure 5a). Of note, this protocol requires exposure of the NPCs to SHH to generate ventral identity progenitors that correspond to medial ganglionic eminence interneuron progenitors41, which account for 35% of the human cortical interneurons51. CFC and control cultures displayed similar expression of the GABAergic progenitor markers GABA, NKX2.1, and GAD65/67 (Figure 5b, 5c, and 5d), corroborating the hypothesis that an increase in the number of GABA+ cells in the CFC mixed neural cultures is associated with the depletion of the progenitor cell pool (Figure 1c and 4a). In the morphology analysis of GABA+GFP+ cells (Supplementary Figure 4m), CFC GABAergic interneurons had a significant decrease in the number of neurites per cell (BRAFWT=2.94±1.45, vs. BRAFQ257R=2.44±1.25, p=0.045; Figure 5e and 5g), in contrast to the morphology of the glutamatergic neurons. Unlike what was observed in CFC glutamatergic neurons, the CFC GABAergic interneurons showed an increase in the phosphorylation of residue S445 of BRAF (1.55-fold increase expression of p-BRAF, p=0.01) accompanied by a decrease in the activation of PI3K (83% reduction in the expression of p-AKT, p=0.03) (Figure 5l and 5m).
Figure 5. CFC GABAergic interneuron cultures show decreased number of neurites per cell and increased phosphorylation of BRAF.
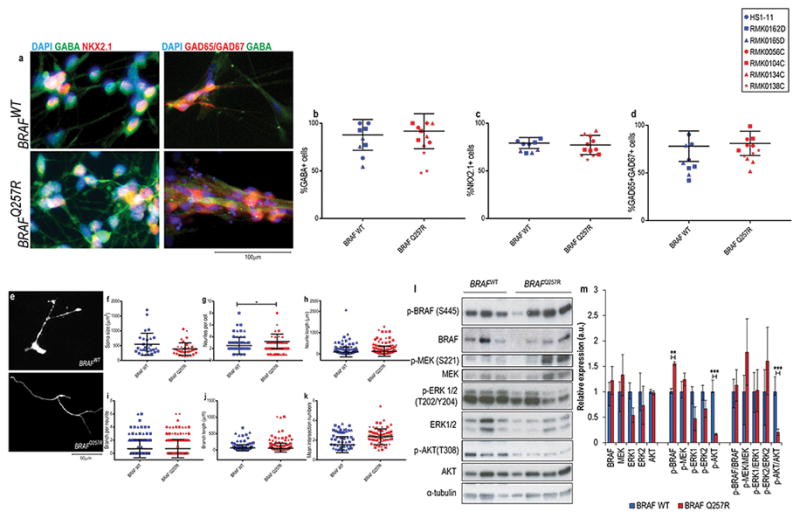
(A) Immunostaining of GABA and NKX2.1 at week 1 of GABAergic interneuron differentiation. Quantification of these stainings is shown as the percentage of cells expressing GABA (B), NKX2.1 (C). (D) Immunostaining of GABA and GAD67 at week 5 of GABAergic interneuron differentiation and quantification of these stainings shown as the percentage of cells expressing GAD65/GAD67. For the above immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. (E) Fluorescent microscopy analysis of GFP-labelled GABA+ cells in five-week old control-derived and CFC-derived neurons. For the morphometric analysis, 20 GFP+GABA+ neurons per coverslip from three independent experiments were counted. Image J analysis of soma size (F), neurites per cell (G), neurite length (H), branches per neurite (I), branch length (J), and Sholl analysis of the number of intersecting neurites (K). Western blot analysis of the components of the Ras pathway and its phosphorylation status (L), and quantification of the blots (M) from protein lysates extracted from five-week old CFC and control GABAergic interneuron cultures. The results were normalized with their corresponding α-tubulin values (assigned a value of 1) and are mean±SD of two independent experiments. For all immunostainings depicted, at least three fields per coverslip from three independent experiments were counted. For all scatter plots illustrated, values represent mean±SD per condition, calculated with repeated measures ANOVA, accounting for biological and technical replicates. Number of subclones and independent experiments performed are detailed in Supplementary Table 3. Values for mean and standard deviation of each control and CFC subject for each experiment are detailed in Supplementary Table 7. p≤0.05 (*), p≤0.01 (**), p≤0.001 (***).
Overall, by performing two distinct neuronal type specific differentiations and analyses, our results suggested that imbalance in the number of different types of neural cells in CFC is associated with the depletion of progenitor cells. Moreover, BRAF could have additional cell type specific regulation during neurodevelopment, as we observed that CFC glutamatergic neurons and GABAergic interneurons had opposite morphological changes compared to the respective control neurons, as well as different levels of phosphorylation of BRAF.
Discussion
The earliest difference we established between CFC and control iPSC derived cultures was the decrease in the progenitor cell pool not associated with cell death. The depletion of the progenitor cell pool in CFC cultures was followed by early expression of mature neuronal markers (Figure 2a–i), imbalance in different neural populations (Figure 2j–o), and increased excitability in CFC cultures (Figure 3). Taken together, these results imply that the BRAFQ257R mutation is associated with accelerated maturation. Moreover, the loss of progenitor cells was followed by the disproportionate representation of glutamatergic cortical layer neurons, increase in the number of GABAergic interneurons, and marked decrease in the number of astroglial cells, all of which can be paralleled to clinical observations in CFC. In a few CFC patients, white matter atrophy (1.5%) and defects of myelination (7.5%) have been reported. However, these findings are only discernible upon completion of myelination, and only a limited number of CFC subjects have undergone a brain MRI. Therefore it is possible that these abnormalities are more common than reported in literature14, 52. We observed an increase in glutamatergic cortical layer VI neurons and a decrease in layer II/III neurons, where cortical projection neurons primarily reside (Figure 2j)53, which could result in a decline in the proportion of callosal projection neurons and increase in the number of cells that project to subcortical structures. Accordingly, corpus callosum abnormalities, including agenesis of corpus callosum, are found in CFC subjects14. The high prevalence of seizure and ASD traits in CFC6, 11 suggests an imbalance of excitation and inhibition, which could be due to a decrease in glutamatergic excitatory signaling, or to increased GABAergic signaling. CFC neural cultures showed an increase in GABA+ neurons, favoring the latter hypothesis.
Our neuronal type-specific results show that without progenitor pool depletion and with specific cues, CFC and control lines give rise to similar percentages of different subtypes of glutamatergic neurons as well as GABAergic interneurons. Even so, the CFC glutamatergic and GABAergic neurons differed in morphology and Ras/MAPK and PI3K/AKT pathway activation profiles, implying that the BRAFQ257R mutation affects cell type specification and cell type morphological development independently. However, we cannot rule out that the differences in the percentage of GABAergic interneurons in the neural and directed differentiation could be because we are looking at different populations of interneurons. In humans, there is a dual origin of cortical interneurons: 65% of cortical GABAergic interneurons are derived from the Dlx1/2+/Mash1+ lineage situated in the neocortical ventricular and subventricular zones, whereas 35% have a ventral origin from the ganglionic eminence51. The neural differentiation protocol40 could enrich for interneurons of the ventricular and subventricular zones, while the directed differentiation generates the progenitors corresponding to the ganglionic eminence41. Of note, the dual origin of cortical interneurons is not present in rodents, so further human-derived cell studies will be needed to investigate this possibility.
We also observed cell context dependent Ras/MAPK and PI3K/AKT pathway activation profiles in CFC. It is thought that the cysteine rich domain of BRAF – where mutation p.Q257R resides – inhibits basal catalytic RAF activity and mediates membrane docking upon activation54. While BRAFQ257R led to enhanced activation of ERK signaling in NIH3T3 cells upon transient transfection25, the function of this mutation for Ras/MAPK signaling in NPC and neural maturation is unknown. Although RASopathies are usually described as syndromes with overactivation of the Ras/MAPK pathway5, 55, 56, the study of the BRAF phosphorylation and dimerization suggest that RASopathy BRAF mutations can cause impaired kinase activity57, 58. Our data support that abnormal activation of the Ras/MAPK pathway is cell context dependent; e.g., the same mutation in BRAF is associated with decreased phosphorylation of BRAF in glutamatergic neurons and increase in p-BRAF in GABAergic interneurons. The cell type dependent role of BRAF is in line with recent work that several RASopathy-associated MEK mutations both increased and reduced the levels of Ras pathway activation in vivo; furthermore, the divergent effects were dependent on cellular context59.
By exposing control NPCs to a PI3K inhibitor, phosphorylation of AKT and ERK1/2 were decreased, and we were able to emulate the progenitor cell depletion and partially recapitulate the imbalance in cell types observed in CFC lines. Conversely, activation of AKT by SC79 in CFC lines was able to attenuate the CFC phenotypes of the cell lines with the BRAF mutation. Therefore, we supported that loss of the progenitor pool in CFC is associated with the decreased activation of AKT. The progenitor pool size determines the eventual size and morphology of the brain. Rapid depletion of the neural progenitor cell pool can contribute to reduced cortical thickness60, which could be linked to cortical atrophy in CFC subjects14, and might also be relevant to cortical atrophy in schizophrenia15, 16. Decreasing activation of the kinases AKT, ERK1, and ERK2 also recapitulates the early increase in MAP2+ and NEUN+ cells in CFC cultures. However, in the longer term, brief one-time initial Wortmannin treatment of control lines is not sufficient to fully recapitulate the CFC cellular phenotype. On the other hand, SC79 may activate AKT irreversibly49 and was able to reduce the progenitor pool depletion and the early expression of MAP2, alleviate the decrease in GFAP+ cells, increase in FOXP2+ neurons and decrease in CUX1+ neurons (Figure 3), which suggests that a sustained decrease in activation of AKT, rather than at a particular time point in development, is associated with an observable phenotype. Our results imply that, besides the ERK pathway (which is most commonly associated with RASopathies5, 59), AKT activation also has a role as a mediator of the CFC neuronal phenotype. That is not surprising, given that there are several cross-feedback loops between Ras/MAPK and PI3K pathways58, 61.
The morphological and biochemical phenotype of glutamatergic neurons and GABAergic interneurons harboring the BRAFQ257R mutation was also cell type dependent. Compared to the same control neuronal types, glutamatergic CFC neurons had more neurites and branching per cell accompanied by a decrease in p-BRAF, while GABAergic CFC interneurons had fewer neurites per cell, increase in p-BRAF, and decrease in p-AKT. This difference could enhance the excitation-inhibition imbalance in CFC. Moreover, it has been shown that the same gene can cause ASD or schizophrenia by affecting distinct neuronal populations62. This difference highlights the importance of taking the cellular context into consideration when studying neurodevelopmental disorders.
Mutation p.G12S in the HRAS gene causes Costello syndrome, also a RASopathy, and in iPSC-derived neurons, it extended the progenitor phase33. This observation is the opposite of the phenotype observed in CFC iPSC-derived neurons. HRASG12S is a constitutively active form of HRAS63. Therefore, the BRAFQ257R mutation decreases activation of ERK 1/2 in NPC cells while the HRASG12S mutation increases phosphorylation of ERK1/2 32, 33, explaining the different neuronal phenotypes. Moreover, the neuronal phenotype in each case corresponds to the clinical phenotype: Costello syndrome shows postnatal progressive overgrowth of the brain 34 that could be due to the extended progenitor phase; cortical atrophy found in some CFC subjects can be linked to the rapid depletion of the progenitor pool. These results call for caution when proposing a general drug therapy or treatment for neurodevelopmental disorders, even when the mutations are in the same pathway.
