

Abstract
Optic neuropathies such as glaucoma are characterized by the degeneration of retinal ganglion cells (RGCs) and the irreversible loss of vision. In these diseases, focal axon injury triggers a propagating axon degeneration and, eventually, cell death. Previous work by us and others identified dual leucine zipper kinase (DLK) and JUN N-terminal kinase (JNK) as key mediators of somal cell death signaling in RGCs following axonal injury. Moreover, others have shown that activation of the DLK/JNK pathway contributes to distal axonal degeneration in some neuronal subtypes and that this activation is dependent on the adaptor protein, sterile alpha and TIR motif containing 1 (SARM1). Given that SARM1 acts upstream of DLK/JNK signaling in axon degeneration, we tested whether SARM1 plays a similar role in RGC somal apoptosis in response to optic nerve injury. Using the mouse optic nerve crush (ONC) model, our results show that SARM1 is critical for RGC axonal degeneration and that axons rescued by SARM1 deficiency are electrophysiologically active. Genetic deletion of SARM1 did not, however, prevent DLK/JNK pathway activation in RGC somas nor did it prevent or delay RGC cell death. These results highlight the importance of SARM1 in RGC axon degeneration and suggest that somal activation of the DLK/JNK pathway is activated by an as-yet-unidentified SARM1-independent signal.
Keywords: sterile alpha and TIR motif containing 1 (SARM1), dual leucine zipper kinase (DLK), axon degeneration, retinal ganglion cell (RGC), neurodegenerative disease
1. Introduction
Glaucoma is an optic neuropathy characterized by the degeneration of retinal ganglion cells (RGCs), projection neurons that are responsible for processing and transmitting visual information to the brain. A key early pathophysiological event is the injury of RGC axons at the lamina cribrosa of the optic nerve head. In order to study axon injury signaling, RGC axons can be experimentally severed in the optic nerve crush (ONC) model. In response to axotomy, RGC axons degenerate and cell death ensues. It is known that this cell death program uses a multi-tiered somal signaling cascade involving dual leucine zipper kinase (DLK) and leucine zipper kinase (LZK), mitogen-activated protein kinase (MAPK) kinases 4 and 7 (MKK4/MKK7), JUN N-terminal kinases 1-3 (JNK1-3), JUN and activating transcription factor 2 (ATF2) and, ultimately, BAX (Fernandes et al., 2012; Fernandes et al., 2014; Li et al., 2000; Libby et al., 2005; Watkins et al., 2013; Welsbie et al., 2017; Welsbie et al., 2013). Interestingly, the role of other BH3 family members like PUMA/BBC3 is more enigmatic with in vitro data supporting a role for PUMA downstream of DLK signaling but with Puma-null RGCs showing little protection from ONC in vivo (Harder and Libby, 2011b, 2013; Simon et al., 2016).
The pathways responsible for cell death partially overlap with those signaling cascades responsible for axonal degeneration. For example, inhibition of MAPK signaling or BH3 proteins like BAX and PUMA delays axon degeneration in various neuronal cell types (Howell et al., 2007; Libby et al., 2005; Nikolaev et al., 2009; Simon et al., 2016; Yang et al., 2015). DLK may also play a minor role in axon degeneration in some models of axon injury (Ghosh et al., 2011; Miller et al., 2009), although we have previously shown that targeted disruption of Dlk did not affect distal axon degeneration in the ONC model (Fernandes et al., 2014). Two of the most robust mediators of axon degeneration appear to be sterile alpha and TIR motif containing 1 (SARM1), a toll-like receptor adaptor family member, and death receptor 6 (DR6), a tumor necrosis factor receptor superfamily member (Gamage et al., 2017; Gerdts et al., 2013; Nikolaev et al., 2009; Osterloh et al., 2012)
SARM1 was initially identified in a forward genetic screen for Drosophila mutants that showed long-term axon survival after axotomy (Osterloh et al., 2012), suggesting SARM1 has a role in axonal degeneration. SARM1 is found in axons and is necessary for axonal degeneration in multiple neuronal cell types and in response to a wide range of axonal insults (Geisler et al., 2016; Osterloh et al., 2012; Turkiew et al., 2017). Forcing SARM1 multimerization is sufficient to activate MAPK signaling and induce axonal degeneration, suggesting that SARM1 functions upstream of the MAPK pathway (Gerdts et al., 2013; Yang et al., 2015). Consistent with this hypothesis, inhibition of MAPK signaling reverses the SARM1-dependent effect on hippocampal neuron dendritic complexity (Chen et al., 2011). However, others have recently shown that MAPK signaling modulates SARM1 activity by degrading the SARM1 inhibitor, nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2), potentially placing SARM1 downstream of MAPK signaling. Moreover, SARM1 can directly consume the axon survival-promoting molecule, nicotinamide adenine dinucleotide (NAD+) (Gerdts et al., 2015 and Essuman et al., 2017), further supporting a role for SARM1 as a downstream effector. In retina, SARM1 mRNA and protein are selectively expressed by RGCs and genetic disruption of Sarm1 delays the degeneration of RGCs axons following direct axotomy or an excitotoxic challenge (Yang et al., 2015, Massoll et al., 2013). Interestingly, several studies have also demonstrated a role for SARM1 in promoting neuronal cell death. SARM1 deficiency has been shown to reduce neuronal cell death induced by oxygen-glucose deprivation (Kim et al., 2007) and viral infection (Mukherjee et al., 2013). Additionally, SARM1 gain of function mutants have been shown to cause neuronal cell death (Gerdts et al., 2013; Yang et al., 2015). Recently, SARM1 was shown to be critical for Sarmaptosis, a novel form of cell death induced by mitochondrial dysfunction (Summers et al., 2014). Given the overlap between axon degeneration and cell death pathways, the unclear relationship between MAPK signaling and SARM1 and the known role of SARM1 in RGC axon degeneration, it is important to investigate whether SARM1 is necessary for the MAPK activation and cell death that follow axon injury.
DR6 (TNFRSF21) is another molecule that has been shown to be involved in axonal degeneration. It was initially identified to be critical for axonal degeneration of dorsal root ganglion neurons following trophic-factor deprivation (Nikolaev et al., 2009). Cleaved β-amyloid precursor protein (APP) was shown to activate DR6 and trigger axonal degeneration by activating a downstream caspase signaling cascade (Nikolaev et al., 2009). Collectively, these previous studies provide strong support for SARM1 and DR6 as critical regulators of axon degeneration, making them attractive targets to assess in the context of glaucomatous axonal degeneration. In this report, we use Sarm1 and DR6 knockout animals to explore the role of these genes in RGC somal and axonal degeneration.
