

Abstract
Retinoic acid inducible gene (RIG-I)-mediated innate immunity plays a pivotal role in defence against virus infections. Previously we have shown that Sendai virus (SeV) defective interfering (DI) RNA functions as an exclusive and potent RIG-I ligand in DI-RNA-rich SeV-Cantell infected cells. To further understand how RIG-I is activated during SeV infection, we used a different interferon (IFN)-inducing SeV strain, recombinant SeVΔC, which, in contrast to SeV-Cantell is believed to stimulate IFN production due to the lack of the SeV IFN antagonist protein C. Surprisingly, we found that in SevΔC-infected cells, DI RNAs also functioned as an exclusive RIG-I ligand. Infections with wild-type SeV failed to generate any RIG-I-associated immunostimulatory RNA and this correlated with the lack of DI genomes in infected cells, as well as with the absence of cellular innate immune responses. Supplementation of the C protein in the context of SeVΔC infection led to a reduction in the number of DI RNAs, further supporting the potential role of the C protein as a negative regulator of DI generation and/or accumulation. Our findings indicate that limiting DI genome production is an important function of viral IFN antagonist proteins.
Keywords: defective interfering RNA, RIG-I, Sendai, C protein
Introduction
Effective innate immune responses are critical for the control and clearance of pathogenic infections. Antiviral innate immunity is composed of specialized immune cells, such as macrophages and NK cells, as well as cell-intrinsic antiviral mechanisms that can be found in all cell types. Cell-intrinsic responses depend on the recognition of viral pathogen-associated molecular patterns (PAMPs) within target cells and the initiation of signalling cascades, leading to the establishment of an antiviral state and the production of many pro-inflammatory cytokines, including type I interferon (IFN). Viral recognition within target cells is accomplished by pattern-recognition receptors (PRRs), proteins that recognize conserved structures within an invading pathogen, allowing for self versus non-self discrimination [1]. Two major families of PRRs are responsible for the detection of RNA viruses and activation of type I IFN: toll-like receptors (TLRs) and RIG-I-like receptors (RLRs). While antiviral TLRs are commonly found in the endosomal compartments of immune cells, RLRs are ubiquitously expressed in the cytoplasm of the majority of cells. As such, RLR receptors are positioned to activate the very first wave of immune responses to an invading viral pathogen. The RLR family is composed of three PRRs: RIG-I, MDA5 and LGP2. Both RIG-I and MDA5 initiate innate immune signalling upon recognition of viral RNA in the cytoplasm and generate the primary inflammatory response to viral infections [2]. RIG-I is activated by binding to its viral RNA ligand – an RNA molecule containing a 5′-tri/diphosphate with an adjacent blunt-ended, double-stranded RNA region [3–10].
During Sendai virus (SeV) infection, RIG-I becomes activated through binding to the SeV copy-back defective interfering (cbDI) RNA [11]. Generated by the viral polymerase during replication, DI RNAs are composed of a sub-genomic RNA with intact promoters, and although they are unable to replicate on their own, DIs interfere with viral replication by sequestering the viral replication machinery [12]. Virus stocks rich in DI RNAs are well known to induce a high IFN response during infection [13, 14]. The cbDI structure, with a 5′-ppp and a perfectly complementary dsRNA region directly adjacent to it, possesses all the structural elements required for robust RIG-I activation. In previous studies we used a strain of SeV, SeV-Cantell, that is rich in DI particles, to identify RIG-I ligands, as this virus has been extensively characterized as a RIG-I-dependent potent IFN inducer [15, 16]. Identification of SeV DI RNA as an exclusive RIG-I ligand in a DI-rich infection naturally led to the question of what RNA molecule(s) may activate RIG-I during the course of infection with a DI-free virus. Wild-type viruses with functional IFN antagonists stay hidden from the immune system for an extensive period of time [17]. Viruses that are known to strongly and quickly activate IFN pathways tend to fall into two distinct categories, those with high DI composition and those lacking an IFN antagonist [18]. Since our previous work was performed with a virus containing high amounts of DIs, we wanted to extend our studies to a virus deficient in its ability to suppress IFN activation. SeV lacking the C protein (SeVΔC) has previously been shown to strongly activate the IFN response through the RIG-I pathway [19]. Accordingly, we utilized this virus to identify RNA molecules which act as RIG-I ligands during infection with a virus defective in IFN antagonism.
The C ORF of SeV is encoded within the P gene, which contains overlapping reading frames for viral V and C proteins. The V protein of SeV is generated through mRNA editing and inhibits IFN activation by binding to MDA5 [20]. The SeV C protein is synthesized from the P mRNA through utilization of alternative ribosomal initiation sites. A nested set of four proteins are expressed from the C ORF: C′, C, Y1, and Y2. The C proteins have been shown to have multiple roles in the viral lifecycle, including regulation of viral replication and transcription [21–26], suppression of apoptosis [27], enhancement of viral budding and assembly [28–30], and inhibition of IFN production and signalling [19, 31–33]. The role of the C proteins in inhibition of IFN responses has been attributed to their ability to inactivate STAT1 [32] as well as RIG-I [19, 33].
To our surprise, we found that despite growth conditions which normally inhibit DI genome accumulation, such as plaque purification and passaging at low multiplicity of infection, SeVΔC nevertheless contained a high abundance of cbDI RNAs and these molecules were again found to function as exclusive RIG-I ligands during infection [34]. Trans-supplementation of SeV C protein during infection led to significant reduction in DI genome levels, supporting the hypothesis that the C protein has a role in suppressing the generation and/or accumulation of cbDI RNAs.
Results
RIG-I binds SeVΔC cbDI RNA
To isolate RIG-I RNA ligands generated during SeVΔC infection, A549 cells were infected at a multiplicity of infection (m.o.i) of 10 and the infection was allowed to proceed for 24 h. At this point the cells were lysed and endogenous RIG-I/RNA complexes were isolated as previously described [11]. The immunoprecipitated RNA was tested for its immunostimulatory activity through transfection into a 293T-IFN-sensitive response element (ISRE)-Fluc reporter cell line, expressing an ISRE promoter-driven firefly luciferase (Fluc) construct (Fig. 1a). Transfection of RIG-I-associated RNA resulted in robust activation of the Fluc reporter compared to the control immunoprecipitated (IP) RNA. To assess the biochemical nature of the RIG-I ligand, we subjected the isolated nucleic acid to RNAse A digestion and calf alkaline phosphatase (CIP) treatment (Fig. 1b). Both treatments resulted in complete loss of immunostimulatory activity, confirming the nature of the RIG-I ligand as an RNA molecule with a functionally important phosphate group.
Fig. 1.
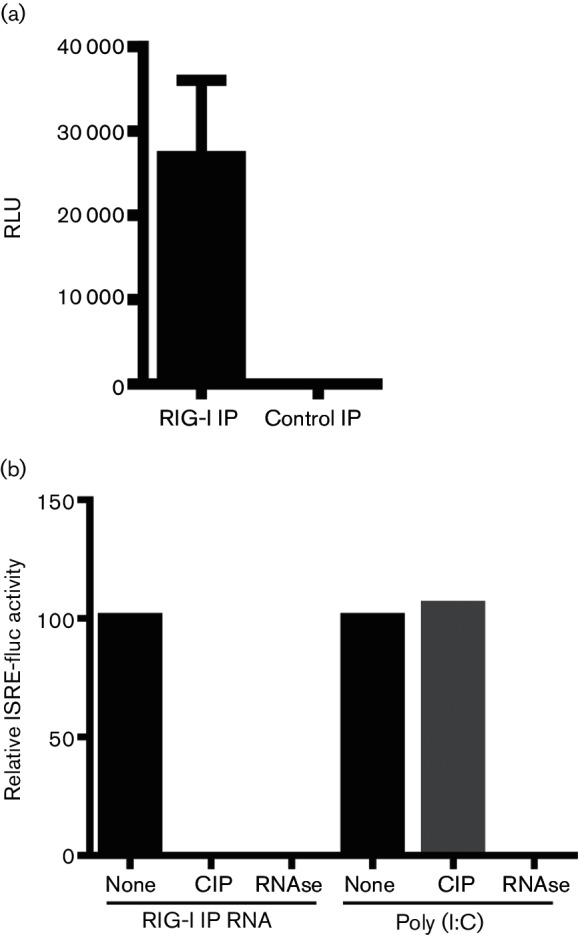
Immunostimulatory activity of RIG-I-associated RNA. (a) RIG-I-associated RNA from SeVΔC-infected A549 cells or control IP RNA was transfected into 293T ISRE-Fluc cells. Data from three individual experiments with SD are shown. (b) ISRE-Fluc reporter activation by RIG-I IP RNA or poly(I:C) treated with calf alkaline phosphatase (CI) and RNAse A.
To identify the precise nature of the RNA species interacting with RIG-I we performed deep sequencing analysis on total IP RNA from both RIG-I and control IPs. The RNA was sequenced on the Illumina HiSeq platform and sequencing reads were aligned to the Sendai-Z virus genome (Fig. 2). Analysis of RNAs specifically enriched in RIG-I IPs versus control IPs revealed two distinct regions mapping to the 5′ end of the SeV genome (Fig. 2, bottom two panels). The high abundance of sequencing reads mapping to the 5′ end of the viral genome was characteristic of high levels of copy-back DI RNA (cbDI), which we previously identified as a RIG-I ligand [11]. As with our previous report with SeV-Cantell, we did not find any RIG-I-specific enrichment within the rest of the SeV genome (Fig. 2, panel 2). To confirm the nature of RIG-I-associated RNA as cbDIs, we performed deep sequencing analysis on viral RNA isolated from purified SeVΔC stock as well as virus stocks of several other Sendai viruses (Table 1). Since SeV DIs are known to be packaged into virus particles, we wanted to confirm that the RNA species that we identified as RIG-I ligands behaved as classical DIs and not another type of aberrant viral replication product. In order to identify sequencing reads belonging to cbDI molecules we developed a computational approach based on the identification of reads spanning a fusion junction within the cbDI. Our method relies on identification of DI-specific sequencing reads which map to the unique point in the cbDI where the virus polymerase makes a junction between the 3′ and 5′ ends of the RNA. This junction corresponds to the portion of the cbDI at the meeting point between the single-stranded and double-stranded stretch and is not found in the full-length genome or L gene mRNA, allowing the identification of DI-specific sequencing reads. By utilizing an algorithm which identifies these types of junctions in RNA-Seq data, we can identify the abundance, exact position and size of cbDI molecules without any a priori knowledge about their sequence. Furthermore, this computational approach allows us to identify the presence of extremely rare DI molecules as long as even a single sequencing read spanning their junction point has been generated. In order to test the robustness and accuracy of our approach we performed sequencing of viral RNA from SeV-Cantell, a strain that contains a well-characterized cbDI species. Indeed, our computational method was able to identify the exact cbDI molecule known to be associated with SeV-Cantell. Table 1 shows the exact position of the fusion junction, as well as the cbDI relative abundance, and the length of the associated cbDI molecules, together with the length of the corresponding dsRNA stretch. For all identified DIs we confirmed that the resulting RNA molecule conforms to the rule of six, a hallmark of SeV genome replication [35]. When we performed the same analysis on the SeVΔC stock we identified two molecules that were identical to those identified from our RIG-I pulldown data. These corresponded to 14258/15280 (DI2) and 14984/15304 (DI1) fusion points. DI1 was approximately 15 times more abundant than DI2, closely mirroring the ratio of these two RNAs in infected cells, and indicating packaging at similar efficiency (Fig. 3). Comparison of the relative RIG-I enrichment of DI1 and DI2 revealed that DI1 is enriched more than 30-fold above background, compared with a 7-fold enrichment for DI2 (Fig. 4). It is not clear whether the greater enrichment for DI1 is due to preferential binding of this RNA to RIG-I, or is simply based on increased abundance of this RNA in infected cells. Although DI1 is 15-fold more abundant in infected cells than DI2, we only saw a fourfold increase in its RIG-I binding compared to DI2. It is possible that DI2 may present a better ligand for RIG-I, based on the longer stretch of the dsRNA region compared to DI1 (104 nt compared to 80 nt). However, many other aspects of virus replication could play a role in RIG-I binding, such as the physical availability of the RNA for interaction with RIG-I. Further analysis of cbDI sequences found in SeV stocks revealed the presence of several other cbDI species in both SeV-Cantell and SeVΔC. Interestingly, the most abundant SeV-Cantell cbDI (14931/15291) was also found in stocks of SeVΔC, although at minimal amounts (Table 1). Vice versa, we were able to identify the prominent SeVΔC DI (DI1) in stocks of SeV-Cantell, but again at very low levels. To eliminate the possibility that the low level of DI sequences common to all viral strains was the result of cross-contamination between the samples, we checked the sequence of the 14984/15302 DI fusion reads from SeV-Cantell and SeV-Z. Strain-specific sequence variation at positions 15 032–15 034 allowed us to differentiate between fusion reads originating from Cantell versus Z strains. We could not perform the same analysis on the predominant Cantell DI species (14 931/15 291) because the genomic sequence encompassed by the fusion reads from that DI were identical between the Cantell and Z strains. The identification of identical cbDIs within two different strains of SeV indicates that cbDIs are prone to arise de novo at specific positions within the virus genome, likely through the influence of the underlying genomic sequence, which is identical at the fusion point between these two strains. The subsequent expansion of select cbDI species likely establishes a prominent DI associated with a particular SeV strain. In addition to these two species we found several other cbDIs present in low amounts within both SeV-Cantell and SeVΔC stocks. Out of all the identified cbDIs, the 14258/15280 (DI2) associated with SeVΔC represented the longest species at 1230 nt. We performed a Western blot to detect the protein C expression in cells infected with the different viruses analysed in the experiments (Fig. 4b).
Fig. 2.

SeVΔC RNAs enriched in RIG-I IPs. Total RNA isolated with RIG-I (red) or control (blue) IP was subjected to deep sequencing. RNA-Seq reads are mapped to their corresponding position within the SeV genome. The top panel illustrates the entire SeV genome. The bottom three panels zoom in on highlighted regions of the SeV genome. The number of RNA-Seq reads is given on the Y-axis. RIG-I-specific enrichment, corresponding to DI RNAs, can be seen in the bottom two panels. The bottom panel encompasses reads mapping to both DI1 and DI2. The second from the bottom panel corresponds to reads mapping to DI2.
Table 1. Description of cbDIs associated with various virus stocks and identified through RNA-Seq analysis.
RNA-Seq fusion reads were used to deduce the exact nature of the cbDIs associated with each listed virus. The exact fusion point of the cbDI, its overall length, and the length of the dsRNA panhandle are listed. The percentage of reads supporting fusion is reflective of the overall prevalence of a particular cbDI within the total viral RNA pool.
| Virus | Fusion point | DI length (nt) | dsRNA length (nt) | No. of fusion reads | No. of total viral reads | Percentage of reads supporting fusion point |
|---|---|---|---|---|---|---|
| SeV-Cantell | 14931/15291 | 546 | 92 | 121 248 | 23 510 595 | 0.5 |
| 14984/15304 | 480 | 80 | 148 | 0.0006 | ||
| 14950/15278 | 540 | 106 | 336 | 0.001 | ||
| SeV-ΔC | 14258/15280 (DI1) | 1230 | 104 | 8435 | 16 361 704 | 0.05 |
| 14931/15291 | 546 | 92 | 49 | 0.0003 | ||
| 14984/15304 (DI2) | 480 | 80 | 127 405 | 0.8 | ||
| 15178/15284 | 306 | 100 | 869 | 0.005 | ||
| SeV-GFP-C | 14931/15291 | 546 | 92 | 38 | 16 040 655 | 0.0003 |
| 14984/15304 | 480 | 80 | 59 | 0.0004 | ||
| SeV-GFP | 14931/15291 | 546 | 92 | 24 | 5 770 180 | 0.0004 |
| 14984/15304 | 480 | 80 | 19 | 0.0003 | ||
| SeV-GFP-P6 | 14931/15291 | 546 | 92 | 49 | 17 478 494 | 0.0002 |
| 14984/15304 | 480 | 80 | 33 | 0.0002 |
Fig. 3.

RNA-Seq profiles of SeVΔC-infected cells and virus stock. Reads from RNA-Seq were mapped to the SeV genome. DI RNA can be visualized as a strong peak on the 5′ end of the genome in both RNA from SeVC-infected cells and RNA from purified virus stock (top two panels). No DI RNA is apparent in SeV-Z-infected cells (bottom panel).
Fig. 4.
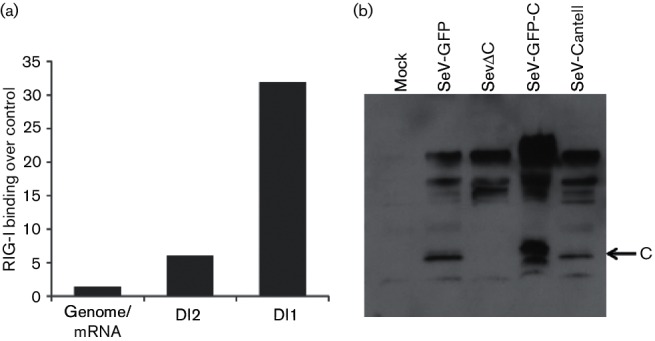
(a) Quantitative enrichment of RIG-I binding by SeV RNA species. Ratios of RIG-I-associated reads over control IP reads were calculated. (b) 293 T cells were infected with the various Sendai viruses and total cell lysates were analysed by Western blot with SeV C protein antiserum 24 h post-infection. Bands corresponding to C protein, as well as P and V proteins, can be seen.
DI-free SeV fails to activate RIG-I
Our identification of DI RNA as a ligand for RIG-I in SeVΔC-infected cells led us to postulate that other recombinant viruses lacking functional IFN antagonists may induce the IFN response through DI accumulation. Since we wanted to examine which, if any, RNA molecules would interact with RIG-I during infection with a virus free of DI RNA, we decided to utilize a virus with wild-type-like growth kinetics and IFN suppression which should be free of DI molecules. To this end we chose to perform experiments with SeV-GFP, a recombinant virus originating from the same Z strain as SeVΔC and shown to behave in a way identical to a parental wild-type virus [36]. In order to confirm the DI-free nature of SeV-GFP we utilized the same computational approach as previously described. Analysis of viral RNA from SeV-GFP stock revealed a very low presence of both DI species associated with SeV-Cantell and SeVΔC (Fig. 4a). Interestingly, when we performed the same analysis on SeV-GFP, which was passaged under conditions associated with DI accumulation, (i.e. six undiluted serial passages in embryonated eggs), we did not find any enrichment in cbDI levels, indicating that this particular virus is extremely resistant to DI accumulation. To find which RNA molecules may associate with RIG-I during SeV-GFP infection we performed RIG-I IPs in SeV-GFP-infected cells. Since levels of endogenous RIG-I in the absence of IRF3/7 activation or IFN signalling are very low, we decided to perform the immunoprecipitations in the context of RIG-I overexpression to make sure that any negative results would not be due to limited amounts of the endogenous RIG-I protein. Infections with SeV-GFP and SeVΔC were performed in parallel with an m.o.i. of 1 and RIG-I/RNA complexes were isolated in the same manner as above, with the exception of using an anti-HA antibody instead of an anti-RIG-I antibody. The isolated RNA was again tested for immunostimulatory activity in 293T-ISRE-Fluc cells (Fig. 5a). Unlike infections with SeVΔC, infections with SeV-GFP failed to produce any RIG-I-associated immunostimulatory RNA, suggesting that a wild-type SeV lacking DI RNA does not normally produce any RIG-I-specific PAMPs. Consistent with this observation, infection of the 293T ISRE-RFP cells with SeV-GFP fails to elicit any ISRE activation, unlike with infection with SeVΔC (Fig. 5b). Although this phenomenon could be attributed solely to the inhibitory activity of viral IFN antagonist proteins, it is also plausible that infection with a wild-type-like virus simply does not produce RIG-I PAMPs in sufficient quantity to activate measurable ISRE activity. In agreement with this hypothesis is a recent study showing that, even in the absence of an IFN antagonist protein, most virus-infected cells still do not activate IFN production [37]. Indeed, during infections with SeV-GFP only minimal ISRE-RFP expression can be seen over the course of several days of infection (Fig. 5c). Cells positive for both red fluorescent protein (RFP) and GFP signal are very rare, indicating that ISRE activation in infected cells is a rare event requiring extensive virus replication (Fig. 5d).
Fig. 5.

DI-free SeV is a weak activator of ISRE reporter. (a) Immunostimulatory activity of HA-RIG-I associated RNA from 293 T cells infected with either SeVΔC or SeV-GFP. In the right panel, a Western blot shows the expression levels of HA-RIG-I in both infections. (b) 293T ISRE-RFP reporter cells were infected with either SeVΔC or SeV-GFP and infections were allowed to proceed for 24 h. Red fluorescent protein (RFP) expression correlates to the immunostimulatory potential of each virus. (c) Infection of ISRE-RFP cells with SeV-GFP results in minimal RFP expression isolated to a few cells over the first 72 h of infection. (d) Individual RFP plaques surrounding SeV-GFP-infected cells at 96h post-infection. The bottom panel is a magnified view of the images in the top panel. The single yellow cell in the middle of the merged image represents a double-positive cell.
Knocking down C protein leads to activation of innate immune responses
Identification of cbDI as a RIG-I ligand in infections with SeVΔC led us to question how innate immune responses would be activated if we abrogated the function of the C protein with a small interfering (siRNA) knockdown approach post-infection. In this scenario, in contrast to initiating infections with a virus already rich in DIs, we would be infecting cells with a DI-free virus and subsequently removing the function of the C protein. Since the C ORF is expressed from the same mRNA as the P ORF, we had to take advantage of a recombinant virus which uncouples expression of P and C proteins. SeV-GFP-C contains a stop codon within the C ORF and a GFP-C protein fusion inserted between the M and F genes. Thus expression of C protein in this virus can be suppressed by siRNA targeting of the GFP ORF. Analysis of viral RNA from SeV-GFP-C stock revealed minimal presence of cbDIs, with overall levels similar to those in SeV-GFP stock, confirming that this virus, unlike SeVΔC, does not favour accumulation of DIs during passaging (Fig. 4a). To assess the effect of C protein knockdown on the replication of the SeV-GFP-C and the immune response of the infected cells we transfected siRNA targeting GFP or control scrambled siRNA into HeLa cells and infected the cells 18 h post-transfection with SeV-GFP-C. Following infection we performed in-depth RNA-Seq analysis on both SeV and cellular RNA. Analysis of viral RNA sequences revealed that despite the loss of the C protein, the overall level of virus replication, as well as the relative levels of viral mRNAs, were remarkably similar between the siGFP and siCNTRL treatments (Fig. 6a). This is surprising, since the C protein has been shown to play a role in inhibition of virus replication [26]. We could see an approximately twofold reduction in numbers of RNA sequences which mapped to the GFP mRNA; however the reduction in GFP protein expression was much more dramatic, likely indicating that the knockdown was occurring through translation suppression (Fig. 6b). In contrast to its minimal effect on virus replication, the loss of C protein had a dramatic effect on the cellular transcriptome. Despite similar levels of virus infection, we could see a clear activation of innate immune responses in siGFP-treated SeV-infected cells. Genome-wide transcriptome analysis revealed a great enrichment of innate immune-related gene cells, including IFNB1, confirming the established role of the C protein in suppressing innate immune responses (Fig. 6c). To determine whether cbDI accumulation was associated with the loss of the C protein we performed computational analysis on viral RNA from knockdown cells. We were unable to identify any reads associated with any cbDI RNAs in either siGFP- or siCNTRL-treated samples. This was not surprising, since the overall levels of viral RNA sequencing reads were much lower than those obtained from sequencing virus stocks, as cellular mRNA represented the majority of the RNA that was sequenced. To enrich for genomic viral RNA, as well as DI RNA, we performed pulldowns from siRNA-treated and SeV-GFP-C-infected cells with an antibody targeting SeV NP protein. Since all viral RNA genomes, including DI RNAs, are reported to be encapsidated within NP-covered ribonucleoprotein complexes, we postulated that isolating NP-bound RNA will likely result in the enrichment of viral genomic and DI RNAs. Sequencing profiles from NP pulldowns confirmed that we were enriching for genomic RNA species. However, we were again unable to identify any fusion RNA-Seq reads belonging to any cbDI molecules. It is possible that the DI RNAs generated during infection are present at too low a level to be detected with this approach. It is also possible that standard sequence preparation methods bias against isolation of DI RNA species, since they are optimized to detect longer RNAs, such as cellular mRNAs. The lack of isolation of any cbDI-containing reads made it impossible for us to conclude whether these RNAs were generated in C-protein-deficient cells and were associated with an activated immune response. In addition to utilizing the RNA-Seq approach, we attempted to detect cbDI RNA associated with SeVΔC through quantitative reverse transcription PCR (qRT-PCR). Although this assay is very effective at detecting cbDI RNAs when they are present in high amounts, it failed to do so in siGFP-treated samples, likely because the levels of cbDI are very low compared to full-length genomic RNA.
Fig. 6.

Knockdown of SeV C protein in infected cells. HeLa cells were transfected with siGFP or siCNTRL and infected with SeV-GFP-C (which expresses a GFP-C fusion protein). (a) Sendai RNA-Seq reads from siRNA-transfected and SeV- or mock-infected cells. The top two panels highlight very similar viral profiles in the presence or absence of C protein. (b) Comparison of loss of GFP RNA versus GFP protein, illustrating that the majority of the GFP protein is removed in siGFP cells. (c) RNA-Seq reads from siRNA-treated and infected cells were mapped to the human IFNB gene. Very strong IFNB expression can be seen in infected cells with GFP-C knockdown.
Exogenous C protein limits levels of DI RNA
The identification of DI RNA as a RIG-I ligand in SeVΔC infections was surprising, since this virus was propagated under DI-free growth conditions, such as plaque purification and low m.o.i. inoculation. Therefore we speculated whether the lack of the C protein inherently led to generation and/or accumulation of DIs. To this end, we measured the levels of DI1 during infection with SeVΔC in the presence or absence of exogenously expressed C protein. We saw a significant decrease in DI1 RNA levels, as well as genomic RNA, in the presence of C (Fig. 7). Since we could not generate a DI-rich stock from SeV-GFP, we postulated that perhaps the Cantell-strain C protein was deficient in its ability to limit DI replication. Analysis of C protein from both Z and Cantell strains, however, revealed no strain-specific differences in the ability of this protein to limit DI accumulation. The ability of exogenously expressed C protein to limit genome and DI RNA levels supports the hypothesis that DI RNA accumulation seen in the SeVΔC mutant could be due to the lack of C function(s).
Fig. 7.
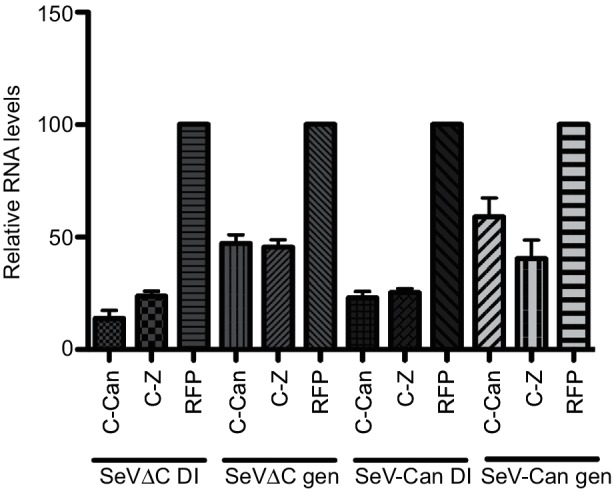
Exogenous C protein limits production of cbDI as well as genomic RNA. Relative expression of SeV cpDI or genomic RNA in the presence or absence of SeV C protein. SeV-Can or SeV-Z C protein-expressing plasmids were transfected into HeLa cells subsequently infected with either SeVΔC or SeV-Cantell. Viral RNA was measured at 24 h post-infection by q-RT-qPCR. Viral RNA levels in RFP-transfected control cells were set to 100 %. Normalized data from three individual experiments are shown with sd.
Discussion
Activation of the RIG-I antiviral pathway by viral PAMPs represents an important early event in mounting an effective antiviral immune response. Although a large number of RNA molecules, both artificial and virus-generated, have been shown to be able to activate RIG-I signalling, the question of which RNAs activate RIG-I during natural infections is more complex. We previously showed that during infection with a Sendai virus rich in DI RNA the DI species functioned as the sole RIG-I PAMP, and we did not see any interaction between RIG-I and other viral RNAs. This left an open question: how does a DI-free virus activate a RIG-I-dependent immune response? In the current study we wanted to extend our work to a Sendai virus that is known to induce an IFN response by a different mechanism, through the absence of its IFN antagonist protein C. Surprisingly, we found that despite DI-free growth conditions, SeVΔC was generating high amounts of DI RNAs, and that these RNAs again served as exclusive ligands for RIG-I during infection. Furthermore, analysis of a wild-type-like DI-free Sendai virus failed to detect any RIG-I-specific ligands. Therefore, it is possible that in the absence of DIs or other aberrant RNA generation, the RIG-I pathway may only be poorly activated. This phenomenon would fit well with the observation that only a very small portion of infected cells activate their IFN responses, which may represent those cells in which DIs or other abnormal virus replication products are generated [38–41].
The high accumulation of DI RNA in the absence of the SeV C protein is intriguing, and the mechanisms responsible for this phenotype are unknown. Interestingly, infection with SeV lacking C protein has previously been shown to lead to accumulation of dsRNA in infected cells, with concurrent PKR activation [42]. A similar observation has been made with a parainfluenza virus lacking the C ORF [43]. Since these studies did not specifically look for DI RNA in their experiments, it is possible that the detected dsRNA species belonged to the double-stranded regions found within the cbDI molecules. Identification of dsRNA in other ΔC paramyxoviruses may indicate that the ability of the C protein to limit DI levels is a more general phenomenon, and not limited exclusively to SeV. It is unclear whether the lack of normal C protein function results in a polymerase complex with a greater propensity to generate DIs, or if the C protein has a more direct role in controlling DI levels. It is also possible that C protein interaction with a host factor ultimately leads to control of DI generation or accumulation. A recent report examining SeV deficient in all four C ORF proteins 4C(−) concluded that the dsRNA species generated by this virus were not DIs, based on their lack of encapsidation and their ability to be detected by the J2-dsRNA-specific antibody versus the cpDI of SeV-Cantell, which cannot be detected by the J2 antibody [44]. Interestingly, in our studies we could not obtain a signal with J2 staining in either SeV-Cantell- or SeVΔC-infected cells, further supporting our conclusion that the IFN-inducing RNA species in SeVΔC-infected cells are DIs. The reason for the discrepancy between our study and that of Yoshida et al. [44] is unclear. The C proteins of these two SeVs were eliminated differently – mutation of the individual ribosomal start codons for 4C(−), and the inclusion of three consecutive stop codons within the C ORF shortly downstream of the Y2 start codon for deltaC – but this difference in itself does not explain the discrepancy.
Another recent study examining measles virus C protein reported accumulation of DI RNA in the absence of measles C, and furthermore the authors reported the accumulation of A-to-G editing, which is indicative of ADAR1 activity [45]. Examination of our own RNA-Seq data from SeVΔC-infected cells or viral stocks did not reveal any A-to-G editing events within the DI RNA sequences.
Our findings, as well as several other recent reports examining C-protein-deficient virus mutants, bring up the interesting possibility that mutant viruses which lack the ability to suppress the IFN response may do so not only through altered protein-mediated mechanisms, but also through accumulation of RNA products that function as cellular PAMPs [45, 46]. In fact, a number of reports indicate that manipulation of the viral genome may unintentionally lead to DI accumulation, irrespective of virus growth conditions [14, 47]. Therefore, it may be prudent to assess the make-up of viral RNA populations when examining phenotypes associated with a particular virus mutant.
The precise mechanisms responsible for DI generation remain unknown, but they are believed to be caused by errors in viral polymerase function. Historically associated with laboratory virus propagation conditions (e.g. high MOI infections), the existence of DI RNAs in natural infections remains a topic that has not been addressed in great detail. However, a number of recent studies indicate that these RNAs may be abundant in natural infections [48–52]. Advances in sequencing technology that allow global, in-depth analysis of viral populations are likely to reveal a great heterogeneity of viral RNA species, including DI RNAs, in infected hosts. The potential role that these RNAs may play in the activation of antiviral innate immunity in vivo is a topic of great interest and importance. This knowledge is especially critical for the design of effective vaccines and antivirals, whose efficacy often hinges on their ability to efficiently stimulate innate immune pathways. Currently, despite intensive investigation, we know very little about precisely how innate immunity is activated during virus infections.
Methods
Viruses
All Sendai viruses with the exception of SeV-Cantell originated from SeV strain Z (SeV-Z). All SeV stocks were grown in the allantoic cavity of 10-day-old embryonated chicken eggs. Viruses were grown under conditions associated with minimal DI accumulation, i.e inoculation at high dilution and plaque purification. Virus present in the allantoic fluid was analysed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining after virus pelleting. Virus titres were determined by plaque formation on LLC-MK2 cells. The construction and rescue of recombinant SeVΔC and SeV-GFP have been described previously [19, 36]. SeVΔC was generated by the inclusion of three consecutive stop codons downstream of the Y2 ATG. Therefore, this virus is lacking in expression of all four C proteins. This virus was rescued by three or four blind passages, with no evidence of viruses on Coomassie blue staining with purified virus from allantoic fluid. By passage six the virus reached a plateau at 107 to 108 p.f.u. ml−1; passages six to eight were used for experiments. rSeV-GFP-C contains a GFP-C fusion insert between M and F ORFs, inserted within MluI sites in a backbone with a stop codon in endogenous C ORF.
RIG-I IPs
Endogenous RIG-I co-IPs were performed as previously described [11]. Briefly, 293 T cells were infected with viruses at an m.o.i. of 10 and the infections were allowed to proceed for 24 h. Cells were lysed in polysome lysis buffer and endogenous RIG-I/RNA complexes were isolated with an anti-RIG-I mouse monoclonal antibody generated by the Mount Sinai School of Medicine hybridoma facility. RIG-I-associated RNA was extracted with a phenol/chloroform extraction and ethanol precipitation. Co-IPs with exogenous RIG-I were performed by transfecting a plasmid expressing HA-tagged RIG-I protein (pCAGGS-HA-RIG-I) into 293 T cells 1 h post-infection. Cells were lysed 24 h post-infection and RIG-I/RNA complexes were isolated with an anti-HA antibody (H3663; Sigma). RNA was isolated by phenol/chloroform extraction and tested for immunostimulatory activity by transfection into 293T ISRE-Fluc cells. Control IPs were performed with a mouse monoclonal IgG1 anti-GFP antibody (ab1218; Abcam). The efficiency of the IPs was verified through Western blot analysis.
RNA deep sequencing and analysis
Isolated RNA was prepared for deep sequencing as previously described [11]. Briefly, ribosomal RNA was depleted with a human Ribo-Zero kit (Epicentre). The remaining RNA was then prepared into a sequencing library with an Illumina RNA-Seq kit, with the omission of the polyA-enrichment step. The resultant library was sequenced on the Illumina HiSeq platform at the Mount Sinai Genomics Center. Sequencing reads were processed and analysed with the assistance of the Mount Sinai genomics facility. RNA from virus stocks and from siRNA-treated SeV-infected cells was prepared for sequencing with the Illumina TruSeq Stranded Total RNA kit and sequenced as single-end 100nt reads on Illumina HiSeq at the Rockefeller Genomics facility. To identify cbDI RNA sequences from virus RNA-Seq data, we utilized open-source RNA-Seq analysis tools. The quality of the FastQ sequencing reads was initially assessed through FastQC and high-quality sequences were selected with the FASTQ Quality Trimmer. High-quality reads were aligned to either SeV-Z or SeV-Cantell genomes with TopHat2. Fusion reads were identified by utilizing the fusion search function within TopHat2. Viral alignments were performed with the following TopHat2 settings: max. edit distance – 4, final read mismatches – 4, max. insertion length – 3, max. deletion length – 3, maximum number of alignments – 20, minimum intron length – 5, maximum intron length – 15 000, number of mismatches allowed in each segment for reads mapped independently – 2, minimum length of read segments – 15, fusion search anchor length – 10, fusion search minimum distance – 5, fusion search read mismatches – 3, fusion search multireads – 5 and fusion search multipairs – 2.
