

Abstract
Protein–ATP interactions are ubiquitous in a wide variety of biological processes. Correctly locating ATP binding sites from protein information is an important but challenging task for protein function annotation and drug discovery. However, there is no method that can optimally identify ATP binding sites for different proteins. In this study, we report a new composite predictor, ATPbind, for ATP binding sites by integrating the outputs of two template-based predictors (i.e., S-SITE and TM-SITE) and three discriminative sequence-driven features of proteins: position specific scoring matrix, predicted secondary structure, and predicted solvent accessibility. In ATPbind, we assembled multiple support vector machines (SVMs) based on a random undersampling technique to cope with the serious imbalance phenomenon between the numbers of ATP binding sites and of non-ATP binding sites. We also constructed a new gold-standard benchmark data set consisting of 429 ATP binding proteins from the PDB database to evaluate and compare the proposed ATPbind with other existing predictors. Starting from a query sequence and predicted I-TASSER models, ATPbind can achieve an average accuracy of 72%, covering 62% of all ATP binding sites while achieving a Matthews correlation coefficient value that is significantly higher than that of other state-of-the-art predictors.
Graphical Abstract
INTRODUCTION
Interactions between proteins and ligands are indispensable for biological activities and play important roles in a wide variety of biological processes.1–3 Hence, accurately locating the protein–ligand binding sites or pockets is of significant importance for both analyzing protein function and designing novel drugs.4–7 Tremendous wet-lab efforts have been made to uncover the intrinsic mechanisms of protein–ligand interactions, and thousands of protein–ligand interaction structure complexes have been deposited into the PDB.8 However, identifying protein–ligand binding sites via wet-lab experimental technologies is often cost-intensive and time-consuming. Due to the importance of protein–ligand interactions and the difficulty of experimentally identifying the binding sites, the development of efficient and automatic computational methods for the fast prediction of protein–ligand binding sites has become an increasingly important problem in bioinformatics, especially when faced with the large-scale protein sequences of the postgenomic era.9,10
Many computational methods have emerged for predicting protein–ligand binding sites during the past decades.9–12 These methods can be generally grouped into two categories: general-purpose methods and ligand-specific methods. In the early stage, general-purpose predictors, which predict ligand binding sites (or pockets) regardless of the ligand types, dominated the field of protein–ligand binding site prediction, including (to name a few) LIGSITE,13 CASTp,14 SURFNET,15 POCKET,16 Fpocket,17 Q-SiteFinder,18 SITEHOUND,19 and 3DLigandSite.20 Recently, another general-purpose predictor, COACH,9 a meta-server approach to protein–ligand binding site prediction, was designed. In COACH, two template-based predictors, i.e., TM-SITE and S-SITE, were first proposed for complementary binding site prediction based on binding-specific substructure comparison and sequence profile alignment, respectively; the binding models from TM-SITE and S-SITE were then combined with results from other general-purpose predictors, such as COFACTOR,21 FINDSITE,22 and ConCavity,11 to obtain the final ligand binding site prediction.9
Since different ligands tend to bind diverse types of residues with prominent specificities due to the specific roles, sizes, and distributions of protein–ligand interactions,23 the second type of ligand-specific predictors, which are designed to predict binding sites (or pockets) for specific ligand types, have been increasingly of interest. Such predictors include NsitePred,24 TargetS,10 TargetSOS,25 and TargetNUCs26 for nucleotides; FINDSITE-metal27 and CHED28 for metal; DNABR29 and MetaDBSite30 for DNA; and IonCom31 for metal and acid radical ion binding site predictions. These studies demonstrated that ligand-specific binding site predictors are often superior to general-purpose binding site predictors due to the added consideration of the physicochemical features of specific ligand–protein interactions.
Among the many ligand-specific binding predictors, adenosine-5′-triphosphate (ATP) is of particular interest. ATP is a nucleotide, also called a nucleoside triphosphate, which is a small molecule that is used in cells as a coenzyme and plays an essential role in membrane transport, cellular motility, muscle contraction, signaling, the transcription and replication of DNA, and various metabolic processes.32 It interacts with proteins through protein–ATP binding sites and provides chemical energy to proteins via the hydrolysis of ATP.33 The proteins can then perform various biological functions using the chemical energy. Additionally, ATP binding sites are valuable drug targets for antibacterial and anticancer chemotherapy.34 Hence, accurately localizing the protein–ATP binding sites is of significant importance for both protein function annotation and drug discovery.
ATPint32 is one of the first custom-designed computational predictors for identifying ATP-specific binding sites, and it was trained with a position specific scoring matrix (PSSM) and several other sequential descriptors, using a data set consisting of 168 nonredundant ATP interacting proteins. ATPsite35 was later proposed by Kurgan and co-workers; this system combined PSSM and an SVM and was trained on a larger data set with 227 nonredundant ATP interacting proteins. However, the imbalanced learning problem36 embedded in protein–ATP binding site prediction, i.e., that the number of non-ATP binding sites is much larger than the number of ATP binding sites, is a problem that could decrease the final prediction performance and is ignored by ATPint and ATPsite. To solve the imbalanced learning problem and enhance the prediction accuracy, we recently proposed TargetATPsite37 by integrating random undersampling (RUS) and AdaBoost36 ensemble algorithms. Except for the above three ATP-specific predictors, many nucleotide-specific binding site predictors, e.g., NsitePred,24 TargetS,33 and TargetNUCs,26 which also contain an ATP binding site prediction model, can be used to predict the ATP binding sites. These existing approaches have the advantage of generating predictions from sequence alone, but the Matthews correlation coefficient (MCC) of the predictions is low (typically approximately 0.580 at 52% sensitivity) because sequence information cannot directly show protein function.
Despite the progress made in the ATP binding prediction, most predictors are based on protein sequence information, but protein structure information, which has demonstrated a significant advantage in other ligand binding studies,9,21 has not been utilized. In this study, we aim to systematically examine the impact of the employment of structure-based features on ATP binding prediction by developing a new meta ATP binding site predictor called ATPbind, which integrates the outputs of two template-based predictors, i.e., S-SITE and TM-SITE, with three sequence-based features, i.e., the position specific scoring matrix, the predicted secondary structure, and the predicted solvent accessibility. Here, S-SITE is a sequence-template-based predictor, and TM-SITE is a structure-template-based predictor. To ensure that the proposed ATPbind is an ATP-specific predictor, we extend S-SITE and TM-SITE to the ATP-specific predictors and rename them as S-SITEatp and TM-SITEatp, respectively. A new gold-standard benchmark data set consisting of 429 nonredundant ATP binding proteins was collected from the PDB database and will be used to systematically examine the strengths and weaknesses of such a composite combination of sequence and structure information for ATP binding predictions. In particular, given the significant imbalance feature of the ATP binding data, we introduced a mean-ensemble-based method to integrate multiple support vector machines (SVMs) based on the random undersampling technique.
MATERIALS AND METHODS
Benchmark Data Sets
We constructed a data set of 2144 ATP binding protein chains, named PATP-2144, which had clear target annotations and had been deposited into the Protein Data Bank (PDB)8 before November 5, 2016. We further removed the redundant sequences using CD-hit software38 with sequence identity <40%, yielding a total of 429 nonredundant protein sequences. Next, we divided the 429 nonredundant sequences into a training data set (PATP-388) and an independent test data set (PATP-TEST). PATP-388 consists of 388 protein sequences that had all been deposited into the PDB before November 5, 2014, and PATP-TEST includes 41 protein chains that were deposited into the PDB after November 5, 2014. More specifically, PATP-388 consists of 5657 ATP binding residues (i.e., positive samples) and 142 086 non-ATP binding residues (i.e., negative samples), and PATP-TEST consists of 674 positive and 14 159 negative samples. Table 1 summarizes the detailed statistical composition of PATP-388 and PATP-TEST. To illustrate the low homology between sequences among training and independent validation data sets, we further list the maximum sequence identity of each protein in PATP-TEST against all proteins in PATP-388 in Table S1. A detailed list of the ATP–protein binding samples is available at http://zhanglab.ccmb.med.umich.edu/ATPbind/Dataset_list.pdf.
Table 1.
Statistical Composition of the Training and Independent Validation Data Sets
| data set | no. of sequences | numPa | numNb | ratioc |
|---|---|---|---|---|
| PATP-388 | 388 | 5,657 | 142,086 | 25.12 |
| PATP-TEST | 41 | 674 | 14,159 | 21.01 |
numP represents the number of positive samples.
numN represents the number of negative samples.
ratio = numN/numP.
Feature Representation
Protein–ATP binding site prediction is a traditional binary classification problem in the machine-learning field. How to encode ATP-specific binding sites with discriminative features is one of the most crucial steps in constructing a machine-learning-based prediction model. The discriminative features used in this study can be categorized into five groups, position specific score matrix (PSSM), predicted secondary structure (PSS), predicted solvent accessibility (PSA), the prediction result of S-SITEatp (an ATP-specific S-SITE9), and the prediction result of TM-SITEatp (an ATP-specific TM-SITE9). Here, S-SITEatp and TM-SITEatp are both template-based methods that can predict the ATP-specific binding probability of each query residue.
Position Specific Scoring Matrix
For each query protein sequence, its PSSM profile is generated by using PSI-BLAST39 to search against the Swiss-Prot database40 through three iterations, with 0.001 used as the E-value cutoff for multiple sequence alignment. The normalization logistic function is then utilized to rescale the score of each element, denoted as x, in a PSSM profile in the interval (0, 1):
| (1) |
To extract the PSSM-view feature of each residue, a sliding window with size W (centered at the target residue) is utilized to search the rescaled PSSM.32 Inspired by ATPint,32 we set W = 17 in this study. This causes the dimensionality of the PSSM-view feature to be 17 × 20 = 340.
Predicted Secondary Structure
For a given protein sequence, we obtain its PSS information by applying PSIPRED,41 which predicts the probabilities belonging to three secondary structure classes (coil (C), helix (H), and strand (E)) of each residue. Thus, for a protein with N residues, we obtain an N × 3 probability matrix, which contains the predicted secondary structure information on the protein. Again, a sliding window of size 17 is used to present the PSS-view feature of each residue and the dimensionality of the extracted feature is 17 × 3 = 51.
Predicted Solvent Accessibility
The PSA characteristics of each residue can be obtained by feeding the corresponding sequence to the standalone SANN program,42 downloaded from http://lee.kias.re.kr/~newton/sann/. For each query sequence, SANN precisely predicts its PSA profile (in N rows and 3 columns, where N is the length of the query sequence), which includes the probabilities of three solvent accessibility classes (i.e., buried (B), intermediate (I), and exposed (E)) for each residue. A sliding window with size 17 is also employed to extract the PSA-based feature of each residue. Accordingly, the dimensionality of the PSA-view feature is 17 × 3 = 51.
S-SITEatp-Based Feature
S-SITE is a template-based method with good performance that detects protein templates and general-purpose binding sites using sequence profile–profile comparisons.9 However, S-SITE can be time-consuming for predicting ATP binding sites, and its predicted results cannot directly tell us whether the predicted binding sites are ATP binding sites. The main reason may be that S-SITE needs to search all protein sequences in the BioLip library whether the protein is an ATP binding one.9 In this study, we extended S-SITE to an ATP-specific S-SITE (named S-SITEatp). In S-SITEatp, the Swiss-Prot database40 is used to generate the multiple sequence alignment profile of the query protein sequence. PATP-2144 is utilized to replace the BioLip library43 for searching the template protein sequences. For a given protein sequence, S-SITEatp predicts the ATP binding probability value for each residue in the protein. It is worth noting that all homologous templates with a sequence identity >30% to the query sequence are excluded in S-SITEatp. Again, a sliding window (W = 17) is used to represent the S-SITEatp-based feature of each residue. The S-SITEatp-based feature dimensionality is 17. The comparison results between S-SITEatp and S-SITE can be found in Text S1 in the Supporting Information (SI).
TM-SITEatp-Based Feature
TM-SITE is a structure-template-based method that is also designed to derive the general-purpose binding sites by structurally comparing the query protein with the template proteins.9 Similar to S-SITE, it is time-consuming to use TM-SITE to detect the ATP-specific binding sites. In this study, we extended TM-SITE to be ATP-specific, named it TM-SITEatp, by replacing the BioLip library43 to an ATP-specific database (PATP-2144 in this paper). For a given protein with a known structure, TM-SITEatp can directly predict the ATP binding probability for each target residue. For a given protein sequence without a 3D structure, we first construct its modeling structure by I-TASSER.44 The ATP binding probability of each residue is then detected by TM-SITEatp. To avoid homologous contamination, homologous templates with a sequence identity >30% to the query have been excluded from both I-TASSER and ATP binding libraries. Finally, the TM-SITEatp-based feature (whose dimensionality is 17) is gained based on a sliding window with size 17. To show the performance of TM-SITEatp, we compare TM-SITEatp and the original TM-SITE in Text S2 in the SI.
Learning from Imbalanced Data
Protein–ATP binding site prediction is a standard imbalanced learning problem, where the number of the majority class (non-ATP binding residues) is larger than that of the minority class (ATP binding residues).37 From Table 1, we see that the imbalanced ratio between the number of non-ATP binding residues and the number of ATP binding residues is larger than 21. Compared to the minority class, the majority class contains lots of redundant information in the original data set, which can decrease the prediction performance and increase the training and testing time. To overcome this hurdle, random under-sampling (RUS)36 and mean ensemble schemes (ME) are combined to solve the imbalanced learning problem. RUS is used to reduce the number of the majority class in this study. However, RUS comes with the disadvantage of potentially losing useful information.36 To overcome this disadvantage caused by RUS, we employ ME to enhance the final prediction accuracy.
More specifically, in the training stage, we randomly sample the majority class T times (in the present study, T = 10) with RUS and thus obtained T majority training subsets. The T majority training subsets each in addition to the minority class (ATP binding sites) training set constitute T new training data sets. Then, a kind of machine learning model (SVM in this study) is trained on each of the T new training data sets. In the prediction stage, for each residue in a given protein sequence, its probability of belonging to the ATP binding site class is predicted by each of the T prediction models. Then, the T probabilities of the residue belonging to the ATP binding site class are fused by ME. The details of ME is described as follows.
Let be the T predicted probabilities, where pt means the probability output of the tth prediction model. The equation of ME can then be represented as follows.
| (2) |
where pME is the average probability.
Support Vector Machine
SVM45 is utilized to construct the base classifiers. We use LIBSVM,46 which is freely available at http://www.csie.ntu.edu.tw/~cjlin/libsvm/, to implement the SVM algorithm. Here, a radial basis function is chosen as the kernel function. The kernel width parameter σ and the regularization parameter γ, which are the two most important parameters, are optimized over a 5-fold cross-validation using a grid search strategy in the LIBSVM tool. The details of cross-validation can be found in Text S3 in the SI.
