

Abstract
There is an exponential increase in biological complexity as initial gene transcripts are spliced, translated into amino acid sequence and post-translationally modified. Each protein can exist as multiple chemical or sequence-specific “proteoforms”, and each has the potential to be a critical mediator of a physiological or pathophysiological signaling cascade. Here, we provide an overview of how different proteoforms come about in biological systems, and how they are most commonly measured using mass spectrometry based proteomics and bioinformatics. Our goal is to present this information at a level accessible to every scientist interested in mass spectrometry and its application to proteome profiling. We will specifically discuss recent data linking various protein post-translational modifications (PTMS) to cardiovascular disease, and conclude with a discussion for enablement and ‘democratization’ of proteomics across the cardiovascular and scientific community. The aim is to inform and inspire the readership to explore a larger breadth of proteoform, particularity PTMs, related to their particular areas of expertise in cardiovascular physiology.
Subject Terms: Genetics
Keywords: proteomics, post-translational regulation, mass spectrometry, proteoforms
Introduction
The capability to sequence whole genomes has led to an explosion in the basic and translational understanding about how nucleotide sequences are associated with or predictive of a multitude of cardiovascular phenotypes.1 Knowledge of DNA sequence variants, however, provides only a partial and often static understanding of how differences in genetic code translate to the reality of physiology and pathophysiology. Linking other intrinsic environmental and lifestyle factors acting at the level of transcriptional, translational, co- and post-translational physiology is a key aim to provide an integrative framework for systemic analysis of cellular homeostasis and disease. Collectively, these variants of amino acid sequences and their chemical modifications are referred to as the ‘proteoforms’ of any single primary nucleotide sequence encoded in the genome.2 Thus, proteoforms exist as both structural variants and/or post-translationally modified variations. There has been recent attention given to the importance of considering splicing, cleavage, and other structural isoforms in cardiovascular physiology including a profiling of apolipoprotein isoforms,3 LOX1 isoforms as they relate to atherosclerosis,4 the role of RBM20 and sarcomeric splice isoforms during dilated cardiomyopathy,5 and profiling myofilament isoforms6 as well as their phosphorylation state as they change after myocardial infarction.7 Since any one or a combination of proteoforms could be the critical diagnostic or therapeutic target for a specific cardiovascular condition, we contend that attention should be given to the profiling, quantifying, and understanding of proteoform complexity within cardiovascular systems. In this review, we will 1) focus on one type of proteoforms, protein post-translational modification (PTM), 2) describe how biochemistry and mass spectrometry (MS) are harnessed to profile, detect and quantify proteins and PTMs, and 3) provide examples of PTMs, including understudied PTMs already shown to be clinically relevant in cardiovascular disease. It is our goal to enhance the understanding of the current ‘state of the art’ for PTMs-proteomics among basic and clinical cardiovascular scientists. We hope that these efforts will lead to a broader appreciation of the need to embrace the complexity inherent to protein biochemistry, and its relevance to disease diagnosis and therapeutics.
Proteoforms – Definition and Scope
Proteoforms represent all possible variants of a single gene product, and include both differences in amino acid sequence (e.g., isoforms and cleavage products) as well as chemical modifications of amino acid residues. They represent an important mechanism for diversifying and regulating the cellular proteome, and can dramatically increase the chemical diversity of a protein. Knowledge and quantification of protein substrates and their PTM sites is key to dissecting PTM-mediated cellular processes, including complex regulatory networks. Despite their importance, the comprehensive identification and discovery of PTMs in complex biological samples has continued to pose challenges for proteomics technologies.8
A PTM is a covalent process resulting from a proteolytic cleavage or from the addition of a modifying group to a particular amino acid. The chemical nature and function of these modifications is diverse. They include covalent additions of particular chemical groups (e.g. phosphoryl), lipids (e.g. palmitic acid), carbohydrates (e.g. glucose) or even entire proteins (e.g. ubiquitin) to amino acid side chains.9 PTMs frequently affect certain groups of amino acid residues, for instance, phosphorylation affects serine (S), threonine (T), and tyrosine (Y); acetylation and ubiquitination occur on lysine (K) and methylation occurs on lysine (K) and arginine (R) residues. Moreover, many amino acid residues can have more than one type of PTM (Figure 1). Some PTMs involve reversible reactions mediated by chemical moieties (e.g. cysteine oxidation) while others, occur via systems of enzymes that recognize short linear motifs in substrate proteins, to add and remove the PTM (e.g. the specific kinases and phosphatase that regulate phosphorylation). Other PTMs are irreversible and cycle only with de novo synthesis of the protein (e.g. citrullination). Regardless of the mechanism of PTM regulation, the results of these processes are that the proteome of a cell or organism has vastly more components than there are genes encoding them. To date there are more than 600 different PTMs assigned in the UniProt database; with greater than 100 different modifications reported in human proteins.10–13
Figure 1.
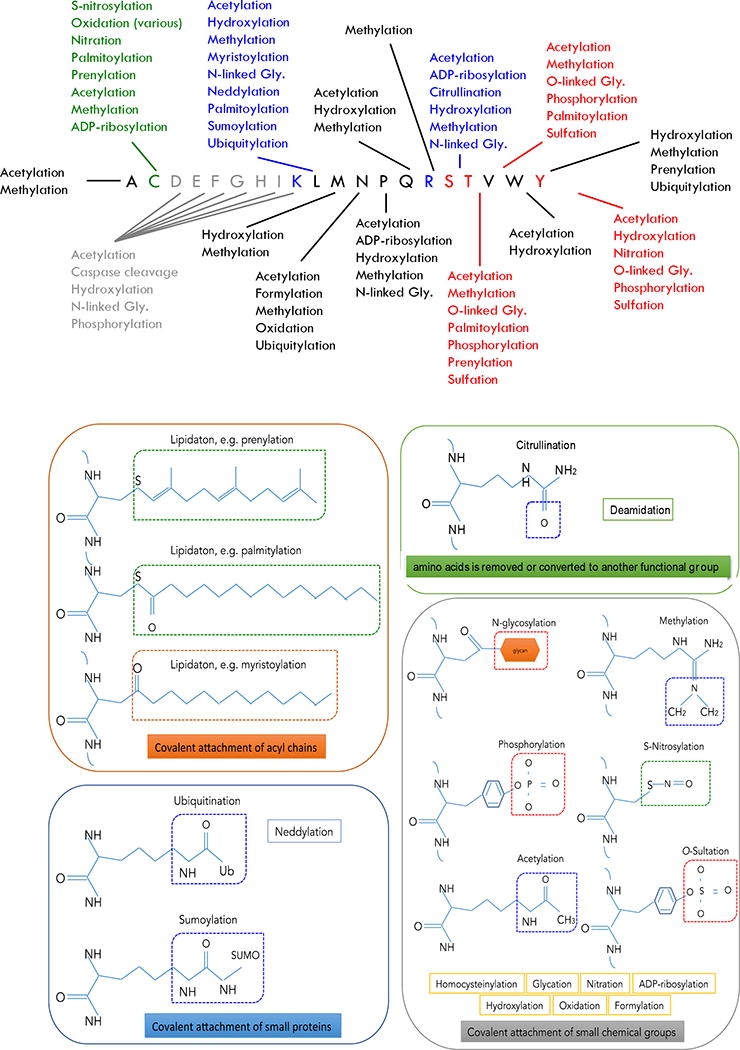
Examples of PTMs, the amino acids they target and their categories.175–177
In addition to the considerable diversity of characterized PTMs, it has become clear that some PTMs can exist in a reciprocal or associational relationship within a protein’s amino acid sequence to further regulate its function.14–17 The role of this PTM ‘crosstalk’ is still not well understood, in part due to a lack of tools. However, successful attempts in developing in silico algorithms that can reliably predict various PTMs in a given protein based on residues surrounding a modification site can help to understand the co-occurrence of the different PTM on the same protein.18,19 In fact, crosstalk has also been observed between PTM located on interacting proteins, however deciphering timing and extent of possible PTM combinations has been challenging.15,20 A further challenge in the large-scale proteomic approach to identifying and meaningfully interpreting PTMs biologically is that many of the sites discovered in the last decade have not yet been integrated in any specific pathway.
In the following sections, we describe some of the available MS methods, online platforms and their contribution in the study of PTMs. This effort is an attempt to establish a stronger link between the path to comprehensive proteoform analysis and the biological functions they confer.
Analysis of PTMs and proteomes by mass spectrometry
Over the last 30 years, experimental techniques used for mapping and quantifying PTMs have undergone impressive progress. Still there are many PTMs that do not have easy to use tools nor any understanding of their regulation or role in biology. In the following section we will outline the several different mass spectrometry approaches that have emerged to identify and quantify peptides and their modified forms by mass spectrometry (MS).21 PTMs can be studied using two basic mass spectrometry strategies; a top-down approach, which measures the mass of intact proteins or a bottom-up approach, which measures proteolytically-derived peptides. While the sample preparation and the data interpretation differentiate the top-down and bottom-up approaches, the basic concepts of how ions are surveyed, selected, and fragmented and how this process can be optimized for PTM discovery is somewhat generalizable across approaches, and we will begin this section with a brief overview of these technical concepts and consideration.
The combination of liquid chromatography (LC) and tandem MS/MS is now a standard technology frequently applied to high-throughput peptide and protein identification and quantification.22–24 Tandem mass spectrometers perform two basic measurements as peptides are being eluted from the LC column: First, in the parent or MS1 scan the mass is determined for each peptide present in a sampling of the eluting intact peptides from the chromatophic separation at a given point in time. Second, in the MS/MS, sometimes referred to as MS2 scan, one or more parent peptides are split into pieces using one of a number of different strategies to break the chemical bonds between amino acids (e.g., collision with inert gases or transfer of electrons). This results in generation of multiple fragments of the original peptide that are again detected and quantified in the mass spectrometer (e.g., the fragment masses). The combination of the parent mass and its associated fragment masses can be used to determine the sequence of the peptide, amino acid localization of its PTMs, and ultimately match its sequence to a protein gene product from a genomic database. This basic process is the foundation for the three common variants of bottom-up proteomics: Data Dependent Acquisition (DDA); Data Independent Acquisition (DIA); and Targeted Acquisition (which can consist of Multiple Reaction Monitoring, Selective Reaction Monitoring or Parallel Reaction Monitoring: MRM, SRM, PRM, respectively). Figure 2 and Table 1 illustrates the defining characteristics and provide a summary of the strengths and weakness of each approach, respectively.
Figure 2.
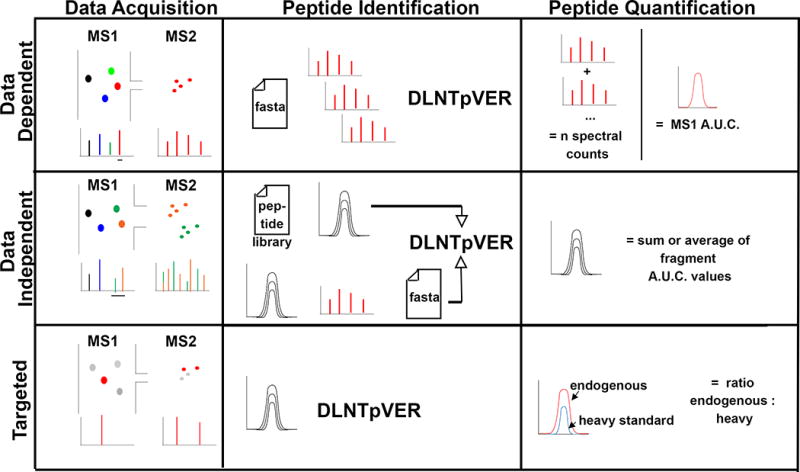
Schematic representation of different LC-MS/MS methods, with principal steps in the workflow for PTM analysis including data acquisition, peptides identification and quantification by mass spectrometer working on DDA, DIA and target (MRM or PRM) mode.
Table 1.