Knockdown of C protein in SeV-C-GFP-infected cells
HeLa cells were transfected with an siRNA targeting the GFP ORF (Thermo Fisher AM4626) or scrambled control siRNA. At 18 h post-transfection the cells were either infected with SeV-C-GFP at an m.o.i. of 10 or mock infected. The knockdown efficiency was monitored by examining the levels of GFP expression in infected cells 24 h post-infection by fluorescence microscopy. Total RNA from infected cells was collected 24 h post-infection and prepared for RNA-Seq analysis with the Illumina TruSeq Stranded Total RNA kit. Sequencing data were analysed in a similar manner to that described above. Sequencing reads were aligned to both Sendai virus and human genomes with TopHat2. Human genome alignments were performed against 1000 genomes (project build 37;GRCh37). Individual gene expression levels were visualized using the Integrative Genomics Viewer (IGV). Reads mapping to the human genome were further subjected to differential expression analysis with HTSeq (Python) followed by EdgeR (R). Statistically significant (P<0.05) differentially expressed genes were analysed for functional enrichment using the NIH-based david Functional Annotation tool (v 6.7). SeV NP IPs were performed with an anti-SeV NP antibody from siRNA-treated and SeV-C-GFP-infected cells in the same manner as was described for RIG-I IPs. NP-associated RNA was prepared for sequencing and analysed in the same manner as total RNA from SeV-C-GFP-infected cells.
C protein complementation
293 T cells were transfected with plasmids expressing SeV C protein (pEBS-SeV-C) [53] or a control (RFP-expressing) plasmid. At 24 h post-transfection, the cells were infected with SeVΔC and infections were allowed to proceed for 24 h. At this time, total RNA was isolated from the infected cells and subjected to TaqMan quantitative PCR analysis for DI1 RNA and genomic RNA levels. TaqMan analysis was performed with the following primer/probe combinations. DI detection: SeV-Z-di1-FWD CAAGACTTCCAGGTACAAA AGAG, SeV-Z-di1-REV ACGCGATCCGGATTACACT and Sen-DI-hyb-probe (5′ FAM, 3′ quencher) CAAGATTGGTA ACTGGGTCATTC. SeV genome detection: Sen-gen-TQM-FWD GGGAGGAGGTGCTGTTATCC, Sen_gen_TQM_REV TAGCCGGACTTCCGTTTGTA and Roche universal probe #no. 82. One-step qRT-PCR was performed with Roche LightCycler 480 RNA Master Hydrolysis Probes (04991885001) kit and GAPDH housekeeping control multiplex assay (Roche 05190541001). Analysis was performed on the Roche LightCycler 480.
Funding information
This work was partially supported by NIAID grant U19AI083025 to A. G.-S.
Acknowledgements
We thank Richard Cadagan and Osman Lizardo for excellent technical assistance.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: cbDI, copy-back defective interfering; CIP, calf alkaline phosphatase; DI, defective interfering; Fluc, firefly luciferase; ISRE, IFN-sensitive response element; NK, natural killer; PAMP, pathogen-associated molecular pattern; PRR, pattern RIG-I-like receptors; qRT-PCR, quantitative reverse transcription PCR; RFP, red fluorescent protein; RIG-I, retinoic acid inducible gene; RLR, RIG-I-like receptors; SeV, Sendai virus; si, small interfering; TLR, toll-like receptor; IP, immuno-precipitated.
References
- 1.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Baum A, García-Sastre A. Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids. 2010;38:1283–1299. doi: 10.1007/s00726-009-0374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 4.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 5.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, et al. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure. 2010;18:1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowalinski E, Lunardi T, Mccarthy AA, Louber J, Brunel J, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci USA. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baum A, Sachidanandam R, García-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci USA. 2010;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazzarini RA, Keene JD, Schubert M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell. 1981;26:145–154. doi: 10.1016/0092-8674(81)90298-1. [DOI] [PubMed] [Google Scholar]
- 13.Marcus PI, Sekellick MJ. Defective interfering particles with covalently linked [±] RNA induce interferon. Nature. 1977;266:815–819. doi: 10.1038/266815a0. [DOI] [PubMed] [Google Scholar]
- 14.Shingai M, Ebihara T, Begum NA, Kato A, Honma T, et al. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J Immunol. 2007;179:6123–6133. doi: 10.4049/jimmunol.179.9.6123. [DOI] [PubMed] [Google Scholar]
- 15.Strahle L, Garcin D, Kolakofsky D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology. 2006;351:101–111. doi: 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 16.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 17.Moltedo B, López CB, Pazos M, Becker MI, Hermesh T, et al. Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity. J Immunol. 2009;183:3569–3573. doi: 10.4049/jimmunol.0900091. [DOI] [PubMed] [Google Scholar]
- 18.Tapia K, Kim WK, Sun Y, Mercado-López X, Dunay E, et al. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog. 2013;9:e1003703. doi: 10.1371/journal.ppat.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strähle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, et al. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J Virol. 2007;81:12227–12237. doi: 10.1128/JVI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Childs K, Stock N, Ross C, Andrejeva J, Hilton L, et al. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 21.Cadd T, Garcin D, Tapparel C, Itoh M, Homma M, et al. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J Virol. 1996;70:5067–5074. doi: 10.1128/jvi.70.8.5067-5074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curran J, Boeck R, Kolakofsky D. The Sendai virus P gene expresses both an essential protein and an inhibitor of RNA synthesis by shuffling modules via mRNA editing. EMBO J. 1991;10:3079–3085. doi: 10.1002/j.1460-2075.1991.tb07860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran J, Marq JB, Kolakofsky D. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology. 1992;189:647–656. doi: 10.1016/0042-6822(92)90588-G. [DOI] [PubMed] [Google Scholar]
- 24.Tapparel C, Hausmann S, Pelet T, Curran J, Kolakofsky D, et al. Inhibition of Sendai virus genome replication due to promoter-increased selectivity: a possible role for the accessory C proteins. J Virol. 1997;71:9588–9599. doi: 10.1128/jvi.71.12.9588-9599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurotani A, Kiyotani K, Kato A, Shioda T, Sakai Y, et al. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells. 1998;3:111–124. doi: 10.1046/j.1365-2443.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 26.Latorre P, Cadd T, Itoh M, Curran J, Kolakofsky D. The various Sendai virus C proteins are not functionally equivalent and exert both positive and negative effects on viral RNA accumulation during the course of infection. J Virol. 1998;72:5984–5993. doi: 10.1128/jvi.72.7.5984-5993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koyama AH, Irie H, Kato A, Nagai Y, Adachi A. Virus multiplication and induction of apoptosis by Sendai virus: role of the C proteins. Microbes Infect. 2003;5:373–378. doi: 10.1016/S1286-4579(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 28.Hasan MK, Kato A, Muranaka M, Yamaguchi R, Sakai Y, et al. Versatility of the accessory C proteins of Sendai virus: contribution to virus assembly as an additional role. J Virol. 2000;74:5619–5628. doi: 10.1128/JVI.74.12.5619-5628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irie T, Nagata N, Yoshida T, Sakaguchi T. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology. 2008;371:108–120. doi: 10.1016/j.virol.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 30.Irie T, Shimazu Y, Yoshida T, Sakaguchi T. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J Virol. 2007;81:2263–2273. doi: 10.1128/JVI.02218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu T, Takeuchi K, Yokoo J, Gotoh B. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology. 2004;325:137–148. doi: 10.1016/j.virol.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 32.Garcin D, Marq JB, Strahle L, Le Mercier P, Kolakofsky D. All four Sendai virus C proteins bind Stat1, but only the larger forms also induce its mono-ubiquitination and degradation. Virology. 2002;295:256–265. doi: 10.1006/viro.2001.1342. [DOI] [PubMed] [Google Scholar]
- 33.Strähle L, Garcin D, Le Mercier P, Schlaak JF, Kolakofsky D. Sendai virus targets inflammatory responses, as well as the interferon-induced antiviral state, in a multifaceted manner. J Virol. 2003;77:7903–7913. doi: 10.1128/JVI.77.14.7903-7913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang AS. Defective interfering viruses. Annu Rev Microbiol. 1973;27:101–117. doi: 10.1146/annurev.mi.27.100173.000533. [DOI] [PubMed] [Google Scholar]
- 35.Calain P, Roux L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol. 1993;67:4822–4830. doi: 10.1128/jvi.67.8.4822-4830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mottet-Osman G, Iseni F, Pelet T, Wiznerowicz M, Garcin D, et al. Suppression of the Sendai virus M protein through a novel short interfering RNA approach inhibits viral particle production but does not affect viral RNA synthesis. J Virol. 2007;81:2861–2868. doi: 10.1128/JVI.02291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Killip MJ, Young DF, Ross CS, Chen S, Goodbourn S, et al. Failure to activate the IFN-β promoter by a paramyxovirus lacking an interferon antagonist. Virology. 2011;415:39–46. doi: 10.1016/j.virol.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Short JA, Young DF, Killip MJ, Schneider M, et al. Heterocellular induction of interferon by negative-sense RNA viruses. Virology. 2010;407:247–255. doi: 10.1016/j.virol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senger K, Merika M, Agalioti T, Yie J, Escalante CR, et al. Gene repression by coactivator repulsion. Mol Cell. 2000;6:931–937. doi: 10.1016/S1097-2765(05)00081-X. [DOI] [PubMed] [Google Scholar]
- 40.Zawatzky R, De Maeyer E, de Maeyer-Guignard J. Identification of individual interferon-producing cells by in situ hybridization. Proc Natl Acad Sci USA. 1985;82:1136–1140. doi: 10.1073/pnas.82.4.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J, Sealfon SC, Hayot F, Jayaprakash C, Kumar M, et al. Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res. 2007;35:5232–5241. doi: 10.1093/nar/gkm557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeuchi K, Komatsu T, Kitagawa Y, Sada K, Gotoh B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol. 2008;82:10102–10110. doi: 10.1128/JVI.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boonyaratanakornkit J, Bartlett E, Schomacker H, Surman S, Akira S, et al. The C proteins of human parainfluenza virus type 1 limit double-stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J Virol. 2011;85:1495–1506. doi: 10.1128/JVI.01297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida A, Kawabata R, Honda T, Tomonaga K, Sakaguchi T, et al. IFN-β-inducing, unusual viral RNA species produced by paramyxovirus infection accumulated into distinct cytoplasmic structures in an RNA-type-dependent manner. Front Microbiol. 2015;6:804. doi: 10.3389/fmicb.2015.00804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, et al. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J Virol. 2015;89:7735–7747. doi: 10.1128/JVI.01017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfaller CK, Radeke MJ, Cattaneo R, Samuel CE. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J Virol. 2014;88:456–468. doi: 10.1128/JVI.02572-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komarova AV, Combredet C, Sismeiro O, Dillies MA, Jagla B, et al. Identification of RNA partners of viral proteins in infected cells. RNA Biol. 2013;10:943–956. doi: 10.4161/rna.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li D, Lott WB, Lowry K, Jones A, Thu HM, et al. Defective interfering viral particles in acute dengue infections. PLoS One. 2011;6:e19447. doi: 10.1371/journal.pone.0019447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pesko KN, Fitzpatrick KA, Ryan EM, Shi PY, Zhang B, et al. Internally deleted WNV genomes isolated from exotic birds in New Mexico: function in cells, mosquitoes, and mice. Virology. 2012;427:10–17. doi: 10.1016/j.virol.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forrester NL, Guerbois M, Adams AP, Liang X, Weaver SC. Analysis of intrahost variation in venezuelan equine encephalitis virus reveals repeated deletions in the 6-kilodalton protein gene. J Virol. 2011;85:8709–8717. doi: 10.1128/JVI.00165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol. 2013;87:8064–8074. doi: 10.1128/JVI.00240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petterson E, Stormoen M, Evensen Ø, Mikalsen AB, Haugland Ø. Natural infection of Atlantic salmon (Salmo salar L.) with salmonid alphavirus 3 generates numerous viral deletion mutants. J Gen Virol. 2013;94:1945–1954. doi: 10.1099/vir.0.052563-0. [DOI] [PubMed] [Google Scholar]
- 53.Garcin D, Marq JB, Iseni F, Martin S, Kolakofsky D. A short peptide at the amino terminus of the Sendai virus C protein acts as an independent element that induces STAT1 instability. J Virol. 2004;78:8799–8811. doi: 10.1128/JVI.78.16.8799-8811.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]