2. Materials and Methods
2.1 Mice
Mice carrying a null allele of Sarm1 (B6.129X1-Sarm1tm1Aidi/J) were obtained from the Jackson Laboratories (Stock Number: 018069). The Wallerian Degeneration Slow (WldS) allele (Lunn et al., 1989; Mack et al., 2001) was backcrossed into C57BL/6J > 20 generations. DR6−/− mice (Tnfrsf21tm2Gne) were obtained from Genentech (Zhao et al., 2001). To genetically label RGC axons for histological assessment of morphological signs of axon degeneration, Thy1-CFP mice (B6.Cg-Tg(Thy1-CFP)23Jrs/J, Stock Number: 003710) which express CFP in ~80% of RGCs(Bernstein et al., 2007) were crossed to Sarm1−/− and WldS mice. Sarm1−/− and WldS mice were crossed to generate Sarm1−/− WldS double mutants. SpCas9 knockin mice (Rosa26 locus) were obtained from Jackson labs (stock 026179). Either littermate controls that did not carry any deleted allele or the WldS allele or C57BL6/J mice were used as experimental wildtype (WT) controls. Mice were housed in a 12-hour light dark cycle and were fed chow and water ad libitum. All experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology’s statement on the use of animals in ophthalmic research and were approved by the University of Rochester’s Committee on Animal Resources and the Johns Hopkins University School of Medicine Institutional Animal Care and Use Committee.
2.2 Optic nerve injury
ONC was performed as previously described (Harder and Libby, 2011b). Briefly, mice were anaesthetized and the optic nerve was exposed. The optic nerve was crushed for approximately 5 seconds about 0.5 mm from the globe using a pair of self-closing forceps (Roboz RS-5027). Contralateral eyes that were unmanipulated or had a sham surgery performed (where the optic nerve was exposed but not crushed) were used as controls. Mice were harvested at the indicated time-points (5, 7, 14 or 35 days) following ONC.
2.3 Histology
Following fixation in 4% paraformaldehyde (PFA), eyes or optic nerves were stored in 1X PBS at 4 °C until processing. For cryosectioning, the anterior segment of each eye was removed and the posterior eye cup was cryoprotected by consecutive immersion in sucrose (10%, 20%, 30%). 14 μm retinal sections or 10 μm longitudinal optic nerve sections were obtained on a cryostat.
2.4 Immunohistochemistry and Cell Counts
Eyes were processed as previously described (Fernandes et al., 2012; Harder and Libby, 2011a; Libby et al., 2005). For retinal section staining, the primary antibodies used were rabbit anti-pJNK (Cell Signaling; 4668, 1:200) and rabbit anti-pJUN (Abcam, ab40766, 1:200). For whole mount immunostaining, the primary antibodies used were rabbit anti-cCASP3 (RD AF835: 1:1000) and mouse anti-βIII tubulin (TUJ1; Biolegend; 801202; 1:1000). Since RGC density varies with retinal location, for cell counts images were obtained from eight 20× fields (for cCASP3+ cell counts) or eight 40× fields (for TUJ1+ cell counts) around the peripheral retina (two from each quadrant) for each whole mounted retina. Each field was approximately 220 μm from the peripheral edge of the retina. The numbers of cCASP3+ or TUJ1+ cells in each image were quantified using the cell-counter tool in ImageJ.
2.5 Electrophysiology
Electrophysiology recordings were performed as previously described (Fernandes et al., 2014). Briefly, optic nerves were harvested on the day of the electrophysiology recordings at the indicated time points after ONC. Optic nerve segments used in the recordings were obtained by transecting the nerve close to the eye and just anterior to the optic chiasm. Immediately following their dissection, optic nerves were allowed to incubate for at least 1 hour in artificial cerebrospinal fluid (ACSF) aerated with 95% oxygen/5% CO2 at room temperature. Suction capillary electrodes filled with ACSF were used to draw the optic nerve in at the retinal (stimulating) and chiasm (recording) ends. The nerve was stimulated with a 2 mA / 50 μsec pulse from a stimulus isolation unit driven by a computer. Prior to recording from the nerve, the resistance of the seal made between the recording pipette and the chiasm end of the nerve was determined. To do this, the optic nerve was first drawn in the stimulating pipette, following which the resistance of the recording pipette both before (R1) and after (R2) the optic nerve was inserted at the chiasm end was determined (Stys et al., 1991). By altering the level of suction, R2 could be varied, which in turn altered the electrical seal of the nerve in the pipette. As a way to normalize data across optic nerves, and to ensure that the amplitude of the signal was independent of the specific glass pipette used, all compound action potential (CAP) measurements were made with R2/R1 =1.7. Recordings were performed at 25 °C, following which records were digitized and analyzed off-line.
2.6 Primary RGCs
Retinas were isolated from postnatal day 0–3 mice and dissociated with papain. Microglia were immunodepleted with CELLection Dynabeads (Invitrogen) conjugated to anti-CD11b (BD Pharmingen, 554859). The suspension of retinal cells was immunopanned on plates pre-conjugated with anti-Thy1.2 antibodies (BioRad, MCA02R) and goat anti-mouse IgM (Jackson Immunoresearch, 115-001-020) at room temperature (RT). After washing, retinal ganglion cells (RGCs) were released from the plate with trypsin (Sigma T9201), counted, and seeded at a density of 3500 cells per well in 384-well plates (Nunclon plates, Poly-D-lysine and laminin coated). Growth medium was composed of Neurobasal (Life Technologies) supplemented with NS21, Sato, L-glutamine, penicillin/streptomycin, N-acetyl-cysteine, insulin, sodium pyruvate, triiodothyronine (T3), forskolin, BDNF, CNTF, and GDNF (Chen et al., 2008). Transfection of sgRNAs was performed at the time of isolation, using NeuroMag magnetic nanoparticles (OZ Biosciences, Marseille).
2.7 Statistical Analyses
For all experiments involving quantitation, the experimenter was masked to genotype and/or experimental group. At least three retinas/optic nerves were assessed for each genotype and experimental conditions. Experiments with two groups were analyzed for statistical differences using the unpaired Student’s t-test. Experiments with three or more groups were compared using a one-way ANOVA followed by the Tukey post-hoc test. Experiments with two or more genotypes and experimental conditions were subjected to statistical analyses using two-way ANOVA with significance determined at P values < 0.05, followed by the Tukey post hoc test for group comparisons.
3. Results
3.1 SARM1 is not required for RGC somal degeneration after axonal injury
In order to explore whether SARM1 has a role in RGC cell death, we first tested whether targeted deletion of Sarm1 increased the survival of primary RGCs isolated from Sarm1-null mice as compared to WT controls. We have previously demonstrated that the activity of RGC-intrinsic genes in this model is highly correlated with activity in vivo in the mouse ONC model (Welsbie et al., 2017; Welsbie et al., 2013). Primary RGCs were isolated from P0-P3 WT or Sarm1-null mice, challenged with 1 μM colchicine at 48 hours and then survival was measured after an additional 48 hours with CellTiter Glo, an ATP-based luminescent assay (Fig. 1A). While combined DLK/LZK inhibition with 1 μM tozasertib was robustly survival-promoting, the loss of SARM1 activity did not increase RGC survival. As a complementary approach that obviates the need for comparing RGCs from different isolations, we turned to a clustered regularly interspaced short palindromic repeat (CRISPR) model using S. pyogenes Cas9 (SpCas9)-expressing knockin animals (Cong et al., 2013; Platt et al., 2014). RGCs were isolated from SpCas9 mice and transfected with guide RNAs (gRNAs) targeting Dlk/Lzk or two independent sequences targeting Sarm1 (Fig. 1B). Again, while inhibition of DLK/LZK increased survival, acute disruption of SARM1 had no effect. Taken together, these results suggested that, unlike the case with DLK/LZK, inhibition of SARM1 was unable to prevent RGC cell death in vitro.
Figure 1. Targeted disruption of SARM1 does not prevent primary RGC cell death.
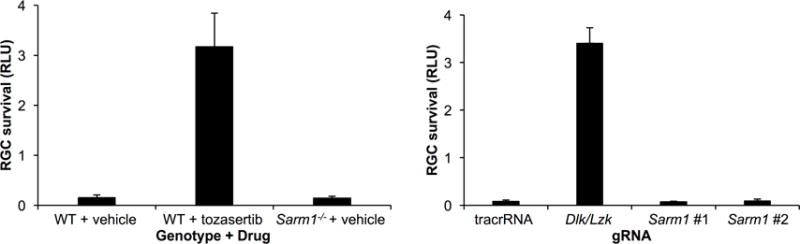
A. CellTiter Glo assay (±SD; n = 4 per sample) of primary RGCs isolated from WT and Sarm1-null mice, treated with either vehicle or the DLK/LZK inhibitor tozasertib (1 μM), 48 hours after a challenge with 1 μM colchicine. As opposed to DLK/LZK inhibition, loss of SARM1 activity did not increase survival of RGCs in vitro. RLU, relative luciferase units. B. CellTiter Glo assay (±SD; n = 4 per sample) of primary RGCs isolated from SpCas9-expressing mice and transfected with either of two gRNAs targeting Sarm1, a 50:50 mix of gRNAs targeting Dlk/Lzk or a non-targeting control, 48 hours after a challenge with 1 μM colchicine.
We next sought to confirm the hypothesis that SARM1 was not necessary for RGC cell death in a rodent model of RGC axon injury. Using the ONC model, we assayed the numbers of cleaved caspase3 (cCASP3) labeled dying RGCs and the numbers of surviving RGCs labeled with the monoclonal β-III-tubulin antibody, TUJ1 (Cui et al., 2003). In the ganglion cell layer of the mouse, TUJ1 has previously been shown to specifically label the vast majority of RGCs (Robinson and Madison, 2004). To confirm TUJ1’s specificity, retinas from C57BL/6J mice were double labeled with TUJ1 and antibodies against RPBMS. RPBMS has been shown to specifically label the vast majority of RGCs in the ganglion cell layer of the rodent retina, including in the mouse (Kwong et al., 2010; Rodriguez et al., 2014). TUJ1 and RPBMS had a large overlap with 96.0% of TUJ1+ cells colabeled with RBPMS and 93.9% of RBPMS+ cells colabeled with TUJ1 (N=3). Consistent with the in vitro data, SARM1 deficiency did not reduce RGC death five days after ONC (Fig. 2A). The number of TUJ1-labeled surviving RGCs 14 days after ONC was also not significantly reduced by SARM1 deficiency (Fig. 2B-C).
Figure 2. Neither SARM1 deficiency or WldS expression alter RGC death after ONC.
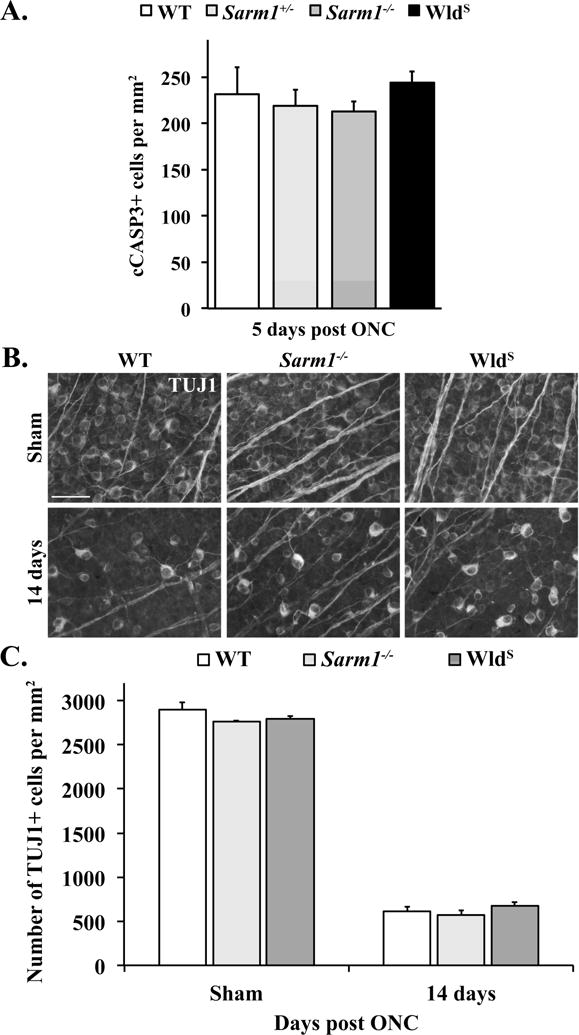
A. Cell counts of cleaved caspase 3+ (cCASP3+) cells 5 days after ONC. Neither Sarm1 deficiency (Sarm1+/− or Sarm1−/−) nor WldS expression altered the number of dying RGCs compared to WT mice (n ≥ 6, All genotypes; P>0.05 for all comparisons). Note, cCasp3+ cells were not found in uninjured mice of any genotype (n ≥ 4 for each genotype). B. Representative images of flat mounted retinas labeled with TUJ1 from WT, Sarm1−/− and WldS mice. C. Cell counts of the number of TUJ1+ RGCs in sham-injured and ONC-injured conditions. Long-term RGC survival 14 days after ONC was not altered by SARM1 deficiency (n ≥ 5 for each genotype; P>0.05 for all comparisons; error bars, SEM).
As a complementary approach to test the role of SARM1 cascade in cell death, we turned to the WldS mouse. WldS is an autosomal dominant mutation and WldS nerves express an in-frame chimeric fusion protein consisting of the first 70 amino acids of Ube4b, a ubiquitin ligase, and NMNAT1, an NAD+ synthesizing enzyme (Mack et al., 2001). WldS activity is required locally in the axon following injury to prevent axon degeneration (Cohen et al., 2012; Wang et al., 2015a). Multiple mechanisms have been proposed for how axonal NMNATs antagonize SARM1 and prevent axonal degeneration, including the production of NAD+, consumption of nicotinamide mononucleotide (NMN) and a direct effect on SARM1 (Di Stefano et al., 2015; Gerdts et al., 2015; Sasaki et al., 2016). As with the SARM1 experiment, WldS mice or WT controls were subjected to ONC and the number of cCASP3-positive RGCs at day five (Fig. 2A) or the number of surviving TUJ-labeled RGCs at day 14 (Fig. 2C) was assayed. Consistent with previous findings, WldS did not alter RGC death after ONC (Beirowski et al., 2008). Collectively, these data indicate that SARM1 signaling does not play a major role in the proximal axon injury response that drives RGC death after ONC.
3.2 Activation of JNK signaling in retinal ganglion cell somas is not altered by SARM1 deficiency
Given that SARM1 has been shown to function upstream of MAPK signaling in some models of neuronal injury and our previous work showing the importance of DLK/JNK signaling proximal in the RGC somal response to axonal injury (Fernandes et al., 2012; Fernandes et al., 2014), we sought to determine if SARM1 inhibition affects proximal JNK signaling. JNK activation was characterized in WT and Sarm1−/− retinas five days after axon injury. In WT retinas, accumulation of activated, phosphorylated JNK (pJNK) was observed in RGC somas five days after ONC, but not in sham-injured retinas (Fig. 3A). Similarly, robust accumulation of pJNK was observed in RGCs somas in Sarm1−/− retinas 5 days after ONC. Additionally, the canonical JNK substrate, JUN was phosphorylated in both WT and Sarm1−/− retinas 5 days after ONC (Fig. 3B). Collectively these data show that SARM1 deficiency does not prevent DLK/JNK activation after ONC.
Figure 3. SARM1 disruption does not prevent JNK activation in RGC somas after axon injury.
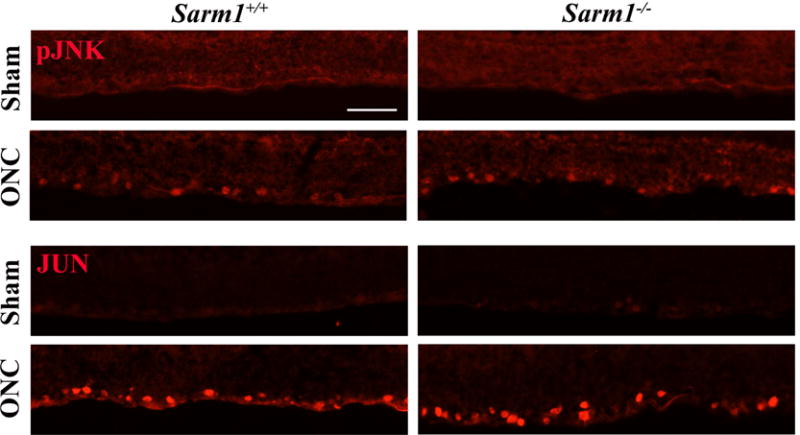
A. 5 days after ONC, JNK is active (phosphorylated, pJNK) in RGCs. SARM1 deficiency does not prevent activation of JNK after ONC. B. Similarly, the canonical target of JNKs, JUN, accumulates in RGCs 5 days after ONC and SARM1 deficiency does not prevent JUN accumulation. These data suggest, unlike in other systems, SARM1 is not required for JNK signaling. At least 3 sections from 3 different eyes were assessed for each genotype.
3.3 SARM1 deficiency is as protective as WldS expression in reducing axon degeneration after optic nerve crush
Using morphological assessment of axon degeneration, Yang et. al. previously showed that AAV delivered Sarm1 shRNA reduced RGC axon degeneration after optic nerve crush (Yang et al., 2015). Since we did not see an effect of Sarm1 disruption on somal survival, we next tested whether Sarm1 knockout would phenocopy the shRNA results and delay axonal degeneration. Thy1-CFP animals, in which RGC axons are easily visualized by their fluorescent protein expression were crossed with Sarm1-null and WldS animals to produce Thy1-CFP23.Sarm1−/− and Thy1-CFP23.WldS mice and the control Thy1-CFP.WT mice. Mice were then subjected to ONC and optic nerves were harvested after five days. Morphological signs of axon degeneration such as axon beading and axon fragmentation were readily detected in WT (Fig. 4A). In contrast, fewer signs of axon degeneration were observed in Sarm1−/− and WldS optic nerves after crush (Fig. 4A). To test whether these remaining axons were functional, compound action potentials (CAPs) were recorded from optic nerves from WT, WldS and Sarm1-null animals five days after ONC. As previously reported with WldS mice (Fernandes et al., 2014), CAP amplitude recorded five days after ONC was significantly higher than control animals (Fig. 4B-C, P=0.0008, n ≥ 5 for each genotype). The maximal amplitude of the CAP was also significantly higher in Sarm1-null optic nerves compared to WT nerves (Fig. 4B-C, P=0.0061, n ≥ 5 for each genotype), validating that SARM1 inhibition was able to delay the loss of function in injured axons. No significant differences were observed in the CAPs recorded from Sarm1−/− or WldS optic nerves five days after crush (Figure 1B), suggesting that Sarm1 deficiency and WldS expression provide similar levels of protection. Collectively these data show that loss of SARM1 activity is as protective as WldS in reducing axon degeneration after ONC and that the remaining axons are capable of propagating action potentials.
Figure 4. SARM1 deficiency protects RGCs from axon degeneration after ONC.
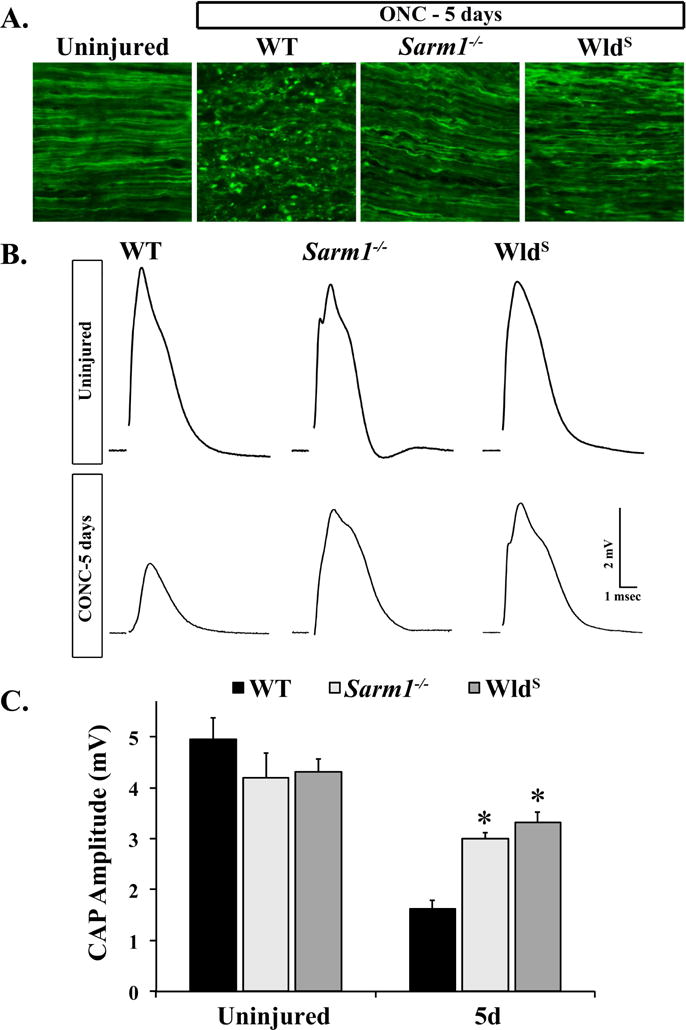
A. Representative images of longitudinal optic nerve sections from mice of the indicated genotypes in which RGC axons were labeled with CFP by crossing the Thy-CFP transgene into the strain (CFP+). Histological signs of axon degeneration such as axon beading and fragmentation were evident in WT optic nerves 5 days after ONC. However, fewer morphological signs of axon degeneration were observed in Sarm1−/−.CFP+ and WldS.CFP+ optic nerves 5 days after ONC (n = 3 for each genotype). B. Representative traces of the CAP recorded from WT, Sarm1−/− and WldS optic nerves 5 days after crush. C. SARM1 deficiency significantly reduced RGC axon degeneration 5 days after crush. The CAP amplitude recorded from Sarm1−/− and WldS nerves was significantly higher than WT nerves 5 days after ONC. Interestingly, the CAP amplitude was not significantly different in Sarm1−/− and WldS nerves 5 days after ONC (n ≥ 5 for each genotype and condition; error bars, SEM). Note, Sarm1+/+ and WldS littermate controls that did not carry the mutation were grouped together in the WT group.
