

Abstract
Energy is required to sustain life and enable stress adaptation. At the cellular level, energy is largely derived from mitochondria – unique multifunctional organelles with their own genome. Four main elements connect mitochondria to stress: (1) Energy is required at the molecular, (epi)genetic, cellular, organellar, and systemic levels to sustain components of stress responses; (2) Glucocorticoids and other steroid hormones are produced and metabolized by mitochondria; (3) Reciprocally, mitochondria respond to neuroendocrine and metabolic stress mediators; and (4) Experimentally manipulating mitochondrial functions alters physiological and behavioral responses to psychological stress. Thus, mitochondria are endocrine organelles that provide both the energy and signals that enable and direct stress adaptation. Neural circuits regulating social behavior – as well as psychopathological processes – are also influenced by mitochondrial energetics. An integrative view of stress as an energy-driven process opens new opportunities to study mechanisms of adaptation and regulation across the lifespan.
Keywords: Mitochondrion, Brain, ATP, Mitochondrial signaling, Mitokine, mtDNA, Chronic stress, CORT, HPA axis, Stress pathophysiology
“Without the vital force the material organism is unable to feel, or act, or maintain itself […]. Without the vital force the body dies.”
Samuel Hahnemann (1833)
1. Introduction
Life emerges when biological structures are animated by energy. Energy is defined as a fundamental entity of nature that is transferred between parts of a system in the production of physical change within the system, and usually regarded as the capacity for doing work (Merriam-Webster, 2017). Without energy, there is no life – molecules alone do not interact in meaningful ways, and complex structures do not assemble nor replicate. A crucial factor that distinguishes the breathing-living organism from the inanimate body (i.e., cadaver) is the flow of energy. There are no intrinsic differences in the molecular composition of dead and living organisms. But it is a required quality of living organisms to experience a constant flow of energy through and between their different parts. This flow of energy sustains the movement, chemical reactions, and dynamic changes in the position and organization of molecules that is required to think, feel, move, and execute every element of the stress response. Without energy, stress adaptation is not possible, and the body dies.
This review discusses the concept of stress as an energy-dependent and coordinated process, with an emphasis on the key role that mitochondria plays in these processes. With a broad view of energy, we take a multi-level approach exploring the role of mitochondria in glucocorticoid and catecholamine metabolism, and the effects of stress hormones on energy substrate distribution within the body. We also consider the reciprocal action of steroid hormones on mitochondria. The influence of stress on food/energy-seeking behavior and emerging evidence regarding the influence of mitochondrial functions on mental health, social behaviors, and physiological stress reactivity are also discussed. An energetic view of stress adaptation and pathophysiology opens new possibilities to rationalize and investigate the integrated adaptive and maladaptive effects of stress on the brain and body.
2. Mitochondria transform energy
In human/mammalian cells, a substantial fraction of energy flow occurs through a specific endosymbiotic organelle, the mitochondrion. Mitochondria are the only organelles that contain their own genome – the mitochondrial DNA (mtDNA). The mtDNA encodes proteins essential to electron flow through a series of protein complexes called the respiratory chain (also known as electron transport chain, or ETC) (Wallace, 2015). As its name implies, the respiratory chain consumes oxygen. Mitochondrial respiration channels high-energy molecular intermediates through a series of enzymatic reactions, transferring chemical energy from food substrates and oxygen into a trans-membrane electrochemical potential (ΔΨm) (Nicholls and Fergusson, 2013). This stored energy is then used to power various mitochondrial functions, including adenosine triphosphate (ATP) synthesis, calcium uptake, protein and molecular import, biosynthesis of macromolecules and hormones, among others. Thus, the breath provides oxygen to the mitochondria, sustaining membrane potential to ensure energy flow through the cell and the whole organism. Mitochondrial energy production then powers growth, healing, as well as the complex processes required for adaptation to the changing environment. Although mitochondria perform several “non-energetic” functions (Picard et al., 2016; Chandel, 2015), this article focuses on their role in energy metabolism.
In cases where there are defects in the energy flow, life is disrupted and shortened by disease. For example, individuals with genetic mitochondrial disorders due to inherited mtDNA mutations, which impair respiratory chain function, suffer from debilitating and often lethal symptoms (Gorman et al., 2016). Organisms with excessive mitochondrial dysfunction are incapable to sustain life and to adapt to dietary and physiological perturbations.
Stress – defined as a brain and body response aimed at promoting adaptation on the face of real or imagined threats to the organism’s homeostasis – cannot occur without energy. As discussed below, every aspect of the stress response requires energy: energy-dependent enzymatic reactions, transcription and translation enabling gene expression and protein synthesis, neurotransmitter release and reuptake, hormone biosynthesis, sympathetic activation, behavioral adaptations, and long-term structural remodeling of organs and tissues are all “powered”, and to some extent regulated, by cellular energy levels. Even though, in fact, basic life-sustaining biological functions also require energy for their maintenance, the energy requirement for stress responses is above the basal needs of the organisms; hence, the emphasis here on the link between stress and energy.
3. Defining stress, allostasis, and allostatic load/overload
“Stress” is a term often used loosely given the difficulties experienced by scientists from the stress field to arrive at a consensus on its definition. Some authors have proposed to restrict the use of the term to conditions in which environmental demands exceed the natural regulatory capacity of an organism, particularly when the stressor is unpredictable and uncontrollable (Koolhaas et al., 2011). Other authors – particularly those concerned with the impact of stress on human mental and physical health – make a distinction between positive and negative (‘toxic’) stress conditions (Shonkoff and Garner, 2012; Johnson et al., 2013). Here, and from a practical point of view, we propose to distinguish between good, tolerable, and toxic stress.
“Good stress” can arise from taking a chance on doing something one wants, like interviewing for a job, or giving a talk before strangers, and feeling rewarded when one is successful. There, stress mediators like cortisol and adrenalin promote adaptation during the challenge because they are turned on when needed and turned off when the challenge is over. “Tolerable stress” means that something bad happens, like losing a job, the end of a relationship, or the death of a loved one, but where the individual has the personal resources and support systems to “weather the storm” an be resilient. There, stress mediators are turned on and may stay turned on or are repeatedly turned on and off to promote adaptation; yet, by being chronically or repeatedly active, they may also promote potentially pathophysiological processes leading to cardiovascular, immune and metabolic dysregulation and changes in brain circuits involved in emotional regulation (McEwen, 1998). However, the individual’s internal resources and external support systems minimize this aspect and eventually create a resolution that limits the long-term pathophysiological consequences.
“Toxic stress” also means that something bad happens, like those events in “tolerable stress”, but where the individual lacks internal resources or external support systems, and, as a result, there is a lack of sense of control that leads to a chronic physiological dysregulation that promotes pathophysiology and results in something called “allostatic load” and “overload”, described below. Lack of control has been one of the defining features of psychological stress that leads to disease (Cohen et al., 2007). As a result, when toxic stress situations are prolonged, mental and physical health disorders develop over time. In this review, we use “stress” as an overall term for the body’s need to adapt to life experiences, particularly those that fall into the categories of “tolerable” and “toxic” stressors that differ in duration, severity, and degree of perceived control leading to some degree of allostatic load or overload.
The concept of “allostatic load” focuses on the paradox that the same mediators that help the body and brain adapt can also cause pathophysiology when overused and dysregulated. This terminology is more inclusive of life events than “stress”. “Homeostasis” represents the physiological state which the body maintains to keep us alive – that is, body temperature, pH, and blood oxygen levels are kept within a narrow physiological range. In order to maintain homeostasis, our body triggers hormone secretion and activates the autonomic and central nervous systems (we call these “mediators” like cortisol, adrenalin, the immune system, and metabolism) to help us adapt. For example, these systems are activated when we get out of bed in the morning, walk up a flight of stairs, have a conversation. These systems are also activated to varying degrees when we are surprised by something unexpected, or get into an argument, or run to catch a train. We may colloquially refer to some of these experiences as “stressful”, but for other experiences we do not, reflecting the imprecision of the term. Likewise, in research, “stress” is often used to denote molecular damage or dysfunction that occurs from a challenge, overuse, or even from toxins. But in a precise sense, any ever so slight perturbation to the system (such as standing up from the sitting position) induces a myriad of biological changes that aim to preserve homeostasis. So using the word “stress” does not really describe all of the underlying biology.
Rather, the term allostasis refers to the active process by which the “mediators” of the neuroendocrine, autonomic, metabolic, and immune systems help us adapt, as long as they are turned on in a balanced way when we need them and then turned off again when the challenge is over (Sterling et al., 1988). In other words, allostasis is what allows certain physiological parameters (e.g., blood glucose) to remain constant through changes in other parameters (e.g., insulin) – stability through change. When allostatic mediators are not turned off, these same mediators can cause unhealthy changes in brain and body. This is also the case when the mediators are not produced in an orchestrated and balanced manner – for example, too much or too little cortisol or an elevated or too low blood pressure. When dysregulation of these systems continues over weeks and months, we call it allostatic load, which refers to the wear and tear on the body that results from the chronic overuse and imbalance of the “mediators” (McEwen, 1998; McEwen and Stellar, 1993). Allostatic load also includes the consequences of the health-damaging behaviors that often accompany a stressful lifestyle or are present in society, like and unhealthy diet, alcohol, smoking, inadequate sleep, lack of exercise, social isolation. Accumulation of belly fat is an example of allostatic load, as is the development of chronic hypertension.
When the wear and tear is strongest, we call it allostatic overload and this is what is occurring in toxic stress and accompanying health-damaging behaviors and what, hopefully, can be minimized in tolerable stress (McEwen and Wingfield, 2003). An example is when hypertension leads to coronary artery blockade, and the belly fat which contributes pro-inflammatory cytokines that accelerate the coronary artery blockade and cause insulin resistance in Type 2 diabetes (Despres and Lemieux, 2006). Note, however, that we are talking, not about one mediator, like cortisol, but a host of mediators that are all released in allostasis in a coordinated, and in an energy-dependent manner, to help us adapt but which can also cause damage when overused and dysregulated. Below we consider the role of energy and mitochondria in the molecular, cellular, and organismal processes that orchestrate allostasis.
4. Energy is required for life and stress adaptation
Two billion years ago, life on earth was limited to unicellular life forms. Complex life arose from a symbiotic relationship whereby the ancestral eukaryotic cell engulfed an aerobic (i.e., capable of using oxygen to produce energy) bacterium (Margulis and Bermudes, 1985). This union of cell and bacterium, which eventually evolved into mitochondria populating the cell cytoplasm, likely constituted the igniting point for the evolution of complex life (Lane and Martin, 2010). The larger amount of energy afforded by hundreds of oxygen-consuming mitochondria thus enabled the regulation of the human genome comprising of > 25,000 genes, eventually culminating in the development of different cell types, multi-cellular tissues, organs, and breathing bodies (Wallace, 2010). Every cell of the human body contains a variable content of 100–1000's of mitochondria, determined by energy demand of each cell type. Thus, the structure and function of the human body is closely linked to energy metabolism in general, and to mitochondrial function in particular.
Stress adaptation, or allostasis, also requires energy. Biologically, energy is necessary to allow plasticity both during development and to enable adaptation and remodeling of mature tissues and organs. The requirement for energy to enable change is a conserved principle well beyond biology. As a simple analogy, imagine a blacksmith attempting to alter the shape of a metallic object. The blacksmith must infuse considerable energy, in the form of heat, into the piece of metal only to make it responsive to the force of the hammer. Here, two different forms of energy are at play: one that contains little information but makes the structure malleable or plastic (i.e., the heat), and one more directed form that informs the transformation (i.e., the velocity and direction of the hammer). In stress biology, both mitochondrial oxidative phosphorylation and glycolysis contribute the “heat” that makes the organism malleable, providing the basic energy supply necessary to enable adaptation, or plasticity; whereas circulating neuroendocrine hormones and other factors interact with cellular-level epigenetic and molecular factors to direct adaptation.
In the presence of “heat” energy, circulating neuroendocrine factors both initiate and guide adaptation. For example, acutely glucocorticoids and catecholamines interact with their respective receptors to trigger processes that engage cellular plasticity mechanisms within neurons and specific brain regions (McEwen, in press). Other circulating factors such as sex hormones, inflammation, and the metabolic state may in turn influence the rate and extent to which stress mediators induce plasticity (McEwen, in press). These factors, which modify the susceptibility of the brain and other systems to given stressors, manifest physiologically as vulnerability or resilience factors. On the other hand, systemic signals must interact with molecular and cellular factors contained within cells including age, genes, the epigenome, and other molecular mechanisms responsible for registering information from past experiences. Together, the information carried by circulating and cellular factors must be integrated, in an energy-dependent process, to direct coordinated cellular and physiological responses aimed at promoting adaptation in the face of stress (Fig. 1).
Fig. 1.
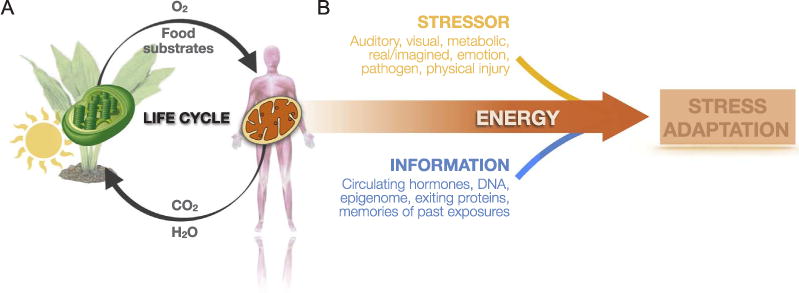
Mitochondria sustain life and enable stress adaptation. (A) Within mammalian cells, mitochondria perform exactly the opposite reaction as the plant chroloroplasts. Powered by solar energy, plants produce oxygen and food substrates (carbohydrates, lipids), which are used within mitochondria to power oxidative phosphorylation and ATP synthesis. In this process, mitochondria release carbon dioxide (CO2) and water (H2O), the substrates required by plants, thus sustaining the cycle of life. (B) Stressors interact with information contained within the organism, such as genetically encoded biological constitution, memories of past events, and the current psycho/physiological state reflected by molecular, neuroendocrine, immune, and metabolic factors. Together, this generates unique adaptive stress responses and behaviors. Without energy, stressors would have no effect on the organism. But in the presence of heat and chemical energy, stressors and information interact in meaningful ways to enable stress adaptation.
4.1. The role of mitochondria in the epigenetic embedding of information
In order for a complex system like the human body to adapt, stress signals from the environment must be transduced into both short- and long-term biological changes that modify cellular composition and functions. A fundamental process at the origin of widespread cellular adaptations is transcriptional regulation of genomic elements, or changes in gene expression. This involves either the activation or repression of specific genes, or group of genes. Genes encode proteins, the basic building blocks that determine and maintain cellular specificity and function. But in complex multicellular organisms, function must be matched to the available supply of energy – a cell should only attempt to divide if there are sufficient levels of the right energy substrates. Given the role of mitochondria in evolution of complex life forms, it is logical that a cell’s gene expression, and in particular basic cellular operations such as cell division, growth, and size regulation (Picard et al., 2014), and even cell death, would be coupled to mitochondrial metabolic signals (Chandel, 2015; Kasahara and Scorrano, 2014; Picard, 2015).
To achieve coupling of energetic environment and cellular behavior, information about the cellular environment are transformed, through signal transduction mechanisms, into metabolic intermediates (Acetyl CoA, α-ketoglutarate, succinate, NAD+) and reactive oxygen species (ROS) that reach the nucleus (Shaughnessy et al., 2014). Once in the nucleus, mitochondrial metabolic intermediates interact with the genome and the chromatin – the genes and associated regulatory proteins. There, metabolic signals contribute to activate or repress gene expression in two main ways.
First, they influence the interaction of transcription factors and coactivators with specific genetic elements, which influence transcript (messenger RNA, mRNA) formation (Hao and O’Shea, 2011). For example, during hypoxia, mitochondria relocate near the nucleus where they produce an oxidized environment via ROS production, and which contributes to the activation of hypoxia-inducible factor 1 α (HIF1α) (Al-Mehdi et al., 2012). Energy deficiency also activates specific cytoplasmic energy sensors (mTORC1, AMPK) and their downstream transcriptional coactivators (e.g., PGC-1α) and transcription factors (e.g., PPARγ) that have widespread effects on nuclear gene expression.
The second and longer-lasting set of mechanisms to transduce metabolic signals into transcriptional changes involves epigenetic (from the greek “epi”, meaning “on top of”) modifications. Combinations of epigenetic modifications of the DNA itself and on the histone tails, including but not limited to acetylation, methylation, phosphorylation, succinylation, ubiquitination, and other, form the basis of the epigenetic “code” (Jenuwein and Allis, 2001; Linghu et al., 2013). Chemical modifications of histone tails at specific loci on the genome can either repress or facilitate the expression of given genes, depending on the nature, location, and specific combinations of epigenetic marks.
Interestingly, metabolic intermediates that are the substrates or cofactors for epigenetic modifications are all derived from the Krebs cycle and other metabolic pathways within mitochondria (reviewed in (Matilainen et al., 2017; Gut and Verdin, 2013). For example, citrate, which is produced by the mitochondrial enzyme citrate synthase, is exported from mitochondria to the cytoplasm. There, citrate is transformed into Acetyl-CoA by ATP citrate lyase, and thereafter serves as a substrate for histone acetylation (Wellen et al., 2009). Normal mitochondrial metabolism is required for histone acetylation, as well as HIF-1α function and cell proliferation (Martinez-Reyes et al., 2016). On the other hand, the removal of epigenetic marks also requires mitochondrial substrates. Histone and DNA demethylation reactions can only proceed in the presence of the cofactor α-ketoglutarate (Klose and Zhang, 2007), also a metabolic intermediate of the Krebs cycle.
Thus, both the addition and removal of epigenetic marks are metabolically – or mitochondrially – regulated. Moreover, in addition to the shared chemical “language” between mitochondria and the epigenome, mitochondria are in close physical proximity to the nucleus and nuclear pores where molecular information transit in and out of the nucleus (Picard, 2015). This presumably facilitates the diffusion of metabolic signals that reflect the integrated state of mitochondrial metabolism to the epigenetic machinery. Combined, these metabolic-to-epigenetic transduction systems may explain how, in an in vitro system, mitochondria regulate the expression of over half of the genes in the human genome (Picard et al., 2014). Likewise, these systems provide a basis to explain how in complex organisms the epigenetic embedding of stress exposure may be regulated by mitochondrial metabolism.
5. Stress increases energy demand
Energy is present in two main forms in living organisms: (i) as heat, which permeates all structures of living organisms; and (ii) as chemical energy, in the form of chemical intermediates such as ATP that fuels specific enzymatic or biophysical reactions. Heat is the energy associated with the random (brownian) motion of molecules. The origin of heat in the human body is the free energy released during the chemical breakdown of molecules. In mammals, a substantial amount of such reactions occurs in the mitochondrial matrix, a compartment filled with enzymes and insulated by a double-membrane layer. As a result, mitochondria is a site of cellular thermogenesis (Okabe et al., 2012). In mammalian cells, where enzymatic activity of the electron transport chain complexes is maximal, mitochondria are estimated to effectively function at temperatures around 50 °C (Chretien et al., 2018).
Mitochondria are therefore cellular sources of heat and account in part for the warm bloodedness of mammalian cells and organisms. In fact, the main mechanism of heat production and thermoregulation in infants, as well as in rodents, consist in uncoupling chemical reactions in the mitochondrial matrix from ATP synthesis, a phenomenon called “mitochondrial uncoupling”. Chemical heat-releasing reactions thus occur unobstructed and thus increase core body temperature (Chouchani et al., 2016). Chemically uncoupling mitochondria in vitro also results in increased cellular temperature (Hayashi et al., 2015). Although a minor leak or uncoupling is ubiquitous in all mitochondria, physiological mitochondrial uncoupling occurs mostly in a specialized tissue – brown fat – through the action of uncoupling protein 1 (UCP1), which inserts into and makes the inner mitochondrial membrane leaky to protons, thus dissipating ΔΨm (Nedergaard et al., 2001).
When the inner mitochondrial membrane is intact, the transmembrane gradient generated can be efficiently used to power the ATP synthesis with minimal heat release. In humans, only ∼25% of the consumed oxygen is transduced into mechanical work during walking for example (Margaria et al., 1963), indicating that this process is not completely efficient. This is also evidenced by the rise in body temperature during exercise, reflecting the loss of energy as heat during movement. The portion of energy that is eventually transferred to ATP synthesized within mitochondria is then exported into the cytoplasm. In the cytoplasm, mitochondria-derived ATP provides the activation of energy for a large number of enzymes and chemical reactions that sustain cellular life and enable allostasis, including gene expression and protein synthesis.
During gene expression in the cell nucleus, the RNA polymerase (RNA Pol II) initiates and sustains transcription in an ATP-dependent manner (Kopytek and Peterson, 1998; Yan and Gralla, 1997). Subsequently, the synthesis of polypeptides by ribosomes also requires four ATP/GTP molecules for each amino acid incorporated into the nascent proteins (Jewett et al., 2009), making protein synthesis a highly energy-demanding process (Kafri et al., 2016). Furthermore, these and many other reactions essential to life occur in the cytoplasm, whose chemical and electrical properties must be kept distinct from those of the extracellular environment. This is accomplished in part through the action of the ‘energivorous’ Na+/K+ ATPase pumps (Clarke et al., 1827). ATP hydrolysis by the NA+/K+ ATPase is also a substantial source of heat thought to contribute to thermogenesis (Rolfe and Brown, 1997). Thus, maintaining the basic conditions for cellular life, from protein synthesis to the electrochemical cellular membrane potential, depends on the constant transformation of energy and heat release.
Beyond the cell, allostasis also involves widespread energy-demanding physiological changes. Consider for example the increase in heart rate associated with the stress response. The heart is a collection of different cell types where each contraction involves the hydrolysis of billions of ATP molecules, which are required to unidirectionally transmit the wave of depolarization/repolarization, for the actin-myosin cross-bridging that provide the power stroke of contraction during systole, and for Ca2+ pumping enabling relaxation during diastole (Suga et al., 1993). Psychological stress alone increases heart rate and blood pressure by > 10–20% (Schubert et al., 2009), which would correspond to an equivalent increase in cardiac energy demand. In turn, this increase in cellular energy demand also results in systemic changes such as increased breathing rate and minute ventilation, which also further increase total energy consumption. These systems-level physiological changes ultimately reflect – or are subservient, to some extent – the increase in ATP-consuming reactions within cells, and the corresponding increase in oxygen consumption within the mitochondria of the contracting heart. The same is also true of other metabolically active organs such as the brain.
Similarly, complex stimuli such as social interactions and other stressors are associated with behavioral responses which incur elevation in energy demand, also met by mitochondrial energy transformation. Locomotion, fighting, vocalization, licking and grooming, and any associated cognitive processes necessarily incur increased energy consumption at the cellular level (Magistretti and Allaman, 2015). Stress in particular increases cerebral energy demand including oxidation of glucose and oxygen consumption (Bryan, 1990), reflecting increased mitochondrial activity within the brain. Interestingly, the stress- induced increase in cerebral energy demand may require adrenergic signaling by catecholamines (Bryan, 1990; Carlsson et al., 1977), indicating the interaction of stress mediators and mitochondrial metabolism (see Section 9).
Overall, biological processes starting from the basic sustenance of vital functions, acutely responding to daily stressors, all the way to the permanent adaptation to chronic stress, require substantial amount of energy. In humans and other mammals, the energetic demand of allostasis is largely met by mitochondrial respiration. Mitochondria transform circulating energy substrates into ATP and metabolic signals that endow biological structures with the necessary heat for plasticity, and interact with the information stored and communicated across organ systems to direct integrated, adaptive stress responses. This is in part achieved by the production of broad acting hormones like glucocorticoids and catecholamines.
6. Mitochondria synthesize and metabolize glucocorticoids and catecholamines
To match increases in mitochondrial energy demand, physiological mechanisms must be activated to ensure the availability of sufficient energy substrates systemically. This occurs via the action of hormones that increase circulating concentrations of glucose and lipids. Before we describe the effects of hormones on circulating levels of metabolic substrates and mitochondrial functions, we consider the role of mitochondria in the synthesis of these hormones and in their metabolism.
Mitochondria, which have so far been described as the major source of cellular energy, are also the site of synthesis for all steroid hormones (Bose et al., 2002). This includes progestogens (e.g., progesterone), mineralocorticoids (e.g., aldosterone), glucocorticoids (e.g., cortisol and corticosterone), androgens (e.g., testosterone), and estrogens (e.g., estriol) (reviewed in (Midzak and Papadopoulos, 2016).
Glucocorticoids synthesis takes place in the zona fasciculata of the adrenal cortex, and occurs via a series of molecular reactions catalyzed within the mitochondrion Fig. 2. The first step in steroidogenesis is the import of cholesterol through the mitochondrial membranes by the steroidogenic acute regulatory protein StAR (Clark, 2016). StAR is activated by a protein kinase A (PKA)-dependent phosphorylation event induced by ACTH signaling, and is a rate-limiting step in steroid synthesis (Bose et al., 2002). Once in the mitochondrial matrix, the first enzymatic reaction transforms cholesterol into pregnanolone, catalyzed by the cytochrome P450 side chain cleavage enzyme (P450SCC). Pregnanolone is then exported to an adjacent organelle, the endoplasmic reticulum (ER), where a series of enzymatic reactions convert it to deoxycorticosterone (mostly in rodents) or 11-deoxycortisol (mostly in humans). The ER is closely associated with mitochondria in a number of tissues, where inter-organelle contacts are important for the function of mitochondria (Klecker et al., 2014; Booth et al., 2016). The terminal reaction, catalyzed by the mitochondrial enzyme 11β-hydroxylase (11βH) then generates cortisol (human) or corticosterone (rodent) in the mitochondrial matrix. How glucocorticoids are released from mitochondria is not well defined, but presumably driven by the concentration gradient from the mitochondrial matrix, the cytoplasm, and systemic circulation (blood), facilitated by the hydrophobic nature of steroid hormones allowing them to diffuse through lipid membranes.
Fig. 2.
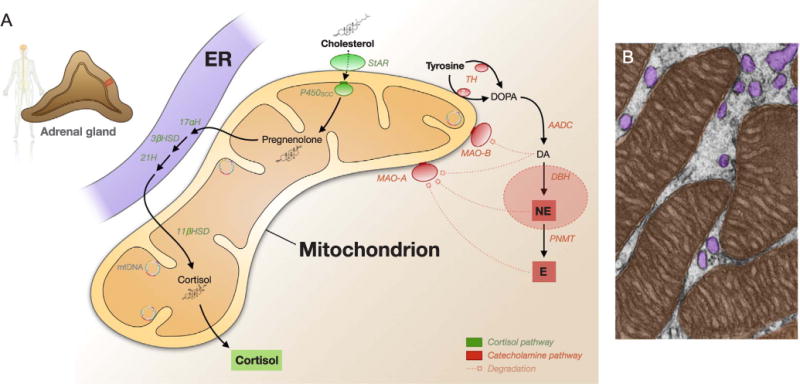
Cortisol and catecholamine metabolism. (A) Steroidogenesis takes place in mitochondria. The first step involves the rate-limiting import of cholesterol into mitochondria by the steroidogenic acute regulatory (StAR) protein, followed by the side chain cleavage to pregnenolone by P450SCC, three enzymatic reactions in the endoplasmic reticulum, and the final 11-β-hydroxylase reaction catalyzing cortisol synthesis in the mitochondrial matrix. Mitochondria are particularly enriched in the adrenal cortex where glucocorticoids are synthesized in response to ACTH. Also shown is the catecholamine pathway which mostly occurs in other cell types, including tyrosine hydroxylase, which may become associate with mitochondrial membranes under some conditions (see text for discussion), and the mitochondria-anchored monoamine oxidases A and B, which degrade catecholamines in specific tissues. Degradation products are not shown. (B) Electron micrograph of the zona fasciculata of the hamster adrenal cortex with pseudocolored mitochondria (orange) and endoplasmic reticulum (purple). Picture modified from (Fawcett, 1981). ER: Endoplasmic reticulum; StAR: Steroidogenic acute regulatory protein; P450SCC: Cytochrome P450 side chain cleavage (Cyp11a1, inner mitochondrial membrane bound); 17αH: 17-alpha hydroxylase; 3βHSD: 3β-hydroxysteroid dehydrogenase; 21H: steroid 21-hydroxylase; 11βHSD: 11-β-hydroxylase; TH: Tyrosine hydroxylase; AADC: Aromatic L-amino acid decarboxylase; DBH: Dopamine β-hydroxylase; PNMT: Phenylethanolamine N-methytransferase; MAO-A/B: Monoamine oxidase A/B.
Another important class of hormones released in response to certain stressors are catecholamines, particularly norepinephrine (NE) and epinephrine (E). Both are derived from the neurotransmitter dopamine, itself generated from the amino acid tyrosine. Interestingly, the biosynthetic enzymes involved in catecholamine degradation, the monoamine oxidases MAO-A and MAO-B are anchored to the outer mitochondrial membrane (Binda et al., 2011). Some reports also suggest that TH may be tethered to the mitochondrial surface (Wang et al., 2009; Baumann et al., 2016), although this may be tissue specific and inducible under certain conditions; more work is needed to examine this potential functional association of mitochondria with catecholamine synthesis and degradation.
Clinically, in relation to glucocorticoids, it has also been shown that primary mitochondrial dysfunction due to mutations in genes encoding mitochondrial proteins, can cause steroidogenesis defects. For example, NNT (nicotinamide nucleotide transhydrogenase) is a mitochondrial protein located in the inner mitochondrial membrane where it uses the transmembrane potential to regenerate NADPH and the intra-mitochondrial antioxidant system and impact energy metabolism (Fisher-Wellman et al., 2015). Mutations of its gene cause severe familial hypocortisolemia (Meimaridou et al., 2012; Meimaridou et al., 2013). In mice lacking NNT, corticosterone is also lower and its release during acute psychological stress blunted by > 50% (Picard et al., 2015), demonstrating that normal mitochondrial function is required for general steroidogenesis and stress-induced corticosterone release. It remains unclear why the organism has evolved in such a way to position the key steps of steroidogenesis, including glucocorticoids and sex hormones, inside mitochondria. One possibility is that the mitochondrial matrix offers the optimal biochemical environment – both ionic and redox state – for the side chain cleavage and hydroxylation reactions that transform cholesterol into glucocorticoids (Hanukoglu et al., 1981). This would seem consistent with the deleterious effects of NNT deficiency-associated redox imbalance on corticosterone/cortisol synthesis in mice and humans. Another possibility is that since these hormones are major regulators of energy metabolism (see below), coupling their synthesis to the major site of energy transformation in the organism (i.e., mitochondria) might have created some physiologically advantageous conditions – enhanced coupling of energy metabolism to endocrine responses – thus maximizing adaptation, and as a result, selection of mitochondrial steroid synthesis during evolution.
7. Stress hormones mobilize energy substrates
Mitochondria fuel the stress response in two main ways. As described above, they use substrates (glucose, lipids, amino acids) and oxygen to provide energy intracellularly, via the transformation of energetic substrates and oxygen into ATP. In a related way, as described above, they also contribute to the synthesis of stress hormones, which mobilize these same energetic substrates into the circulation.
Glucocorticoids (glucose, adrenal cortex, steroid) were originally named based on their ability to increase blood glucose concentration. Adrenalectomized animals are incapable to synthesize CORT, as are individuals with adrenocortical deficiency (Addison’s disease), and as a result suffer from persistent hypoglycemia (Auron and Raissouni, 2015). In humans, glucocorticoids elevates circulating levels of glucose and lipids metabolites within minutes (De Feo et al., 1989), indicating their widespread role in the regulation of systemic metabolism.
Glucocorticoids increase circulating glucose levels by acting simultaneously on the liver, skeletal muscles, and adipose tissue (reviewed in (Magomedova and Cummins, 2016). In these target tissues, glucocorticoids signal via the action of the glucocorticoid receptor (GR). In the liver, GR activation has pleiotropic effects where it induces chromatin remodeling (Wang et al., 2004) and transcriptionally induces key genes that encode for rate-limiting enzymes for gluconeogenesis (i.e., the synthesis of glucose from other carbon sources) (Magomedova and Cummins, 2016; Vander Kooi et al., 2005). This leads to net hepatic glucose synthesis and output into the blood. In mice, inactivation of GR specifically in hepatocytes is sufficient to cause hypoglycemia (Opherk et al., 2004), demonstrating the major role of GR signaling and liver glucose output in glycemic control.
Outside the liver, in skeletal muscle, GR activation acts in two main ways. First, it prevents glucose entry into myocytes by preventing the translocation of insulin-sensitive glucose transporters GLUT4 (Weinstein et al., 1998). Second, GC activity antagonizes several elements of insulin signaling and inhibits the uptake of pyruvate, a breakdown product of glucose oxidation, by mitochondria (Magomedova and Cummins, 2016). The mechanism underlying this effect involves the transcription of pyruvate dehydrogenase kinase isoform 4 (Pdk4) by GC (Connaughton et al., 2010). Because skeletal muscles are normally a major glucose “sink”, GC-induced inhibition of skeletal muscle glucose uptake and its oxidation by skeletal muscle mitochondria cause a substantial accumulation of glucose in the blood, thus making it available for the brain, heart, and other organ systems most in need during stress. In adipocytes also, where fat is stored, glucocorticoids also antagonize insulin signaling and inhibits GLUT4 translocation to the plasma membrane, preventing glucose uptake and thus, as for skeletal muscle, passively raising blood glucose (Caperuto et al., 2006). Overall, glucocorticoids plays a number of roles on target tissues aimed at increasing circulating glucose levels. Although not discussed in details here, glucocorticoids also regulate multiple processes related to lipid metabolism that culminate in increasing their systemic levels.
In the absence of real stressors and without the need to engage in physically demanding behavioral responses such as running away or fighting, stress hormones can dysregulate metabolism. In humans, individuals with higher circulating levels of cortisol under resting (non-stressed) conditions also have higher levels of glucose and triglycerides, and a higher score reflecting insulin resistance and a pre-diabetic state (Phillips et al., 1998) Fig. 3A and B). Likewise, in mice, chronic glucocorticoid administration results in elevated triglycerides, glucose intolerance, and weight gain (Karatsoreos et al., 2010). These metabolic changes are associated with higher levels of insulin and leptin hormones, indicating cross-talk between these systems. And in addition to their effects on metabolism, chronic glucocorticoid administration can also induce physical inactivity and depressive-like behavior (Karatsoreos et al., 2010; Gourley and Taylor, 2009).
Fig. 3.
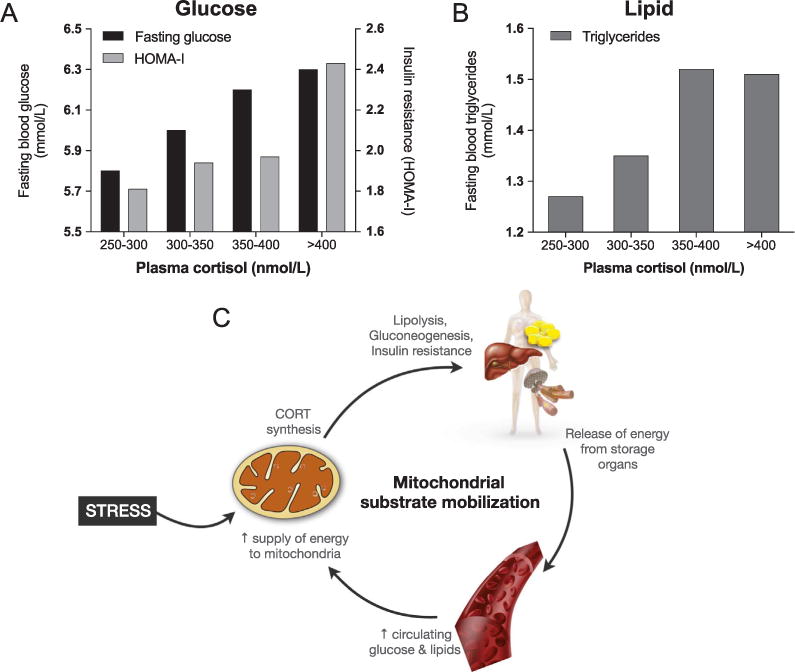
Interplay of primary stress mediators, glucose and lipids, and mitochondria. (A) Fasting blood glucose levels and the homeostatic model assessment of insulin resistance (HOMA-IR) index reflecting insulin resistance, and (B) blood triglycerides as a function of resting plasma cortisol concentration categories. These data reflect dose-response association between glucocorticoids and metabolic stress, in line with the glucose-mobilizing properties of cortisol (CORT). Data are from 286 individuals, adapted from (Phillips et al., 1998). (C) Mitocentric model of mitochondrial substrate mobilization to serve their bioenergetic needs in target tissues. Mitochondria in the adrenal cortex are the source of CORT, whereas mitochondria in other tissues are the recipient of resulting increases in circulating energy substrates used for oxidative phosphorylation, ATP synthesis, and metabolic signaling.
These coordinated physiological changes cooperate to increase circulating levels of energetic substrates to fuel the mitochondria of metabolically active organs, such as the brain and the heart, most in need during stress. In this emerging mitometabolic picture, mitochondria-derived hormones from the adrenal cortex, a specialized tissue, calls upon energy stores and reprograms metabolism in other tissues. This represents a form of “mitocrine” signaling – systemic endocrine signaling by a mitochondria-derived hormone Fig. 3C. Mitochondria-derived molecules produced in certain cells thus affect the function of other cells and their mitochondria in a cell non-autonomous manner (Schinzel and Dillin, 2015). Therefore, glucocorticoid hormones synthesized in mitochondria can be regarded as mitokines.
Mitochondria also produce and secrete other mitokines, such as humanin, mitochondrial open reading frame of the 12S rRNA-c (MOTS-c), and small humanin-like peptides (SHLP1-6), which influence systemic energy homeostasis and regulation of whole organisms functions (reviewed in (Kim et al., 2017). Together with their role as site of steroid hormone synthesis, these discoveries are positioning mitochondria as important endocrine organelles particularly active during stress.
8. Glucocorticoids: Not just “stress”
Glucocorticoid secretion has important functions other than responding to stressors, namely, coordinating waking and sleeping functions during the diurnal cycle. A defining characteristic differentiating wakefulness and sleep states is the metabolic demand, which along with body temperature, decreases significantly > 15–30% during sleep (Ryan et al., 1989; Jung et al., 2011). Cortisol levels are lowest in the early phase of sleep, and undergo an elevation before wakening, promoting increased locomotor activity and appetite (McEwen et al., 1993). A flat glucocorticoid diurnal rhythm leads to a sluggish stress-induced increase of ACTH (Jacobson et al., 1988; Akana et al., 1988).
Consistent with the role of glucocorticoids on the brain, plasticity of the brain extends to the day-night (diurnal) cycle of waking and sleeping. Some, but not all, synapses in many parts of the cerebral cortex turn over during the diurnal cycle due to the ultradian fluctuation of glucocorticoids, and interfering with that cycle by elevated glucocorticoids at the wrong time of day interferes with motor learning, like learning to play golf (Liston et al., 2013). The formation of new spines (Li et al., 2004), dynamic regulation of synaptic strength (Kwon et al., 2016; Sun et al., 2013), and terminal axon branching (Courchet et al., 2013).
Considering how many ways modern humans interfere with the natural day-night rhythm, for example, by turning on a light in the middle of the night, this is a lesson to all to give the “wisdom of the body” a better chance to help us. Another way we interfere with the natural cycle is through shift work and jet lag. An animal model of shift work caused dendrites in the prefrontal cortex to shrink and the animal to become cognitively rigid when challenged with a memory task that required changing the rules (Karatsoreos et al., 2011). The prefrontal cortex governs “self-regulation” and the ability to regulate emotions and impulses as well as working memory.
Moreover, the “shift work” animals became fatter and insulin resistant, signs of pre-diabetes and depressive-like behavior (Karatsoreos et al., 2011). Shift work in our own species is associated with greater obesity, diabetes, cardiovascular disease and mental health problems (McEwen and Karatsoreos, 2015; Bowles et al., 2017). This story is relevant to the health effects of shift work in transit workers (Bowles et al., 2017). One of the mediators for circadian disruption-related obesity, acting peripherally, are endocannabinoids acting via the type-1 cannabinoid receptor CB1 (Bowles et al., 2015). Disrupted patterns of glucocorticoids are linked to circadian disruption induced obesity, and CB1 activity is necessary for the obesogenic effects of glucocorticoids (Bowles et al., 2015). The CB1 receptor, which localize at the plasma membrane of the presynaptic terminal, is also found in mitochondria (Benard et al., 2012). Upon activation, mitochondrial CB1 can decrease mitochondrial oxidative capacity and modulate memory in a mitochondria-dependent manner (Hebert-Chatelain et al., 2016). Thus, as discussed in the next section, glucocorticoids and other systemic signaling molecules may converge upon mitochondria to induce systemic changes in the metabolism of the brain and multiple other tissues.
9. Glucocorticoids influence mitochondrial functions
In an intriguing reciprocal connection, mitochondria are not only the source of glucocorticoids and other systemic signaling molecules, but are also affected by them. Here we specifically focus on the effects of glucocorticoids on mitochondria and the some of the underlying mechanisms. Mitochondria have emerged as major players in steroid hormone actions and containment of excitotoxicity because of their ability to generate free radicals and to sequester Ca2+ ions to contain that process, as well as to express genes from its own genome and receive nuclear coded genes that, together, regulate a host of important cell functions (Picard et al., 2016).
Mitochondria are also known to respond to GR translocated into them (Moutsatsou et al., 2001; Psarra and Sekeris, 2009). In cultured neurons, the synthetic glucocorticoid dexamethasone exerts biphasic effects on mitochondrial function in which physiological levels of glucocorticoids promote Ca2+ sequestration, moderate oxidation and maintain mitochondrial membrane potential while supra-physiological glucocorticoid levels start to do so but fail after 24 h and lead to a failure of all three measures (Du et al., 2009). In other cell types, chronic glucocorticoid treatment can reduce the activity of specific mitochondrial electron transport chain complexes and increase mitochondrial ROS production (Tome et al., 2012; He et al., 2017). The underlying mechanisms for these effects are not fully understood, but there are three glucocorticoid response elements (GREs) on the circular mtDNA (Psarra and Sekeris, 2009), where GR likely bind and influence mtDNA gene expression (Psarra and Sekeris, 1813; Hunter et al., 2016).
It is also important not to ignore estradiol, and possibly other steroid hormones, which regulates mitochondrial respiratory capacity and oxidative stress, since estradiol receptor beta is also translocated into mitochondria and has neuroprotective actions (Rettberg et al., 2014). In particular, after a surgical menopause, as shown in rhesus monkeys, cortical neurons presynaptic terminals contain donut-shaped mitochondria that are negatively correlated with working memory (Hara et al., 2014; Picard and McEwen, 2014), where estradiol treatment may be particularly important as a neuroprotective strategy. The GR is ubiquitously expressed across tissues, and the mechanisms regulating how mitochondria in different cell types respond to glucocorticoid stimulation remains to be elucidated.
10. Effects of psychological stressors on food- and energy-seeking behaviors
Stress increases energy demand at the cellular level and activates systemic mitocrine and endocrine processes to sustain this increased demand. Based on the increased energetic needs associated with stress responses and allostasis, and the energy-mobilizing effect of mitochondria-derived glucocorticoids, it is logical to expect that stress would be coupled to changes in food- and energy-seeking behaviors. Indeed, there is evidence, described below, that psychological stress triggers eating behavior, shifts macronutrient preferences to denser calories, and shift fat storage to the intraabdominal (i.e., visceral) fat stores, a form of energy storage with more rapidly mobilized free fatty acids than subcutaneous fat depots.
Historically, in animal studies, stressor exposure led to anhedonia, lack of interest in food, and weight loss. However, these conclusions were based on loss of interest in the standard lab chow. Once studies started making highly palatable food (high fat, high sugar) accessible, the opposite effect of stress on food consumption was observed. In rodent models, when animals are given intermittent access to calorically dense junk food, they tend to overeat and gain weight, likely due to changes to the reward system (Boggiano and Chandler, 2006). However, if on top of this, they are also exposed to psychological stress, they develop excessive drive to eat, high cortisol, and binge eating (Boggiano and Chandler, 2006). When rats are exposed to stress, particularly subordination stress, and given the opportunity to consume lard or sugar, they binge eat (Razzoli et al., 2017) and develop excessive abdominal fat (Dallman, 2010), a highly reliable source of energy for times of stress.
In humans, there are also well documented relationships between exposure to both acute and chronic stress with greater food seeking and food intake, particularly of comfort food (Masih et al., 2017; Adam and Epel, 2007). A classic study also showed that when administered glucocorticoids men had increased energy expenditure but also a disproportionate increased appetite, resulting in weight gain (Tataranni et al., 1996). In a double-blind, randomized, cross-over study assessing metabolic responses to a high fat meal in relation to stress, previous day stress was associated with lower fat oxidation as well as higher insulin production (Kiecolt-Glaser et al., 2015), both physiological processes that favor “energy storage”. Together, these studies show that stress and glucocorticoids in particular may lead to energy seeking and replenishment behavior. This can help maintain allostatic balance both acutely and chronically by replenishing adipose energy stores over periods of stress exposure.
Although this has not been examined in clinical studies of feeding behavior, it may be that mitochondria, via their dynamic processes of fusion with each other and their interaction with other organelles, play a role in sensing circulating levels of energy substrates. Mitochondria can respond within minutes to levels of glucose and lipids by undergoing fusion, leading to their elongation and branching of smaller mitochondria (Gomes et al., 2011; Rambold et al., 2011); or fission, leading to the fragmentation of longer mitochondria into shorter globular organelles (Shenouda et al., 2011; Yu et al., 2008) (reviewed in (Liesa and Shirihai, 2013). In turn, changes in mitochondrial shape or morphology are associated with functional recalibrations linked to mitochondrial signaling (Picard et al., 2013). Based mostly on evidence from in vitro systems and animal models, chronic inhibition of mitochondrial fusion leading to persistent fragmentation of mitochondria might contribute to the adverse and pro-aging effects of metabolic stress (Picard and Turnbull, 2013). In physiological conditions, changes in mitochondrial shape enable neurons of the hypothalamus to sense glucose (Ramirez et al., 2017) and lipid (Schneeberger et al., 2013; Benani et al., 2007), triggering satiety and regulating insulin and energy balance. Thus, dynamic mitochondrial responses to circulating energy levels may be involved in regulating allostasis systemically.
The evidence reviewed in this section suggest that stress and the associated mitochondrial-derived glucocorticoids play a role in furthering energy seeking behavior. Combined with evidence presented earlier, there are therefore three main ways by which mitochondria contribute to the metabolic aspect of stress responses: (i) mitochondria provide energy in the form of ATP and metabolic signals to power cellular events during stress; (ii) they produce stress hormones that coordinately ensure adequate circulating levels of energy substrates; and (iii) mitochondria in specialized tissues sense and monitor circulating levels of metabolic substrates and generate signals that direct food seeking behavior. These levels are obviously nested within one another, where eating behavior supply food substrates for the circulation, which provides the fuel for mitochondria to convert into ATP. From an evolutionary perspective, stress-induced energy/food-seeking behaviors likely aim to replenish energy stores that are depleted during the active coping process.
Thus, mitochondria in different tissues execute different roles but collectively, via communication among each other, ensure the efficient sensing, integration, and signaling of metabolic information. This multi-site stress regulation of metabolism is reminiscent of distributed control networks, recognized to optimize the function of complex systems in complex environments (Lian et al., 2002). This intriguing set of mitochondrial roles – from cellular to behavior – lead us to consider the potential involvement of these multifunctional organelles as a link between metabolic dysregulation and psychopathology.
11. Mitochondria link metabolic dysregulation and depression
Mitochondria play a key role in the allostasis of neurotransmission and depression. Dysfunctional mitochondria may promote oxidative stress and the inflammatory tone that contribute to the depressive state (Raison et al., 2009). A major determinant of mitochondrial function and of the signals they generate is the energy substrates that they metabolize. Within the brain and other tissues, mitochondrial fatty acid metabolism is facilitated by esterification with carnitine (Fritz and McEwen, 1959). A short-chain acylcarnitine; acetyl-L-carnitine (LAC), not only protects and enhances mitochondrial function (Rosca et al., 2009) but also acts as an acetyl donor for metabolism and for epigenetic modification of histones (Nasca et al., 2013) and for mitochondrial proteins in a biphasic manner (Kerner et al., 2015; Hirschey et al., 2011). Under biphasic control by glucocorticoids, mitochondria sequester calcium released by NMDA receptor activation and glutamate release (Du et al., 2009). But glutamate is also a mitochondrial substrate, and synergizes with glucocorticoids to promote shrinkage of dendrites and turnover of spine synapses (McEwen, 1999). The metabotrophic glutamate receptor mGlu2, which is regulated epigenetically via acetylation of lysine at position 27 on histone H3 (H3K27) (Nasca et al., 2013), presynaptically contains and restricts spontaneous glutamate release. Down-regulation of this receptor leads to spillover of glutamate that inhibits neurogenesis and promotes dendritic shrinkage and spine synapse loss (McEwen, 1999; Cameron et al., 1998).
This condition, involving a depressive-like behavioral state, is associated with LAC deficiency. In animal models, depression can be rapidly ameliorated within 3–5 days by oral LAC supplementation, while classical antidepressants, if efficient at all, will show effects after several weeks. As an acetyl donor via acetyl-CoA (Takahashi et al., 2006) for H3K27acetylation, LAC epigenetically upregulates mGlu2 expression and reduces the glutamate overflow (Nasca et al., 2013; Nasca et al., 2015). LAC itself may promote mitochondrial biogenesis and enhance mitochondrial oxidative capacity in the context of aging and depression (Rosca et al., 2009). In lower organisms like worms, mitochondrial dysfunction alone is sufficient to induce histone acetylation and methylation (Merkwirth et al., 2016). This suggests that mitochondrial remodeling, via one or may potential mechanisms including calcium buffering and reducing ROS generation, mitochondrial signaling reducing inflammatory tone, or donating acetyl groups for histone acetylation, may collectively contribute to the antidepressant-like actions of LAC (Raison et al., 2009).
Moreover, LAC deficiency in animal models is associated with metabolic dysregulation, including insulin resistance, elevated triglycerides and leptin that, like the depressive-like behavior, is rapidly corrected by LAC treatment (Bigio et al., 2016). Connecting this to LAC and its actions, research shows that a ketogenic diet that relies on and activates mitochondrial oxidative metabolism ameliorates insulin resistance and Type 2 diabetes, and also increases LAC levels (Berry-Kravis et al., 2001). Furthermore, a ketogenic metabolite, beta-hydroxybutyrate, is an histone deacetylase inhibitor (Newman and Verdin, 2014) that blocks removal of acetyl groups from lysine residues on histones and has the same rapid effects on mGlu2 as LAC. Taken together, mitochondrial dysregulation appears to play a key role in the progression from metabolic dysregulation to depression and dementia, and thus connects systemic dysregulation with central nervous system allostatic load (Rasgon and McEwen, 2016).
12. Mitochondrial function in the brain influences social behaviors
A link between mitochondrial function and social behaviors has started to emerge in recent years. Notably, an increasing number of studies are reporting mitochondrial dysfunction in Autism Spectrum Disorder (ASD) (Gu et al., 2013; Tang et al., 2013), a neurodevelopmental disorder involving core alterations in social behaviors. Key recent evidence includes reports indicating redox metabolism abnormalities in autistic children associated with mitochondrial disease (Frye et al., 2013) and genetic evidence for pathogenic mtDNA mutations as a potential cause for ASD (Wang et al., 2016). Beyond autism, a recent study has identified an association between a mitochondrial DNA (mtDNA) single nucleotide polymorphism (SNP), which affects the regulation of mitochondrial calcium levels related to energy production, with aggression and leadership in children with attention deficiency and hyperactivity disorder (ADHD) (Hwang et al., 2017).
While the reported clinical data provides correlational data, preclinical studies in rodents are starting to provide evidence that metabolic alterations contribute to social dysfunctions. For example, treatment with a ketogenic diet – a high-fat low-carbohydrate diet that enhances mitochondrial function – was able to ameliorate impaired mitochondrial respiration and deficits in social behaviors in a valproic acid model of autism in rats (Ahn et al., 2014). In another study, acute infusion in rodents of resveratrol, a compound that activates AMPK-induced mitochondrial biogenesis, counteracted diminished sociability induced by peripubertal stress (Poirier et al., 2014). Similarly, treatment with the antioxidant N-acetyl cysteine reversed impairment in social isolation and accumulated striatal ATP levels observed in a rat model of social isolation rearing stress in rats (Moller et al., 2013).
Further insights for a key role of mitochondrial function in specific brain regions in influencing complex social behaviors have been provided by a series of studies in rats relating anxiety with diminished social competitiveness (Hollis et al., 2015; van der Kooij et al., 2017), a phenomenon that has also been highlighted in humans (Goette et al., 2015). In rats, lower mitochondrial function in the nucleus accumbens was observed in high-anxious animals compared to their less anxious littermates, and was causally implicated in their low social competitiveness (Hollis et al., 2015). More specifically, high-anxious animals that are prone to become subordinate during a social encounter exhibited reduced mitochondrial respiratory capacity, decreased ATP levels, and increased ROS production in the nucleus accumbens. Furthermore, micro-infusing specific mitochondrial electron transport chain inhibitors into the nucleus accumbens reduced social rank, recapitulating the low probability to become dominant in anxious animals. Conversely, intraaccumbal infusion of nicotinamide, an amide form of vitamin B3 known to enhance brain energy metabolism, prevented the development of a subordinate status in anxious rats (Hollis et al., 2015). Additionally, treatment with a low dose of the anxiolytic diazepam was effective to facilitate social dominance, ameliorating both the competitive disadvantage and low mitochondrial function in the nucleus accumbens displayed by high-anxious rats (van der Kooij et al., 2017).
Interestingly, homecage dominant and subordinate male inbred C57BL/6J mice were found to display differences in energy metabolites in the nucleus accumbens. Data from 1H NMR spectroscopy indicated that subordinates show lower levels of energy-related metabolites than dominant mice (Larrieu et al., 2017). When exposed to chronic social defeat, dominant males were the ones that specifically showed vulnerability to display depression-like behaviors. While levels of brain energy metabolites increased in ‘resilient’, subordinate mice, metabolites in dominant, ‘vulnerable’ animals tended to be reduced following exposure to chronic social defeat (Larrieu et al., 2017).
While these studies strongly point out at a relevant role for mitochondria in organizing complex behaviors, future work should elucidate how mitochondrial function and energy metabolism contributes to neuronal and circuit properties that eventually regulate specific social behaviors and vulnerability/resilience to stress. One related synergistic process by which mitochondria may regulate stress vulnerability/resilience is by regulating the reactivity of neuroendocrine, immune, and metabolic axes to stressors.
13. Mitochondria regulate physiological stress reactivity
The ability to generate whole-body physiological response to environmental and physiological challenges is critical to allostasis and was likely a diving force behind natural selection (Weiner, 1992). The integrated energy-demanding stress responses, which involve transcriptional regulation, the secretion of various stress hormones such as glucocorticoids and catecholamines, acute metabolic changes, inflammatory mediators, neural plasticity, and many others, determine an organisms’ ability to thrive and adapt, or become ill in the face of stressful situations (McEwen, 2012). In the context of this review, and in an attempt to explain individual differences in vulnerability to stress is, a critical question is whether differences in mitochondrial energy metabolism influence how the organism responds to complex stressors.
The evidence reviewed above has described pathways and mechanisms whereby mitochondria directly contribute to gene expression regulation through direct and epigenetic mechanisms, the synthesis of important stress mediators, metabolic regulation, and neural circuits that orchestrate complex behaviors. To examine the role of mitochondria in the physiological stress response, we tested the hypothesis that abnormal mitochondrial functions would differentially modulate the organism’s multisystemic response to a psychological stressor. In mice, it is possible to genetically modify different aspects of mitochondrial function: mitochondrial respiratory chain function, energy exchange, or the intramitochondrial redox status. In a study of five different mouse lines with different mitochondrial defects, we found that mitochondrial dysfunctions altered hippocampal gene expression, the hypothalamic-pituitaryadrenal axis, sympathetic adrenal-medullary activation and catecholamine levels, the inflammatory cytokine IL-6, circulating glucose and lipids in response to stress Fig. 4) (Picard et al., 2015). Each mitochondrial defect generated a distinct whole-body stress response signature. Thus mitochondria are stress-response modulators, with implications for understanding the mechanisms of stress pathophysiology and mitochondrial diseases (Picard and McEwen, 2018a). In a ‘non-linear’ way reminiscent of the transcriptional reprogramming in the cells with mtDNA mutations (Picard et al., 2014; Chae et al., 2013), this study found that each mitochondrial defect caused a unique multisystemic stress response signature sometimes opposite to each other (see Fig. 4D–E), depending upon the nature of the mitochondrial defect (Picard et al., 2015).
Fig. 4.
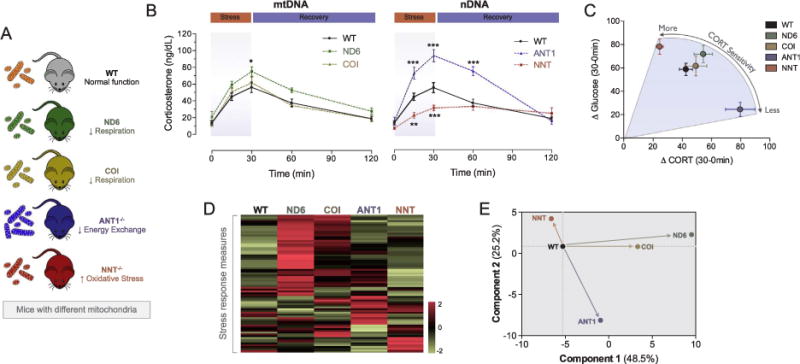
Mitochondrial defects cause unique stress response patterns in mice. (A) Mice with normal mitochondria (wild type, WT) were compared with mice with mitochondrial defects, including four different genes involved in energy production (ND6: respiratory chain Complex I, ND6 subunit; COI: respiratory chain Complex IV, MT-COI subunit), ANT1−/−: energy transfer (adenine nucleotide translator 1, ANT1), and NNT−/−: mitochondrial oxidative stress (nicotinamine nucleotide transhydrogenase, NNT). All mice were exposed to 30 min of restraint stress with sequential tail blood collections. (B) Hypothalamic-pituitary-adrenal (HPA) axis response kinetic indicated by corticosterone (CORT) increase during stress and recovery. (C) This acute stressor also caused hyperglycemia, as expected from the glucose mobilizing properties of CORT. This graph shows the juxtaposition of the stress-induced CORT levels over the first 30 min and the associated increase in circulating blood glucose. Note that the mitochondrial defect causing excess CORT release simultaneously causes the lowest glucose increase, whereas the NNT defect that blunts CORT release causes the highest glucose response. These results indicate an uncoupling of neuroendocrine and metabolic allostasis by mitochondria. (D) Each mitochondrial defect produced a unique stress response signature, here illustrated as a heatmap showing results of an unsupervised hierarchical clustering analysis of gene expression, neuroendocrine, inflammatory, and metabolic measurements (n = 77 parameters). (E) Principal component analysis illustrating qualitatively and quantitatively distinct whole-body stress response patterns for each mitochondrial defect. Figures adapted from (Picard et al., 2015).
Psychological or physical stress triggers neuroendocrine, inflammatory, metabolic, and transcriptional perturbations that ultimately predispose to disease via allostatic load/overload (Juster et al., 2010). The systemic neuroendocrine consequences of mitochondrial defects have implications for our understanding of the pathogenic mechanism underlying mitochondrial disease onset and progression. Stress influences the biology of multiple diseases including cancer growth and metastasis (Thaker et al., 2006; Cole et al., 2015); diabetes (Faulenbach et al., 2012); neurodegenerative disorders (Schon and Przedborski, 2011; Picard and McManus, 2016), as well as cellular aging (Epel et al., 2004) and age-related physical and cognitive decline (Juster et al., 2010). Thus, combining the notions that mitochondria regulate the activation of stress responses and release of allostatic mediators, and that stress response mediators influence disease trajectory, an emerging possibility is that mitochondrial dysfunction – or mitochondrial allostatic load (MAL) – may contribute to translating stressful experiences into pathophysiological processes (Picard et al., 2014). In humans, much research is still needed to ascertain to what extent mitochondria regulate physiological responses to both positive and negative psychological states, whether they partly underlie the drive for energy intake, and whether mitochondrial stress transduction contribute to stress pathophysiology and maladaptation.
14. Sexual dimorphism in mitochondria and stress physiology
It must be noted that the female and male organisms have qualitatively different mitochondria, but that little is known about the interaction with stress. Indeed, even if all mitochondria and the mtDNA are uniquely inherited from the mother (Giles et al., 1980), differences in protein composition and function are acquired during development and as a result, multiple facets of mitochondrial biology differ by sex in adult animals (Ventura-Clapier et al., 2017). This is, in part, due to the fact that sex hormones regulate mitochondrial function and biogenesis (Gaignard et al., 2017). Metabolomic signatures also, which indirectly reflect mitochondrial and cellular metabolism of the whole organism, show that up to one-third of metabolites at baseline differ between women and men (Krumsiek et al., 2015). In relation to stress, a recent meta-analysis of the literature on the effects of induced stress on mitochondrial structure and function revealed that all studies to date have been conducted in male rodents (Picard and McEwen, 2018b). The studies of social behavior and stress regulation described in the above sections have also been conducted exclusively in male animals. In humans, the opposite trend is beginning to appear, with women being predominantly studied (Picard and McEwen, 2018b).
Two main factors might have contributed to this unbalanced state of the field: (i) the need for larger and more complex study designs when including both sexes, and (ii) the largely false belief that hormonal fluctuations in female animals introduces additional variability that weakens conclusions and is unnecessary to the progress of science. The former argument is valid, as studying both sexes with equal power necessarily doubles sample sizes and increases costs. However, the argument about female animals being more variable has been demonstrated not to be accurate for behavioral, morphological, physiological and molecular outcomes in neuroscience (Prendergast et al., 2014). In fact, inclusion of both sexes in basic research may even drive discovery (Klein et al., 2015), as well as the production of more directly translatable pre-clinical and clinical research. Therefore, based on known sex differences in mitochondrial biology, in stress physiology, and disease risk in humans, future studies would benefit from an equal inclusion of both sexes.
15. Conclusions section
Mitochondria were essential to the development of complex multicellular life forms, and only recently have we started to develop a scientific understanding of the interplay between specific aspects of mitochondrial biology and stress responses. The mechanistic pre-clinical findings reviewed above provide converging evidence that mitochondrial energetics may directly impact neuroendocrine and metabolic responses to psychological stress, epigenetic regulation within the brain, food and energy seeking behaviors, psychological states such as depression, and complex social behaviors. It will be a fascinating challenge for behavioral neuroscientists to determine whether mitochondrial energy production capacity or other mitochondrial signals influence psychological states and social behaviors in humans also, and if so, whether interventions targeted at mitochondria can influence complex behaviors and promote adaptation and health.
Much attention in the past decades has been devoted to mapping the contribution of specific anatomical structures within and outside the brain to the stress response, including neuroendocrine mediators and the immune system. The brain has been regarded as a central organ of adaptation (McEwen, 2006). The work reviewed above implicates mitochondria as a central organelle of adaptation, operating at multiple levels within the stress response cascade. Via their effects on neuroendocrine mechanisms, mitochondria drive adaptive signaling processes and behaviors. Simultaneously, mitochondria sense changes in energy demand, and rapidly respond – in shape and function – to energy-mobilizing glucocorticoids and catecholamine hormones. It is enlightening to consider that evolution has routed the production of glucocorticoids and sex hormones within the mitochondria of specialized steroid-secreting cells (adrenal cortex, gonads). In turn, mitochondria in other tissues like the brain, muscles, and liver sense and functionally respond to glucocorticoids via the action of GR and other “nuclear” receptors on the mtDNA. Mitochondria are both mediators and targets of the main stress axes. Therefore, glucocorticoids be regarded as mitochondria-derived hormones, or mitokines mediating mitochondria-to-mitochondria communication among distant sites throughout the organism. Much remains to be discovered about the mechanisms and implications of mitocrine signaling.
Mitochondria play vital role in sustaining basic life processes and those that specifically become activated during allostasis. An “energetic” scientific perspective of stress and the downstream (mal) adaptive consequences invites a focus on the communication systems and mechanisms that ensure and direct the flow of energy towards adaptive processes. Energy and mitochondria are primary drivers of these processes. Moving away from a predominantly anatomically-directed scientific framework, we must consider a model of stress focused on understanding how information and energy in their different forms are exchanged between the environment, the organism, organelles, and the plastic epigenome. Mapping the mechanisms underlying these exchanges will likely open the door to next generation approaches to identify and target maladaptive responses early before they manifest as disease, and to foster physiological responses underlying successful adaptation and resilience.
Acknowledgments
The authors are grateful to Eugene Mosharov, Mary-Elizabeth Sutherland, Judyann McNamara, and members of the Picard Lab for stimulating discussions.
Funding
This work was partly supported by the Wharton Fund and the National Institutes of Health (R35GM119793, R21MH113011) to MP; Hope for Depression Research Foundation to BSM; National Institutes of Health (R24AG048024) to ESE; and the Swiss National Foundation (31003A-176206; and NCCR Synapsy grant no. 51NF40-158776) to CS.
References
- Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behav. 2007;91:449–458. doi: 10.1016/j.physbeh.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Ahn Y, Narous M, Tobias R, Rho JM, Mychasiuk R. The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev Neurosci. 2014;36:371–380. doi: 10.1159/000362645. [DOI] [PubMed] [Google Scholar]
- Akana SF, Jacobson L, Cascio CS, Shinsako J, Dallman MF. Constant corticosterone replacement normalizes basal adrenocorticotropin (ACTH) but permits sustained ACTH hypersecretion after stress in adrenalectomized rats. Endocrinology. 1988;122:1337–1342. doi: 10.1210/endo-122-4-1337. [DOI] [PubMed] [Google Scholar]
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auron M, Raissouni N. Adrenal insufficiency. Pediatr Rev. 2015;36:92–102. doi: 10.1542/pir.36-3-92. quiz 103, 129. [DOI] [PubMed] [Google Scholar]
- Baumann A, Jorge-Finnigan A, Jung-Kc K, Sauter A, Horvath I, Morozova-Roche LA, Martinez A. Tyrosine hydroxylase binding to phospholipid membranes prompts its amyloid aggregation and compromises bilayer integrity. Scientific Rep. 2016;6:39488. doi: 10.1038/srep39488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benani A, Troy S, Carmona MC, Fioramonti X, Lorsignol A, Leloup C, Casteilla L, Penicaud L. Role for mitochondrial reactive oxygen species in brain lipid sensing: redox regulation of food intake. Diabetes. 2007;56:152–160. doi: 10.2337/db06-0440. [DOI] [PubMed] [Google Scholar]
- Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria-Gomez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutierrez S, Martin-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, Lopez-Rodriguez ML, Grandes P, Rossignol R, Marsicano G. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat Neurosci. 2012;15:558–564. doi: 10.1038/nn.3053. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Booth G, Sanchez AC, Woodbury-Kolb J. Carnitine levels and the ketogenic diet. Epilepsia. 2001;42:1445–1451. doi: 10.1046/j.1528-1157.2001.18001.x. [DOI] [PubMed] [Google Scholar]
- Bigio B, Mathe AA, Sousa VC, Zelli D, Svenningsson P, McEwen BS, Nasca C. Epigenetics and energetics in ventral hippocampus mediate rapid antidepressant action: Implications for treatment resistance. Proc Natl Acad Sci USA. 2016;113:7906–7911. doi: 10.1073/pnas.1603111113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C, Mattevi A, Edmondson DE. Structural properties of human monoamine oxidases A and B. Int Rev Neurobiol. 2011;100:1–11. doi: 10.1016/B978-0-12-386467-3.00001-7. [DOI] [PubMed] [Google Scholar]
- Boggiano MM, Chandler PC. Binge eating in rats produced by combining dieting with stress. Curr Protoc Neurosci. 2006 doi: 10.1002/0471142301.ns0923as36. Chapter 9 Unit9 23A. [DOI] [PubMed] [Google Scholar]
- Booth David M, Enyedi B, Geiszt M, Várnai P, Hajnóczky G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell. 2016;63:240–248. doi: 10.1016/j.molcel.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose HS, Lingappa VR, Miller WL. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature. 2002;417:87–91. doi: 10.1038/417087a. [DOI] [PubMed] [Google Scholar]
- Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood JA, Li Z, Tamashiro KL, Schwartz GJ, Makriyannis AM, Kunos G, Hillard CJ, McEwen BS, Hill MN. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc Natl Acad Sci USA. 2015;112:285–290. doi: 10.1073/pnas.1421420112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles NP, McEwen BS, Boutin-Foster C. Trouble in transit: organizational barriers to workers' health. Am J Ind Med. 2017;60:350–367. doi: 10.1002/ajim.22701. [DOI] [PubMed] [Google Scholar]
- Bryan RM., Jr Cerebral blood flow and energy metabolism during stress. Am J Physiol. 1990;259:H269–H280. doi: 10.1152/ajpheart.1990.259.2.H269. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Tanapat P, Gould E. Adrenal steroids and N-methyl-D-aspartate receptor activation regulate neurogenesis in the dentate gyrus of adult rats through a common pathway. Neuroscience. 1998;82:349–354. doi: 10.1016/s0306-4522(97)00303-5. [DOI] [PubMed] [Google Scholar]
- Caperuto LC, Anhe GF, Amanso AM, Ribeiro LM, Medina MC, Souza LC, Carvalho OM, Bordin S, Saad MJ, Carvalho CR. Distinct regulation of IRS proteins in adipose tissue from obese aged and dexamethasone-treated rats. Endocrine. 2006;29:391–398. doi: 10.1385/ENDO:29:3:391. [DOI] [PubMed] [Google Scholar]
- Carlsson C, Hagerdal M, Kaasik AE, Siesjo BK. A catecholamine-mediated increase in cerebral oxygen uptake during immobilisation stress in rats. Brain Res. 1977;119:223–231. doi: 10.1016/0006-8993(77)90102-0. [DOI] [PubMed] [Google Scholar]
- Chae S, Ahn BY, Byun K, Cho YM, Yu MH, Lee B, Hwang D, Park KS. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci Signal. 2013;6:rs4. doi: 10.1126/scisignal.2003266. [DOI] [PubMed] [Google Scholar]
- Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, Robinson AJ, Gygi SP, Spiegelman BM. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532:112–116. doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chretien D, Benit P, Ha HH, Keipert S, El-Khoury R, Chang YT, Jastroch M, Jacobs HT, Rustin P, Rak M. Mitochondria are physiologically maintained at close to 50 degrees C. PLoS Biology. 2018;16:e2003992. doi: 10.1371/journal.pbio.2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BJ. ACTH action on StAR biology. Front Neurosci. 2016;10:547. doi: 10.3389/fnins.2016.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Catauro M, Rasmussen HH, Apell HJ. Quantitative calculation of the role of the Na(+), K(+)-ATPase in thermogenesis. BBA. 1827;2013:1205–1212. doi: 10.1016/j.bbabio.2013.06.010. [DOI] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563–572. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol. 2010;315:159–167. doi: 10.1016/j.mce.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchet J, Lewis TL, Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via pre-synaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF. Stress-induced obesity and the emotional nervous system. Trends Endocrinol Metab. 2010;21:159–165. doi: 10.1016/j.tem.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feo P, Perriello G, Torlone E, Ventura MM, Fanelli C, Santeusanio F, Brunetti P, Gerich JE, Bolli GB. Contribution of cortisol to glucose counter-regulation in humans. Am J Physiol. 1989;257:E35–E42. doi: 10.1152/ajpendo.1989.257.1.E35. [DOI] [PubMed] [Google Scholar]
- Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci USA. 2009;106:3543–3548. doi: 10.1073/pnas.0812671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci USA. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulenbach M, Uthoff H, Schwegler K, Spinas GA, Schmid C, Wiesli P. Effect of psychological stress on glucose control in patients with Type 2 diabetes. Diabet Med. 2012;29:128–131. doi: 10.1111/j.1464-5491.2011.03431.x. [DOI] [PubMed] [Google Scholar]
- Fawcett DW. The Cell. W. B. Saunders Company; Philadelphia: 1981. The Cell, Chapter 7: Mitochondria. [Google Scholar]
- Fisher-Wellman KH, Lin CT, Ryan TE, Reese LR, Gilliam LA, Cathey BL, Lark DS, Smith CD, Muoio DM, Neufer PD. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J. 2015;467:271–280. doi: 10.1042/BJ20141447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz E, McEwen BS. Effect of carnitine on fatty-acid oxidation by muscle. Science. 1959;128:334. doi: 10.1126/science.129.3345.334. [DOI] [PubMed] [Google Scholar]
- Frye RE, Delatorre R, Taylor H, Slattery J, Melnyk S, Chowdhury N, James SJ. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Translational Psychiatr. 2013;3:e273. doi: 10.1038/tp.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaignard P, Liere P, Therond P, Schumacher M, Slama A, Guennoun R. Role of sex hormones on brain mitochondrial function, with special reference to aging and neurodegenerative diseases. Front Aging Neurosci. 2017;9:406. doi: 10.3389/fnagi.2017.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goette L, Bendahan S, Thoresen J, Hollis F, Sandi C. Stress pulls us apart: anxiety leads to differences in competitive confidence under stress. Psychoneuroendocrinology. 2015;54:115–123. doi: 10.1016/j.psyneuen.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Taylor JR. Recapitulation and reversal of a persistent depression-like syndrome in rodents. Curr Protocol Neurosci. 2009:32. doi: 10.1002/0471142301.ns0932s49. Chapter 9 Unit 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Chauhan V, Kaur K, Brown WT, LaFauci G, Wegiel J, Chauhan A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl Psychiatr. 2013;3:e299. doi: 10.1038/tp.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hanukoglu I, Privalle CT, Jefcoate CR. Mechanisms of ionic activation of adrenal mitochondrial cytochromes P-450scc and P-45011 beta. J Biol Chem. 1981;256:4329–4335. doi: 10.1016/S0021-9258(19)69437-8. [DOI] [PubMed] [Google Scholar]
- Hao N, O’Shea EK. Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat Struct Mol Biol. 2011;19:31–39. doi: 10.1038/nsmb.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci USA. 2014;111:486–491. doi: 10.1073/pnas.1311310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Fukuda N, Uchiyama S, Inada N. A cell-permeable fluorescent polymeric thermometer for intracellular temperature mapping in mammalian cell lines. PLoS One. 2015;10:e0117677. doi: 10.1371/journal.pone.0117677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang L, Zhu Z, Xiao A, Yu H, Gan X. Blockade of cyclophilin D rescues dexamethasone-induced oxidative stress in gingival tissue. PLoS One. 2017;12:e0173270. doi: 10.1371/journal.pone.0173270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Pagano Zottola AC, Delamarre A, Cannich A, Vincent P, Varilh M, Robin LM, Terral G, Garcia-Fernandez MD, Colavita M, Mazier W, Drago F, Puente N, Reguero L, Elezgarai I, Dupuy JW, Cota D, Lopez-Rodriguez ML, Barreda-Gomez G, Massa F, Grandes P, Benard G, Marsicano G. A can-nabinoid link between mitochondria and memory. Nature. 2016;539:555–559. doi: 10.1038/nature20127. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:267–277. doi: 10.1101/sqb.2011.76.010850. [DOI] [PubMed] [Google Scholar]
- Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Canto C, Sandi C. Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci USA. 2015;112:15486–15491. doi: 10.1073/pnas.1512653112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RG, Seligsohn M, Rubin TG, Griffiths BB, Ozdemir Y, Pfaff DW, Datson NA, McEwen BS. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc Natl Acad Sci USA. 2016;113:9099–9104. doi: 10.1073/pnas.1602185113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IW, Hong JH, Kwon BN, Kim HJ, Lee NR, Lim MH, Kwon HJ, Jin HJ. Association of mitochondrial DNA 10398 A/G polymorphism with attention deficit and hyperactivity disorder in Korean children. Gene. 2017;630:8–12. doi: 10.1016/j.gene.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122:1343–1348. doi: 10.1210/endo-122-4-1343. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Miller ML, Chen Y, Swartz JR. Continued protein synthesis at low [ATP] and [GTP] enables cell adaptation during energy limitation. J Bacteriol. 2009;191:1083–1091. doi: 10.1128/JB.00852-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SB, Riley AW, Granger DA, Riis J. The science of early life toxic stress for pediatric practice and advocacy. Pediatrics. 2013;131:319–327. doi: 10.1542/peds.2012-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CM, Melanson EL, Frydendall EJ, Perreault L, Eckel RH, Wright KP. Energy expenditure during sleep, sleep deprivation and sleep following sleep deprivation in adult humans. J Physiol. 2011;589:235–244. doi: 10.1113/jphysiol.2010.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juster RP, McEwen BS, Lupien SJ. Allostatic load biomarkers of chronic stress and impact on health and cognition. Neurosci Biobehav Rev. 2010;35:2–16. doi: 10.1016/j.neubiorev.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Kafri M, Metzl-Raz E, Jona G, Barkai N. The cost of protein production. Cell Rep. 2016;14:22–31. doi: 10.1016/j.celrep.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatsoreos IN, Bhagat SM, Bowles NP, Weil ZM, Pfaff DW, McEwen BS. Endocrine and physiological changes in response to chronic corticosterone: a potential model of the metabolic syndrome in mouse. Endocrinology. 2010;151:2117–2127. doi: 10.1210/en.2009-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci USA. 2011;108:1657–1662. doi: 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Kerner J, Yohannes E, Lee K, Virmani A, Koverech A, Cavazza C, Chance MR, Hoppel C. Acetyl-L-carnitine increases mitochondrial protein acetylation in the aged rat heart. Mech Age Dev. 2015;145:39–50. doi: 10.1016/j.mad.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Habash DL, Fagundes CP, Andridge R, Peng J, Malarkey WB, Belury MA. Daily stressors, past depression, and metabolic responses to high-fat meals: a novel path to obesity. Biol Psychiatry. 2015;77:653–660. doi: 10.1016/j.biopsych.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Xiao J, Wan J, Cohen P, Yen K. Mitochondrially derived peptides as novel regulators of metabolism. J Physiol. 2017;595:6613–6621. doi: 10.1113/JP274472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klecker T, Bockler S, Westermann B. Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol. 2014;24:537–545. doi: 10.1016/j.tcb.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Klein SL, Schiebinger L, Stefanick ML, Cahill L, Danska J, de Vries GJ, Kibbe MR, McCarthy MM, Mogil JS, Woodruff TK, Zucker I. Opinion: sex inclusion in basic research drives discovery. Proc Natl Acad Sci USA. 2015;112:5257–5258. doi: 10.1073/pnas.1502843112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flugge G, Korte SM, Meerlo P, Murison R, Olivier B, Palanza P, Richter-Levin G, Sgoifo A, Steimer T, Stiedl O, van Dijk G, Wohr M, Fuchs E. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev. 2011;35:1291–1301. doi: 10.1016/j.neubiorev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Kopytek SJ, Peterson DO. ATP-mediated activation of RNA polymerase II transcription complexes. Gene Expr. 1998;7:75–86. [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J, Mittelstrass K, Do KT, Stuckler F, Ried J, Adamski J, Peters A, Illig T, Kronenberg F, Friedrich N, Nauck M, Pietzner M, Mook-Kanamori DO, Suhre K, Gieger C, Grallert H, Theis FJ, Kastenmuller G. Gender-specific pathway differences in the human serum metabolome. Metabolomics. 2015;11:1815–1833. doi: 10.1007/s11306-015-0829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SK, Sando R, Lewis TL, Hirabayashi Y, Maximov A, Polleux F. LKB1 regulates mitochondria-dependent presynaptic calcium clearance and neurotransmitter release properties at excitatory synapses along cortical axons. PLoS Biology. 2016;14(7):e1002516. doi: 10.1371/journal.pbio.1002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- Larrieu T, Cherix A, Duque A, Rodrigues J, Lei H, Gruetter R, Sandi C. Hierarchical status predicts behavioral vulnerability and nucleus accumbens metabolic profile following chronic social defeat stress. Curr Biol. 2017;27:2202–2210 e6. doi: 10.1016/j.cub.2017.06.027. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto KI, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lian FL, Moyne J, Tilbury D. Network design consideration for distributed control systems. IEEE Trans Cont Syst Tech. 2002;10:297–307. [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linghu C, Zheng H, Zhang L, Zhang J. Discovering common combinatorial histone modification patterns in the human genome. Gene. 2013;518:171–178. doi: 10.1016/j.gene.2012.11.038. [DOI] [PubMed] [Google Scholar]
- Liston C, Cichon JM, Jeanneteau F, Jia Z, Chao MV, Gan WB. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat Neurosci. 2013;16:698–705. doi: 10.1038/nn.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- Magomedova L, Cummins CL. Glucocorticoids and metabolic control. Handb Exp Pharmacol. 2016;233:73–93. doi: 10.1007/164_2015_1. [DOI] [PubMed] [Google Scholar]
- Margaria R, Cerretelli P, Aghemo P, Sassi G. Energy cost of running. J Appl Physiol. 1963;18:367–370. doi: 10.1152/jappl.1963.18.2.367. [DOI] [PubMed] [Google Scholar]
- Margulis L, Bermudes D. Symbiosis as a mechanism of evolution: status of cell symbiosis theory. Symbiosis. 1985;1:101–124. [PubMed] [Google Scholar]
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, Zhao Y, DeBerardinis RJ, Chandel NS. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masih T, Dimmock JA, Epel ES, Guelfi KJ. Stress-induced eating and the relaxation response as a potential antidote: a review and hypothesis. Appetite. 2017;118:136–143. doi: 10.1016/j.appet.2017.08.005. [DOI] [PubMed] [Google Scholar]
- Matilainen O, Quiros PM, Auwerx J. Mitochondria and epigenetics – crosstalk in homeostasis and stress. Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2017.02.004. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci. 2006;8:367–381. doi: 10.31887/DCNS.2006.8.4/bmcewen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci USA. 2012;109(Suppl 2):17180–17185. doi: 10.1073/pnas.1121254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Karatsoreos IN. Sleep Deprivation and Circadian Disruption: Stress, Allostasis, and Allostatic Load. Sleep Med Clin. 2015;10:1–10. doi: 10.1016/j.jsmc.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease Arch Int Med. 1993;153:2093–2101. [PubMed] [Google Scholar]
- McEwen BS, Sakai RR, Spencer RL. Adrenal Steroid Effects on the Brain: Versatile Hormones with Good and Bad Effects. In: Schulkin J, editor. Hormonally-Induced Changes in Mind and Brain. Academic Press; San Diego: 1993. pp. 157–189. [Google Scholar]
- McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Redefining neuroendocrinology: Epigenetics of brain-body communication over the life course. Front Neuroendocrinol. doi: 10.1016/j.yfrne.2017.11.001. [in press]. [DOI] [PubMed]
- Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, Nurnberg P, Mann NP, Banerjee R, Saka HN, Chapple JP, King PJ, Clark AJ, Metherell LA. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet. 2012;44:740–742. doi: 10.1038/ng.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimaridou E, Hughes CR, Kowalczyk J, Guasti L, Chapple JP, King PJ, Chan LF, Clark AJ, Metherell LA. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol. 2013;371:195–200. doi: 10.1016/j.mce.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam-Webster, editor. Merriam-Webster.com. Energy. Oct 16, 2017. n.d. [Google Scholar]
- Midzak A, Papadopoulos V. Adrenal mitochondria and steroidogenesis: from individual proteins to functional protein assemblies. Front Endocrinol (Lausanne) 2016;7:106. doi: 10.3389/fendo.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller M, Du Preez JL, Viljoen FP, Berk M, Emsley R, Harvey BH. Social isolation rearing induces mitochondrial, immunological, neurochemical and behavioural deficits in rats, and is reversed by clozapine or N-acetyl cysteine. Brain Behav Immun. 2013;30:156–167. doi: 10.1016/j.bbi.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Moutsatsou P, Psarra AM, Tsiapara A, Paraskevakou H, Davaris P, Sekeris CE. Localization of the glucocorticoid receptor in rat brain mitochondria. Arch Biochem Biophys. 2001;386:69–78. doi: 10.1006/abbi.2000.2162. [DOI] [PubMed] [Google Scholar]
- Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathe AA, Pittaluga A, Lionetto L, Simmaco M, Nicoletti F. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA. 2013;110:4804–4809. doi: 10.1073/pnas.1216100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasca C, Zelli D, Bigio B, Piccinin S, Scaccianoce S, Nistico R, McEwen BS. Stress dynamically regulates behavior and glutamatergic gene expression in hippo-campus by opening a window of epigenetic plasticity. Proc Natl Acad Sci USA. 2015;112:14960–14965. doi: 10.1073/pnas.1516016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Golozoubova V, Matthias A, Asadi A, Jacobsson A, Cannon B. UCP1: the only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. BBA. 2001;1504:82–106. doi: 10.1016/s0005-2728(00)00247-4. [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Fergusson SJ. Bioenergetics. Academic Press; 2013. [Google Scholar]
- Okabe K, Inada N, Gota C, Harada Y, Funatsu T, Uchiyama S. Intracellular temperature mapping with a fluorescent polymeric thermometer and fluorescence lifetime imaging microscopy. Nat Commun. 2012;3:705. doi: 10.1038/ncomms1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- Phillips DI, Barker DJ, Fall CH, Seckl JR, Whorwood CB, Wood PJ, