

Abstract
Insulin is known to have neurotrophic properties and loss of insulin support to sensory neurons may contribute to peripheral diabetic neuropathy (PDN). Here, genetically-modified mice were generated in which peripheral sensory neurons lacked the insulin receptor (SNIRKO mice) to determine whether disrupted sensory neuron insulin signaling plays a crucial role in the development of PDN and whether SNIRKO mice develop symptoms of PDN due to reduced insulin neurotrophic support. Our results revealed that SNIRKO mice were euglycemic and never displayed significant changes in a wide range of sensorimotor behaviors, nerve conduction velocity or intraepidermal nerve fiber density. However, SNIRKO mice displayed elevated serum insulin levels, glucose intolerance, and increased insulin content in the islets of Langerhans of the pancreas. These results contribute to the growing idea that sensory innervation of pancreatic islets is key to regulating islet function and that a negative feedback loop of sensory neuron insulin signaling keeps this regulation in balance. Our results suggest that a loss of insulin receptors in sensory neurons does not lead to peripheral nerve dysfunction. The SNIRKO mice will be a powerful tool to investigate sensory neuron insulin signaling and may give a unique insight into the role that sensory neurons play in modifying islet physiology.
Keywords: insulin, insulin receptor, sensory neuron, dorsal root ganglia, mouse models, pancreas, beta cell, neuropathy, gene deletion, islet
Introduction
The pathogenesis of peripheral diabetic neuropathy (PDN) is complex but involves complications of hyperglycemia and reduced neurotrophic support (Singh et al., 2014). In addition to its actions on glucose, insulin acts as a neurotrophic factor for sensory neurons. Increasing evidence suggests direct neuronal insulin signaling impacts sensory neuron function and disruption of neuronal insulin signaling may contribute to PDN (Kim and Feldman, 2012). Studies of insulin action on sensory neurons in vivo face inherent difficulties due to insulin’s broad systemic actions, making it difficult to identify direct actions of insulin on sensory neurons. With current PDN rodent models, neither hyperglycemia nor reductions in peripheral nervous system (PNS) insulin signaling can be isolated to establish the pathogenesis arising from either insulting factor. For example, STZ-induced diabetic mice are hyperglycemic and hypoinsulinemic and ob/ob mice are hyperglycemic and insulin resistant; thus, both models have elevated glucose levels and reduced insulin signaling that complicate studies direct actions of insulin on sensory neurons (Grote et al., 2013a; Grote et al., 2011).
The generation of tissue specific insulin receptor knockout mice has greatly increased our understanding of the physiological role of insulin (Kahn, 2003). The purpose of this study was to assess systemic metabolic and sensorimotor consequences generated by absent insulin signaling selectively in sensory neurons by generating sensory neuron insulin receptor knockout (SNIRKO) mice. Our prediction was that SNIRKO mice would develop neuropathy symptoms like other mouse models of PDN due to the absent neurotrophic support from insulin. Our results suggest that reductions in sensory neuron insulin signaling alone do not contribute to the symptoms of PDN. However, our results reveal a potential and surprising feedback mechanism may exist between sensory neuron insulin signaling and beta cell insulin production.
Methods
Animals and Genotyping
AdvillinCre/+ mice were a generous gift of Dr. Fan Wang (Hasegawa et al., 2007) and have been previously characterized to demonstrate sensory neuron specific cre recombinase activity (da Silva et al., 2011; Hasegawa et al., 2007; Minett et al., 2012; Zhou et al., 2010; Zurborg et al., 2011) and this was confirmed in our lab using a fluorescent reporter line Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (Tomato) (Jackson Laboratories, Bar Harbor, ME). Mice tissues were fixed via intracardial perfusion with Zamboni’s fixative (4% paraformaldehyde and 15% picric acid) prior to dissection. Tissue sections were cut using a Leica CM 1950 cryostat and placed on slides in serial sections. Sections were then covered with PBS and cover slipped. Images were acquired using a Nikon Eclipse 90i microscope. Exposure times were kept constant between experimental groups. (Supplemental Figure S1).
Mice with loxp sites flanking exon 4 of the insulin receptor gene (IRlox/lox) were purchased from Jackson Laboratories (Bar Harbor, ME). In the presence of cre recombinase, exon 4 is deleted creating a frameshift mutation resulting in a stop codon. The resultant product would be a nonfunctional 308 amino acid truncated peptide. Male AdvillinCre/+ mice were bred to female IRlox/lox to generate heterozygous AdvillinCre/+, IRlox/+ mice. Male AdvillinCre/+, IRlox/+ were bred to female IRlox/lox to produce SNIRKO mice with an AdvillinCre/+, IRlox/lox genotype. Mice were genotyped via tail clip. Primers used for genotyping AdvillinCre/+ were:
p1: 5′-CCCTGTTCACTGTGAGTAGG-3′,
p2: 5′-AGTATCTGGTAGGTGCTTCCAG-3′,
p3: 5′-GCGATCCCTGAACATGTCCATC-3′.
A wildtype allele produced a 500bp fragment and a cre-expressing allele produced a 180bp fragment. Primers used for genotyping IRlox/lox were:
p1: 5′-GATGTGCACCCCATGTCTG-3′,
p2: 5′-CTGAATAGCTGAGACCACAG-3′.
A wildtype allele produced a 279bp fragment and an allele with loxp insertion produced a 313bp fragment. All primers were added to a supermix to genotype SNIRKO mice and the PCR product was amplified for 35 cycles (94°C for 15 sec, 62°C for 15 sec, 72°C for 90 sec) in a 40μL reaction (Supplemental Figure S2). IRlox/lox were used as controls for all experiments. All experiments were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
Insulin Receptor Quantification
RT-PCR
Total RNA was isolated from control and SNIRKO DRG to assess Cre/loxP recombination as described previously (Guerra et al., 2001). A reverse primer specific for exon 6 of the insulin receptor was used for reverse transcription: 5′-GTGATGGTGAGGTTGTGTTTGCTC-3′. The reaction was carried out using an iScript select kit (Bio-Rad) at 42 degrees for 30 min, followed by 85 degrees for 5 min. The generated cDNA was then used for PCR template. Primers to exon 3, 5′-GCTGCACAGCTGAAGGCCTGT-3′, and exon 5, 5′-CTCCTCGAATCAGATGTAGCT-3′ were used to amplify the region corresponding to exon 4. PCR conditions were 94° for 30 secs followed by 35 cycles of 94° for 30 secs, 58° for 30 secs, 72 °for 1 min and a final extension at 72° for 7 min. A 585bp fragment indicates an intact insulin receptor and a 435bp fragment indicates cre/loxp recombination and deletion of the 150bp exon 4.
Western blots
Insulin receptor protein expression was quantified in gastrocnemius muscle and DRG using Western blot analysis. Samples were homogenized in Cell Extraction Buffer (Invitrogen, Carlsbad, CA) containing 55.55 μl/ml protease inhibitor cocktail, 200mM Na3VO4, and 200mM NaF. Protein concentration of the supernatant was measured with a Bradford assay (Bio-Rad, Hercules, CA). Samples were boiled with Lane Marker Reducing Sample Buffer (Thermo Scientific, Waltham, MA) for 3 minutes. Thirty μg of protein was loaded per lane and samples were separated on a 4–15% gradient tris-glycine gel (Bio-Rad). After gel electrophoresis, samples were transferred to a nitrocellulose membrane and blocked in 5% milk. Following incubation with primary (insulin receptor β subunit (Santa Cruz), actin (Millipore) and secondary antibodies, bands were visualized with film and analyzed with ImageJ (NIH).
Akt activation was assessed using western blots in muscle and DRG following an intraperitoneal injection of insulin at 10.0 U/Kg or vehicle control as previously described (Grote et al., 2013b). Mice were fasted 3 hours prior to insulin injection. Thirty minutes after insulin stimulation mice were sacrificed and tissues were harvested. Western samples were then prepared and blots were probed with total Akt and p-(Ser473)Akt (Cell Signaling, Danvers, MA).
Metabolic Parameters
Metabolic parameters were monitored throughout the course of SNIRKO development and testing. Mice weights were recorded at 3, 5, 6, 7, 8, 16, 22, and 28 weeks of age.
Glucose and Hemoglobin A1C
Blood glucose levels were determined using a glucose diagnostic assay (Sigma-Aldrich, St. Louis, MO). Mice were fasted 3 hours prior to glucose measurement and blood was collected via tail snip at 6, 10, 16, 22, and 29 weeks of age. In addition, long term glucose levels were assessed by determining hemoglobin A1C levels immediately prior to sacrifice at 29 weeks of age using A1CNow+ Meter (Bayer, Leverkusen, Germany).
Insulin and IGF-1
After a 3-hour fast, whole blood was collected via tail snip and allowed to clot on ice for 30 minutes. Samples were then centrifuged at 3000g for 15 minutes. The resultant serum supernatant was used for analysis. Serum insulin and IGF-1 levels were measured with ELISAs from ALPCO (Salem, NH). Serum insulin levels were measured alongside glucose at 6, 10, 16, 22, and 29 weeks of age and IGF-1 levels were determined at sacrifice.
Intraperitoneal Glucose Tolerance Test (IPGTT)
At 28 weeks of age, both IRlox/lox and SNIRKO glucose tolerance was analyzed with an IPGTT. After a 6-hour fast, mice were administered a glucose bolus of 2g/kg body weight via IP injection. Blood glucose measurements were taken immediately prior to glucose stimulation and at 15, 30, 60 and 120 minutes thereafter.
Sensorimotor Behavior and Neuron Analysis
Thermal sensitivity
Mice were acclimated to the behavior facility and equipment for a minimum of 2 days. On test days, mice were acclimated to the behavior facility for 30 minutes and subsequently to the Hargreaves table for 30 minutes prior to data collection. The table surface was maintained at 30°C and mice were housed in individual clear plastic cages. A 4.0V radiant heat source was applied to the mid plantar surface of the hind paw, and time to withdrawal was measured (Hargreaves et al., 1988). Four trials were recorded for each hind paw, alternating paws between trials. Data was presented as the average latency to withdrawal across both paws.
Mechanical sensitivity
Mice were acclimated to the mesh grid in clear individual plastic cages. The up-down method was used to test paw mechanical sensitivity (Dixon, 1980). A set von Frey monofilaments (Stoelting, Wood Dale, IL) capable of exerting forces of 0.0045, 0.02, 0.068, 0.158, 0.178, 1.2, 2.041, and 5.5 g were applied to the right hind paw of each mouse. The duration of each stimulus was approximately 1 sec and the inter-stimulus interval was approximately 30–60 secs. The 50% threshold was calculated for each mouse (Chaplan et al., 1994).
Beamwalk
Two days before testing, mice were acclimated to the behavior facility and trained to traverse a wooden beam with a length 1.0 m and a diameter of 1.2 cm (Guilford et al., 2011). On test day, mice were recorded with a digital video camera as they walked across the beam. Each mouse had 3 trials per testing period. Videos were analyzed and the number of times the left or right hind paw slipped of the beam was counted as a foot slip and foot slips were averaged across all 3 trials.
Rotorod
Mice were placed on a rotating rod to test motor coordination and balance (Lalonde et al., 1995). The speed of rotation was increased from 4.0 to 40 rpm over a 5-minute period and the latency to fall was recorded. If a mouse did not fall throughout the testing period, the rod was stopped and latency was recorded as 5 minutes. Mice were acclimated for one 5-minute period before being tested in 3 separate trials with 5 minutes between trials. The average latency to fall across all 3 trials is reported.
Nerve Conduction Velocity
Nerve conduction velocity was conducted as previously described (Jack et al., 2012). Briefly, mice were anesthetized with 200 mg/kg Avertin (1.25% v/v tribromoethanol, 2.5% tert-amyl alcohol, dH2O). Mouse body temperature was monitored with rectal probe and maintained at 37°C via feed-back controlled heating pad. Motor nerve conduction velocities (MNCV) were obtained by measuring compound muscle action potentials using 9.9 mA stimulation at the ankle distally and at the sciatic notch proximally. MNCV were reported as the average of 3 independent recordings. Sensory nerve conduction velocity (SNCV) was measured behind the medial malleolus with a 2.4 mA stimulation of the second toe digital nerve. SNCV was reported as the average of 10 recordings.
Intraepidermal Nerve Fiber Density (IENFD)
At sacrifice, the skin of the hind paw footpad was removed from IRlox/lox and SNIRKO mice and fixed in Zamboni’s fixative for 1 hour. Hind paw footpad skin was cut in sagittal sections with a Leica CM 1950 cryostat and placed on slides in serial sections. Slides were blocked at room temperature for 1 hour with pre-incubation solution (1.5% Normal Goat or Donkey Serum, 0.5% Porcine Gelatin, 0.5% Triton X-100, and 450 μL Superblock (Thermo Scientific)). Primary antibody to PGP 9.5 (Chemicon, Temecula, CA) and donkey anti-rabbit Alexa-488 conjugated secondary antibody (Molecular Probes) were then used to label and visualize epidermal nerve fibers. Images were acquired with a Nikon Eclipse 90i microscope. Fibers that crossed the dermal-epidermal border were counted in 3 regions per section and in 3 sections per mouse. The length of the epidermal region was measured with NIH Image J software and the IENFD was expressed as number of fibers/millimeter of epidermis.
Pancreas Morphology
Tissue Preparation
Pancreata were removed and fixed in 4% paraformaldehyde. Tissue was embedded in paraffin using an automated vacuum tissue processor Leica ASP300S (Leica Microsystems Inc., Bannockburn, IL). Tissue sections of 8 μm thickness were cut on a RM2255microtome (Leica Microsystems Inc.) and mounted directly on Superfrost/Plus microscope slides (Fisher, Pittsburgh, PA, #12-550-12). Slides were dried at +40°C overnight in and stored at +4°C.
Paraffin embedded sections were deparaffinized/rehydrated in xylene followed by ethanol and PBS serial rehydration. Antigen retrieval was completed in a steamer using 0.01M citrate buffer, pH 6.2, with 0.002M EDTA, for 30 min. Slides were permeabilized in 1% Triton X-100 in PBS for 30 min. Sections were blocked in 10% normal donkey serum (NDS), 1% bovine serum albumin (BSA), and 0.03% Triton X-100.
Immunofluorescence (IF)
Blocked sections were incubated with the primary antibody mix at +4°C overnight and with secondary antibody for 2 hours at room temperature. DAPI (4′6-diamindino-2-phenylindole; 0.5 μg/ml; Molecular Probes, Eugene, OR, # D1306) staining was performed for 5 min at room temperature following the first wash after secondary antibody exposure. After incubation with secondary antibodies, slides were mounted with anti-fading agent Gel/Mount (Biomeda, Foster City, CA).
The following primary antibodies were used to stain the pancreas: anti-insulin (1:200, Abcam, Cambridge, MA, # ab7842), anti-glucagon (1:300, Abcam, # ab10988), anti-somatostatin (1:300, Abcam, # ab53165). Secondary antibodies were conjugated with DyLight 488 (1:400, Jackson ImmunoResearch Laboratories Inc., West Grove, PA, # 706-485-148), Alexa 555 (1:400, Molecular Probes, Eugene, OR, # A31570), or Alexa 647 (1:400, Molecular Probes, # A31573).
Images were captured on a Nikon C1Si or C1 Plus confocal microscopes (Nikon Instruments Inc, Melville, NY). Images were analyzed using Nikon software EZ-C1 3.90 Free Viewer. The cellular composition of islets was measured by counting the individual types of cells (β-cells labeled with anti-insulin, α-cells with anti-glucagon and δ-cells with anti-somatostatin) in each islet and dividing the number of each cell type by the total number of all labeled cells per islet.
Immunohistochemistry (IHC)
Anti-insulin (1:100, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, # sc-9168) or anti-glucagon (1:200, Santa Cruz Biotechnology, # sc-13091) primary antibodies were used. Staining was developed using Histostain-Plus Broad Spectrum (AEC) Kit (Invitrogen, Frederick, MD, # 859943). The IHC procedure was conducted per manufacturer instructions and slides were counterstained with hematoxylin.
After staining, slides were rinsed in deionized water and placed on coverslips in Clear Mount mounting medium (Electron Microscopy Sciences, Hatfield, PA, #17985-12). The staining was observed using a Nikon Eclipse 80i light microscope (Nikon Instruments Inc, Melville, NY.). Images were analyzed using Adobe Photoshop CZ4 extended software. The relative insulin content was measured based on the intensity of staining of pancreatic sections with anti-insulin. The average pixel value of staining per cell or per islet was determined and background staining was subtracted from each sample.
Statistical analysis
All data is reported as the mean ± standard error of the mean. Male and female mice were included in analysis, as the pattern of differences between IRlox/lox and SNIRKO mice was consistent across both sexes. Graphpad Prism software was used for all statistical analysis and a P-value less than 0.05 was considered statistically significant.
Results
SNIRKO mice insulin receptor expression in the DRG
Cre/lox recombination of the insulin receptor was confirmed in the DRG using RT-PCR (Fig. 1A). An RT-PCR product of 585bp, suggesting intact insulin receptor RNA, was observed in DRG both from IRlox/lox and SNIRKO mice. A 435bp product suggesting deletion of insulin receptor exon 4 was only observed in DRG from SNIRKO mice. The presence of a 585bp product in the DRG of SNIRKO mice was most likely due to nonneuronal cells in the DRG that may express the insulin receptor, including Schwann cells, transient immune cells, endothelial cells, and satellite cells (Sugimoto et al., 2002). Analysis of DRG and sciatic nerve using AdvillinCre/+; Tomato+/− mice revealed specific Cre expression in DRG neurons and sciatic nerve (Fig. S1). No specific labeling of Cre driver activity was seen in the pancreas (data not shown).
Figure 1. SNIRKO Insulin Receptor Expression and Insulin Stimulation.
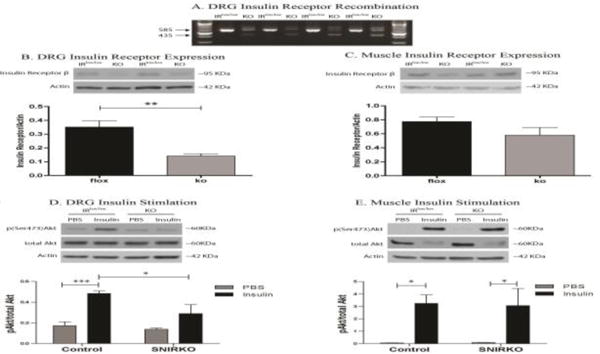
Cre recombinase excision of insulin receptor exon 4 flanked by loxp sites was confirmed in the DRG with RT-PCR (A). A 585bp band was indicative of intact insulin receptor RNA and a 435bp band suggested a recombination event and the excision of the 150bp exon 4. A 435bp band was only observed in DRG from SNIRKO mice. Western blots show a significant decrease in insulin receptor beta expression in the DRG of SNIRKO mice (B), however no significant change was observed in the muscle of SNIRKO mice (C). Results were analyzed with a student’s t-test. n=15 IRlox/lox and n=8 SNIRKO. *=p<0.05, **=p<0.01, ***=p<0.001.
Insulin stimulation failed to significantly activate Akt over baseline in the DRG of SNIRKO mice and insulin-induced activated Akt levels were significantly lower as compared to IRlox/lox (D). Insulin stimulation significantly activated Akt over baseline in the muscle from both IRlox/lox and SNIRKO mice. No significant difference was observed between IRlox/lox and SNIRKO mice insulin-induced Akt activation levels in the muscle (E). Results were analyzed with a 2-way ANOVA and Bonferroni’s post-hoc analysis. n=4 IRlox/lox PBS, n=4 IRlox/lox insulin, n=3 SNIRKO PBS and n=3 SNIRKO insulin.
Western blot analysis was used to determine whether cre/lox recombination resulted in reduced insulin receptor protein levels in the DRG (Fig. 1B). SNIRKO mice had significantly decrease insulin receptor expression in DRG. Insulin receptor levels in SNIRKO mice were approximately 60% lower than in IRlox/lox mice. A similar decrease in insulin receptor expression was not present in the gastrocnemius muscle of SNIRKO mice (Fig. 1C).
To determine if reductions in DRG insulin receptor expression affected insulin signaling transduction, DRG Akt activation was assessed after IP insulin injection. As expected, DRG from IRlox/lox mice showed a significant increase in Akt activation (p(ser473)/totalAkt) in response to insulin stimulation (Fig. 1D). However, Akt activation in the DRG of SNIRKO mice was similar to baseline (PBS) and was significantly lower as compared to IRlox/lox mice. Insulin-induced Akt activation was significantly activated in the muscle from IRlox/lox and SNIRKO mice compared to baseline and there was no significant difference between groups (Fig. 1E).
SNIRKO mice do not display gross changes in development and are euglycemic
Previous mouse models with systemic insulin receptor knockout are born 10% smaller and die within 72 hours from severe diabetic ketoacidosis (Accili et al., 1996). Here, SNIRKO mice were born with expected frequencies and showed no obvious differences in appearance, development, or weight gain through 28 weeks of age (Fig. 2A, B). SNIRKO development was assessed with a modified SHIRPA analysis (Hatcher et al., 2001) and no statistical differences were observed compared to IRlox/lox mice (data not included).
Figure 2. SNIRKO mice do not have gross developmental defects and display euglycemia.
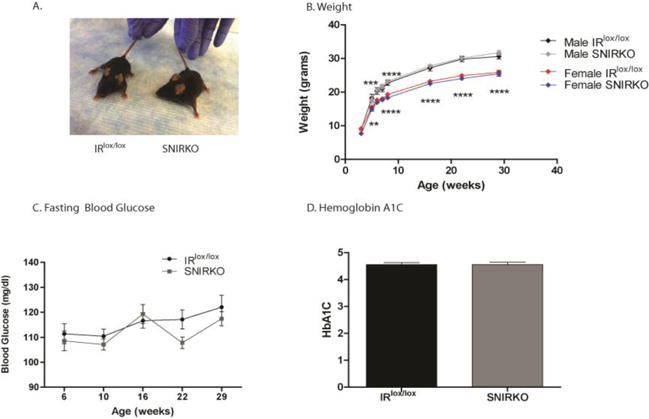
SNIRKO mice did not show signs of major birth defects and appear healthy from birth (A). SNIRKO mice weight was similar to IRlox/lox mice of the same sex (B). Results were analyzed with a repeated measures 2-way ANOVA and Bonferroni’s post-hoc. There were no differences between IRlox/lox and SNIRKO mice. The differences indicated in the figure are between males and females only. n=9 IRlox/lox male, n=10 SNIRKO male, n=8 IRlox/lox female, and n=6 SNIRKO female. Blood glucose levels were measured at 6, 10, 16, 22, and 28 weeks of age. Results were analyzed with a repeated measures 2-way ANOVA and Bonferroni’s post-hoc. No significant differences were observed between male or female IRlox/lox and SNIRKO mice. (C) n=22 IRlox/lox and n=23 SNIRKO. Hemoglobin A1C levels were measured at 29 weeks of age. There were no significant differences noted in males or females between IRlox/lox and SNIRKO groups when data from females and males was combined (D). Results were analyzed with a 2-way ANOVA and Bonferroni’s post-hoc (C) and student’s t-test (D).
Blood glucose levels were monitored for 6 months and no significant differences were observed between SNIRKO and IRlox/lox mice (Fig. 2C). Independent of genotype, female mice had significantly lower blood glucose levels compared to males (not denoted in the figure). In addition, there was no significant difference in hemoglobin A1C levels between groups (Fig. 2D).
SNIRKO mice do not show sensorimotor behavior changes
Hallmarks of PDN in rodent models include changes in mechanical and thermal sensitivity. SNIRKO mice had no significant differences in mechanical sensitivity (Fig. 3A). Analysis of thermal sensitivity with a repeated measures 2-way ANOVA indicated no main effect between IRlox/lox and SNIRKO mice throughout the testing periods (Fig. 3B), however Bonferroni’s post-hoc revealed a significant difference at 13 weeks of age. Interestingly, this appears to be due more to an increase in IRlox/lox threshold unique to this age rather than a decrease in SNIRKO threshold. Two additional tests were performed to assess motor coordination and balance/proprioception. SNIRKO mice showed no significant differences in the number of foot slips on the beamwalk (Fig. 3C) or in the latency to fall on the rotorod (Fig. 3D).
Figure 3. SNIRKO mice do not display sensorimotor deficits characteristic of PDN in murine models.
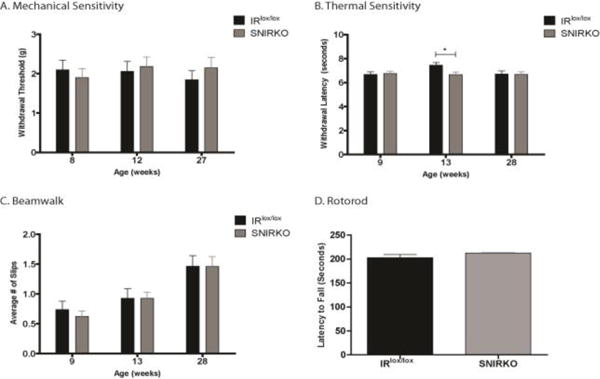
It was hypothesized that the reduction in sensory neuron insulin signaling despite euglycemia would produce a phenotype similar to PDN in mouse models. Similar patterns were observed across males and females between groups and data presented here is combined from males and females. No significant difference between IRlox/lox and SNIRKO was observed in mechanical sensitivity (A). For thermal sensitivity, there was no significant difference of group between IRlox/lox and SNIRKO mice upon analysis with 2-way repeated measures ANOVA (B). However, Bonferroni’s post-hoc does show a significant difference between IRlox/lox and SNIRKO mice at 13 weeks of age. Of note, the thermal threshold for SNIRKO mice appears to remain relatively constant throughout the period tested, yet IRlox/lox mice show an increase in threshold at 13 weeks. No significant difference between IRlox/lox and SNIRKO mice was observed in beamwalk (C) or rotorod (D). Mechanical sensitivity, thermal sensitivity, and beamwalk were analyzed with a repeated measures 2-way ANOVA and Bonferroni’s post-hoc. Rotorod was analyzed with a student’s t-test. n=24 IRlox/lox and n=23 SNIRKO. *=p<0.05.
SNIRKO mice do not show morphologic or physiologic changes consistent with PDN
Axonal innervation of the hind paw epidermis was measured using IENFD. The IENFD in SNIRKO mice was not significantly different compared to IRlox/lox mice (Fig. 4A–C). In addition, SNIRKO mice showed no significant differences in SNCV or MNCV (Fig. 4D, E).
Figure 4. SNIRKO mice do not show morphological or physiological changes characteristic of PDN in murine models.
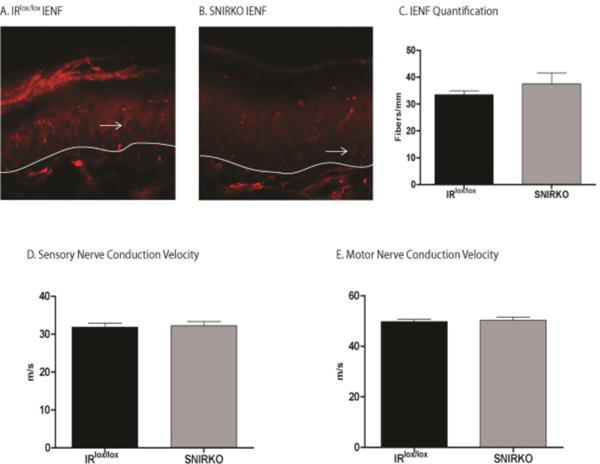
Two characteristic signs of PDN in mice include decreased IENFD and reduced NCV. Nerve fibers (arrows) that cross the dermal-epidermal border (white line) were quantified in both IRlox/lox (A) and SNIRKO mice (B). No significant differences were noted (C). Results were analyzed with a student’s t-test. n=4 IRlox/lox and n=4 SNIRKO. In addition, no differences were observed in either SNCV (D) or MNCV (E). Results were analyzed with a student’s t-test. n=24 IRlox/lox and n=23 SNIRKO.
SNIRKO mice display significantly elevated serum insulin levels and glucose intolerance
SNIRKO mice displayed significantly elevated serum insulin levels (Fig. 5). The difference between IRlox/lox and SNIRKO mice appeared to increase with age. For comparison, IRlox/lox mice had serum insulin levels of 0.93 ng/mL at 6 weeks of age and 1.00 ng/mL at 29 weeks of age. SNIRKO mice had serum insulin levels of 1.14 ng/mL at 6 weeks of age and 2.03 ng/mL at 29 weeks of age. No change was found in serum IGF1 levels or DRG IGF receptor expression at sacrifice (Fig. S2). Analysis of IPGTT revealed that SNIRKO mice maintained significantly higher blood glucose levels upon glucose challenge (Fig. 6A, B) resulting in significantly different areas under the curve (AUC) between SNIRKO and IRlox/lox mice (Fig. 6C, D).
Figure 5. SNIRKO mice have significantly elevated serum insulin levels.
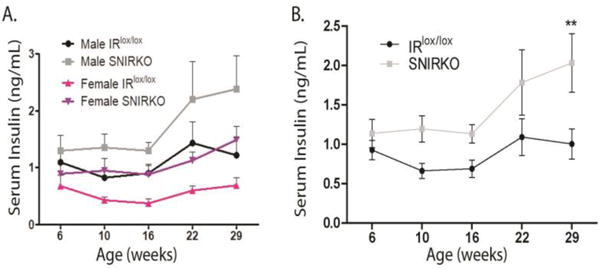
Despite a conditional knockout of the insulin receptor specifically in sensory neurons, SNIRKO mice presented with a systemic increase in serum insulin levels. Male and female SNIRKO mice show elevated insulin levels above IRlox/lox mice of the same sex (A). Combined male and female insulin data show a significant main effect of group (IRlox/lox vs. SNIRKO) upon analysis with repeated measures 2-way ANOVA (p=0.036) (B). Bonferroni’s post-hoc indicates a significant difference at 29 weeks of age. n=22 total IRlox/lox (13 = males, 9 = females) and n=23 SNIRKO (14 = males, 9 = females). **=p<0.01.
Figure 6. SNIRKO mice display glucose intolerance.
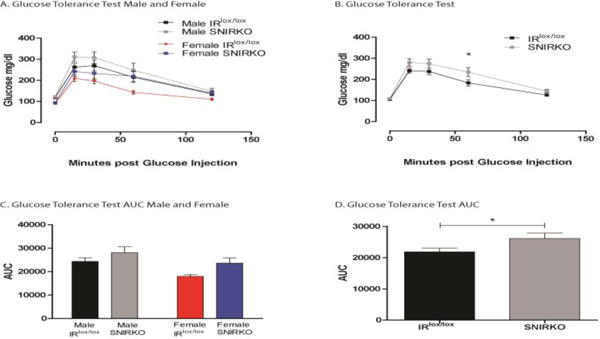
Elevated insulin levels are capable of producing signs of insulin resistance and glucose intolerance. IRlox/lox and SNIRKO mice were subjected to an IPGTT. Male and female SNIRKO mice show elevated glucose levels above IRlox/lox mice of the same sex (A). A combination of data from male and female mice shows that SNIRKO mice appear to maintain elevated glucose level throughout the IPGTT (B). Analysis with a 2-way repeated measure ANOVA indicates the p-value for group (IRlox/lox vs. SNIRKO) was 0.054. Bonferroni’s post-hoc indicates at significant difference at 60 minutes post glucose injection. Calculation of the area under the curve (AUC) shows that male and female SNIRKO mice once again show similar patterns as compared to IRlox/lox mice (C) and the AUC was significantly elevated in SNIRKO mice as compared to IRlox/lox when data from male and females was combined (D). Results were analyzed with a student’s t-test. n=20 IRlox/lox (12 = males, 8 = females) and n=18 SNIRKO (10 = males, 8 = females). *=p<0.05.
SNIRKO mice show normal islet morphology, no insulitis, but increased insulin content
SNIRKO mice showed no significant changes in islet morphology or islet cell composition compared to IRlox/lox mice (Fig. 7A–C). Large islets are known to produce less insulin than small islets (MacGregor et al., 2006). Islet size was quantified in IRlox/lox and SNIRKO mice, but no significant differences were noted (Fig. 7D). Finally, insulitis (islet inflammation) and insulin content was assessed using immunohistochemistry. SNIRKO mice showed no signs of islet lymphocyte infiltration (Fig. 8A, B). However, analysis of insulin staining in individual islets using IHC islets revealed a significant increase of insulin immunoreactivity in SNIRKO mice compared to IRlox/lox mice (Fig. 8C).
Figure 7. SNIRKO islet of Langerhans cell composition.
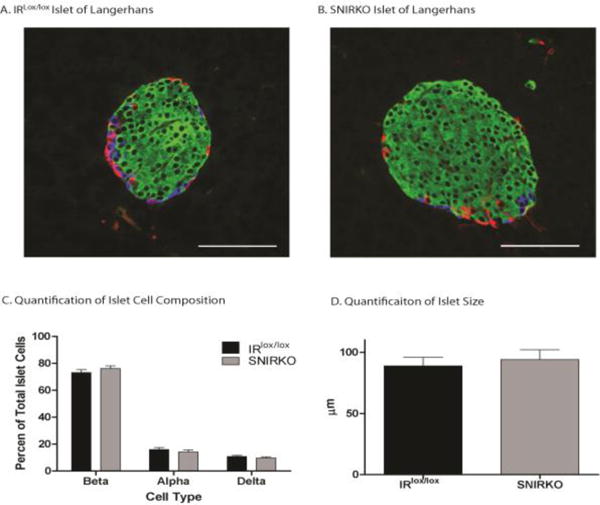
Endocrine cells of the islets of Langerhans were labeled (green=beta cells, red=alpha cells, blue=delta cells) and cell composition was quantified (C). IRlox/lox (A) and SNIRKO (B) mice display similar islet of Langerhans cell composition, with no significant difference between beta, alpha, or delta cells. Data were analyzed with a 1-way nested ANOVA on ranks and Dunn’s pairwise comparison. 42 islets from 3 IRlox/lox mice and 45 islets from 3 SNIRKO mice were analyzed. (D) There was also no statistically significant difference in islet size. Data were analyzed with student’s t-test. n=42 islet IRlox/lox n= 45 islets SNIRKO. Scale bar=100μm.
Figure 8. Islets of Langerhans from SNIRKO mice have increased insulin content and no lympocytic infiltrate.
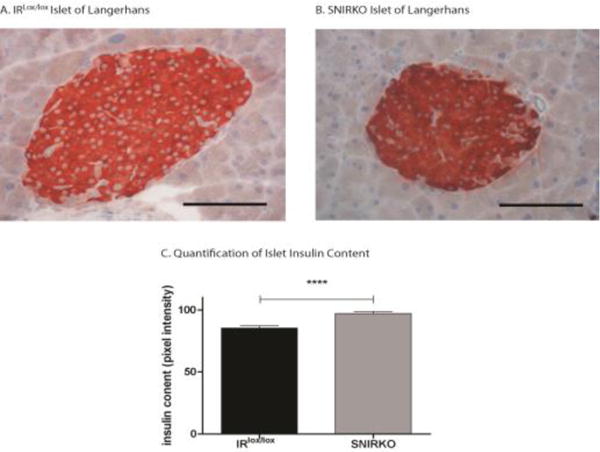
Insulitis was analyzed by assessing samples stained with hematoxylin for immune cell infiltration. No significant insulitis was observed in either islets from IRlox/lox (A) or SNIRKO (B) mice. Islet insulin content was analyzed by quantifying insulin staining intensity. Islets from SNIRKO mice have significantly elevated insulin content as compared to islets from IRlox/lox mice (C). Results were analyzed with a student’s t-test. n=45 islets from 3 IRlox/lox mice and n=33 islets from 3 SNIRKO mice. Scale bar=100μm.
Discussion
While several studies have demonstrated the neurotrophic potential of insulin and have suggested that reduced sensory neuron insulin support may be a major contributor to the pathogenesis of diabetic neuropathy (Kim and Feldman, 2012), approaches to specifically test sensory neuron insulin signaling in vivo without changes in other variables has proven difficult. Here, we developed sensory neuron insulin receptor knockout mice to investigate how insulin signaling impacts sensory neuron function. While the insulin receptor has been conditionally deleted in other tissues that include muscle (Bruning et al., 1998), liver (Michael et al., 2000), pancreas (Kulkarni et al., 1999), and the central nervous system (Bruning et al., 2000), this study is the first to generate mice with a conditional knockout of the insulin receptor in sensory neurons.
Our results reveal that SNIRKO mice do not display behavioral, physiological, or morphological signs of PDN. Importantly, this supports the view that absent insulin signaling in sensory neuron insulin is not sufficient to induce sensory abnormalities or neuropathy. There are a number caveats that need to be considered when using tissue specific null mutant mice, including compensatory pathway upregulation, insufficient knockdown, remnant IR receptor expression in non-neuronal support cells, and developmental compensatory changes. That SNIRKO mice did not develop signs of neuropathy or sensory dysfunction speaks to complexity of mechanisms driving PDN. Other insults on DRG neurons and the PNS, including vascular, neuronal, and immunologic all likely contribute important components to PDN. We recognize that SNIRKO mice have limitations in understanding insulin’s role in the PNS model the reduced PNS insulin support present in diabetes, as global reductions in insulin support (reduced circulating levels or reduced signaling), to Schwann cells satellite glia, endothelial cells, immune cells and peripheral tissues impact these cell types involved in PDN. Based on our results, an improved approach should eliminate insulin signaling in both sensory neurons and Schwann cells as well as use an inducible knockout model to avoid developmental compensatory changes. Because the best prevention of PDN still appears to be strict glycemic control (Pop-Busui and Martin, 2016), our results support the view that hyperglycemia may be the major causal factor in the pathogenesis of PDN and other mechanisms like altered insulin support may play minor, additive roles. Another limitation of the SNIRKO mice is the potential role of insulin signaling through IGF receptors, or alternatively, compensation through IGF support of these neurons (Pires et al., 2017; Taguchi and White, 2008). Models that eliminate signaling through the insulin receptor and the IGF receptors and have additional temporal control of insulin and/or IGF receptor signaling will be necessary to resolve this question.
The mechanisms of PDN include other toxic insults and signaling pathways associated with hyperglycemic injury. It has been shown that advanced glycation endproducts (AGEs) and the upregulation of the polyol pathway is unrepairable due to the reduced repair/regeneration capacity resulting from changes in neurotrophic factors (Guo et al., 2011; Toth et al., 2006) or vascularity (Tesfaye et al., 2005). Accordingly, SNIRKO mice may require additional hyperglycemic or other injuries (high fat diet, nerve injury models, or noxious agents) to reveal a direct role for insulin on sensory neuron function.
One intriguing observation from these studies was the elevated insulin levels apparent in SNIRKO mice. Recent publications suggest that the efferent actions of sensory neurons modify beta cell insulin production/release via a negative feedback mechanism involving sensitization of TRPV1 (Razavi et al., 2006; Tsui et al., 2007), and our observation that SNIRKO mice display hyperinsulinemia, both in the serum and within the pancreatic islets, is supportive of this feedback loop. We have previously shown that insulin production within individual islets can be modified by exercise, and increased islet production within individual islets in the SNIRKO mice is consistent with the ability of islets to modify insulin production (Huang et al., 2011). In 2005, Razavi et. al. demonstrated that TRPV1 positive sensory neurons are critically involved in the pathogenesis of type 1 diabetes in non-obese diabetic (NOD) mice. NOD mice express a hypofunctional TRPV1 and embryological ablation of TRPV1 with capsaicin or substance P treatment could correct insulitis and diabetes in NOD mice (Razavi et al., 2006). Furthermore, several studies have demonstrated that insulin can sensitize and potentiate TRPV1 signaling by lowering the threshold for activation and increasing membrane translocation (Lilja et al., 2007; Sathianathan et al., 2003; Van Buren et al., 2005). In addition, the neuropeptides CGRP and substance P have been demonstrated to have a trophic effect on beta cells (Fu and Sun, 1989; Hermansen and Ahren, 1990; Tsui et al., 2007) and treatment with CGRP and/or substance P causes normalization of insulin secretion and resolution of diabetes (Khachatryan et al., 1997; Razavi et al., 2006). It was also reported that capsaicin treatment can significantly reduce blood glucose levels in a mouse model of late phase type 1 diabetes and type 2 diabetic Zucker rats (Gram et al., 2007; Gram et al., 2005; Radu et al., 2013).
The demonstration that multiple interventions associated with the insulin-TRPV1-neuropeptide-diabetes model results in similar and predictable outcomes is strong support for the existence of such a system in vivo and SNIRKO mice could be an important model to delineate these interactions. Future experiments in SNIRKO mice could assess neuropeptide levels of islets and the innervating sensory neurons, as well as assess TRPV1 sensitivity. Additionally, it will be important to determine whether CGRP and/or substance P treatment can reverse the observed hyperinsulinemia in SNIRKO mice.
Collectively, our results suggest that that a loss of insulin receptors in sensory neurons does not lead to peripheral nerve dysfunction and contribute to the growing idea that peripheral neurons play a role in metabolism and insulin regulation (Grabauskas et al., 2010; Grabauskas et al., 2013). Further research into these areas may help determine the insulin-related mechanisms affecting diabetes and its complications.
Supplementary Material
Highlights.
Mice were generated in which peripheral sensory neurons lacked the insulin receptor (SNIRKO mice)
A lack of insulin receptors on sensory neurons does not lead to peripheral nerve dysfunction
SNIRKO mice display elevated insulin and impaired glucose tolerance
Disruption of insulin signaling in sensory neurons may impact insulin and glucose regulation
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01NS43313 (DEW) and P20 GM103418 from the IDeA Network of Biomedical Research Excellence (INBRE) Program and by the Kansas IDDRC, Kansas IDDRC, U54 HD 090216. CGW and DEW designed the experiments and were responsible for the overall completion of the study. NW, NKK, BLG, JMR, NL, and LS contributed significantly to data collection, interpretation, and editing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
All authors declare no conflicts of interest.
References
- Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nature genetics. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science (New York, NY) 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Molecular cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- da Silva S, Hasegawa H, Scott A, Zhou X, Wagner AK, Han BX, Wang F. Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3395–3400. doi: 10.1073/pnas.1014411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annual review of pharmacology and toxicology. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fu XW, Sun AM. Stimulative effect of substance P on insulin secretion from isolated rat islets under normobaric oxygen incubation. Zhongguo yao li xue bao = Acta pharmacologica Sinica. 1989;10:69–73. [PubMed] [Google Scholar]
- Grabauskas G, Song I, Zhou S, Owyang C. Electrophysiological identification of glucose-sensing neurons in rat nodose ganglia. The Journal of physiology. 2010;588:617–632. doi: 10.1113/jphysiol.2009.182147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabauskas G, Zhou SY, Lu Y, Song I, Owyang C. Essential elements for glucosensing by gastric vagal afferents: immunocytochemistry and electrophysiology studies in the rat. Endocrinology. 2013;154:296–307. doi: 10.1210/en.2012-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gram DX, Ahren B, Nagy I, Olsen UB, Brand CL, Sundler F, Tabanera R, Svendsen O, Carr RD, Santha P, Wierup N, Hansen AJ. Capsaicin-sensitive sensory fibers in the islets of Langerhans contribute to defective insulin secretion in Zucker diabetic rat, an animal model for some aspects of human type 2 diabetes. The European journal of neuroscience. 2007;25:213–223. doi: 10.1111/j.1460-9568.2006.05261.x. [DOI] [PubMed] [Google Scholar]
- Gram DX, Hansen AJ, Deacon CF, Brand CL, Ribel U, Wilken M, Carr RD, Svendsen O, Ahren B. Sensory nerve desensitization by resiniferatoxin improves glucose tolerance and increases insulin secretion in Zucker Diabetic Fatty rats and is associated with reduced plasma activity of dipeptidyl peptidase IV. European journal of pharmacology. 2005;509:211–217. doi: 10.1016/j.ejphar.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Grote C, Groover A, Ryals J, Geiger P, Feldman E, Wright D. Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathologica Communications. 2013a;1:15. doi: 10.1186/2051-5960-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote CW, Morris JK, Ryals JM, Geiger PC, Wright DE. Insulin receptor substrate 2 expression and involvement in neuronal insulin resistance in diabetic neuropathy. Exp Diabetes Res. 2011;2011:212571. doi: 10.1155/2011/212571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote CW, Ryals JM, Wright DE. In vivo peripheral nervous system insulin signaling. Journal of the peripheral nervous system: JPNS. 2013b;18:209–219. doi: 10.1111/jns5.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Navarro P, Valverde AM, Arribas M, Bruning J, Kozak LP, Kahn CR, Benito M. Brown adipose tissue-specific insulin receptor knockout shows diabetic phenotype without insulin resistance. The Journal of clinical investigation. 2001;108:1205–1213. doi: 10.1172/JCI13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilford BL, Ryals JM, Wright DE. Phenotypic changes in diabetic neuropathy induced by a high-fat diet in diabetic C57BL/6 mice. Exp Diabetes Res. 2011;2011:848307. doi: 10.1155/2011/848307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Kan M, Martinez JA, Zochodne DW. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiology of disease. 2011;43:414–421. doi: 10.1016/j.nbd.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Abbott S, Han BX, Qi Y, Wang F. Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J Neurosci. 2007;27:14404–14414. doi: 10.1523/JNEUROSCI.4908-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher JP, Jones DN, Rogers DC, Hatcher PD, Reavill C, Hagan JJ, Hunter AJ. Development of SHIRPA to characterise the phenotype of gene-targeted mice. Behavioural brain research. 2001;125:43–47. doi: 10.1016/s0166-4328(01)00275-3. [DOI] [PubMed] [Google Scholar]
- Hermansen K, Ahren B. Dual effects of calcitonin gene-related peptide on insulin secretion in the perfused dog pancreas. Regulatory peptides. 1990;27:149–157. doi: 10.1016/0167-0115(90)90213-g. [DOI] [PubMed] [Google Scholar]
- Huang HH, Farmer K, Windscheffel J, Yost K, Power M, Wright DE, Stehno-Bittel L. Exercise increases insulin content and basal secretion in pancreatic islets in type 1 diabetic mice. Exp Diabetes Res. 2011;2011:481427. doi: 10.1155/2011/481427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack MM, Ryals JM, Wright DE. Protection from diabetes-induced peripheral sensory neuropathy–a role for elevated glyoxalase I? Experimental neurology. 2012;234:62–69. doi: 10.1016/j.expneurol.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn CR. Knockout mice challenge our concepts of glucose homeostasis and the pathogenesis of diabetes. Exp Diabesity Res. 2003;4:169–182. doi: 10.1155/EDR.2003.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachatryan A, Guerder S, Palluault F, Cote G, Solimena M, Valentijn K, Millet I, Flavell RA, Vignery A. Targeted expression of the neuropeptide calcitonin gene-related peptide to beta cells prevents diabetes in NOD mice. J Immunol. 1997;158:1409–1416. [PubMed] [Google Scholar]
- Kim B, Feldman EL. Insulin resistance in the nervous system. Trends in endocrinology and metabolism: TEM. 2012;23:133–141. doi: 10.1016/j.tem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Bensoula AN, Filali M. Rotorod sensorimotor learning in cerebellar mutant mice. Neuroscience research. 1995;22:423–426. doi: 10.1016/0168-0102(95)00916-h. [DOI] [PubMed] [Google Scholar]
- Lilja J, Laulund F, Forsby A. Insulin and insulin-like growth factor type-I up-regulate the vanilloid receptor-1 (TRPV1) in stably TRPV1-expressing SH-SY5Y neuroblastoma cells. J Neurosci Res. 2007;85:1413–1419. doi: 10.1002/jnr.21255. [DOI] [PubMed] [Google Scholar]
- MacGregor RR, Williams SJ, Tong PY, Kover K, Moore WV, Stehno-Bittel L. Small rat islets are superior to large islets in in vitro function and in transplantation outcomes. American journal of physiology Endocrinology and metabolism. 2006;290:E771–779. doi: 10.1152/ajpendo.00097.2005. [DOI] [PubMed] [Google Scholar]
- Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Molecular cell. 2000;6:87–97. [PubMed] [Google Scholar]
- Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M, Wood JN. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nature communications. 2012;3:791. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires KM, Buffolo M, Schaaf C, David Symons J, Cox J, Abel ED, Selzman CH, Boudina S. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. Journal of molecular and cellular cardiology. 2017;113:39–50. doi: 10.1016/j.yjmcc.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop-Busui R, Martin C. Neuropathy in the DCCT/EDIC-What Was Done Then and What We Would Do Better Now. Int Rev Neurobiol. 2016;127:9–25. doi: 10.1016/bs.irn.2016.02.020. [DOI] [PubMed] [Google Scholar]
- Radu BM, Iancu AD, Dumitrescu DI, Flonta ML, Radu M. TRPV1 Properties in Thoracic Dorsal Root Ganglia Neurons are Modulated by Intraperitoneal Capsaicin Administration in the Late Phase of Type-1 Autoimmune Diabetes. Cellular and molecular neurobiology. 2013;33:187–196. doi: 10.1007/s10571-012-9883-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi R, Chan Y, Afifiyan FN, Liu XJ, Wan X, Yantha J, Tsui H, Tang L, Tsai S, Santamaria P, Driver JP, Serreze D, Salter MW, Dosch HM. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell. 2006;127:1123–1135. doi: 10.1016/j.cell.2006.10.038. [DOI] [PubMed] [Google Scholar]
- Sathianathan V, Avelino A, Charrua A, Santha P, Matesz K, Cruz F, Nagy I. Insulin induces cobalt uptake in a subpopulation of rat cultured primary sensory neurons. The European journal of neuroscience. 2003;18:2477–2486. doi: 10.1046/j.1460-9568.2003.03004.x. [DOI] [PubMed] [Google Scholar]
- Singh R, Kishore L, Kaur N. Diabetic peripheral neuropathy: current perspective and future directions. Pharmacological research. 2014;80:21–35. doi: 10.1016/j.phrs.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. Journal of the peripheral nervous system: JPNS. 2002;7:44–53. doi: 10.1046/j.1529-8027.2002.02005.x. [DOI] [PubMed] [Google Scholar]
- Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annual review of physiology. 2008;70:191–212. doi: 10.1146/annurev.physiol.70.113006.100533. [DOI] [PubMed] [Google Scholar]
- Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, Witte DR, Fuller JH. Vascular risk factors and diabetic neuropathy. The New England journal of medicine. 2005;352:341–350. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- Toth C, Brussee V, Martinez JA, McDonald D, Cunningham FA, Zochodne DW. Rescue and regeneration of injured peripheral nerve axons by intrathecal insulin. Neuroscience. 2006;139:429–449. doi: 10.1016/j.neuroscience.2005.11.065. [DOI] [PubMed] [Google Scholar]
- Tsui H, Razavi R, Chan Y, Yantha J, Dosch HM. ‘Sensing’ autoimmunity in type 1 diabetes. Trends in molecular medicine. 2007;13:405–413. doi: 10.1016/j.molmed.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Van Buren JJ, Bhat S, Rotello R, Pauza ME, Premkumar LS. Sensitization and translocation of TRPV1 by insulin and IGF-I. Molecular pain. 2005;1:17. doi: 10.1186/1744-8069-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurborg S, Piszczek A, Martinez C, Hublitz P, Al Banchaabouchi M, Moreira P, Perlas E, Heppenstall PA. Generation and characterization of an Advillin-Cre driver mouse line. Molecular pain. 2011;7:66. doi: 10.1186/1744-8069-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.