3.4 SARM1 and WldS function in same pathway
We next questioned whether there might be any additive benefit to targeting the axon degeneration pathway with combined SARM1 deficiency and WldS expression. To test this hypothesis, we generated mice with both alleles and assayed for improved electrophysiological integrity of the optic nerves two weeks after ONC. The CAP recorded from Sarm1−/−WldS double mutants 14 days after crush did not, however, appear higher than the CAP recorded from either Sarm1−/− or WldS optic nerves (Fig. 5). The failure to observe any complementation in the Sarm1−/− WldS double mutants suggest that Sarm1−/− and WldS ultimately converge on the same axon degeneration pathway.
Figure 5. SARM1 loss and WldS function in the same axon degeneration pathway.
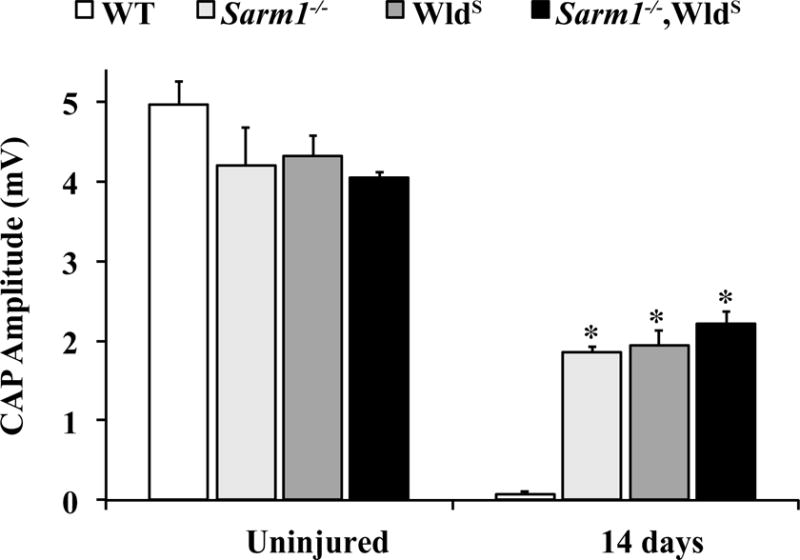
To determine if SARM1 and WLDS function in the same axon degeneration pathway, mice expressing WldS in a Sarm1-null background were generated. Axon degeneration was assessed by quantifying the maximal amplitude of the CAP 14 days after ONC. The data for Sarm1−/− and WldS mice are the same as from Figure 1. The CAP amplitude of all genotypes was significantly reduced 14 days after ONC, however, and Sarm1−/−, WldS, and Sarm1−/−;WldS mice all had significantly higher CAPs compared to WT controls. There was no difference in CAPs from Sarm1−/−;WldS mice compared to WldS or Sarm1−/− mice (n ≥ 5 for each genotype and condition).
3.5 DR6 is not necessary for RGC somal or axonal degeneration after axonal injury
DR6 has been shown to promote neuronal cell death in several different animal models of neurodegeneration. Antagonizing DR6 function has been shown to promote motor neuron survival in ALS models (Huang et al., 2013) and attenuate Aβ-induced cortical neuronal death (Hu et al., 2013). Interestingly, DR6 has also been shown to activate JNK signaling after a variety of insults (Hu et al., 2014; Pan et al., 1998; Zhao et al., 2001), suggesting that DR6 could function in the proximal axon injury signaling cascade that leads to RGC death after axon injury. To test the involvement of DR6 in axonal injury induced RGC death, the numbers of cleaved caspase-3 positive cells was assessed in WT and DR6−/− retinas. DR6 deficiency did not significantly reduce RGC death 5 days after ONC (Fig. 6A-B; P=0.1403), indicating that DR6 is not a major regulator of axonal injury induced RGC death. DR6 has also been shown to regulate axon degeneration during developmental axon pruning (Nikolaev et al., 2009; Olsen et al., 2014) as well as axon degeneration induced by various pathological insults (Wang et al., 2015b). The ligand for DR6, APP, was shown to accumulate both proximally and distally to the site of insult in RGCs in ocular hypertensive models of glaucoma (Chidlow et al., 2012). Interestingly, APP accumulates near the lamina (Chidlow et al., 2011; Chidlow et al., 2012), a region where morphological signs of axon damage are first observed in ocular hypertensive models of glaucoma (Howell et al., 2007). Thus, it appears that DR6 could play a role in RGC axon degeneration after axon injury. To test this possibility, CAP amplitudes were measured from DR6−/− or WT optic nerves 5 days after ONC. Surprisingly, DR6 deficiency did not lessen the decline of CAPs compared to WT mice (Fig. 6C-D). Collectively, these results indicate that DR6 does not play a major role in the somal or axonal degeneration pathways controlling RGC degeneration after axonal injury.
Figure 6. DR6 deficiency does not protect RGCs from somal or axonal degeneration after axonal injury.
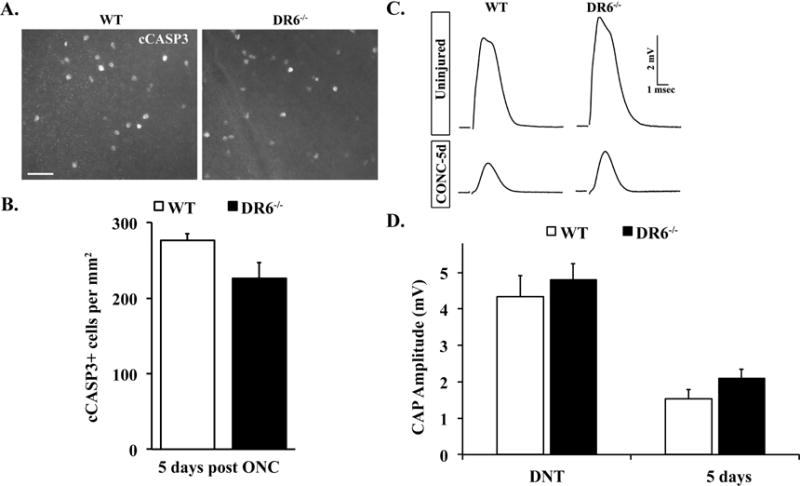
A. Representative images from WT and DR6−/− flat-mounted retinas stained with anti-cleaved caspase 3 (cCASP3) 5 days after ONC. B. Cell counts of the number of cCASP3 positive cells 5 days after crush (n ≥ 5 for each genotype) showed no significant difference between WT and DR6−/− mice (P>0.05). C. Representative traces of the compound action potential (CAP) recorded from WT and DR6−/− optic nerves 5 days after crush. D. The CAP amplitude was significantly reduced in both WT and DR6−/− optic nerves 5 days after crush. However, the reduction of CAP amplitude observed in DR6 deficient mice was similar to that observed in control mice (P>0.05; (n ≥ 5 for each genotype and condition), suggesting DR6 is not an important component of the axonal degeneration pathway in adult RGCs. Scale bar: A, 50 m; error bars, SEM).