Architecture of ATPbind
Figure 1 illustrates the architecture of the proposed ATPbind for ATP binding site prediction. For a given protein, ATPbind can extract the above five different view features for each target residue by calling the corresponding programs and applying the sliding window technique. In the training phase, after extracting the features of all proteins in PATP-388, we can obtain the extremely unbalanced training sample set. We then employ RUS T (T = 10 in this study) times to construct a set of multiple training subsets. One prediction model is trained for each subset using SVM. The ensemble prediction model is finally created using the ME method by integrating the outputs of the different models. In the prediction phase, for a protein to be predicted, the ensemble prediction model can be utilized to give the probability output for each residue of being an ATP binding residue. We also propose a sequence-based ATP binding predictor, named ATPseq, that only utilizes information from the protein sequence. The only difference between ATPseq and ATPbind is that ATPseq does not use TM-SITEatp-based feature.
Figure 1.
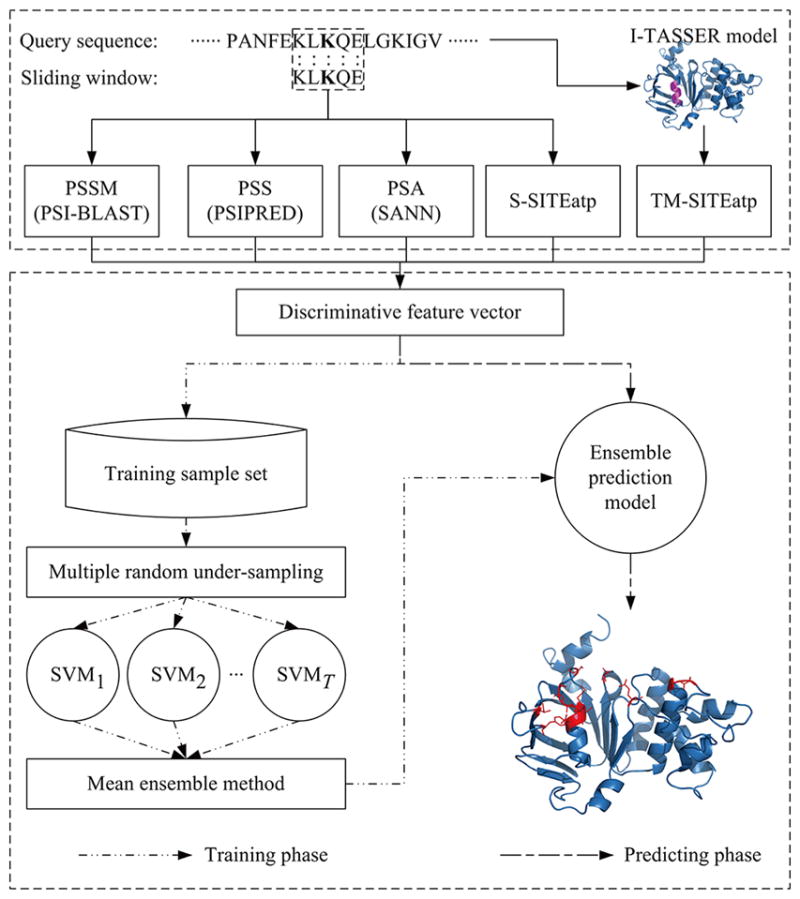
Architecture of ATPbind.
Assessing Predictive Ability
Five evaluation indexes that are routinely used in this field, i.e., sensitivity (Sen), specificity (Spe), accuracy (Acc), precision (Pre), and the Matthews correlation coefficient (MCC), are utilized to evaluate predictive ability, as follows:
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where TN, TP, FN, and FP are abbreviations for true negatives, true positives, false negatives, and false positives, respectively. The MCC (ranging from −1 to 1) evaluates the overall predictive quality. A higher MCC value means a better prediction performance. 0 represents that all residues are predicted as nonbinding (or binding). The reported threshold, which can maximize the MCC value, is then chosen to calculate the values of Sen, Spe, Acc, Pre, and MCC. Furthermore, the area under the receiver operating characteristic (ROC) curve (termed AUC), which increases in direct proportion to the overall prediction performance, is employed to assess the overall predictive ability.
RESULTS AND DISCUSSION
Assessment of the Quality of the I-TASSER-Modeled Structures
Since the quality of the modeled structure of the protein has an impact on TM-SITEatp, we compare the accuracy of the current predictions using I-TASSER44 with the experimental structures on the PATP-TEST data set, where the protein lengths range from 115 to 863, in terms of the TM-score and RMSD evaluation indexes.47 For each given protein, the standard I-TASSER program, which excludes all homologous template proteins with sequence identity >30% to the given sequence, generates its structural model from the query protein sequence with iterative fragment assembly simulations. We then calculate TM-score and RMSD of the I-TASSER-modeled structures (ITAMSs) for the 41 testing proteins. The results are compiled in Figure 2. From Figure 2, it is easily found that the majority of the testing proteins (≈87.8%) can be modeled by I-TASSER with a correct fold, i.e., TM-score >0.5, and 32 proteins (78.05% in PATP-TEST) have a RMSD below 4 Å. The overall average TM-score and RMSD for the testing proteins is 0.721 and 3.502 Å, respectively. These represent that the quality of the ITAMSs is acceptable for ATP binding site prediction.
Figure 2.

TM-score and RMSD distributions of the I-TASSER-modeled structures on PATP-TEST.
To further check the ITAMS quality, we compare the quality of the ITAMSs and the MODELER-modeled structures (MODMSs), which are modeled by MODELER software,48 on PATP-TEST. The detailed comparison results can be found in Table S4 and Figure S1. The overall average TM-score and RMSD of the ITAMSs are 0.721 and 3.502 Å, respectively, which are approximately 0.116 and 0.695 Å better than those of the MODMSs. Concretely, there are 26 testing proteins (≈63.41%) for which the ITAMSs have a higher TM-score than the MODMSs. Meanwhile, there are 58.54% proteins in PATP-TEST for which the ITAMSs have lower RMSD than the MODMSs. These results indicate that the quality of the ITAMSs outperforms the quality of the MODMSs for ATP binding site prediction.
Do S-SITEatp-Based and TM-SITEatp-Based Features Help ATP Binding Site Prediction?
In this section, the discriminative performances of the three combination features, i.e., PSSM+PSS+PSA (PPP), PSSM+PSS+PSA+S-SITEatp (PPPS), and PSSM+PSS+PSA+S-SITEatp+TM-SITEatp (PPPST), will be investigated for measuring whether S-SITEatp-based and TM-SITEatp-based features help ATP binding site prediction. Here, the TM-SITEatp-based feature is extracted by the ground-truth 3D structure. Each feature is evaluated by a 5-fold cross-validation test on the training data set PATP-388 with a single SVM classifier. In each training phase of the cross-validation test, we first employ RUS to make the sample number of the majority class equal to that of the minority class, and we then train a single SVM model. Table 2 summarizes the discriminative performance comparison of the three features on PATP-388 over 5-fold cross-validation tests with the single SVM classifier.
Table 2.