In conclusion, our results provide insight into ways that the BRAFQ257R mutation may contribute to common neurological and psychiatric symptoms of CFC. Our findings at a cellular level – progenitor pool depletion, decrease in layer II/III neurons, and increase in GABAergic interneurons – can be correlated with neurological findings in the CFC subjects – cortical atrophy, corpus callosum abnormalities, and ASD and seizures, respectively. Hence, our data indicate that iPSC-derived cells are an informative model to study human neurodevelopment and underlying cellular pathologies resulting from abnormal signaling. Through directed differentiation into specific neuronal lineages, we showed morphological differences of BRAFQ257R neurons, which are cell type dependent. Although the use of iPSCs as a platform to study neurodevelopment has become widespread, most of the effort to date has focused on specific neuronal lineages and failed to investigate different neuronal types simultaneously. This traditional narrow focus could generate seemingly contradictory or non-comparable results across studies examining different lineages. Finally, our results, alongside a growing body of evidence in the literature, suggest a complex and intertwined crosstalk between two signaling pathways associated with major psychiatric disorders: Ras/MAPK and PI3K/AKT pathways. Our study suggests that for future screening of potential therapeutic agents to be successful, these pathways must be considered in depth and that cellular context should be taken into account.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health New Innovator (1DP2OD007449 to LAW), Simons Foundation Autism Research Initiative (to LAW and EMU), National Alliance for Research on Schizophrenia and Depression Young Investigator Grant from the Brain & Behavior Research Foundation (to EY), Staglin Family/ International Mental Health Research Organization Assistant Professorship (to LAW), University of California San Francisco Resource Allocation Program (to LAW and WZ), the LeJeune Foundation (to LAW and EY), the City College of San Francisco Bridges to Stem Cell Program (to ZYW, FMC and CT), National Institutes of Health (T32 EY007120 to DQD), Research to Prevent Blindness - Walt and Lilly Disney Award for Amblyopia Research (to DQD) and National Institutes of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (5RO1AR062165 to KAR). We thank all of the participants in our study and their families; Dina Bseiso and Dr. Alinoë Lavillaureix for collecting and reviewing the patients’ clinical data; Brigid Adviento and Dr. Keren Messing-Guy for preparing and shipping the skin biopsies; Dr. Michela Traglia for help with repeated measures ANOVA; Dr. Arnold Kriegstein’s lab (UCSF) and, specifically, Dr. Alex Pollen for kindly providing the control iPSC line HS1–11; Dr. Jody Baron (UCSF) for allowing us to use the QuantStudio 6 Flex Real-Time PCR System; Dr. Susan M. Voglmaier’s lab (UCSF) and, specifically Dr. Magda Santos for generously providing anti-GAD65 and anti-GAD67 antibodies; Liorimar R. Medina and John Paul Kwak for technical assistance; and Jody Williams, MA, for revision of the manuscript and department assistance. We also thank NF, Inc., Children’s Tumor Foundation, Noonan Foundation, CFC International, Costello Syndrome Family Support Network, Costello Kids, and RASopathy Network for their contribution to our recruitment efforts.
Footnotes
Conflict of interest:
The authors declare that they have no competing interests.
References
- 1.World Health Organization. The global burden of disease: 2004 update. 2008. [Google Scholar]
- 2.Cross-Disorder Group of the Psychiatric Genomics C. Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nature genetics. 2013;45(9):984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Network Pathway Analysis Subgroup of the Psychiatric Genomics C, International Inflammatory Bowel Disease Genetics C, International Inflammatory Bowel Disease Genetics Consortium I. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature neuroscience. 2015;18(2):199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krencik R, Zhang SC. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nature protocols. 2011;6(11):1710–1717. doi: 10.1038/nprot.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tidyman WE, Rauen Ka. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Current opinion in genetics & development. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adviento B, Corbin IL, Widjaja F, Desachy G, Enrique N, Rosser T, et al. Autism traits in the RASopathies. Journal of medical genetics. 2014;51(1):10–20. doi: 10.1136/jmedgenet-2013-101951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adachi M, Abe Y, Aoki Y, Matsubara Y. Epilepsy in RAS/MAPK syndrome: two cases of cardio-facio-cutaneous syndrome with epileptic encephalopathy and a literature review. Seizure. 2012;21(1):55–60. doi: 10.1016/j.seizure.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508(7495):199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. Journal of medical genetics. 2008;45(4):249–254. doi: 10.1136/jmg.2007.054460. [DOI] [PubMed] [Google Scholar]