4. Discussion
4.1 Distinct signaling cascades for somal and axonal degeneration
Our data support previous findings by Osterloh et. al., and showed that SARM1 deficiency was as protective as WldS in preventing axon degeneration. However, sarmaptosis does not contribute to RGC death after mechanical axon injury. Collectively, these data show that SARM1 is critical for axonal but not somal degeneration of RGCs after ONC. These results are consistent with previous studies that suggest that distinct signaling pathways control the degeneration of different cellular compartments of RGCs (Fernandes et al., 2012; Fernandes et al., 2014; Howell et al., 2007; Whitmore et al., 2005). Interestingly, while SARM1 has been shown to be upstream of JNK activation in other neuronal systems, our data show that SARM1 deficiency does not prevent JNK activation in RGC somas after ONC. The finding that SARM1 is not critical for JNK activation proximal to the site of axon injury suggests that axon injury signaling in the somal and axonal compartments utilize different mechanisms. Consistent with this, while DLK disruption prevented JNK activation in RGC somas proximal to the site of insult (Watkins et al., 2013; Welsbie et al., 2013), it was not a major regulator of axon degeneration of RGCs after ONC (Fernandes et al., 2014). Interestingly, it may be that DLK cooperates with different combinations of MAP3Ks to promote axonal versus somal degeneration. For instance, we have shown that LZK synergizes with DLK to trigger somal degeneration, while Yang et al. have shown that DLK interacts with mixed lineage kinase 2 (MLK2) and MAPK/ERK kinase kinase 4 (MEKK4) to promote axonal degeneration (Welsbie et al., 2017; Yang et al., 2015). These data highlight the complexity and both the specificity and redundancy at the level of the MAP3Ks regulating somal and axonal degeneration. Moreover, as the signaling response may vary depending on the nature of the axonal injury, it underscores the need to determine which MAP3Ks are responsible for somal and axonal degeneration after an ocular hypertensive insult.
4.2 Degenerative pathways differ in development and disease
It is interesting to note the differences between developmental and pathological cell death and axon degeneration. For instance, we have shown that DLK and JUN are critical for somal degeneration after ONC while both Jun- and Dlk-null animals have normal levels of RGC programmed cell death during development (Fernandes et al., 2012; Fernandes et al., 2014; Herzog et al., 1999; Syc-Mazurek et al., 2017a; Syc-Mazurek et al., 2017b; Watkins et al., 2013; Welsbie et al., 2017; Welsbie et al., 2013). Conversely, apoptotic mediators like PUMA play a role in developmental, but not pathological, RGC cell death (Harder and Libby, 2011b). A similar story seems to exist for developmental versus pathological axon degeneration. Both Sarm1−/− and WldS protect axons from axon injury-induced degeneration following a variety of insults, but neither are required for axon degeneration in models of developmental axon pruning (Hoopfer et al., 2006; Osterloh et al., 2012). Conversely, DR6 deficiency has previously been shown to impair developmental axonal pruning of RGC axons (Nikolaev et al., 2009) (Olsen et al., 2014), while our data show that DR6 is not critical for adult axonal degeneration after ONC. This is consistent with previous studies showing that distinct molecular mechanisms regulate developmental axon pruning versus adult axon degeneration (Cusack et al., 2013). Though, for both somal and axonal degeneration experiments, there was a trend towards protection, so we cannot exclude the possibility that DR6 might have a minor role in RGC degeneration after axonal injury.
4.3 Axon degeneration pathways as a target for glaucoma
Using Sarm1−/− WldS double mutants, we show that there is no additional functional protection from axon degeneration in comparison to the single mutants (Sarm1−/− or WldS alone) indicating that SARM1 and WldS ultimately participate in the same RGC axon degeneration pathway. Supporting this, SARM1-mediated NAD+ depletion has been shown to contribute to axon degeneration (Sasaki et al., 2016), while several studies have shown that WldS expression compensates for NAD+ depletion after axon injury (Araki et al., 2004; Wang et al., 2005). Collectively, these studies point to a convergence of SARM1- and WldS-dependent pathways at the point of regulating NAD+ levels in axons after mechanical axonal injury. It remains to be seen, however, whether interventions which target this pathway will be protective in glaucoma models. For instance, in the DBA/2J mouse model of ocular hypertension-induced RGC degeneration, expression of WldS reduced the frequency of optic nerves with severe axon damage (Howell et al., 2007) and increased somal survival in those eyes with axon protection. However, 40% of optic nerves from 12-month old DBA/2J mice expressing WldS had axonal damage, suggesting that endogenous axon degeneration pathway(s) could still be activated in these mice (Howell et al., 2007). Moreover, in the rat laser glaucoma model, axonal degeneration occurs (albeit modestly delayed) despite the presence of the WldS allele (Beirowski et al., 2008). These data highlight the importance of better characterizing the endogenous axon degeneration pathways that are critical for glaucomatous axon degeneration.
5. Conclusion
After mechanical axonal injury (ONC) the degeneration pathways controlling somal and axonal degeneration appear to be molecularly distinct (Fig. 7). Proximal to the site of injury, DLK/LZK, JNK2/3 and JUN dependent MAPK signaling have been shown to be important for a BAX-dependent RGC somal degeneration after ONC (Fernandes et al., 2012; Fernandes et al., 2014; Li et al., 2000; Libby et al., 2005; Watkins et al., 2013; Welsbie et al., 2017; Welsbie et al., 2013). Moreover, ER stress and other factors are also important mediators of RGC somal degeneration (Fernandes et al., 2015; Hu, 2016; Hu et al., 2012; Syc-Mazurek et al., 2017b; Yang et al., 2016), although it is generally unclear how the activation of these pathways relates to MAPK axon injury signaling. Here we show that SARM1 does not appear to be important in somal degeneration after ONC. In contrast, SARM1 does have a major role in axonal degeneration distal to the site of ONC injury. And while MAPK signaling has been implicated in axon degeneration, the precise contributors to this appear to be different than in the soma as neither DLK nor JNK2/3 appear to play a major role in RGC axonal degeneration after ONC injury (Fernandes et al., 2012; Fernandes et al., 2014). We cannot rule out, however, that more complete inhibition (e.g. combined DLK/LZK or JNK1-3 inhibition) is required to see a protective phenotype. Interestingly, MAPK signaling has been shown to be both upstream and downstream of SARM1 (Geden and Deshmukh, 2016; Walker et al., 2017; Yang et al., 2015). Thus, the importance of MAPK signaling in axonal degeneration in RGCs remains to be determined. A comprehensive neuroprotective strategy may need to target members of both degenerative cascades for sustained, functional protection of RGCs after axonal injury. It is important to note that ONC was used to induce axonal injury in RGCs to study axonal injury signaling and the resultant degeneration. In the future, it will be necessary to test the importance of these molecules in an ocular hypertensive model of glaucoma which may vary in terms of magnitude and types of insults that trigger axonal and somal degeneration. Furthermore, ocular hypertension may very well have different signaling pathways controlling RGC death compared to mechanical axonal injury.
Figure 7. Summary diagram of somal and axonal degeneration pathways controlling RGC somal and axonal degeneration after mechanical optic nerve injury.
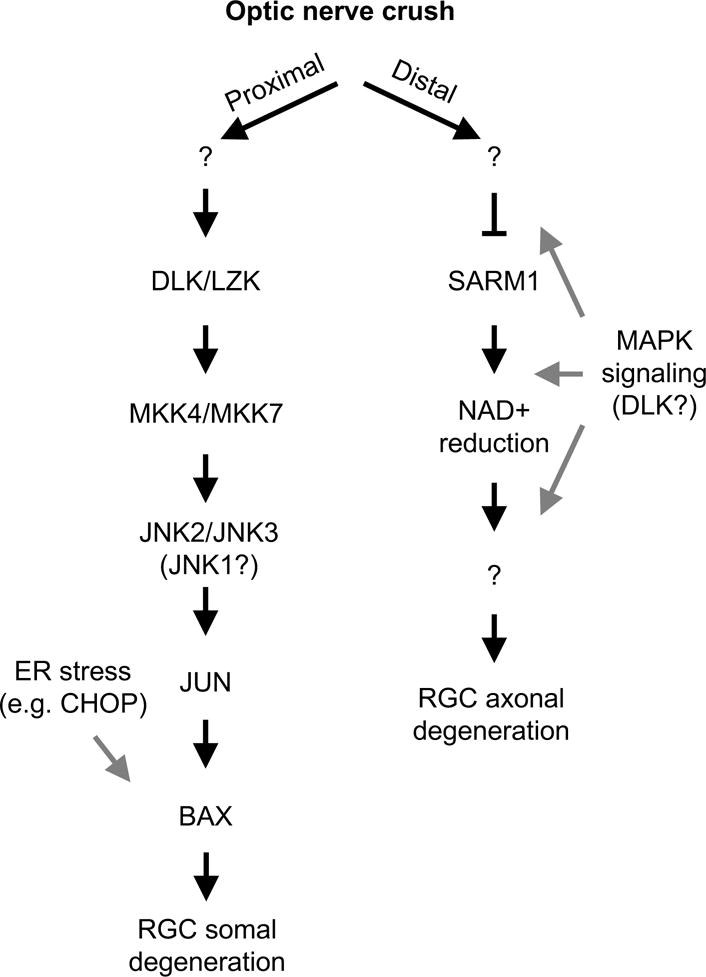
After mechanical optic nerve injury (ONC) distinct injury signaling pathways appear to control somal (proximal to the site of injury) and axonal (distal to the site of injury) degeneration. Proximal to the site of injury a MAPK pathway that includes DLK/LZK and JNK2/3 activation activates the transcription factor JUN. JUN activation, presumably through altering the transcription of injured RGCs, leads to BAX activation and somal apoptosis. Also, an ER stress pathway and other factors have been shown to regulate RGC death after axonal injury. Axonal degeneration distal to the site of axonal injury is less well defined. After ONC injury, SARM1 deficiency lessons axonal degeneration; thus, the presence of SARM1 facilitates axonal degeneration presumably by consuming NAD+. The MAPK pathway, particularly the MAP2Ks (MKK4 and MKK7) and DLK have also been implicated in axonal degeneration. It is unclear (gray arrows) how important these molecules are for axonal degeneration in RGCs and whether they are upstream or downstream of SARM1 dependent events. “?” represents a presumed step(s) in the pathway that involve an unknown molecule(s).
Supplementary Material
Highlights.
SARM1, but not DR6, deficiency delays distal axonal degeneration in RGCs
Neither SARM1 nor DR6 deficiency increases RGC survival following optic nerve crush
DLK/JNK signaling in RGC somas is unaffected by genetic disruption of SARM1
Acknowledgments
The authors would like to thank Aaron DiAntonio and Genentech for generously providing the Sarm1 and Dr6 mice, respectively. This work was supported by The Glaucoma Research Foundation (RTL), The E. Matilda Ziegler Foundation, Research to Prevent Blindness, Guerrieri Family Foundation, EY018606 (RTL), EY023754 (DJZ), and Research to Prevent Blindness unrestricted grants to the Departments of Ophthalmology at the University of Rochester Medical Center, University of California, San Diego and the Johns Hopkins University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci. 2008;28:1166–1179. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- Bernstein SL, Guo Y, Slater BJ, Puche A, Kelman SE. Neuron stress and loss following rodent anterior ischemic optic neuropathy in double-reporter transgenic mice. Invest Ophthalmol Vis Sci. 2007;48:2304–2310. doi: 10.1167/iovs.06-0486. [DOI] [PubMed] [Google Scholar]
- Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J Cell Biol. 2011;193:769–784. doi: 10.1083/jcb.201008050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow G, Ebneter A, Wood JP, Casson RJ. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011;121:737–751. doi: 10.1007/s00401-011-0807-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow G, Wood JP, Ebneter A, Casson RJ. Interleukin-6 is an efficacious marker of axonal transport disruption during experimental glaucoma and stimulates neuritogenesis in cultured retinal ganglion cells. Neurobiol Dis. 2012;48:568–581. doi: 10.1016/j.nbd.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Ghosh AK, Kim HJ, Jeon NL, Jaffrey SR. Chemical genetic-mediated spatial regulation of protein expression in neurons reveals an axonal function for wld(s) Chem Biol. 2012;19:179–187. doi: 10.1016/j.chembiol.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yip HK, Zhao RC, So KF, Harvey AR. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol Cell Neurosci. 2003;22:49–61. doi: 10.1016/s1044-7431(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013;4:1876. doi: 10.1038/ncomms2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JR, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, Magni G, Coleman MP, Conforti L. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015;22:731–742. doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron. 2017;93:1334–1343. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, John SW, Libby RT. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46:393–401. doi: 10.1016/j.nbd.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, John SW, Shrager P, Libby RT. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol Dis. 2014;69:108–116. doi: 10.1016/j.nbd.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Williams PA, Rausch RL, Kiernan AE, Nair KS, Anderson MG, John SW, Howell GR, Libby RT. Using genetic mouse models to gain insight into glaucoma: Past results and future possibilities. Exp Eye Res. 2015;141:42–56. doi: 10.1016/j.exer.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage KK, Cheng I, Park RE, Karim MS, Edamura K, Hughes C, Spano AJ, Erisir A, Deppmann CD. Death Receptor 6 Promotes Wallerian Degeneration in Peripheral Axons. Curr Biol. 2017;27:890–896. doi: 10.1016/j.cub.2017.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geden MJ, Deshmukh M. Axon degeneration: context defines distinct pathways. Curr Opin Neurobiol. 2016;39:108–115. doi: 10.1016/j.conb.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain. 2016;139:3092–3108. doi: 10.1093/brain/aww251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, Diantonio A, Milbrandt J. Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011a;6:50. doi: 10.1186/1750-1326-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011b;6:50. doi: 10.1186/1750-1326-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Libby RT. Deficiency in Bim, Bid and Bbc3 (Puma) do not prevent axonal injury induced death. Cell Death Differ. 2013;20:182. doi: 10.1038/cdd.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog KH, Chen SC, Morgan JI. c-jun Is dispensable for developmental cell death and axogenesis in the retina. J Neurosci. 1999;19:4349–4359. doi: 10.1523/JNEUROSCI.19-11-04349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O’Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Du Q, Yin X, Li J, Wang T, Zhang L. Agonist antibody activates death receptor 6 downstream signaling involving TRADD recruitment. FEBS Lett. 2014;588:401–407. doi: 10.1016/j.febslet.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Hu Y. Axon injury induced endoplasmic reticulum stress and neurodegeneration. Neural Regen Res. 2016;11:1557–1559. doi: 10.4103/1673-5374.193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Lee X, Shao Z, Apicco D, Huang G, Gong BJ, Pepinsky RB, Mi S. A DR6/p75(NTR) complex is responsible for beta-amyloid-induced cortical neuron death. Cell Death Dis. 2013;4:e579. doi: 10.1038/cddis.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Park KK, Yang L, Wei X, Yang Q, Cho KS, Thielen P, Lee AH, Cartoni R, Glimcher LH, Chen DF, He Z. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73:445–452. doi: 10.1016/j.neuron.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Lee X, Bian Y, Shao Z, Sheng G, Pepinsky RB, Mi S. Death receptor 6 (DR6) antagonist antibody is neuroprotective in the mouse SOD1G93A model of amyotrophic lateral sclerosis. Cell Death Dis. 2013;4:e841. doi: 10.1038/cddis.2013.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C, Iadecola C, Nathan C, Ding A. My D885 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JM, Caprioli J, Piri N. RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:1052–1058. doi: 10.1167/iovs.09-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–213. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Massoll C, Mando W, Chintala SK. Excitotoxicity upregulates SARM1 protein expression and promotes Wallerian-like degeneration of retinal ganglion cells and their axons. Invest Ophthalmol Vis Sci. 2013;54:2771–2780. doi: 10.1167/iovs.12-10973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity. 2013;38:705–716. doi: 10.1016/j.immuni.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Olsen O, Kallop DY, McLaughlin T, Huntwork-Rodriguez S, Wu Z, Duggan CD, Simon DJ, Lu Y, Easley-Neal C, Takeda K, Hass PE, Jaworski A, O’Leary DD, Weimer RM, Tessier-Lavigne M. Genetic analysis reveals that amyloid precursor protein and death receptor 6 function in the same pathway to control axonal pruning independent of beta-secretase. J Neurosci. 2014;34:6438–6447. doi: 10.1523/JNEUROSCI.3522-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, Zuchner S, Freeman MR. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 1998;431:351–356. doi: 10.1016/s0014-5793(98)00791-1. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GA, Madison RD. Axotomized mouse retinal ganglion cells containing melanopsin show enhanced survival, but not enhanced axon regrowth into a peripheral nerve graft. Vision Res. 2004;44:2667–2674. doi: 10.1016/j.visres.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Rodriguez AR, de Sevilla Muller LP, Brecha NC. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol. 2014;522:1411–1443. doi: 10.1002/cne.23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife. 2016;5 doi: 10.7554/eLife.19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tesic Mark M, Molina H, Tessier-Lavigne M. Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell. 2016;164:1031–1045. doi: 10.1016/j.cell.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 1991;546:18–32. doi: 10.1016/0006-8993(91)91154-s. [DOI] [PubMed] [Google Scholar]
- Summers DW, DiAntonio A, Milbrandt J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci. 2014;34:9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syc-Mazurek SB, Fernandes KA, Libby RT. JUN is important for ocular hypertension-induced retinal ganglion cell degeneration. Cell Death Dis. 2017a;8:e2945. doi: 10.1038/cddis.2017.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syc-Mazurek SB, Fernandes KA, Wilson MP, Shrager P, Libby RT. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol Neurodegener. 2017b;12:71. doi: 10.1186/s13024-017-0214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, Hoke A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J Peripher Nerv Syst. 2017;22:162–171. doi: 10.1111/jns.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife. 2017;6 doi: 10.7554/eLife.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Vargas ME, Barres BA. Local axonal protection by WldS as revealed by conditional regulation of protein stability. Proc Natl Acad Sci U S A. 2015a;112:10093–10100. doi: 10.1073/pnas.1508337112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao D, Pan B, Song Z, Shah SZ, Yin X, Zhou X, Yang L. Death Receptor 6 and Caspase-6 Regulate Prion Peptide-Induced Axonal Degeneration in Rat Spinal Neurons. J Mol Neurosci. 2015b;56:966–976. doi: 10.1007/s12031-015-0562-1. [DOI] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Mitchell KL, Jaskula-Ranga V, Sluch VM, Yang Z, Kim J, Buehler E, Patel A, Martin SE, Zhang PW, Ge Y, Duan Y, Fuller J, Kim BJ, Hamed E, Chamling X, Lei L, Fraser IDC, Ronai ZA, Berlinicke CA, Zack DJ. Enhanced Functional Genomic Screening Identifies Novel Mediators of Dual Leucine Zipper Kinase-Dependent Injury Signaling in Neurons. Neuron. 2017;94:1142–1154 e1146. doi: 10.1016/j.neuron.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Jr, Fuller J, Fu J, Cao LH, Han B, Auld D, Xue T, Hirai S, Germain L, Simard-Bisson C, Blouin R, Nguyen JV, Davis CH, Enke RA, Boye SL, Merbs SL, Marsh-Armstrong N, Hauswirth WW, DiAntonio A, Nickells RW, Inglese J, Hanes J, Yau KW, Quigley HA, Zack DJ. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24:639–662. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 2015;160:161–176. doi: 10.1016/j.cell.2014.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li S, Miao L, Huang H, Liang F, Teng X, Xu L, Wang Q, Xiao W, Ridder WH, 3rd, Ferguson TA, Chen DF, Kaufman RJ, Hu Y. Rescue of Glaucomatous Neurodegeneration by Differentially Modulating Neuronal Endoplasmic Reticulum Stress Molecules. J Neurosci. 2016;36:5891–5903. doi: 10.1523/JNEUROSCI.3709-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yan M, Wang H, Erickson S, Grewal IS, Dixit VM. Impaired c-Jun amino terminal kinase activity and T cell differentiation in death receptor 6-deficient mice. J Exp Med. 2001;194:1441–1448. doi: 10.1084/jem.194.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.