Performance Comparison between PPP, PPPS, and PPPST Features on PATP-388 over 5-Fold Cross-Validation Tests with a Single SVM Classifier
| feature type | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| PPP | 51.86 | 97.64 | 95.88 | 46.64 | 0.470 | 0.895 |
| PPPS | 58.05 | 98.11 | 96.58 | 55.04 | 0.547 | 0.919 |
| PPPST | 65.49 | 98.25 | 96.99 | 59.74 | 0.610 | 0.935 |
From Table 2, it is found that the PPPS and PPPST features consistently outperform the PPP feature concerning the six evaluation indexes. Comparing PPPS and PPP, the Sen, MCC, and AUC of PPPS are 58.05%, 0.547, and 0.919, which are approximately 11.94%, 16.38%, and 2.68% better than that of PPP, respectively. The Sen, MCC, and AUC of PPPST are superior to those of the other two features, i.e., PPP and PPPS, and the improvements of 12.82%, 11.52%, and 1.74% are achieved, respectively, compared with the second-best feature PPPS. The P-values of student’s t-test for the difference in MCC scores between PPPST and the other two features are both less than 10−4. Figure S2 illustrates the corresponding ROC curves. In Figure S2, we can intuitively discover that PPPST is the best one and that PPPS is also better than PPP.
Does the Mean Ensemble Strategy Help ATP Binding Site Prediction?
Table 3 lists the prediction performances of the single SVM classifier and the ensemble classifier, whose prediction model is obtained by using the mean ensemble strategy to integrate T (T = 10) SVM models, with PPPS and PPPST features on PATP-388 over 5-fold cross-validation tests.
Table 3.
Performance Comparison between the Ensemble Classifier and Single SVM Classifier with PPPS and PPPST Features on PATP-388 over 5-Fold Cross-Validation Tests
| feature type | classifier | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC | AUC |
|---|---|---|---|---|---|---|---|
| PPPS | singlea | 58.05 | 98.11 | 96.58 | 55.04 | 0.547 | 0.919 |
| ensembledb | 57.52 | 98.86 | 97.27 | 66.69 | 0.605 | 0.913 | |
| PPPST | singlea | 65.49 | 98.25 | 96.99 | 59.74 | 0.610 | 0.935 |
| ensembledb | 64.04 | 98.88 | 97.55 | 69.57 | 0.655 | 0.932 |
”Single” represents the single SVM classifier.
”Ensembled” represents the ensemble classifier.
From Table 3, we can find that the ensemble classifier consistently outperforms the single SVM classifier on both PPPS and PPPST features in the Spe, Acc, Pre, and MCC evaluation indexes, although the ensemble classifier has a slightly lower Sen and AUC. Taking results with PPPST as an example, the Spe, Acc, Pre, and MCC of the ensemble classifier are 98.88%, 97.55%, 69.57%, and 0.655, which are approximately 0.6%, 0.6%, 16.5%, and 7.4% higher than those of the single SVM classifier, respectively. The Sen and AUC (64.04% and 0.932) of the ensemble classifier are both slightly lower than that (65.49% and 0.935) of the single SVM classifier.
In addition to Table 3, we draw Figures S3 and S4 to show the ROC curves and the variation curves of MCC versus the false positive rate of the ensemble classifier and the single SVM classifier on the PPPS and PPPST features, respectively. Figures S3 and S4 show that the ensemble classifier outperforms the single SVM classifier in the low false positive rate (FPR = FP/(FP + TN)) regions, where the FPR is less than 11.32% on the PPPS feature and 14.60% on the PPPST feature, although the overall AUCs of the ensemble classifier are slightly lower than those of the single SVM classifier for both PPPS and PPPST features. Note that the low FPR region is more important than the high FPR region, especially in the imbalanced data learning problem. Since the maximum MCC values of the two classifiers all lie in the low FPR regions on PPPS and PPPST features, the reported MCCs (0.605 and 0.655) of the ensemble classifier are 10.6% and 7.4% higher than those of the single SVM classifier.
Comparing ATPseq and ATPbind with Existing ATP Binding Site Predictors
In this section, we demonstrate the efficacy of ATPseq and ATPbind by comparing them with other existing ATP binding site predictors over 5-fold cross-validation tests on PATP-388 and independent validation tests on PATP-TEST.
Performance Comparison over Cross-Validation Tests
Table 4 illustrates the performance comparison of ATPbind, ATPseq, TM-SITEatp, and S-SITEatp on PATP-388 over 5-fold cross-validation tests. By observing Table 4, we can find that the proposed ATPbind and ATPseq are both better than TM-SITEatp and S-SITEatp, and ATPbind is superior to the other three predictors. Compared to S-SITEatp, the MCC values of ATPseq and ATPbind for the ATP binding site prediction increase by 32.97% and 43.96%, respectively. Meanwhile, ATPseq and ATPbind achieve improvements of 19.33% and 29.19%, respectively, in the MCC evaluation index compared with TM-SITEatp. It is noted that the results of ATPbind and TM-SITEatp are both obtained by employing the experimental 3D structure information.
Table 4.
Performance Comparison of ATPbind, ATPseq, TM-SITEatp, and S-SITEatp on PATP-388 over 5-Fold Cross-Validation Tests
| predictor | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| S-SITEatp | 69.88 | 94.47 | 93.53 | 33.47 | 0.455 | N/Aa |
| TM-SITEatp | 73.64 | 95.29 | 94.46 | 38.37 | 0.507 | N/A |
| ATPseq | 57.52 | 98.86 | 97.27 | 66.69 | 0.605 | 0.913 |
| ATPbind | 64.04 | 98.88 | 97.55 | 69.57 | 0.655 | 0.932 |
”N/A” means that the corresponding value could not be computed.
Performance Comparison over Independent Validation Tests
Table 5 illustrates the performance comparison of ATPbind, ATPseq, and other existing protein-ATP binding site predictors, including three structure-based predictors (i.e., COACH,9 3DLigandSite,20 TM-SITEatp) and six sequence-based predictors (i.e., TargetNUCs,26 TargetSOS,25 TargetS,33 TargetATPsite,37 NsitePred,24 S-SITEatp) on the independent test data set (PATP-TEST). Figure 3 shows the ROC curves of ATPbind, ATPseq, and the above-mentioned six sequence-based predictors. From Table 5 and Figure 3, it is clear that the proposed ATPbind achieves the best performance on PATP-TEST and that the proposed ATPseq outperforms the other six sequence-based predictors.
Table 5.
Performance Comparison of ATPbind, ATPseq, and Other Existing ATP Binding Site Predictors on the Independent Test Dataset (PATP-TEST)h
| predictor | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC | AUC | |
|---|---|---|---|---|---|---|---|
| NS | S-SITEatp | 67.51 | 92.65 | 91.51 | 30.41 | 0.416 | N/A |
| NsitePreda | 46.74 | 97.70 | 95.39 | 49.22 | 0.456 | 0.852 | |
| TargetATPsiteb | 41.25 | 99.49 | 96.84 | 79.43 | 0.559 | 0.853 | |
| TargetSc | 51.63 | 98.89 | 96.74 | 68.91 | 0.580 | 0.872 | |
| TargetSOSd | 49.26 | 99.46 | 97.18 | 81.37 | 0.620 | 0.863 | |
| TargetNUCse | 46.88 | 99.66 | 97.26 | 86.81 | 0.627 | 0.856 | |
| ATPseq | 54.45 | 99.27 | 97.24 | 78.09 | 0.639 | 0.878 | |
| ITA | TM-SITEatp | 69.73 | 96.09 | 84.89 | 45.90 | 0.541 | N/A |
| 3DLigandSitef | 48.81 | 98.58 | 96.32 | 62.08 | 0.532 | N/A | |
| COACHg | 58.16 | 98.59 | 96.76 | 66.33 | 0.604 | N/A | |
| ATPbind | 62.31 | 98.85 | 97.19 | 72.04 | 0.656 | 0.905 | |
| EXP | TM-SITEatp | 78.78 | 96.27 | 95.48 | 50.14 | 0.607 | N/A |
| 3DLigandSitef | 56.82 | 99.31 | 97.38 | 79.63 | 0.660 | N/A | |
| COACHg | 63.20 | 98.73 | 97.11 | 70.30 | 0.652 | N/A | |
| ATPbind | 63.06 | 99.03 | 97.40 | 75.62 | 0.677 | 0.915 |
Results computed using the NsitePred server at http://biomine.cs.vcu.edu/servers/NsitePred.
Results computed using the TargetATPsite server at http://www.csbio.sjtu.edu.cn/bioinf/TargetATPsite.
Results computed using the TargetS server at http://www.csbio.sjtu.edu.cn/bioinf/TargetS.
Results computed using the TargetSOS server at http://www.csbio.sjtu.edu.cn/bioinf/TargetSOS.
Results computed using the TargetNUCs server at http://csbio.njust.edu.cn/bioinf/TargetNUCs/.
Results computed using the 3DLigandSite server at http://www.sbg.bio.ic.ac.uk/~3dligandsite/ by submitting the I-TASSER-modeled or experimental protein structure.
Results computed using the standalone program COACH which was downloaded at https://zhanglab.ccmb.med.umich.edu/COACH/.
”NS”, “ITA”, and “EXP” mean no structure, I-TASSER-modeled structure, and experimental structure, respectively. “N/A” means that the corresponding value could not be computed.
Figure 3.
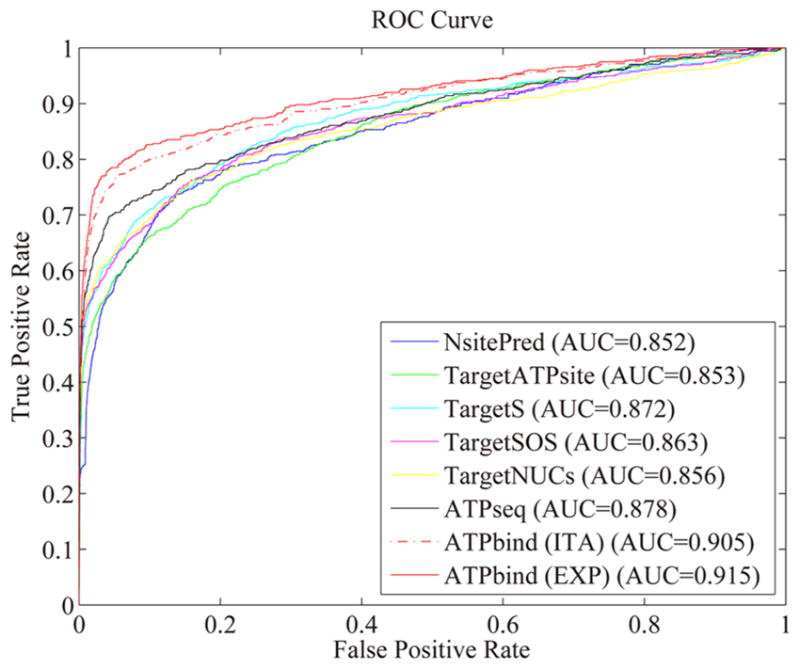
ROC curves of ATPbind (ITA and EXP), ATPseq, TargetNUCs, TargetSOS, TargetS, TargetATPsite, and NsitePred on the independent test data set (PATP-TEST). “ITA” and “EXP” indicate the I-TASSER-modeled and experimental structures, respectively.
The MCC and AUC values of ATPseq are consistently superior to those of all six other sequence-based predictors here considered, and improvements of 1.91% and 2.57%, respectively, are achieved compared with the second-best sequence-based performer, TargetNUCs.26 It has not escaped our notice that TargetNUCs achieves the highest Pre score, 0.8681, and with similar Spe and Acc values; however, its Sen value (0.4688) is much lower than that of ATPseq (0.5445), indicating that more false negatives are incurred during prediction.
The MCC and AUC values of ATPbind are 0.656 and 0.905, respectively, which are 2.66% and 3.08% higher (P-value <0.02 in student’s t-test for the difference in MCC score) than those of ATPseq, which is the best sequence-based predictor. Compared with the best general-purpose ligand binding site predictor, i.e., COACH, ATPbind obtains Sen and MCC improvements of 7.14% and 8.61%, respectively. Furthermore, ATPbind achieves Sen and MCC improvements of 27.61% and 23.32%, respectively, compared to 3DLigandSite, which is the second-best general-purpose predictor. TM-SITEatp’s Sen (0.6973) is higher than that of ATPbind (0.6231), but its Pre (0.459) is significantly lower than that of ATPbind (0.7204), resulting in a low MCC value of 0.541. The differences between ATPbind and COACH, 3DLigandSite, and TM-SITEatp in MCC values are all statistically significant, with P-values <0.05, <10−3, and <10−2, respectively, by student’s t-tests.
In Table 5, we list the prediction results of the structure-dependent predictors when the experimental 3D structure of the query proteins is used. As expected, the performance of all the structure-based methods is enhanced due to the increase of structural accuracy of the query proteins. It is also easily found that ATPbind outperforms the other three structure-based predictors in the MCC evaluation index.
To further compare ATPbind with the existing structure-based ATP binding site predictors, we first divide the independent test data set, PATP-TEST, into two parts, denoted PATP-TEST-easy and PATP-TEST-hard, based on the quality of the I-TASSER-modeled structure of each protein. Concretely, PATP-TEST-easy contains 36 proteins with a high quality of the modeled structure, in which each TM-score between the I-TASSER-modeled structure and the experimental structure is higher than 0.5. PATP-TEST-hard includes 5 proteins with a low quality of the I-TASSER-modeled structure, in which each TM-score between the modeled and experimental structures is lower than 0.5. Note that the protein structure pairs with a TM-score >0.5 are mostly in the same fold;49 the TM-scores of the 41 PATP-TEST proteins can be found in Table S4. Overall, the average TM-score for PATP-TEST-easy and PATP-TEST-hard is 0.765 and 0.400, respectively. Next, we compared the prediction performances of ATPbind, COACH,9 3DLigandSite,20 and TM-SITEatp on the PATP-TEST-easy and PATP-TEST-hard data sets. Tables 6 and 7 present the comparison results on PATP-TEST-easy and PATP-TEST-hard, respectively.
Table 6.
Performance Comparison of ATPbind and Other Existing Structure-Based ATP Binding Site Predictors on PATP-TEST-easy for Which the TM-Score of the I-TASSER Model is >0.5
| predictor | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC |
|---|---|---|---|---|---|
| TM-SITEatp | 71.38 | 96.04 | 94.88 | 47.11 | 0.555 |
| 3DLigandSite | 49.66 | 98.67 | 96.36 | 64.84 | 0.549 |
| COACH | 60.27 | 98.49 | 96.69 | 66.30 | 0.615 |
| ATPbind | 64.14 | 98.83 | 97.20 | 72.99 | 0.670 |
Table 7.
Performance Comparison of ATPbind and Other Existing Structure-Based ATP Binding Site Predictors on PATP-TEST-hard for Which the TM-Score of the I-TASSER Model is <0.5
| predictor | Sen (%) | Spe (%) | Acc (%) | Pre (%) | MCC |
|---|---|---|---|---|---|
| TM-SITEatp | 57.50 | 96.33 | 94.93 | 37.10 | 0.437 |
| 3DLigandSite | 42.50 | 98.07 | 96.06 | 45.33 | 0.419 |
| COACH | 42.50 | 99.20 | 97.15 | 66.67 | 0.519 |
| ATPbind | 48.75 | 98.97 | 97.15 | 63.93 | 0.544 |
From Tables 6 and 7, we can easily find that ATPbind consistently outperforms the other three structure-based predictors, i.e., TM-SITEatp, 3DLigandSite, and COACH, on both the PATP-TEST-easy and PATP-TEST-hard data sets with regard to the MCC evaluation index. The MCCs of ATPbind on PATP-TEST-easy and PATP-TEST-hard are 0.670 and 0.544, respectively, which are 8.9% and 4.8% higher than those of COACH, 22.0% and 29.8% higher than those of 3DLigandSite and 20.7% and 24.5% higher than those of TM-SITEatp, respectively.
Why Does ATPseq Perform Better than COACH and 3DLigandSite?
By revisiting Table 5, we find that the proposed ATPseq, which is a sequence-based predictor, outperforms the two general-purpose structure-based methods (i.e., COACH and 3DLigandSite) when only the I-TASSER-modeled 3D structure information exists, with an MCC value 5.79% better than COACH and 20.11% higher than 3DLigandSite. ATPseq can also obtain a comparable prediction performance with COACH and 3DLigandSite when the ground-truth 3D structure is employed.
There are two potential reasons to explain why the sequence-based predictor ATPseq works better than the two general-purpose structure-based predictors under the I-TASSER-modeled structure. The first one is that the I-TASSER algorithm44 may not correctly generate the local ATP binding sites (or pockets) structure, even though the accuracy of the modeled protein structure is high. Figure 4 shows the comparison between the experimental and I-TASSER-modeled structures of the 4-Coumarate–COA Ligase 2 protein (PDB ID: 5bsmA). As Figure 4 shows, the RMSD and TM-score47 of the I-TASSER-modeled structure are 3.68 Å and 0.769, respectively, and the PS-score (the pocket similarity score) is 0.53, with a P-value <10−5 between the experimental and I-TASSER-modeled ATP binding site structures, as calculated by the APoc algorithm.50 A PS-score = 0.53 means that the I-TASSER-modeled binding site structures contain both useful information and useless noise. Over-reliance on the modeled protein structure must lead to a decline in prediction accuracy. That is why the MCC value (0.604) of COACH,9 which combines the sequence and modeled structural information, is better than that (0.532) of 3DLigandSite, which only employs the modeled structural information. However, COACH also employs many structure-based predictors, i.e., TM-SITE, COFACTOR,21 FINDSITE,22 and ConCavity,11 to extract more information from the modeled structure such that the noise information in the modeled structure is magnified and the predicted performance is decreased.
Figure 4.
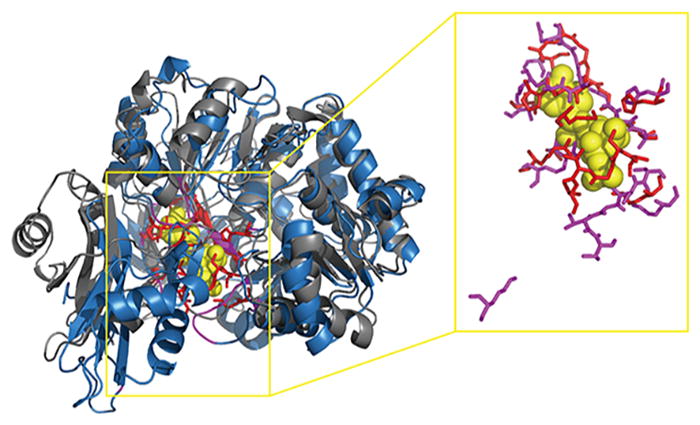
Comparison between the experimental structure (gray cartoon) and I-TASSER-modeled structure (blue cartoon) of the 4-Coumarate–COA Ligase 2 protein (PDB ID: 5bsmA). The RMSD and TM-score are 3.68 Å and 0.769, respectively, which are calculated by the TM-score algorithm. The PS-score between the experimental binding site (red sticks) and the I-TASSER-modeled binding site (magenta sticks) structures is 0.53 with P-value = 6.4 × 10−6, as calculated with the APoc algorithm.
The second reason is that different ligands tend to bind diverse types of residues with prominent specificities, and protein–ligand binding sites (or pockets) vary significantly in their role, size, and distribution for different types of protein–ligand interactions.23,51 Ligand-specific predictors are often superior to general-purpose predictors for a specific ligand type (e.g., ATP) binding site prediction, which is the main reason to explain why the performance of ATPbind is higher than that of other existing general-purpose predictors such as TM-SITEatp, 3DLigandSite, and COACH on the experimental structures. Figure 5 illustrates the prediction results of 3DLigandSite, COACH, ATPseq, and ATPbind on the protein 5bsmA (containing 530 residues), which has 21 ATP binding residues, including S182, S183, G184, T185, T186, K190, H230, A302, A303, P304, Q324, G325, Y326, G327, M328, T329, C353, D413, I425, R428, and K519. From Figure 5, it is easy to find that ATPseq outperforms 3DLigandSite and COACH on 5bsmA using the I-TASSER-modeled structure. ATPseq, which is an ATP-specific predictor, correctly predicted 20 true positives and only 1 false negative. However, 3DLigandSite used 1 COA, 4 Mg2+, 12 AMP, and 1 ATP binding proteins as templates to predict the ATP binding sites of 5bsmA, and the predicted results are 14 true positives, 5 false positives, and 7 false negatives. COACH mainly used numerous SLU and BEZ binding proteins as templates to identify the ATP binding sites of 5bsmA, resulting in 13 true positives, 10 false positives, and 8 false negatives. Interestingly, although the overall prediction performance (MCC = 0.656 and AUC = 0.905) of ATPbind is higher than that of ATPseq (MCC = 0.639 and AUC = 0.878), the performance of ATPseq (20 true positives, 0 false positive, and 1 false negative) is slightly higher than that of ATPbind (19 true positives, 1 false positive, and 2 false negatives) in this example. This result could be explained by the first reason described in the last paragraph; i.e., the local ATP binding sites (or pockets) structure is difficult to model with I-TASSER. Similar to the existing structure-based predictor, the prediction performance of ATPbind is affected by the quality of the I-TASSER-modeled pocket structure.
Figure 5.