- 10.Sabatino G, Verrotti A, Domizio S, Angeiozzi B, Chiarelli F, Neri G. The cardio-facio-cutaneous syndrome: a long-term follow-up of two patients, with special reference to the neurological features. Child’s nervous system: ChNS: official journal of the International Society for Pediatric Neurosurgery. 1997;13(4):238–241. doi: 10.1007/s003810050075. [DOI] [PubMed] [Google Scholar]
- 11.Alfieri P, Piccini G, Caciolo C, Perrino F, Gambardella ML, Mallardi M, et al. Behavioral profile in RASopathies. Am J Med Genet A. 2014;164A(4):934–942. doi: 10.1002/ajmg.a.36374. [DOI] [PubMed] [Google Scholar]
- 12.Cesarini L, Alfieri P, Pantaleoni F, Vasta I, Cerutti M, Petrangeli V, et al. Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am J Med Genet A. 2009;149A(2):140–146. doi: 10.1002/ajmg.a.32488. [DOI] [PubMed] [Google Scholar]
- 13.Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, Rauen KA, et al. Effects of germline mutations in the Ras/MAPK signaling pathway on adaptive behavior: cardiofaciocutaneous syndrome and Noonan syndrome. Am J Med Genet A. 2010;152A(3):591–600. doi: 10.1002/ajmg.a.33268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurological complications of cardio-facio-cutaneous syndrome. Developmental medicine and child neurology. 2007;49(12):894–899. doi: 10.1111/j.1469-8749.2007.00894.x. [DOI] [PubMed] [Google Scholar]
- 15.van Erp TG, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2015 doi: 10.1038/mp.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophrenia bulletin. 2013;39(5):1129–1138. doi: 10.1093/schbul/sbs118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frazier TW, Hardan AY. A meta-analysis of the corpus callosum in autism. Biological psychiatry. 2009;66(10):935–941. doi: 10.1016/j.biopsych.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haar S, Berman S, Behrmann M, Dinstein I. Anatomical Abnormalities in Autism? Cerebral cortex. 2014 doi: 10.1093/cercor/bhu242. [DOI] [PubMed] [Google Scholar]
- 19.Palmen SJ, Hulshoff Pol HE, Kemner C, Schnack HG, Janssen J, Kahn RS, et al. Larger brains in medication naive high-functioning subjects with pervasive developmental disorder. Journal of autism and developmental disorders. 2004;34(6):603–613. doi: 10.1007/s10803-004-5282-2. [DOI] [PubMed] [Google Scholar]
- 20.Chen AP, Ohno M, Giese KP, Kuhn R, Chen RL, Silva AJ. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. Journal of neuroscience research. 2006;83(1):28–38. doi: 10.1002/jnr.20703. [DOI] [PubMed] [Google Scholar]
- 21.Pfeiffer V, Gotz R, Xiang C, Camarero G, Braun A, Zhang Y, et al. Ablation of BRaf impairs neuronal differentiation in the postnatal hippocampus and cerebellum. PloS one. 2013;8(3):e58259. doi: 10.1371/journal.pone.0058259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nature neuroscience. 2007;10(5):598–607. doi: 10.1038/nn1898. [DOI] [PubMed] [Google Scholar]
- 23.Inoue S, Moriya M, Watanabe Y, Miyagawa-Tomita S, Niihori T, Oba D, et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum Mol Genet. 2014;23(24):6553–6566. doi: 10.1093/hmg/ddu376. [DOI] [PubMed] [Google Scholar]
- 24.Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, Wright EM, et al. Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(12):5015–5020. doi: 10.1073/pnas.1016933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154(3):518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krencik R, Zhang SC. Stem cell neural differentiation: A model for chemical biology. Curr Opin Chem Biol. 2006;10(6):592–597. doi: 10.1016/j.cbpa.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Muotri AR. The Human Model: Changing Focus on Autism Research. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 30.Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162(2):375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khalilov I, Le Van Quyen M, Gozlan H, Ben-Ari Y. Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron. 2005;48(5):787–796. doi: 10.1016/j.neuron.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 32.Krencik R, Hokanson KC, Narayan AR, Dvornik J, Rooney GE, Rauen KA, et al. Dysregulation of astrocyte extracellular signaling in Costello syndrome. Science translational medicine. 2015;7(286):286ra266. doi: 10.1126/scitranslmed.aaa5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rooney GE, Goodwin AF, Depeille P, Sharir A, Schofield CM, Yeh E, et al. Human iPS Cell-Derived Neurons Uncover the Impact of Increased Ras Signaling in Costello Syndrome. J Neurosci. 2016;36(1):142–152. doi: 10.1523/JNEUROSCI.1547-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A. 2010;152A(5):1161–1168. doi: 10.1002/ajmg.a.33391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villegas J, McPhaul M. Establishment and culture of human skin fibroblasts. Current protocols in molecular biology / edited by Frederick M Ausubel [et al] 2005;Chapter 28(Unit 28):23. doi: 10.1002/0471142727.mb2803s71. [DOI] [PubMed] [Google Scholar]
- 36.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nature methods. 2011;8(5):409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 37.Mali P, Ye Z, Chou BK, Yen J, Cheng L. An improved method for generating and identifying human induced pluripotent stem cells. Methods in molecular biology. 2010;636:191–205. doi: 10.1007/978-1-60761-691-7_12. [DOI] [PubMed] [Google Scholar]
- 38.Mitra I, Lavillaureix A, Yeh E, Traglia M, Tsang K, Bearden CE, et al. Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. PLoS Genet. 2017;13(1):e1006516. doi: 10.1371/journal.pgen.1006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nature biotechnology. 2001;19(12):1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Liu HS, Sauvey C, Yao L, Zarnowska ED, Zhang SC. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nature protocols. 2013;8(9):1670–1679. doi: 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ, et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33(2):95–103. doi: 10.1016/j.ymeth.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 43.von Bohlen Und Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007;329(3):409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- 44.Izant JG, McIntosh JR. Microtubule-associated proteins: a monoclonal antibody to MAP2 binds to differentiated neurons. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(8):4741–4745. doi: 10.1073/pnas.77.8.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116(1):201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 46.Leach MK, Naim YI, Feng ZQ, Gertz CC, Corey JM. Stages of neuronal morphological development in vitro--an automated assay. Journal of neuroscience methods. 2011;199(2):192–198. doi: 10.1016/j.jneumeth.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 47.Dehay C, Kennedy H, Kosik KS. The outer subventricular zone and primate-specific cortical complexification. Neuron. 2015;85(4):683–694. doi: 10.1016/j.neuron.2014.12.060. [DOI] [PubMed] [Google Scholar]
- 48.Song M, Mohamad O, Chen D, Yu SP. Coordinated development of voltage-gated Na+ and K+ currents regulates functional maturation of forebrain neurons derived from human induced pluripotent stem cells. Stem cells and development. 2013;22(10):1551–1563. doi: 10.1089/scd.2012.0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(26):10581–10586. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perea G, Sur M, Araque A. Neuron-glia networks: integral gear of brain function. Front Cell Neurosci. 2014;8:378. doi: 10.3389/fncel.2014.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature. 2002;417(6889):645–649. doi: 10.1038/nature00779. [DOI] [PubMed] [Google Scholar]
- 52.Papadopoulou E, Sifakis S, Sol-Church K, Klein-Zighelboim E, Stabley DL, Raissaki M, et al. CNS imaging is a key diagnostic tool in the evaluation of patients with CFC syndrome: two cases and literature review. Am J Med Genet A. 2011;155A(3):605–611. doi: 10.1002/ajmg.a.33787. [DOI] [PubMed] [Google Scholar]
- 53.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nature reviews Neuroscience. 2013;14(11):755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. The Journal of investigative dermatology. 2010;130(6):1657–1667. doi: 10.1038/jid.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311(5765):1287–1290. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 56.Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nature genetics. 2006;38(3):294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 57.Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Molecular and cellular biology. 2010;30(3):806–819. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nature reviews Molecular cell biology. 2015;16(5):281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 59.Goyal Y, Jindal GA, Pelliccia JL, Yamaya K, Yeung E, Futran AS, et al. Divergent effects of intrinsically active MEK variants on developmental Ras signaling. Nature genetics. 2017;49(3):465–469. doi: 10.1038/ng.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nature reviews Neuroscience. 2014;15(4):217–232. doi: 10.1038/nrn3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron. 2016;89(1):147–162. doi: 10.1016/j.neuron.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312(5989):71–75. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 64.Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2(3):135–164. doi: 10.18632/oncotarget.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.