Summary of MS acquisition methods DDA, DIA, and MRM/PRM with respect to precursor selection, precision of detection and quantification, reproducibility and PTM analysis.
| Meth od |
Precur sor selecti on |
Fragme nt- ation |
Collision method |
Detectio n |
Precision of peptide quantificati on |
Reproducibilit y of peptide identification |
Analysis (software) |
PTM localization |
Validation of PTM |
|---|---|---|---|---|---|---|---|---|---|
| DDA | Single | Single parent mass of the compound | CID, HCD, ETD, CAD51 | All fragments ions26 | Low/Moderate for spectra counts26 | Low: missing peptides across multiple runs26 | Multiple, straightforward178 | CID: neutral loss ETD: multistage activation HCD: fragment ions |
Predominant method with ETD fragmentation 15,179 |
| DIA | Multiple | All peptides in a given m/z window | Alternating low-energy CID and high-energy CID51 | All fragments ions29 | Accurate: similar to MRM but more vulnerable to variation caused by interference from other peptides29 | Moderate: ~80% peptide overlap across multiple runs180 | Complex and requires deconvolution180,181 | CID: neutral loss | Complex182 |
| MRM | Single | Single parent mass of the compound | HCD51 | High: Selection and detection of single fragment ion 29 | Precise183 | High38 | Multiple, straightforward 184 | MS2 fragmentation analysis185 | Rely on synthetic unlabeled and stable isotope-labeled peptides80 |
| PRM | Single of multiple | Single parent mass of the compound | CID HCD |
All fragment ions | High: Run-to run peptide identification at the upper 85%39 | High186 | Multiple, straightforward | multistage activation fragmentation187 | Useful to study PTMs that are low in abundance188 |
Data Dependent Acquisition (DDA) attempts to isolate and sequence every peptide individually. This method is also referred to as shotgun proteomics and is the most commonly used approach.24 DDA involves cycling between detecting the intact parent peptide (e.g., MS1 ions) and then the instrument selecting a subset of individual peptides (MS1) ions for further fragmentation (e.g., MS/MS). Importantly, this approach requires a ‘decision’ by the instrument in each cycle as to which MS1 ions to select and filter for fragmentation. Despite ever increasing sensitivity and cycling speed of DDA when implemented on modern mass spectrometers,25 its semi-stochastic nature leads to sampling of a slightly different subset of peptides each time a sample is analyzed, resulting in missing peptide identifications and decreased reproducibility across multiple runs or experimental samples. But importantly, it is the only way to find novel proteins that are present in any particular sample.26,27
As an alternative to DDA, DIA is an approach that improves data completeness by not programming the instrument to isolate selected precursors for fragmentation but sweeping through a preset precursor mass range, filtering groups of intact peptides for fragmentation.28–30 Each MS/MS spectra generated produces a convoluted mix of the fragments derived from all peptides isolated within a given mass window at a given time in the LC gradient. Peptide and ultimately protein identifications are determined by comparing the acquired data against a previously established library of expected fragments as well as the discrete time they are expected to elute along the chromatogram (e.g., retention time [RT]).31 This means that only those proteins and their selected peptides will be analyzed. Therefore, you can miss novel proteins that maybe present in samples if they are not included in the library.
In theory, since all peptides are fragmented and thereby ‘sequenced’, the digital record of the complete peptide contents of the sample is created.32 However, this makes it complicated and ensuring the correct peptide identification becomes critical.25 The benefit to this approach is that as every precursor mass range is fragmented in every sample there should be fewer gaps in the data and improved run-to-run reproducibility.33,34 Finally, although DIA has been gradually accepted by the proteomics community, the sensitivity for detecting low abundant protein and PTMs is still challenged by signal-to-noise limitations and library composition and completeness.35,36
Targeted MS is the third category of bottom-up analysis and is more closely related to ELISA and western blots, in that predefined protein(s) is analyzed or quantified. With this method, a sample is queried for the presence and quantity of specific peptides, unique to the protein of interest, and the instrument hones in on only a few characteristic fragments of these peptides. Thus, the mass spectrometer only allows those pre-specified peptides (based on their mass) to be detected. The time the instrument has to dwell on any one peptide is increased compared to the other methods, and this results in a greater sensitivity over the other untargeted acquisition modes.37 For specific, precise quantification, reproducibility, and validation of circulating biomarkers, this targeted mode of LC-MS/MS has been the standard.38.39
A variant of MRM that may be important for the analysis of PTMs is PRM.40 In PRM, the precursor ion is targeted but data are collected on all of the resulting fragment ions. The key advantage over traditional MRM is specificity, as all potential product ions of a peptide are available to unambiguously confirm the identity of the peptide and, in the case of PTMs, potentially also confirm the site of chemical modification (as discussed below).40 Furthermore, monitoring of all transitions, does not require prior knowledge of, or preselect, target transitions before analysis, which minimal upfront development time and straightforward data analysis.
In contrast to these bottom-up approaches, with top-down proteomics intact proteins are first detected in a parent MS1 scan, and subsequently isolated and fragmented as described above in order to identify component peptides, deduce their amino acid sequences, and sites of PTM. Top-down is perhaps the only technique capable of truly detecting all of the combinations of PTMs present in a system at a given sampling time in their relative quantities.41 A close correlate of top-down is ‘middle-down’ in which the protein is digested up into larger fragments than a typical tryptic digest to both improve ionizability yet retain the potential for mapping some of the different combinations of PTMs present on a protein in a given system.42 In addition to multiple, more generalized predecessors,43 a recent and thorough review by Cai, Tuchulski, Gregorich, and Ge elegantly summarizes the concepts, technical considerations, and contributions of top-down proteomics specifically in the understanding of proteoforms and PTMs in cardiovascular biology and disease.41
Regardless of whether top-down or bottom-up strategies are being used, an important additional consideration, particularly in reference to the PTM-proteome, is the mode of peptide (or protein, in the case of top-down) fragmentation employed during MS2 acquisition. While this topic is far too technical for in depth consideration here, a brief review of fragmentation modes and their benefits or drawbacks to PTM discovery is warranted. Fragmentation approaches within the MS include physical methods dependent on collision of ions with inert gases (e.g., Collision induced dissociation (CID), high-energy collision dissociation (HCD)), and energy transfer methods whereby more gentle or controllable electron transfer is used (e.g., electron transfer dissociation (ETD)).
In the CID approach, fragmentation results from collision of the analyte with neutral gas. It is a low-energy process that breaks the peptide backbone at the C-N amide bond to form “b” or “y”-type fragment ions, which are most useful for determining the peptide sequence.44 The downside of the method is that fragmentation of a peptide is size dependent since all the bonds of the peptide absorb the energy of the collision. Thus, in practice, if a peptide is larger than 2500 Da, it is difficult to get enough energy into a single bond to break it. Moreover, labile PTMs like phosphorylation, glycosylation or S-nitrosylation are often the first to absorb the energy and break from the peptide with CID, producing a dominant ‘neutral loss’ ion of the original peptide parent mass and very few subsequent fragment ions for sequence identification. Further, these more labile modifications are thus easily “lost” during CID fragmentation resulting in a lack of product ions that provided definitive evidence for the correct site of modification, ultimately limiting the ability to unambiguously assign the correct site of PTM (see more on this below).45
In ETD, low-energy electrons are captured by peptide cations to form odd-electron peptides which then dissociate to fragments primarily along the N–Cα bonds of the peptide backbone, generating c-and z-type ions.46 Unlike CID processes, ETD cleavage does not increase the analyte’s internal vibrational energy.47 Therefore, the weakest bonds, such as labile phosphate bonds, are maintained. Ideally for peptides with PTMs, ETD can provide both the sequence information for peptide identification and generation of product ions that are diagnostic for the modification site(s).48
In the HCD approach, essentially the same collisional approach to fragmentation is used, but with higher activation energy and shorter activation time comparing the traditional ion trap CID. Higher energy for HCD still generates b- and y-type fragment ions at typical peptide bonds, but can also generate further fragmentation of peptide backbone, between the C-C bond, to form a-ions or smaller species.49 Without the low mass cut-off restriction and with high mass accuracy MS2 spectra, HCD has been successfully applied for de novo peptide sequencing, providing more informative ion series.49
As for PTMs studies, certain diagnostic ions specific for HCD can be recognized for PTMs identification, like the 80 Da ion for phosphopeptides or the 204 Da O-GlcNAc oxonium ion.50 The faster scan rate for HCD is expected to improve the identification coverage and accurate site localization for phosphoproteomics analysis.51. Today, a variety of hybrid mass spectrometers enable multiple types of MS/MS – allowing one to tailor the fragmentation method dynamically during a single experiment.52 The ability of HCD to produce ‘diagnostic’ product ions for a PTM has enabled a serial-fragmentation workflow in which an initial HCD MS/MS run generates fragment data that, if the appropriate diagnostic low mass ion for a given PTM is present will subsequently trigger an additional ETD fragmentation event in order to accurately ascertain sequence and site specificity of the PTM(s) of interest. As an elegant example, this dual fragmentation approach has been employed by Mayr and colleagues to interrogate secreted glycoproteins from in vitro endothelial cells53 and ECM proteins associated with atrial fibrillation in human cardiac tissue.54
Informatic considerations for PTM data analysis: What a basic or clinical cardiovascular scientist should know to evaluate proteomic PTM data
Large-scale, bioinformatic workflows are required to evaluate any data set and to gain new insight about changes in the proteome within a cell or tissue. The magnitude of data generated is the clear benefit but also the challenge of this approach for profiling proteoforms in biological systems. It can be daunting for scientists who don’t have a background in MS-based proteomics or informatics to review and evaluate the quality of the data put forth in these types of studies. Apart from the same basic concepts of experimental design and proper use of positive and negative controls, there are a handful of additional, generalizable concepts that can aid in demystifying the evaluation of proteomic PTM data quality. These include: (1) Understanding the quality of sequence identification(s); (2) Assessment for how peptides were matched to proteins in bottom-up workflows and/or gene products from sequence databases in either bottom-up or top-down MS; (3) Appropriate assurances that modifications are assigned to the correct amino acid and (4) Consideration of the assumptions made during data normalization and quantification. In the following paragraphs, each of these concepts will be briefly discussed. Since these categories apply to traditional, bottom-up workflows, we will then finish with a brief discussion of top-down workflows and their application to the study of PTMs in cardiovascular biology.
Peptide sequencing and protein matching
The biological importance of a presumed PTM is only as strong as the accuracy of its assignment to a given amino acid within a peptide or protein sequence and subsequent translated gene product. For bottom-up peptide sequence matching, each mode of MS data acquisition (e.g., DDA, DIA and targeted) uses slightly different assumptions, approaches, and assurances. In DDA experiments, peptide spectral matches are made by matching an observed fragment spectrum against a genomic sequence database using one of any number of available matching algorithms.55,56 The addition of PTM masses to this search process inflates the ‘search space’ and increases the risk of false positive matches. Similarly, modifications on lysines or arginines (e.g., methylation, acetylation, citrullination) can interfere with trypsin, causing missed cleavages that must also be accounted for informatically and can further increase search space and contribute to analytical processing demand as well as impact the false positive identification rate. To control for these false positives, preliminary matches should then be subject to an additional false discovery rate (FDR) assessment.57,58 Typically, a database of “decoy” sequences is generated by shuffling or reversing the sequences of the genomic database.59 The score distribution of these false, decoy sequences is used to assign the probability that a given peptide spectral match score of a target sequence is correct.
For DIA experiments, peptide sequences are assigned using a pre-existing peptide library that defines both the intact peptide mass, with modification in the case of PTMs, as well as some number of common fragment masses.31,60,61 The co-elution of these two mass sets (e.g., a whole peptide and its fragments) is a fundamental identifying feature. Multiple features of the co-eluting MS1 and MS2 mass sets (e.g., chromatographic behavior, signal-to-noise ratio, fragment intensity) are scored according and assembled into an overall metric that represents the quality of a potential peptide sequence match. These raw scores produced by the initial sequence matching algorithms should be modeled against scores of decoy datasets, and researchers should report the probability and/or false discovery rate values used for assigning cut offs.62
Peptide identification using Targeted Acquisition is, in general, a simpler case as the predefined analysis is limited to only a few proteins (normally <100). Importantly, for optimal accuracy, an isotopically-labeled, internal standard peptide is included for each analyte that serves as both an identification and quantitative reference.63 However, we encourage using internal standard peptides for all MS approaches for at least subsets of biologically important proteins.
In top-down proteomics, tandem MS/MS spectra tend to include larger fragments with higher charge states, and each fragment can be present in multiple isotope isomer forms. The ‘isotopomer envelopes’ of one fragment from the intact protein often overlap with one or more others, and thus deconvolution of these complex spectra to derive the most common form of a given peptide isotope species is required and performed by various algorithms.64–66 Once deconvoluted, the resulting mass lists can be assembled to predict the protein gene product that was selected for fragmentation during the MS1 step using a database search process roughly akin to that used to identify peptide sequences in bottom-up DDA analysis pipelines.67–69 As with bottom-up workflows, searching of the data against a randomized or reversed database in order to derive false discovery rate prediction of a given protein ID by this pipeline is paramount in order to provide confidence limits to the identifications produced from automated, high throughput workflows.70
In all cases, often the final critical step prior to functional analysis is to assign peptide sequences to a specific gene product (e.g., protein). While simple in theory, there is considerable complexity in this process that warrants brief discussion here. Peptide sequences do not always uniquely match to a single gene product, and many closely related genes (e.g., the actin and myosin gene families) have conserved sequences. Similarly, some post transcriptional processing generates splice isoforms of the same gene product that further complicate simple matching of sequence-to-protein. Top-down proteomics is uniquely suited to identify the relative abundances of sequence proteoforms in a given sample, since the protein species are directly observed intact during MS1, and not inferred by their pre-digested peptide components.70 For bottom-up approaches, there is a healthy debate and several methods available for matching peptides to gene products for protein-level quantification that can be reviewed elsewhere.71 In terms of PTMs, since the quantification step is often performed at the peptide level it is less critical to the quantitative conclusions that the peptide be matched to a single proteoform. Making functional inferences as to the meaning of a given PTM-difference between samples can be very difficult if the peptide sequence in question is shared between many different gene products. While there is no single way to best make the protein inferences in bottom-up proteomic workflows, researchers should be very clear as to the assumptions and choices used in this process (e.g., did the genomic sequence database contain isoforms or only canonical sequences, how were peptides matching multiple proteins handled, etc.).
Assurances regarding accurate amino acid assignment of a given PTM
The process of peptide spectral matching in all MS experiments will identify the presence of a given PTM(s) on a peptide sequence. However, if multiple residues within the same peptide are amenable to modification there are additional steps required to assess certainty in PTM-site localization. For example, the phosphorylated activation sequence for ERK1 (VADPDHDHTGFLTEYVATR) has three threonine’s and one tyrosine amenable to phosphorylation (underlined) while only two residues are critical for the protein’s activation, as shown in bold. In all three modes of acquisition, the ability to determine if an individual residue is modified depends on observing fragments that can distinguish one modified form of the peptide from its alternatives. For most methods, choosing the correct peptide fragment that contains the modified residues is required.63 For DIA, there are at least two bioinformatics tools that can determine the confidence of a PTM assignment72,73 while for DDA-experiments there are a handful of algorithms that score the confidence of site designation, including A-Score74 and luciphor75 which assign probabilities of correct site assignment to the identified PTM-peptides. All of these algorithms help increase the confidence ofassigning which amino acid is modified. Top-down workflows utilize a conceptually similar approach, first identifying mass shifts between precursor ions (whole proteins in this case) and then using the presence or absence of a given PTM in various fragments ions from the MS/MS spectra to home in on the amino acid localization of the modification(s) present. This process is predicted from data analysis pipelines, but often verified manually in top-down proteomics and thus for review purposes, representation of the spectral data used to infer site localization may be important in order to validate particularly novel or biologically critical PTM events for the subsequent conclusions to be drawn by the researchers.76
Data normalization and quantification
A final set of considerations when evaluating proteomic PTM data involve how technical variability across samples was evaluated or normalized for and how the quantification of the PTM was done.77 Many PTM workflows, both for bottom-up and top-down approaches, involve biochemical enrichment steps prior to MS analysis (see next section), and these procedures introduce technical errors that can muddy the detection of biological differences between samples.78 The use of isotopic labeling strategies like SILAC or peptide tagging can help to mitigate technical variation by enabling samples to be mixed prior to the enrichment workflow.79 Alternatively, isotopically labeled control peptides or proteins can be spiked into samples to provide an index and normalization factor to account for sample preparation variance. At the very least, assurances that the same amount of sample input was used at the outset of the preparation protocol as well as a calculation of the technical variance inherent to the preparation protocol (e.g., triplicate runs of one sample to determine technical %CV) should be performed and reported. In bottom-up proteomics, the question of whether to quantify the full PTM-peptide species or each of the modified sites individually is an important consideration. Quantifying by site is a simpler approach, but ignores the potential combinatorial relationships between multiple modification sites on the same protein. In bottom-up proteomics, inferring combinatorial relationships between PTM sites across different peptides from the same protein is quite challenging whereas this is a defining strength of top-down approaches.
Finally, whether or not the PTM-peptide should be normalized to the total non-modified protein is a question to consider. For many PTM workflows, biochemical enrichment remains a prerequisite to obtain the sensitivity necessary for detecting and quantifying these typically lower abundant species.80,81 This process of enrichment uncouples the unmodified forms of the protein from the modified form, and this challenge may be a crippling limitation preventing the normalization of many PTM species identified in a discovery workflow. We recommend that the biological goal of the experiment be considered as well. If the goal is to know the signaling status in terms of the PTM-forms present in a system, then knowing how much of the total protein is present is not as important because the active species is represented by the specific PTM-forms. If, however, the goal is to monitor the signaling impact of a perturbation, specifically on increasing or decreasing the PTM-state of the analytes, then knowing whether a change in a given PTM is due to a chemical modification rather than protein synthesis or degradation change is key. In this case, it is mandatory to attempt to quantify total protein alongside PTM-changes.
There are many factors to consider when evaluating the meaningfulness of PTM-proteomic data. Our intention here was to simplify these considerations into key concepts to help basic and clinical cardiovascular scientists who are not experts at MS or proteomics to critically evaluate the data and ask appropriate questions of methodological assumptions.
PTM-and Cardiovascular Physiology
PTMs have been found to exhibit a wide array of regulatory mechanisms in both physiological and pathological scenarios. Interestingly, a large number of PTM are enriched and affected by inherited and somatic disease mutations. These disease-associated mutations change polarity, charge, and hydrophobicity of the wild-type amino acids in PTM sites making PTM variation more likely deleterious.82,83 There are a number of cases in which mutations of the post-translational target sites are directly involved in disease. For example, the Reimand group performed systematic studies of somatic cancer mutations affecting protein phosphorylation. Particularly, they were able to identified hotspots of disease mutation in PTM regions and disordered sequence.84 The group highlighted 152 genes, called PTM-associated disease, where disease mutations, including cardiovascular (LMNA, MYH7), cystic fibrosis (CFTR), diabetes (HNF4A, IRS1) migraine (ATP1A2) and cancer genes significantly accumulate in PTM regions. This study published list of PTM-associated disease genes that potentially, can serve as a starting point for investigating PTM mechanisms in many diseases.82 In several other studies somatic mutation was shown to affect multiple PTM target sites, including methylation, ubiquitination, and O-linked glycosylation, implicating all three modifications in disease.85,86 The discovery and characterization of more PTMs in a variety of physiological and pathological conditions generates valuable new knowledge, druggable targets, and biomarkers. Here, we will discuss a well-studied PTM, phosphorylation, as well as highlight some of the PTMs that have garnered less attention in the cardiovascular field but have no less potential for significant physiological insight. A summary of some of these modifications and the number of proteins they have been detected on that are related to cardiac function and disease is presented in Figure 3.
Figure 3.
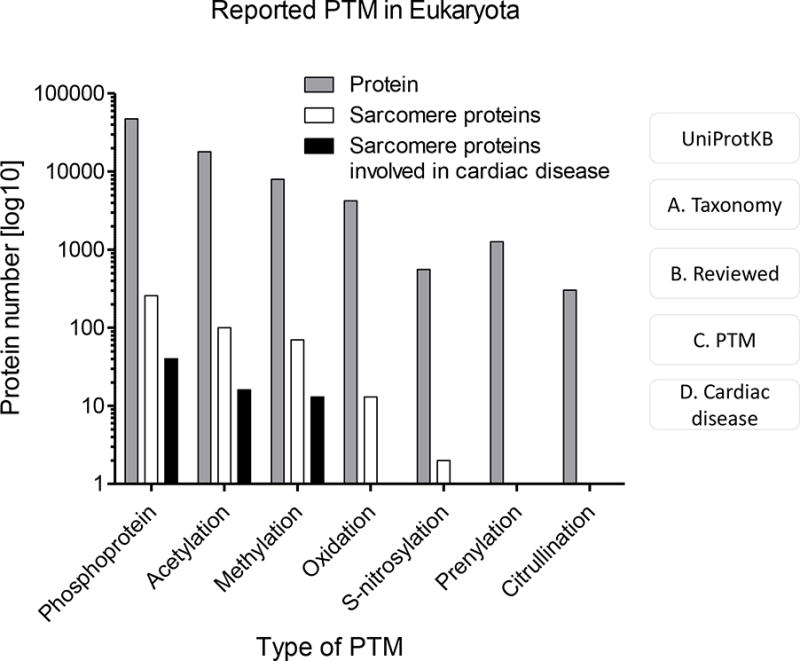
Number of proteins reported to have PTMs in Eukaryota. Summary of the number of proteins modified with the indicated PTMs in Eukaryota (blue bar), the sarcomere (grey bar) or cardiac disease (orange bar) (http://www.uniprot.org). The high number of known PTM proteins is in stark contrast to the limited knowledge about their involvement in disease. PTM: Post-translational modification. Accessing the human PTM proteome from the UniProt web site (http://www.uniprot.org). A. Select taxonomy on the left, for example, ‘Eukaryota’. B. Select ‘Reviewed’ in the ‘Map to’ section on the left. C. Use the search box to specify the PTM type. D. Additional terms can be included in the search ‘and cardiac disease’.
Phosphorylation is arguably the most popular of PTMs analyzed in cardiovascular research, and not without good cause. Phosphorylation is one of the best understood mechanisms for intracellular signaling, its presence drives the activation, inhibition, stabilization or degradation of kinases, enzymes, and other proteins, and it regulates myofilament stiffness, relaxation rate, and contractility.87 For example, phospholamban (PLB) can be phosphorylated at Ser16 by PKA following beta-adrenergic stimulation. This phosphorylation event relieves PLB’s inhibitory effect on sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA) enhancing the SERCA dependent uptake of Ca2+ to the SR.88-89 This modulation in calcium availability results in greater myocardial relaxation and contractility to improve cardiac output. Changes in phosphorylation status have also been linked to cardiovascular disease. Decreased phosphorylation of Troponin I (TnI) at PKA sites (Ser22, Ser 23) and an increased phosphorylation of TnI by PKC (Ser41, Ser43, and Thr142) were shown to be involved in heart failure.90
There are also numerous examples of bottom-up phosphoproteomic studies in cardiovascular physiology and disease. All of these studies utilize a biochemical enrichment strategy to deplete the bulk of non-phosphorylated peptides, reducing sample complexity to devote more MS time to the analyte of interest, and generally improve the signal-to-noise of the PTM-peptide relative to the global proteome.78,91 Phospho-enrichment approaches include affinity-based methods that utilize antibodies generated to generalized modified epitopes (e.g., pan-phosphotyrosine antibodies92) or other chemicals with high affinity for the chemical properties of a given PTM (e.g., metal affinity of TiO2 for binding phosphate groups in acid buffers93,94) although other specific enrichment approaches are discusses below. These studies targeting phosphorylation have revealed a variety of insight related to cardiovascular biology and disease. They include: the relative NO-independency of PDE9A on cGMP signaling by cardiomyocytes;95 traced the beta adrenergic signaling pathway in mouse heart tissue;96 described the signaling response of vascular smooth muscle cells in response to stretch;97 demonstrated novel components and potential therapeutic targets controlling platelet aggregation in response to ADP;98 outlined the early phosphorylation events associated with ischemia-reperfusion injury in the heart;99 profiled the phosphorylation sites present on β-adrenergic receptors in mouse;100 distinguished ischemic from non-ischemic end-stage heart failure in terms of phosphorylated protein signatures;101 identified the mitophagy regulator dynamin related protein 1 (DRP1) as a key mediator of mouse hypertrophic cardiomyopathy.102 Complementary examples from top-down proteomics have identified multiple phosphorylation states present on cardiac Troponin I that were capable of distinguishing between different types103 and clinical stages104 of heart failure; and profiled differences in Myosin Light Chain isoforms and phosphorylation states between atrial and ventricular cardiac regions;105 that truncation of Myosin Binding Protein C results in its altered phosphorylation.106 All this focused investigation has led to a wealth of cardiac specific phospho-site information and the potential for tremendous insight into the regulation of these processes.
Beyond the well-characterized PTM there lies a vast, largely unexplored landscape of other, less appreciated PTMs. One of the critical barriers to this investigation is technical. Without specific and robust tools to detect these other modifications, their study will be impeded and will lead to insight delay. We have chosen to focus on three emerging PTMs: S-nitrosylation, prenylation and citrullination to provide an overview of the workflows currently used to detect them and some examples of the early insights for their importance in cardiovascular physiology.
S-nitrosylation (also known as S-nitrosation, SNO) is a redox-based modification where a nitric oxide group (NO) is added to the thiol moiety of a cysteine residue.107 Although it is a covalent modification, it can be highly labile making it very difficult to study using traditional techniques like western blot and mass spectrometry. In 2001, the Synder group introduced the biotin switch technique.108,109 At the time, this revolutionary new method selectively replaced the SNO modifications with a stable biotin group. This approach had two clear advantages: the labile SNO group was replaced with a more stable modification and the biotin moiety provided a means for selective enrichment. Since this initial innovation, numerous iterations of the technique have been introduced. These tweaks to the protocol have focused on improving mapping the individual cysteine residues that are modified, quantifying the modified site between different samples and examining the extent of SNO occupancy of each modified cysteine under a given condition.110–118 More thorough reviews of the techniques for detecting SNO modifications can be found here.119–121 With the advent of all these technical advancements, the number of characterized SNO sites has increased significantly to over 6500 independent observations in human, mouse and rat tissues representing over 4000 unique sites of modification.122
The energized focus resulting from the technical innovation has resulted some significant insights for the cardiovascular impact of these modifications.123 For example, SNO modified proteins have been found to modulate the Ca2+ handling response in β-adrenergic receptor (β-AR) signaling. β-AR signaling is a critical component to modulate the caridomyocyte’s calcium homeostasis however chronic stimulation can lead to the development of hypertrophy.124 Irie et al. (2015) reported that S-nitrosylation of phospholamban at Cys 36 and 41 could suppress its inhibitory effect on SERCA, resulting in an increase of Ca2+ to the sarcoplasmic reticulum.125 In the same study, the authors also found that S-nitrosylation of troponin C at Cys 84 decreased myocardial sensitivity to Ca2+. They also observed a general decrease in SNO during the development of hypertrophy. These insights have led to a greater appreciation for the synergy between SNO and phosphorylation in this system.125 Another SNO-modified protein is the glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). SNO modification has been previously been found to decrease its enzymatic activity however it has more recently been reported that GAPDH may have a role in the S-nitrosylation of mitochondrial proteins.126,127 Mitochondrial proteins in the heart are known to be S-nitrosylated although the source of the nitric oxide was not clear. Kohr et al. (2014) reported that SNO-modified GAPDH could translocate to the inner mitochondria and to transfer its NO group via transnitrosylation.127 The authors also observed that overexpression of GAPDH resulted in elevated SNO-modification of several mitochondrial proteins. The examples described above, and many others, are all significant insights that have been facilitated by the introduction and continued refinement of a strategy for broad based screening of a PTM that was challenging to study by traditional means.
In contrast to SNO, some PTMs have remained less studied for lack of an effective large-scale identification strategy in a biologically relevant context. A recent example of a PTM with an emerging broad-based approach for identification is prenylation. Prenylation is the covalent addition of a lipid prenyl group to a cysteine residue near the C-terminus of a protein. Prenylation can take two forms; the addition of a farnesyl group (farnesylation) by a farnesyl transferase or one of two possible geranylgerany groups (geranylgeraylation) catalyzed by two different transferases (GGTase I or II). Both involve the addition of an isoprenoid to a C-terminal Cys; farnsylation is composed of 15 carbons (204 Da) and the geranylgernayl group is 20 carbons (272 Da).128 These modifications increase the protein’s hydrophobicity and improve association with the cell membrane. For example, prenylation is required for Ras family GTPases and G-proteins membrane localization.129,130 Modulation of protein prenylation has also been associated with cardiovascular regulation. Changes in prenylation have been linked to cardiac hypertrophy via an mTORC1 mechanism leading to the development of chronic heart failure.131 Conversely, Spindle et al. (2012) found that statin treatment increases lifespan and improves cardiac health in Drosophila by decreasing specific protein prenylation.132 In both of these examples, no broad-based mapping of the changes in protein prenylation has been performed to determine their role in the specific deleterious or improved outcome.
One of the challenges facing the field is the difficulty in reliably identifying sites and discriminating between the two forms. In the past, study of prenylated proteins was performed on an individual proteins basis using primarily radiographic techniques which could take 2-months to visualize. Mass spectrometry analysis has also been challenging. The addition of a lipid group increases the hydrophobicity and reduces the ionization efficiency of a peptide. It has been reported that farnsylation can be observed by a 204 Da neutral loss during CID however this fragmentation has not been robust.133,134 Given these limitations, large-scale, comprehensive proteomic identification of this modification has been difficult indicating there is an opportunity for a technological innovation to significantly advance the field. One possibility could be seen in a recently proof of concept study by Bhawal et al. (2015).134 In their study the authors introduce an epoxidation with mCPBA step to derivatize prenylated peptides prior to MS analysis. They report that the addition of an expoy to the double bonds of the farnsyl or geranylgerany prenyl groups offers several advantages. First, it significantly improves the neutral loss fragmentation in CID. The authors and others found that mono-oxidation of a thio-ether bond resulted in the loss of a specific RSOH fragments.134,135 By evaluating the neutral loss product, it is possible to discriminate between a farnsyl and geranylgerany because of the 68 Da difference between them. The addition of the epoxy groups also significantly reduces the hydrophobicity of the prenylated peptides. This improves peptide solubility and ionization facilitation better MS identifications. Analyzing underivatized lipid modified peptides using a reverse phase LC-MS required very long and high organic gradients. In a sample set of peptides, epoxidation resulted in significantly reduced retention times allowing for shorter separations. The final advantage of this approach is the possibility for an enrichment strategy. An azide can be added to expoy modified prenyl groups; this would allow for a click-chemistry addition of a biotin-alkyne group for specific capture of the prenylated peptides or proteins. While this approach is still in an early phase, this type of advance will lead to greater access to the PTM in vivo which will ultimately lead to a better understanding of its role in health and disease.
Another modification with important role in a variety of biological processes that has been difficult to study is citrullination. This modification results from the deamination of the guanidino group of arginine side chains to form an ureido group and the non-standard amino acid citrulline. It is performed by a small family of enzymes known as peptidylarginine deiminase (PAD) 1, 2, 3, 4, and 6 on the basis of their cDNA nucleotide sequence.136 Detection methods for PAD activity and citrullination are related, because conversion of arginine to citrulline in protein sequence is used as a read-out for PAD activity. There are several assays available for the detection of PAD activity, such as colorimetric assay,137 antibody based assay (with anti-modified citrulline antibodies)138 or even direct MS analysis139 however, none of them is specific, sensitive and efficient enough to truly detect and identify modified residue.140 The main challenge in MS detection of citrullinated proteins/peptides is exactly the same mass increase (0.9802Da) as other PTM, deamidation. Those two modifications cannot be distinguished based on mass difference alone and proper characterization of the exact citrullination site in the MS/MS spectrum, or additional information, is therefore required for peptides containing both potential deamidation and citrullination sites. Unfortunately, there is not currently an algorithm that will prevent false positives assignment based on MS/MS data without manual, time-consuming verification.
Keeping in mind technical difficulties, this modification increases the protein’s hydrophobicity, creates neoantigens and confers immunostimulatory properties.141 For example, the autoimmune response is linked to the pathogenesis of several diseases including rheumatoid arthritis, multiple sclerosis and psoriasis.142–145 It has been also reported that the levels of citrullinated proteins and PAD4 were elevated in patients with various cancers, such as oesophageal cancer, breast cancer and lung adenocarcinoma.146 A more recent work by Geraldino-Pardilla and co-workers reported that anti-citrullinated proteins were detected and associated with left ventricular structure and function in rheumatoid arthritis.147 Fert-Bober et al. identified peptide regions of citrullination in major sarcomeric proteins indicating that this PTM may be functionally significant.148 Studies with knockout mice in an experimental model of cardiac fibrosis, cardiac pressure overload, support important role of PAD4-in neutrophil extracellular trap (NET) formation (Netosis) in aged mice.149 In this study the author reported reduction in fibrosis in the hearts and lungs of aged PAD4−/− mice compared with wild-type mice. Furthermore, they found an increase in left ventricular interstitial collagen deposition and a decline in systolic and diastolic function only in wild-type mice, and not in PAD4−/− mice.150 Contrariwise, Sokolove at al. (2013) found that citrullinated proteins are prevalent within the atherosclerotic plaque, and certain anti-citrullinated protein antibodies are associated with atherosclerotic burden.151,152 In both of these examples, the observations suggested that targeting of citrullinated epitopes or PAD enzyme itself could provide a mechanism for accelerated atherosclerosis.
These finding offers the potential for extensive further investigations attempting to sort out the complex modifications in health and disease.
There are many additional chemical PTMs that may ultimately prove of crucial diagnostic or therapeutic interest for a given pathophysiological event in cardiovascular disease. These include SUMOylation153–155, NEDDylation156,157, ubiquitinylation158, palmitoylation and other lipid modifications in addition to prenylation159, o-glycNAcylation,160 and N- and O-linked glycosylation161,162 As described for the examples above, improving the biochemical approaches for enriching or detecting PTMs, along with improving the ease and accessibility (e.g., democratizing) of MS workflows for their detection and quantification by all cardiovascular scientists will be critical before integration of proteoform knowledge into precision physiology can be truly implemented and translated to clinically relevant discoveries.
Conclusion and moving forward
The concept of democratization of MS means increasing the ability of the broader scientific community to use proteomic data, in part through the reducing the barriers to MS and data analysis software.163 Proteomic analysis of human tissues, has led to the realization that there are subsets of proteins that are closely involved in conserved pathophysiological processes seen across tissue and cell types. To take advantage of this concept we have proposed that one strategy to democratize proteomics is to develop quantitative targeted MS-based assays to these pathways. These assays provide quantification equal to or better than ELISAs and western blots which are commonly used in basic science. Like their antibody-dependent counterparts, however, targeted assays are able to not only accurately quantify a specific protein abundance, but they can also determine the site-specific ratio (stoichiometry) of a particular modified residue due to a SNP or chemical post-translational modification(s) and they can be multiplexed to analyze between tens- and hundreds of peptide analytes within a few (e.g., < 1-5) micrograms of a given sample. Thus, the potential is to be able to measure many proteoforms of interest in a straightforward, standardized, and quantitative method that is reproducible across many laboratories. Today, MS-based protein measurement is perceived as difficult and being owned by experts in academics or in MS-based laboratories.95,164 However, based on the existing proteomic technology, automation and data software, this is doable.
Once technical complexity is resolved, one other barrier to adoption is menu. It is not clear what proteins or pathways should be selected for producing these targeted assays. NCI has targeted a number of cancer pathways165,166 and as well the community has produced standards and white papers for application of targeted assays.33 But, knowing which proteins and pathways to target in other disease areas or if there are conserved (and thus, high valued targets) across diseases is not known. To address this challenge is the essence of initiative by the human proteome organization (HUPO, https://www.hupo.org/B/D-HPP) to identify and then build assays for the most popular proteins used in different diseases. There is currently an ever-growing list of high value proteins targets at https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/proteinListSelector, which will help the individual fields in CVD and related diseases.
Popular proteins are defined as those proteins that have the highest citation number based on PubMed. These assays can be used for clinical translation as well for basic science essentially supplementing ELISA and western blots. The first paper on this concept concentrated on heart167,168 but currently there are popular proteins for 6 organ systems169 and also liver170 with more to come. This brings up the second aspect required for democratization of proteomics. It is important that the broader community be informed about the proteoforms that can for their favorite protein or proteins and the tools available to access this information. For example, Table 2 lists three popular proteins that have been found to be modified by the less popularly studied PTM, citrullination. Here, we have highlighted known sites of modification that are also the sites of SNPs linked to CVD. Similar disruption of critical function is apt to occur whenever a genomic event occurs at the site, or in close enough proximity for a biochemical impact, of a regulatory PTM. Thus, combining well characterized information about a diversity of PTMs with well annotated knowledge of sequence variants and their association with various diseases enables the most comprehensive linkage between sequence variation and its functional impact on all possible gene products. True ‘precision profiling’ of any individuals’ multi-omic risk or treatment response profile will require a comprehensive catalogue of possible PTM sites and their regulatory impact on protein structure and function. A common route for integrated -omic queries such as this is to use Uniprot or NeXtProt. However, new tools are continually being developed to help the user query the rapidly changing landscape of proteoforms to discover and target new aspects of the regulatory potential of their favorite protein or pathway.171–174
Table 2.
Representative citrullinated proteins that have a SNP at the originating Arg residue
| UniProt ID | Cit. (Uniprot) | Natural disease causing variant | Cit (published) | Citrullinated peptide |
|---|---|---|---|---|
|
P02649 APOE |
no | • Hyperlipoproteinemia, R to C or S (residues 154,160,163,176) | yes | SELEEQLTPVAEET
ARLSK SWFEPLVEDMQ QWAGLVEK LEEQAQQIRLQAEAFQA LK |
|
P45379 TNNT2 |
no | • Cardiomyopathy, familial restrictive R to Q,W,L,C, or P(residues102,104,140,288) • Cardiomyopathy, dilated R to W or L (residues 141,151,215) |
yes | VLAIDHLNEDQL
EK YEINVL NRINDNQK |
|
P19429 TNNI3 |
no | • Cardiomyopathy, familial hypertropnic R to Q,G,P, or H (residues 141,145,162,186,204) • Cardiomyopathy, familial restrictive • R to W, or H (residues145,192) |
yes | ENREVGDW
K ESLDL AHLK NIDALSGMEG K VDEE YDIEAK |
Citrullination [Cit or ] = deimidation of the guanidino group of arginine side chains to form an ureido group and the non-standard amino acid citrulline. The modified (Cit) and unmodifed (R) peptides were observed by MS148
It is our goal as proteomics researchers to continue to reduce the technical barriers limiting the study of these complicated biochemical analytes across the greater scientific community. Considerably, more work is required to democratize proteomics including the need for open database sharing and integration alongside with bioinformatics tool for automatically validation of PTMs assignments, particularly from large-scale proteomic studies. These key processes will allow for the establishment of comprehensive databases; discover additional PTMs, chemical derivatives, and sample-specific amino acid variants. In conclusion, proteoforms highly likely exist for every translated gene product. Studying the chemically-derived proteoforms present in a physiological system is a technically challenging process often requiring biochemical enrichment, sophisticated mass spectrometry, and bioinformatics tools. This is an exciting time and it is a time to ensure that all aspect of proteomics together with other “omics” platform will also play a key role in understanding the extensive chemical diversity of biology.
Figure 4.
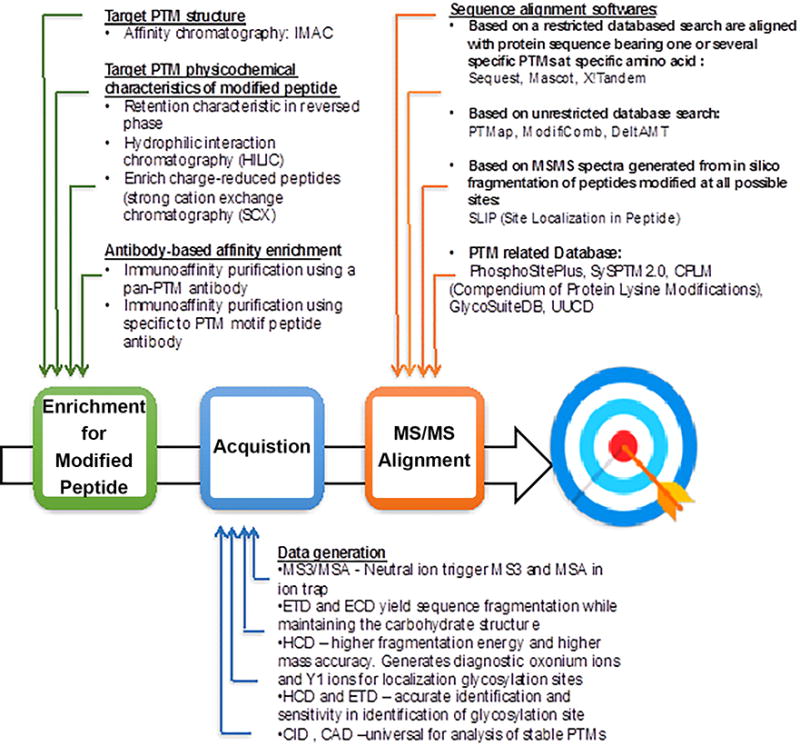
Overview of PTM analysis and challenges. Schematic representation of the well-established enrichment and mapping methods for specific PTMs workflows.
PTM proteins and peptides can be enriched using a single- or multiple-step strategie. The final peptide mixture is selected and further fragmented on MS-MS to obtain informative ions that allow their identification. All fragmentations methods have advantages and disadvantages. Therefore different combinations of fragmentation techniques and mass analyzers can be used in a single analysis. Subsequently, MS/MS spectra (raw data) are processed with commercially available database search tools to identify and quantify PTM residue.
Acknowledgments
Sources of Funding
This work was supported by 1K99HL128787 – 01A1 (SP), and Barbara Streisand Women’s Heart Center (J.V.E.), The Smidt Heart Institute at Cedars Sinai Medical Center (J.V.E) and The Erika Glazer Endowed Chair in Women’s Heart Health (J.V.E).
Footnotes
Disclosures
None.
Nonstandard Abbreviation and Acronyms
None
References
- 1.Morita H. Human genomics in cardiovascular medicine: implications and perspectives. Circ J. 2013;77:876–85. doi: 10.1253/circj.cj-13-0126. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, Ge Y, Gunawardena J, Hendrickson RC, Hergenrother PJ, Huber CG, Ivanov AR, Jensen ON, Jewett MC, Kelleher NL, Kiessling LL, Krogan NJ, Larsen MR, Loo JA, Ogorzalek Loo RR, Lundberg E, MacCoss MJ, Mallick P, Mootha VK, Mrksich M, Muir TW, Patrie SM, Pesavento JJ, Pitteri SJ, Rodriguez H, Saghatelian A, Sandoval W, Schlüter H, Sechi S, Slavoff SA, Smith LM, Snyder MP, Thomas PM, Uhlén M, Van Eyk JE, Vidal M, Walt DR, White FM, Williams ER, Wohlschlager T, Wysocki VH, Yates NA, Young NL, Zhang B. How many human proteoforms are there? Nat Chem Biol. 2018;14:206–214. doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazur MT, Cardasis HL, Spellman DS, Liaw A, Yates NA, Hendrickson RC. Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc Natl Acad Sci U S A. 2010;107:7728–33. doi: 10.1073/pnas.0910776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzacasa B, Morini E, Pucci S, Murdocca M, Novelli G, Amati F. LOX-1 and Its Splice Variants: A New Challenge for Atherosclerosis and Cancer-Targeted Therapies. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rexiati M, Sun M, Guo W. Muscle-Specific Mis-Splicing and Heart Disease Exemplified by RBM20. Genes (Basel) 2018;9 doi: 10.3390/genes9010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kooij V, Venkatraman V, Kirk JA, Ubaida-Mohien C, Graham DR, Faber MJ, Van Eyk JE. Identification of cardiac myofilament protein isoforms using multiple mass spectrometry based approaches. Proteomics Clin Appl. 2014;8:578–589. doi: 10.1002/prca.201400039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng Y, Gregorich ZR, Valeja SG, Zhang H, Cai W, Chen Y-C, Guner H, Chen AJ, Schwahn DJ, Hacker TA, Liu X, Ge Y. Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction. Mol Cell Proteomics. 2014;13:2752–64. doi: 10.1074/mcp.M114.040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen JV, Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 2013;12:3444–52. doi: 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh CT. Post-translational Modifications of Proteins: Expanding Nature’s Inventory. 2005 [Google Scholar]
- 10.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okada M. Post-translational isoprenylation of tryptophan. Biosci Biotechnol Biochem. 2011;75:1413–7. doi: 10.1271/bbb.110087. [DOI] [PubMed] [Google Scholar]
- 12.Nolan EM, Walsh CT. Investigations of the MceIJ-catalyzed posttranslational modification of the microcin E492 C-terminus: linkage of ribosomal and nonribosomal peptides to form “trojan horse” antibiotics. Biochemistry. 2008;47:9289–99. doi: 10.1021/bi800826j. [DOI] [PubMed] [Google Scholar]
- 13.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep. 2011;1 doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–8. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Lothrop AP, Torres MP, Fuchs SM. Deciphering post-translational modification codes. FEBS Lett. 2013;587:1247–57. doi: 10.1016/j.febslet.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng M, Scholten A, Heck AJR, van Breukelen B. Identification of enriched PTM crosstalk motifs from large-scale experimental data sets. J Proteome Res. 2014;13:249–59. doi: 10.1021/pr4005579. [DOI] [PubMed] [Google Scholar]
- 17.Venne AS, Kollipara L, Zahedi RP. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 2014;14:513–24. doi: 10.1002/pmic.201300344. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz D, Chou MF, Church GM. Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol Cell Proteomics. 2009;8:365–79. doi: 10.1074/mcp.M800332-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan G, Walther D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput Biol. 2015;11:e1004049. doi: 10.1371/journal.pcbi.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S-B, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–7. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen B, Lietz CB, Li L. Coupling matrix-assisted ionization with high resolution mass spectrometry and electron transfer dissociation to characterize intact proteins and post-translational modifications. Anal Bioanal Chem. 2018;410:1007–1017. doi: 10.1007/s00216-017-0611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 23.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–65. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Fonslow BR, Shan B, Baek M-C, Yates JR. Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113:2343–94. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riley NM, Coon JJ. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal Chem. 2016;88:74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geromanos SJ, Vissers JPC, Silva JC, Dorschel CA, Li G-Z, Gorenstein MV, Bateman RH, Langridge JI. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics. 2009;9:1683–95. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 27.Matafora V, Corno A, Ciliberto A, Bachi A. Missing Value Monitoring Enhances the Robustness in Proteomics Quantitation. J Proteome Res. 2017;16:1719–1727. doi: 10.1021/acs.jproteome.6b01056. [DOI] [PubMed] [Google Scholar]
- 28.Venable JD, Dong M-Q, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 29.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruderer R, Bernhardt OM, Gandhi T, Xuan Y, Sondermann J, Schmidt M, Gomez-Varela D, Reiter L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol Cell Proteomics. 2017;16:2296–2309. doi: 10.1074/mcp.RA117.000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426–41. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Hüttenhain R, Collins B, Aebersold R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Rev Mol Diagn. 2013;13:811–25. doi: 10.1586/14737159.2013.845089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Hüttenhain R, Koomen JM, Liebler DC, Liu T, MacLean B, Mani DR, Mansfield E, Neubert H, Paulovich AG, Reiter L, Vitek O, Aebersold R, Anderson L, Bethem R, Blonder J, Boja E, Botelho J, Boyne M, Bradshaw RA, Burlingame AL, Chan D, Keshishian H, Kuhn E, Kinsinger C, Lee JSH, Lee S-W, Moritz R, Oses-Prieto J, Rifai N, Ritchie J, Rodriguez H, Srinivas PR, Townsend RR, Van Eyk J, Whiteley G, Wiita A, Weintraub S. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13:907–17. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS, Canterbury JD, Marsh DM, Kellmann M, Zabrouskov V, Wu CC, MacCoss MJ. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10:744–6. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat Methods. 2013;10:1246–53. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 36.Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Müller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics. 2015;15:964–80. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- 37.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham A-JL, Keshishian H, Hall SC, Allen S, Blackman RK, Borchers CH, Buck C, Cardasis HL, Cusack MP, Dodder NG, Gibson BW, Held JM, Hiltke T, Jackson A, Johansen EB, Kinsinger CR, Li J, Mesri M, Neubert TA, Niles RK, Pulsipher TC, Ransohoff D, Rodriguez H, Rudnick PA, Smith D, Tabb DL, Tegeler TJ, Variyath AM, Vega-Montoto LJ, Wahlander A, Waldemarson S, Wang M, Whiteaker JR, Zhao L, Anderson NL, Fisher SJ, Liebler DC, Paulovich AG, Regnier FE, Tempst P, Carr SA. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–41. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52:3797–806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11:1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai W, Tucholski TM, Gregorich ZR, Ge Y. Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev Proteomics. 2016;13:717–30. doi: 10.1080/14789450.2016.1209414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sidoli S, Garcia BA. Middle-down proteomics: a still unexploited resource for chromatin biology. Expert Rev Proteomics. 2017;14:617–626. doi: 10.1080/14789450.2017.1345632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, Ge Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ Cardiovasc Genet. 2011;4:711. doi: 10.1161/CIRCGENETICS.110.957829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wells JM, McLuckey SA. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 2005;402:148–85. doi: 10.1016/S0076-6879(05)02005-7. [DOI] [PubMed] [Google Scholar]
- 45.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJR, Mohammed S. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10:2377–88. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 46.Xie B, Sharp JS. Relative Quantification of Sites of Peptide and Protein Modification Using Size Exclusion Chromatography Coupled with Electron Transfer Dissociation. J Am Soc Mass Spectrom. 2016;27:1322–7. doi: 10.1007/s13361-016-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones AW, Cooper HJ. Probing the mechanisms of electron capture dissociation mass spectrometry with nitrated peptides. Phys Chem Chem Phys. 2010;12:13394–9. doi: 10.1039/c0cp00623h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarbu M, Ghiulai RM, Zamfir AD. Recent developments and applications of electron transfer dissociation mass spectrometry in proteomics. Amino Acids. 2014;46:1625–34. doi: 10.1007/s00726-014-1726-y. [DOI] [PubMed] [Google Scholar]
- 49.Chi H, Sun R-X, Yang B, Song C-Q, Wang L-H, Liu C, Fu Y, Yuan Z-F, Wang H-P, He S-M, Dong M-Q. pNovo: de novo peptide sequencing and identification using HCD spectra. J Proteome Res. 2010;9:2713–24. doi: 10.1021/pr100182k. [DOI] [PubMed] [Google Scholar]
- 50.Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics. 2014;11:8. doi: 10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jedrychowski MP, Huttlin EL, Haas W, Sowa ME, Rad R, Gygi SP. Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics. 2011;10:M111.009910. doi: 10.1074/mcp.M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guthals A, Clauser KR, Frank AM, Bandeira N. Sequencing-grade de novo analysis of MS/MS triplets (CID/HCD/ETD) from overlapping peptides. J Proteome Res. 2013;12:2846–57. doi: 10.1021/pr400173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol Cell Proteomics. 2013;12:956–78. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barallobre-Barreiro J, Gupta SK, Zoccarato A, Kitazume-Taneike R, Fava M, Yin X, Werner T, Hirt MN, Zampetaki A, Viviano A, Chong M, Bern M, Kourliouros A, Domenech N, Willeit P, Shah AM, Jahangiri M, Schaefer L, Fischer JW, Iozzo RV, Viner R, Thum T, Heineke J, Kichler A, Otsu K, Mayr M. Glycoproteomics Reveals Decorin Peptides With Anti-Myostatin Activity in Human Atrial Fibrillation. Circulation. 2016;134:817–32. doi: 10.1161/CIRCULATIONAHA.115.016423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marcotte EM. How do shotgun proteomics algorithms identify proteins? Nat Biotechnol. 2007;25:755–7. doi: 10.1038/nbt0707-755. [DOI] [PubMed] [Google Scholar]
- 56.Verheggen K, Martens L, Berven FS, Barsnes H, Vaudel M. Database Search Engines: Paradigms, Challenges and Solutions. Adv Exp Med Biol. 2016;919:147–156. doi: 10.1007/978-3-319-41448-5_6. [DOI] [PubMed] [Google Scholar]
- 57.Reiter L, Claassen M, Schrimpf SP, Jovanovic M, Schmidt A, Buhmann JM, Hengartner MO, Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–17. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J Proteome Res. 2008;7:47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- 59.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parker SJ, Venkatraman V, Van Eyk JE. Effect of peptide assay library size and composition in targeted data-independent acquisition-MS analyses. Proteomics. 2016;16:2221–37. doi: 10.1002/pmic.201600007. [DOI] [PubMed] [Google Scholar]
- 61.Ting YS, Egertson JD, Bollinger JG, Searle BC, Payne SH, Noble WS, MacCoss MJ. PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat Methods. 2017;14:903–908. doi: 10.1038/nmeth.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teleman J, Röst HL, Rosenberger G, Schmitt U, Malmström L, Malmström J, Levander F. DIANA–algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics. 2015;31:555–62. doi: 10.1093/bioinformatics/btu686. [DOI] [PubMed] [Google Scholar]
- 63.Abbatiello SE, Mani DR, Keshishian H, Carr SA. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 2010;56:291–305. doi: 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11:320–32. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 65.Liu X, Inbar Y, Dorrestein PC, Wynne C, Edwards N, Souda P, Whitelegge JP, Bafna V, Pevzner PA. Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Mol Cell Proteomics. 2010;9:2772–82. doi: 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87:4370–6. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frank AM, Pesavento JJ, Mizzen CA, Kelleher NL, Pevzner PA. Interpreting top-down mass spectra using spectral alignment. Anal Chem. 2008;80:2499–505. doi: 10.1021/ac702324u. [DOI] [PubMed] [Google Scholar]
- 68.Zamdborg L, LeDuc RD, Glowacz KJ, Kim Y-B, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35:W701–6. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Sirotkin Y, Shen Y, Anderson G, Tsai YS, Ting YS, Goodlett DR, Smith RD, Bafna V, Pevzner PA. Protein identification using top-down. Mol Cell Proteomics. 2012;11:M111.008524. doi: 10.1074/mcp.M111.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–8. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005;4:1419–40. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 72.Rosenberger G, Liu Y, Röst HL, Ludwig C, Buil A, Bensimon A, Soste M, Spector TD, Dermitzakis ET, Collins BC, Malmström L, Aebersold R. Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat Biotechnol. 2017;35:781–788. doi: 10.1038/nbt.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keller A, Bader SL, Kusebauch U, Shteynberg D, Hood L, Moritz RL. Opening a SWATH Window on Posttranslational Modifications: Automated Pursuit of Modified Peptides. Mol Cell Proteomics. 2016;15:1151–63. doi: 10.1074/mcp.M115.054478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hansen TA, Sylvester M, Jensen ON, Kjeldsen F. Automated and high confidence protein phosphorylation site localization using complementary collision-activated dissociation and electron transfer dissociation tandem mass spectrometry. Anal Chem. 2012;84:9694–9. doi: 10.1021/ac302364r. [DOI] [PubMed] [Google Scholar]
- 75.Fermin D, Avtonomov D, Choi H, Nesvizhskii AI. LuciPHOr2: site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics. 2015;31:1141–3. doi: 10.1093/bioinformatics/btu788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai W, Guner H, Gregorich ZR, Chen AJ, Ayaz-Guner S, Peng Y, Valeja SG, Liu X, Ge Y. MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol Cell Proteomics. 2016;15:703–14. doi: 10.1074/mcp.O115.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Solari FA, Dell’Aica M, Sickmann A, Zahedi RP. Why phosphoproteomics is still a challenge. Mol Biosyst. 2015;11:1487–93. doi: 10.1039/c5mb00024f. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Y, Jensen ON. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics. 2009;9:4632–41. doi: 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tape CJ, Worboys JD, Sinclair J, Gourlay R, Vogt J, McMahon KM, Trost M, Lauffenburger DA, Lamont DJ, Jørgensen C. Reproducible automated phosphopeptide enrichment using magnetic TiO2 and Ti-IMAC. Anal Chem. 2014;86:10296–302. doi: 10.1021/ac5025842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pagel O, Loroch S, Sickmann A, Zahedi RP. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev Proteomics. 2015;12:235–53. doi: 10.1586/14789450.2015.1042867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thygesen C, Boll I, Finsen B, Modzel M, Larsen MR. Characterizing disease-associated changes in post-translational modifications by mass spectrometry. Expert Rev Proteomics. 2018;15:245–258. doi: 10.1080/14789450.2018.1433036. [DOI] [PubMed] [Google Scholar]
- 82.Reimand J, Wagih O, Bader GD. Evolutionary constraint and disease associations of post-translational modification sites in human genomes. PLoS Genet. 2015;11:e1004919. doi: 10.1371/journal.pgen.1004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A, Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK, Mohmood R, Ramachandra YL, Krishna V, Rahiman BA, Mohan S, Ranganathan P, Ramabadran S, Chaerkady R, Pandey A. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–72. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reimand J, Bader GD. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mort M, Evani US, Krishnan VG, Kamati KK, Baenziger PH, Bagchi A, Peters BJ, Sathyesh R, Li B, Sun Y, Xue B, Shah NH, Kann MG, Cooper DN, Radivojac P, Mooney SD. In silico functional profiling of human disease-associated and polymorphic amino acid substitutions. Hum Mutat. 2010;31:335–46. doi: 10.1002/humu.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–80. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun Z, Hamilton KL, Reardon KF. Phosphoproteomics and molecular cardiology: Techniques, applications and challenges. J Mol Cell Cardiol. 2012;53:354–368. doi: 10.1016/j.yjmcc.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 88.James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature. 1989;342:90–92. doi: 10.1038/342090a0. [DOI] [PubMed] [Google Scholar]
- 89.Wegener A, Simmerman H, Lindemann J, Jones L. Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J Biol Chem. 1989;264:11468. [PubMed] [Google Scholar]
- 90.Murphy AM. Heart failure, myocardial stunning, and troponin: a key regulator of the cardiac myofilament. Congest Heart Fail. 12:32-8–40. doi: 10.1111/j.1527-5299.2006.04320.x. [DOI] [PubMed] [Google Scholar]
- 91.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–7. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhong J, Molina H, Pandey A. Phosphoproteomics. Curr Protoc protein Sci. 2007 doi: 10.1002/0471140864.ps2404s50. Chapter 24: Unit 24.4. [DOI] [PubMed] [Google Scholar]
- 93.Pinkse MWH, Uitto PM, Hilhorst MJ, Ooms B, Heck AJR. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76:3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- 94.Mithoe SC, Boersema PJ, Berke L, Snel B, Heck AJR, Menke FLH. Targeted quantitative phosphoproteomics approach for the detection of phospho-tyrosine signaling in plants. J Proteome Res. 2012;11:438–48. doi: 10.1021/pr200893k. [DOI] [PubMed] [Google Scholar]
- 95.Lee DI, Zhu G, Sasaki T, Cho G-S, Hamdani N, Holewinski R, Jo S-H, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–6. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV. In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- 97.Yang Y-C, Wang X-D, Huang K, Wang L, Jiang Z-L, Qi Y-X. Temporal phosphoproteomics to investigate the mechanotransduction of vascular smooth muscle cells in response to cyclic stretch. J Biomech. 2014;47:3622–9. doi: 10.1016/j.jbiomech.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 98.Beck F, Geiger J, Gambaryan S, Solari FA, Dell’Aica M, Loroch S, Mattheij NJ, Mindukshev I, Pötz O, Jurk K, Burkhart JM, Fufezan C, Heemskerk JWM, Walter U, Zahedi RP, Sickmann A. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood. 2017;129:e1–e12. doi: 10.1182/blood-2016-05-714048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ledee D, Kang MA, Kajimoto M, Purvine S, Brewer H, Pasa-Tolic L, Portman MA. Quantitative cardiac phosphoproteomics profiling during ischemia-reperfusion in an immature swine model. Am J Physiol Heart Circ Physiol. 2017;313:H125–H137. doi: 10.1152/ajpheart.00842.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hayashi K, Gotou M, Matsui T, Imahashi K, Nishimoto T, Kobayashi H. Identification of phosphorylation sites on β1-adrenergic receptor in the mouse heart. Biochem Biophys Res Commun. 2017;488:362–367. doi: 10.1016/j.bbrc.2017.05.054. [DOI] [PubMed] [Google Scholar]
- 101.Schechter MA, Hsieh MKH, Njoroge LW, Thompson JW, Soderblom EJ, Feger BJ, Troupes CD, Hershberger KA, Ilkayeva OR, Nagel WL, Landinez GP, Shah KM, Burns VA, Santacruz L, Hirschey MD, Foster MW, Milano CA, Moseley MA, Piacentino V, Bowles DE. Phosphoproteomic profiling of human myocardial tissues distinguishes ischemic from non-ischemic end stage heart failure. PLoS One. 2014;9:e104157. doi: 10.1371/journal.pone.0104157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chang Y-W, Chang Y-T, Wang Q, Lin JJ-C, Chen Y-J, Chen C-C. Quantitative phosphoproteomic study of pressure-overloaded mouse heart reveals dynamin-related protein 1 as a modulator of cardiac hypertrophy. Mol Cell Proteomics. 2013;12:3094–107. doi: 10.1074/mcp.M113.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dong X, Sumandea CA, Chen Y-C, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP, Ge Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–57. doi: 10.1074/jbc.M111.293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang J, Guy MJ, Norman HS, Chen Y-C, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH, Moss RL, Ge Y. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10:4054–65. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gregorich ZR, Cai W, Lin Z, Chen AJ, Peng Y, Kohmoto T, Ge Y. Distinct sequences and post-translational modifications in cardiac atrial and ventricular myosin light chains revealed by top-down mass spectrometry. J Mol Cell Cardiol. 2017;107:13–21. doi: 10.1016/j.yjmcc.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ge Y, Rybakova IN, Xu Q, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc Natl Acad Sci U S A. 2009;106:12658–63. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 108.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 109.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–7. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 110.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006;103:1012–7. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–9. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou X, Han P, Li J, Zhang X, Huang B, Ruan H-Q, Chen C. ESNOQ, proteomic quantification of endogenous S-nitrosation. PLoS One. 2010;5:e10015. doi: 10.1371/journal.pone.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murray CI, Uhrigshardt H, O’Meally RN, Cole RN, Van Eyk JE. Identification and Quantification of S-Nitrosylation by Cysteine Reactive Tandem Mass Tag Switch Assay. Mol Cell Proteomics. 2012(11):M111.013441–M111.013441. doi: 10.1074/mcp.M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-β activation results in s-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;120:245–254. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1327–H1335. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kohr MJ, Aponte A, Sun J, Gucek M, Steenbergen C, Murphy E. Measurement of S-nitrosylation occupancy in the myocardium with cysteine-reactive tandem mass tags: Short communication. Circ Res. 2012;111:1308–1312. doi: 10.1161/CIRCRESAHA.112.271320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Camerini S, Polci ML, Restuccia U, Usuelli V, Malgaroli A, Bachi A. A novel approach to identify proteins modified by nitric oxide: The HIS-TAG switch method. J Proteome Res. 2007;6:3224–3231. doi: 10.1021/pr0701456. [DOI] [PubMed] [Google Scholar]
- 119.Murray CI, Van Eyk JE. Chasing cysteine oxidative modifications: Proteomic tools for characterizing cysteine redox status. Circ Cardiovasc Genet. 2012;5:1–10. doi: 10.1161/CIRCGENETICS.111.961425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Foster MW. Methodologies for the characterization, identification and quantification of S-nitrosylated proteins. Biochim Biophys Acta. 2012;1820:675–83. doi: 10.1016/j.bbagen.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qu Z, Greenlief CM, Gu Z. Quantitative Proteomic Approaches for Analysis of Protein S-Nitrosylation. J Proteome Res. 2016;15:1–14. doi: 10.1021/acs.jproteome.5b00857. [DOI] [PubMed] [Google Scholar]
- 122.Chen Y-J, Lu C-T, Su M-G, Huang K-Y, Ching W-C, Yang H-H, Liao Y-C, Chen Y-J, Lee T-Y. dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res. 2015;43:D503–11. doi: 10.1093/nar/gku1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, Steenbergen C. Signaling by S-nitrosylation in the heart. J Mol Cell Cardiol. 2014;73:18–25. doi: 10.1016/j.yjmcc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 125.Irie T, Sips PY, Kai S, Kida K, Ikeda K, Hirai S, Moazzami K, Jiramongkolchai P, Bloch DB, Doulias P-T, Armoundas AA, Kaneki M, Ischiropoulos H, Kranias E, Bloch KD, Stamler JS, Ichinose F. S-Nitrosylation of Calcium-Handling Proteins in Cardiac Adrenergic Signaling and Hypertrophy. Circ Res. 2015;117:793–803. doi: 10.1161/CIRCRESAHA.115.307157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Molina y Vedia L, McDonald B, Reep B, Brüne B, Di Silvio M, Billiar TR, Lapetina EG. Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J Biol Chem. 1992;267:24929–32. [PubMed] [Google Scholar]
- 127.Kohr MJ, Murphy E, Steenbergen C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS One. 2014;9:e111448. doi: 10.1371/journal.pone.0111448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brioschi M, Martinez Fernandez A, Banfi C. Exploring the biochemistry of the prenylome and its role in disease through proteomics: progress and potential. Expert Rev Proteomics. 2017;14:515–528. doi: 10.1080/14789450.2017.1332998. [DOI] [PubMed] [Google Scholar]
- 129.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004:RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wedegaertner PB, Wilson PT, Bourne HR. Lipid modifications of trimeric G proteins. J Biol Chem. 1995;270:503–6. doi: 10.1074/jbc.270.2.503. [DOI] [PubMed] [Google Scholar]
- 131.Xu N, Guan S, Chen Z, Yu Y, Xie J, Pan F-Y, Zhao N-W, Liu L, Yang Z-Z, Gao X, Xu B, Li C-J. The alteration of protein prenylation induces cardiomyocyte hypertrophy through Rheb-mTORC1 signalling and leads to chronic heart failure. J Pathol. 2015;235:672–85. doi: 10.1002/path.4480. [DOI] [PubMed] [Google Scholar]
- 132.Spindler SR, Li R, Dhahbi JM, Yamakawa A, Mote P, Bodmer R, Ocorr K, Williams RT, Wang Y, Ablao KP. Statin treatment increases lifespan and improves cardiac health in Drosophila by decreasing specific protein prenylation. PLoS One. 2012;7:e39581. doi: 10.1371/journal.pone.0039581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hoffman MD, Kast J. Mass spectrometric characterization of lipid-modified peptides for the analysis of acylated proteins. J Mass Spectrom. 2006;41:229–41. doi: 10.1002/jms.981. [DOI] [PubMed] [Google Scholar]
- 134.Bhawal RP, Sadananda SC, Bugarin A, Laposa B, Chowdhury SM. Mass spectrometry cleavable strategy for identification and differentiation of prenylated peptides. Anal Chem. 2015;87:2178–86. doi: 10.1021/ac503794s. [DOI] [PubMed] [Google Scholar]
- 135.Chowdhury SM, Munske GR, Ronald RC, Bruce JE. Evaluation of low energy CID and ECD fragmentation behavior of mono-oxidized thio-ether bonds in peptides. J Am Soc Mass Spectrom. 2007;18:493–501. doi: 10.1016/j.jasms.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vossenaar ER, Zendman AJW, van Venrooij WJ, Pruijn GJM. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25:1106–18. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 137.Boyde TR, Rahmatullah M. Optimization of conditions for the colorimetric determination of citrulline, using diacetyl monoxime. Anal Biochem. 1980;107:424–31. doi: 10.1016/0003-2697(80)90404-2. [DOI] [PubMed] [Google Scholar]
- 138.Senshu T, Sato T, Inoue T, Akiyama K, Asaga H. Detection of citrulline residues in deiminated proteins on polyvinylidene difluoride membrane. Anal Biochem. 1992;203:94–100. doi: 10.1016/0003-2697(92)90047-b. [DOI] [PubMed] [Google Scholar]
- 139.Jin Z, Fu Z, Yang J, Troncosco J, Everett AD, Van Eyk JE. Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics. 2013;13:2682–91. doi: 10.1002/pmic.201300064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hensen SMM, Pruijn GJM. Methods for the detection of peptidylarginine deiminase (PAD) activity and protein citrullination. Mol Cell Proteomics. 2014;13:388–96. doi: 10.1074/mcp.R113.033746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mohamed BM, Boyle NT, Schinwald A, Murer B, Ward R, Mahfoud OK, Rakovich T, Crosbie-Staunton K, Gray SG, Donaldson K, Volkov Y, Prina-Mello A. Induction of protein citrullination and auto-antibodies production in murine exposed to nickel nanomaterials. Sci Rep. 2018;8:679. doi: 10.1038/s41598-017-19068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Darrah E, Rosen A, Giles JT, Andrade F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: novel insights into autoantigen selection in rheumatoid arthritis. Ann Rheum Dis. 2012;71:92–8. doi: 10.1136/ard.2011.151712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Holers VM. Autoimmunity to citrullinated proteins and the initiation of rheumatoid arthritis. Curr Opin Immunol. 2013;25:728–35. doi: 10.1016/j.coi.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta. 2013;1829:1126–35. doi: 10.1016/j.bbagrm.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Acharya NK, Nagele EP, Han M, Coretti NJ, DeMarshall C, Kosciuk MC, Boulos PA, Nagele RG. Neuronal PAD4 expression and protein citrullination: possible role in production of autoantibodies associated with neurodegenerative disease. J Autoimmun. 2012;38:369–80. doi: 10.1016/j.jaut.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 146.Jiang Z, Cui Y, Wang L, Zhao Y, Yan S, Chang X. Investigating citrullinated proteins in tumour cell lines. World J Surg Oncol. 2013;11:260. doi: 10.1186/1477-7819-11-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Geraldino-Pardilla L, Russo C, Sokolove J, Robinson WH, Zartoshti A, Van Eyk J, Fert-Bober J, Lima J, Giles JT, Bathon JM. Association of anti-citrullinated protein or peptide antibodies with left ventricular structure and function in rheumatoid arthritis. Rheumatology (Oxford) 2017;56:534–540. doi: 10.1093/rheumatology/kew436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fert-Bober J, Giles JT, Holewinski RJ, Kirk JA, Uhrigshardt H, Crowgey EL, Andrade F, Bingham CO, Park JK, Halushka MK, Kass DA, Bathon JM, Van Eyk JE. Citrullination of myofilament proteins in heart failure. Cardiovasc Res. 2015;108:232–42. doi: 10.1093/cvr/cvv185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13:690–8. doi: 10.1111/acel.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. 2017;214:439–458. doi: 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.van Groeningen CJ, Leyva A, Kraal I, Peters GJ, Pinedo HM. Clinical and pharmacokinetic studies of prolonged administration of high-dose uridine intended for rescue from 5-FU toxicity. Cancer Treat Rep. 1986;70:745–50. [PubMed] [Google Scholar]
- 152.Sokolove J, Brennan MJ, Sharpe O, Lahey LJ, Kao AH, Krishnan E, Edmundowicz D, Lepus CM, Wasko MC, Robinson WH. Brief report: citrullination within the atherosclerotic plaque: a potential target for the anti-citrullinated protein antibody response in rheumatoid arthritis. Arthritis Rheum. 2013;65:1719–24. doi: 10.1002/art.37961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li X-C, Zeng Y, Sun R-R, Liu M, Chen S, Zhang P-Y. SUMOylation in cardiac disorders – a review. Eur Rev Med Pharmacol Sci. 2017;21:1583–1587. [PubMed] [Google Scholar]
- 154.Chang E, Abe J-I. Kinase-SUMO networks in diabetes-mediated cardiovascular disease. Metabolism. 2016;65:623–33. doi: 10.1016/j.metabol.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mendler L, Braun T, Müller S. The Ubiquitin-Like SUMO System and Heart Function: From Development to Disease. Circ Res. 2016;118:132–44. doi: 10.1161/CIRCRESAHA.115.307730. [DOI] [PubMed] [Google Scholar]
- 156.Stastna M, Van Eyk JE. Posttranslational modifications of lysine and evolving role in heart pathologies-recent developments. Proteomics. 2015;15:1164–80. doi: 10.1002/pmic.201400312. [DOI] [PubMed] [Google Scholar]
- 157.Pandey D, Hori D, Kim JH, Bergman Y, Berkowitz DE, Romer LH. NEDDylation promotes endothelial dysfunction: a role for HDAC2. J Mol Cell Cardiol. 2015;81:18–22. doi: 10.1016/j.yjmcc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 158.Martins-Marques T, Ribeiro-Rodrigues T, Pereira P, Codogno P, Girao H. Autophagy and ubiquitination in cardiovascular diseases. DNA Cell Biol. 2015;34:243–51. doi: 10.1089/dna.2014.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ferri N, Paoletti R, Corsini A. Lipid-modified proteins as biomarkers for cardiovascular disease: a review. Biomarkers. 10:219–37. doi: 10.1080/13547500500216660. [DOI] [PubMed] [Google Scholar]
- 160.Wright JN, Collins HE, Wende AR, Chatham JC. O-GlcNAcylation and cardiovascular disease. Biochem Soc Trans. 2017;45:545–553. doi: 10.1042/BST20160164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kailemia MJ, Wei W, Nguyen K, Beals E, Sawrey-Kubicek L, Rhodes C, Zhu C, Sacchi R, Zivkovic AM, Lebrilla CB. Targeted Measurements of O- and N-Glycopeptides Show That Proteins in High Density Lipoprotein Particles Are Enriched with Specific Glycosylation Compared to Plasma. J Proteome Res. 2018;17:834–845. doi: 10.1021/acs.jproteome.7b00604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barallobre-Barreiro J, Baig F, Fava M, Yin X, Mayr M. Glycoproteomics of the Extracellular Matrix: A Method for Intact Glycopeptide Analysis Using Mass Spectrometry. J Vis Exp. 2017 doi: 10.3791/55674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yates JR. Proteomics power to the people! Scientist. 2005;19:30–31. [Google Scholar]
- 164.Shillingford JM, Miyoshi K, Robinson GW, Bierie B, Cao Y, Karin M, Hennighausen L. Proteotyping of mammary tissue from transgenic and gene knockout mice with immunohistochemical markers: a tool to define developmental lesions. J Histochem Cytochem. 2003;51:555–65. doi: 10.1177/002215540305100501. [DOI] [PubMed] [Google Scholar]
- 165.Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, MacLean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJ, Meyer MR, Mesri M, Boja E, Carr SA, Chan DW, Chen X, Chen J, Davies SR, Ellis MJC, Fenyö D, Hiltke T, Ketchum KA, Kinsinger C, Kuhn E, Liebler DC, Liu T, Loss M, MacCoss MJ, Qian W-J, Rivers R, Rodland KD, Ruggles KV, Scott MG, Smith RD, Thomas S, Townsend RR, Whiteley G, Wu C, Zhang H, Zhang Z, Rodriguez H, Paulovich AG. Using the CPTAC Assay Portal to Identify and Implement Highly Characterized Targeted Proteomics Assays. Methods Mol Biol. 2016;1410:223–36. doi: 10.1007/978-1-4939-3524-6_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, MacLean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJ, Meyer MR, Mesri M, Rodriguez H, Clinical Proteomic Tumor Analysis Consortium (CPTAC) Paulovich AG. CPTAC Assay Portal: a repository of targeted proteomic assays. Nat Methods. 2014;11:703–4. doi: 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Abbatiello SE, Mani DR, Schilling B, Maclean B, Zimmerman LJ, Feng X, Cusack MP, Sedransk N, Hall SC, Addona T, Allen S, Dodder NG, Ghosh M, Held JM, Hedrick V, Inerowicz HD, Jackson A, Keshishian H, Kim JW, Lyssand JS, Riley CP, Rudnick P, Sadowski P, Shaddox K, Smith D, Tomazela D, Wahlander A, Waldemarson S, Whitwell CA, You J, Zhang S, Kinsinger CR, Mesri M, Rodriguez H, Borchers CH, Buck C, Fisher SJ, Gibson BW, Liebler D, Maccoss M, Neubert TA, Paulovich A, Regnier F, Skates SJ, Tempst P, Wang M, Carr SA. Design, implementation and multisite evaluation of a system suitability protocol for the quantitative assessment of instrument performance in liquid chromatography-multiple reaction monitoring-MS (LC-MRM-MS) Mol Cell Proteomics. 2013;12:2623–39. doi: 10.1074/mcp.M112.027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lam MPY, Venkatraman V, Cao Q, Wang D, Dincer TU, Lau E, Su AI, Xing Y, Ge J, Ping P, Van Eyk JE. Prioritizing Proteomics Assay Development for Clinical Translation. J Am Coll Cardiol. 2015;66:202–4. doi: 10.1016/j.jacc.2015.04.072. [DOI] [PubMed] [Google Scholar]
- 169.Lam MPY, Venkatraman V, Xing Y, Lau E, Cao Q, Ng DCM, Su AI, Ge J, Van Eyk JE, Ping P. Data-Driven Approach To Determine Popular Proteins for Targeted Proteomics Translation of Six Organ Systems. J Proteome Res. 2016;15:4126–4134. doi: 10.1021/acs.jproteome.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mora MI, Molina M, Odriozola L, Elortza F, Mato JM, Sitek B, Zhang P, He F, Latasa MU, Ávila MA, Corrales FJ. Prioritizing Popular Proteins in Liver Cancer: Remodelling One-Carbon Metabolism. J Proteome Res. 2017;16:4506–4514. doi: 10.1021/acs.jproteome.7b00390. [DOI] [PubMed] [Google Scholar]
- 171.Jagodnik KM, Koplev S, Jenkins SL, Ohno-Machado L, Paten B, Schurer SC, Dumontier M, Verborgh R, Bui A, Ping P, McKenna NJ, Madduri R, Pillai A, Ma’ayan A. Developing a framework for digital objects in the Big Data to Knowledge (BD2K) commons: Report from the Commons Framework Pilots workshop. J Biomed Inform. 2017;71:49–57. doi: 10.1016/j.jbi.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sidiropoulos K, Viteri G, Sevilla C, Jupe S, Webber M, Orlic-Milacic M, Jassal B, May B, Shamovsky V, Duenas C, Rothfels K, Matthews L, Song H, Stein L, Haw R, D’Eustachio P, Ping P, Hermjakob H, Fabregat A. Reactome enhanced pathway visualization. Bioinformatics. 2017;33:3461–3467. doi: 10.1093/bioinformatics/btx441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wu CH, Chen C, editors. Bioinformatics for Comparative Proteomics. Totowa, NJ: Humana Press; 2011. [Google Scholar]
- 174.Ping P, Watson K, Han J, Bui A. Individualized Knowledge Graph: A Viable Informatics Path to Precision Medicine. Circ Res. 2017;120:1078–1080. doi: 10.1161/CIRCRESAHA.116.310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ross KE, Huang H, Ren J, Arighi CN, Li G, Tudor CO, Lv M, Lee J-Y, Chen S-C, Vijay-Shanker K, Wu CH. iPTMnet: Integrative Bioinformatics for Studying PTM Networks. Methods Mol Biol. 2017;1558:333–353. doi: 10.1007/978-1-4939-6783-4_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Murray CI, Chung HS, Papanicolaou KN, Foster DBKMJ. Post-translational Modifications in the Cardiovascular Proteome. In: Agnetti G, Lindsey MFD, editors. Manual of Cardiovascular Proteomics. Springer; Cham: 2016. [Google Scholar]
- 178.Jacob RJ. Bioinformatics for LC-MS/MS-based proteomics. Methods Mol Biol. 2010;658:61–91. doi: 10.1007/978-1-60761-780-8_4. [DOI] [PubMed] [Google Scholar]
- 179.Parker CE, Mocanu V, Mocanu M, Dicheva N, Warren MR. Mass Spectrometry for Post-Translational Modifications. 2010 [PubMed] [Google Scholar]
- 180.Parker SJ, Holewinski RJ, Tchernyshyov I, Venkatraman V, Parker LVEJE. Label-Free Quantification by Data Independent Acquisition Mass Spectrometry to Map Cardiovascular Proteomes. In: Agnetti G, Lindsey MFD, editors. Manual of Cardiovascular Proteomics. Springer; Cham: 2016. [Google Scholar]
- 181.Wang J, Tucholska M, Knight JDR, Lambert J-P, Tate S, Larsen B, Gingras A-C, Bandeira N. MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nat Methods. 2015;12:1106–8. doi: 10.1038/nmeth.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Meyer JG, Mukkamalla S, Steen H, Nesvizhskii AI, Gibson BW, Schilling B. PIQED: automated identification and quantification of protein modifications from DIA-MS data. Nat Methods. 2017;14:646–647. doi: 10.1038/nmeth.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chambers AG, Percy AJ, Yang J, Borchers CH. Multiple Reaction Monitoring Enables Precise Quantification of 97 Proteins in Dried Blood Spots. Mol Cell Proteomics. 2015;14:3094–104. doi: 10.1074/mcp.O115.049957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Colangelo CM, Chung L, Bruce C, Cheung K-H. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods. 2013;61:287–98. doi: 10.1016/j.ymeth.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cheng D, Zheng L, Hou J, Wang J, Xue P, Yang F, Xu T. A new dimethyl labeling-based SID-MRM-MS method and its application to three proteases involved in insulin maturation. Biophys reports. 2015;1:71–80. doi: 10.1007/s41048-015-0012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gallien S, Kim SY, Domon B. Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM) Mol Cell Proteomics. 2015;14:1630–44. doi: 10.1074/mcp.O114.043968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bauer M, Ahrné E, Baron AP, Glatter T, Fava LL, Santamaria A, Nigg EA, Schmidt A. Evaluation of data-dependent and -independent mass spectrometric workflows for sensitive quantification of proteins and phosphorylation sites. J Proteome Res. 2014;13:5973–88. doi: 10.1021/pr500860c. [DOI] [PubMed] [Google Scholar]
- 188.Rauniyar N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int J Mol Sci. 2015;16:28566–81. doi: 10.3390/ijms161226120. [DOI] [PMC free article] [PubMed] [Google Scholar]