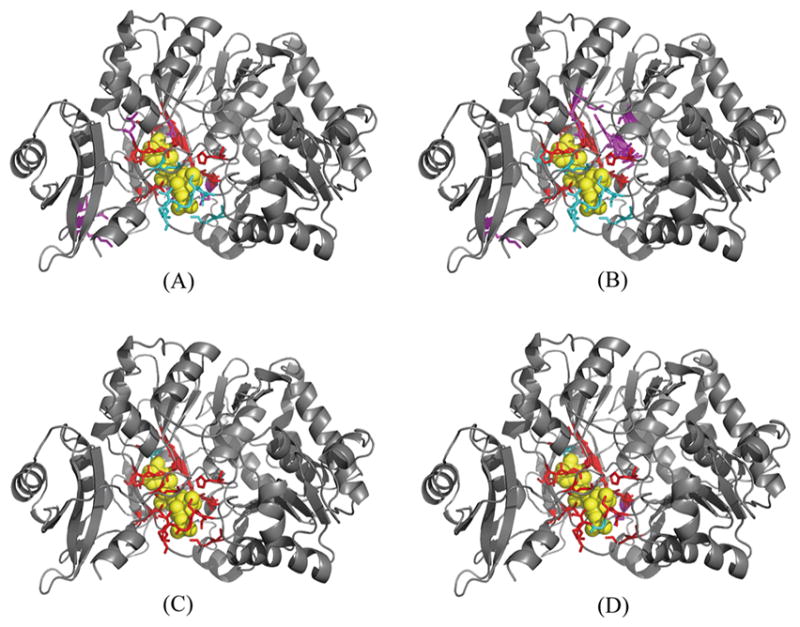
Visualization of prediction results for 5bsmA. (A) 3DLigandSite predicted results based on the I-TASSER-modeled structure of 5bsmA. (B) COACH predicted results based on the I-TASSER-modeled structure of 5bsmA. (C) ATPseq predicted results based on the sequence of 5bsmA. (D) ATPbind predicted results based on the I-TASSER-modeled structure of 5bsmA. The following color scheme is used: ATP in yellow, true positives in red, false positives in magenta, false negatives in cyan. The cartoon protein in the picture is the experimental structure of 5bsmA.
CONCLUSIONS
In this study, we have designed and implemented a new predictor of ATP-specific binding sites named ATPbind based on the sequential and 3D structural information on proteins. Experimental results with a training data set and an independent test data set have demonstrated that the proposed ATPbind outperforms not only general-purpose predictors but also other existing ATP-specific binding site predictors. The superior performance of ATPbind mainly stems from the use of novel custom-designed discriminative features that are based on sequential and 3D structural information, i.e., the position specific scoring matrix profiles, the predicted secondary structure, the predicted solvent accessibility, the S-SITEatp-predicted probability, and the TM-SITEatp-predicted probability. Furthermore, we employ random undersampling and the mean ensemble method to solve the imbalanced learning problem and thereby enhance the prediction performance. ATPbind has already been implemented as a web server that is now available at http://zhanglab.ccmb.med.umich.edu/ATPbind/.
It is noted that ATPbind takes a relatively long computation time to predict the ATP binding sites of each query protein with structural information (approximately 45 m for a protein of 300 residues). The long computational time stems from the fact that ATPbind must perform PSI–PRED, PSIPRED, SANN, S-SITEatp, TM-SITEatp, and LIBSVM to gain discriminative features and predict ATP binding sites. Nevertheless, given the importance of ATP binding site prediction and the overall acceptable CPU range, the strong performance advance is probably sufficient to demonstrate the worthiness of investing the time in running this method. While this method still has room for optimization (e.g., by integrating more programs when available), it represents one of the most accurate tools for ATP binding site prediction using cutting-edge structure modeling and machine-learning training techniques.
Supplementary Material
Acknowledgments
This project was supported in part by the National Natural Science Foundation of China (No. 61772273 and 61373062), the Fundamental Research Funds for the Central Universities (No. 30916011327), China Scholarship Council (No. 201606840087), the National Institute of General Medical Sciences (GM083107 and GM116960), and the National Science Foundation (DBI1564756).
ABBREVIATIONS
- ATP
adenosine-5′-triphosphate
- SVM
support vector machine
- RUS
random undersampling
- MCC
Matthews correlation coefficient
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00397.
Supporting details, Figures S1–S4, and Tables S1–S7 (PDF)
ATPbind is available free of charge as a web server at http://zhanglab.ccmb.med.umich.edu/ATPbind/. Supporting details are also available free of charge at http://zhanglab.ccmb.med.umich.edu/ATPbind/Supplementary-ATPbind.pdf
References
- 1.Gao M, Skolnick J. The Distribution of Ligand-Binding Pockets around Protein-Protein Interfaces Suggests A General Mechanism for Pocket Formation. Proc Natl Acad Sci U S A. 2012;109:3784–3789. doi: 10.1073/pnas.1117768109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kokubo H, Tanaka T, Okamoto Y. Ab Initio Prediction of Protein-Ligand Binding Structures by Replica-Exchange Umbrella Sampling Simulations. J Comput Chem. 2011;32:2810–2821. doi: 10.1002/jcc.21860. [DOI] [PubMed] [Google Scholar]
- 3.Turton DA, Senn HM, Harwood T, Lapthorn AJ, Ellis EM, Wynne K. Terahertz Underdamped Vibrational Motion Governs Protein-Ligand Binding in Solution. Nat Commun. 2014;5:3999. doi: 10.1038/ncomms4999. [DOI] [PubMed] [Google Scholar]
- 4.Schmidtke P, Barril X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J Med Chem. 2010;53:5858–5867. doi: 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- 5.Sirimulla S, Bailey JB, Vegesna R, Narayan M. Halogen Interactions in Protein-Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J Chem Inf Model. 2013;53:2781–2791. doi: 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- 6.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. Virtual Screening Using Protein-Ligand Docking: Avoiding Artificial Enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 7.Amari S, Aizawa M, Zhang J, Fukuzawa K, Mochizuki Y, Iwasawa Y, Nakata K, Chuman H, Nakano T. VISCANA: Visualized Cluster Analysis of Protein-Ligand Interaction Based on The Ab Initio Fragment Molecular Orbital Method for Virtual Ligand Screening. J Chem Inf Model. 2006;46:221–230. doi: 10.1021/ci050262q. [DOI] [PubMed] [Google Scholar]
- 8.Rose PW, Prlić A, Bi C, Bluhm WF, Christie CH, Dutta S, Green RK, Goodsell DS, Westbrook JD, Woo J, et al. The RCSB Protein Data Bank: Views of Structural Biology for Basic and Applied Research and Education. Nucleic Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Roy A, Zhang Y. Protein-Ligand Binding Site Recognition Using Complementary Binding-Specific Substructure Comparison and Sequence Profile Alignment. Bioinformatics. 2013;29:2588–2595. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu DJ, Hu J, Yang J, Shen HB, Tang JH, Yang JY. Designing Template-Free Predictor for Targeting Protein-Ligand Binding Sites with Classifier Ensemble and Spatial Clustering. IEEE/ACM Trans Comput Biol Bioinf. 2013;10:994–1008. doi: 10.1109/TCBB.2013.104. [DOI] [PubMed] [Google Scholar]
- 11.Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting Protein Ligand Binding Sites by Combining Evolutionary Sequence Conservation and 3D Structure. PLoS Comput Biol. 2009;5:e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosc N, Wroblowski B, Meyer C, Bonnet P. Prediction of Protein Kinase-Ligand Interactions through 2.5 D Kinochemometrics. J Chem Inf Model. 2017;57:93–101. doi: 10.1021/acs.jcim.6b00520. [DOI] [PubMed] [Google Scholar]
- 13.Hendlich M, Rippmann F, Barnickel G. LIGSITE: Automatic and Efficient Detection of Potential Small Molecule-Binding Sites in Proteins. J Mol Graphics Modell. 1997;15:359–363. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 14.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: Computed Atlas of Surface Topography of Proteins with Structural and Topographical Mapping of Functionally Annotated Residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laskowski RA. SURFNET: A Program for Visualizing Molecular Surfaces, Cavities, and Intermolecular Interactions. J Mol Graphics. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 16.Levitt DG, Banaszak LJ. POCKET: A Computer Graphies Method for Identifying and Displaying Protein Cavities and Their Surrounding Amino Acids. J Mol Graphics. 1992;10:229–234. doi: 10.1016/0263-7855(92)80074-n. [DOI] [PubMed] [Google Scholar]
- 17.Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinf. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laurie AT, Jackson RM. Q-SiteFinder: An Energy-Based Method for The Prediction of Protein-Ligand Binding Sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 19.Hernandez M, Ghersi D, Sanchez R. SITEHOUND-web: A Server for Ligand Binding Site Identification in Protein Structures. Nucleic Acids Res. 2009;37:W413–W416. doi: 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wass MN, Kelley LA, Sternberg MJ. 3DLigandSite: Predicting Ligand-Binding Sites Using Similar Structures. Nucleic Acids Res. 2010;38:W469. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy A, Yang J, Zhang Y. COFACTOR: An Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 2012;40:W471. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brylinski M, Skolnick J. A Threading-Based Method (FINDSITE) for Ligand-Binding Site Prediction and Functional Annotation. Proc Natl Acad Sci U S A. 2008;105:129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henrich S, Salo-Ahen OM, Huang B, Rippmann FF, Cruciani G, Wade RC. Computational Approaches to Identifying and Characterizing Protein Binding Sites for Ligand Design. J Mol Recognit. 2009;23:209–219. doi: 10.1002/jmr.984. [DOI] [PubMed] [Google Scholar]
- 24.Chen K, Mizianty MJ, Kurgan L. Prediction and Analysis of Nucleotide-Binding Residues Using Sequence and Sequence-Derived Structural Descriptors. Bioinformatics. 2012;28:331–341. doi: 10.1093/bioinformatics/btr657. [DOI] [PubMed] [Google Scholar]
- 25.Hu J, He X, Yu DJ, Yang XB, Yang JY, Shen HB. A New Supervised Over-Sampling Algorithm with Application to Protein-Nucleotide Binding Residue Prediction. PLoS One. 2014;9:e107676. doi: 10.1371/journal.pone.0107676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, Li Y, Yan WX, Yang JY, Shen HB, Yu DJ. KNN-Based Dynamic Query-Driven Sample Rescaling Strategy for Class Imbalance Learning. Neurocomputing. 2016;191:363–373. [Google Scholar]
- 27.Brylinski M, Skolnick J. FINDSITE-metal: Integrating Evolutionary Information and Machine Learning for Structure-Based Metal-Binding Site Prediction at The Proteome Level. Proteins: Struct, Funct Genet. 2011;79:735–751. doi: 10.1002/prot.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babor M, Gerzon S, Raveh B, Sobolev V, Edelman M. Prediction of Transition Metal-Binding Sites from Apo Protein Structures. Proteins: Struct, Funct Genet. 2008;70:208–217. doi: 10.1002/prot.21587. [DOI] [PubMed] [Google Scholar]
- 29.Ma X, Guo J, Liu HD, Xie JM, Sun X. Sequence-Based Prediction of DNA-Binding Residues in Proteins with Conservation and Correlation Information. IEEE/ACM Trans Comput Biol Bioinf. 2012;9:1766–1775. doi: 10.1109/TCBB.2012.106. [DOI] [PubMed] [Google Scholar]
- 30.Si J, Zhang Z, Lin B, Schroeder M, Huang B. MetaDBSite: A Meta Approach to Improve Protein DNA-Binding Sites Prediction. BMC Syst Biol. 2011;5:S7. doi: 10.1186/1752-0509-5-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu X, Dong Q, Yang J, Zhang Y. Recognizing Metal and Acid Radical Ion-Binding Sites by Integrating Ab Initio Modeling with Template-Based Transferals. Bioinformatics. 2016;32:3260–3269. doi: 10.1093/bioinformatics/btw396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chauhan JS, Mishra NK, Raghava GP. Identification of ATP Binding Residues of A Protein From Its Primary Sequence. BMC Bioinf. 2009;10:434. doi: 10.1186/1471-2105-10-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu DJ, Hu J, Tang ZM, Shen HB, Yang J, Yang JY. Improving Protein-ATP Binding Residues Prediction by Boosting SVMs with Random Under-Sampling. Neurocomputing. 2013;104:180–190. [Google Scholar]
- 34.Maxwell A, Lawson DM. The ATP-Binding Site of Type II Topoisomerases as A Target for Antibacterial Drugs. Curr Top Med Chem. 2003;3:283–303. doi: 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- 35.Chen K, Mizianty MJ, Kurgan L. ATPsite: Sequence-Based Prediction of ATP-Binding Residues. Proteome Sci. 2011;9:S4. doi: 10.1186/1477-5956-9-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He H, Garcia EA. Learning from Imbalanced Data. IEEE T Knowl Data En. 2009;21:1263–1284. [Google Scholar]
- 37.Yu DJ, Hu J, Huang Y, Shen HB, Qi Y, Tang ZM, Yang JY. TargetATPsite: A Template-Free Method for ATP-Binding Sites Prediction with Residue Evolution Image Sparse Representation and Classifier Ensemble. J Comput Chem. 2013;34:974–985. doi: 10.1002/jcc.23219. [DOI] [PubMed] [Google Scholar]
- 38.Li W, Godzik A. Cd-hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 39.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving The Accuracy of PSI-BLAST Protein Database Searches with Composition-Based Statistics and Other Refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bairoch A, Apweiler R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones DT. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 42.Joo K, Lee SJ, Lee J. Sann: Solvent Accessibility Prediction of Proteins by Nearest Neighbor Method. Proteins: Struct, Funct Genet. 2012;80:1791–1797. doi: 10.1002/prot.24074. [DOI] [PubMed] [Google Scholar]
- 43.Yang J, Roy A, Zhang Y. BioLiP: A Semi-Manually Curated Database for Biologically Relevant Ligand-Protein Interactions. Nucleic Acids Res. 2012;41:D1096–D1103. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinf. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vapnik VN. Statistical Learning Theory. Wiley-Interscience; New York: 1998. [Google Scholar]
- 46.Chang CC, Lin CJ. LIBSVM: A Library for Support Vector Machines. ACM Trans Intell Syst Technol. 2011;2:1. [Google Scholar]
- 47.Zhang Y, Skolnick J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins: Struct, Funct Genet. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 48.Eswar N, Webb B, Marti-Renom MA, Madhusudhan M, Eramian D, Shen My, Pieper U, Sali A. Comparative Protein Structure Modeling Using Modeller. Current protocols in bioinformatics. 2006:5.6.1. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, Zhang Y. How Significant is A Protein Structure Similarity with TM-score= 0.5? Bioinformatics. 2010;26:889–895. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao M, Skolnick J. APoc: Large Scale Identification of Similar Protein Pockets. Bioinformatics. 2013;29:597. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kasahara K, Shirota M, Kinoshita K. Comprehensive Classification and Diversity Assessment of Atomic Contacts in Protein-Small Ligand Interactions. J Chem Inf Model. 2013;53:241–248. doi: 10.1021/ci300377f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.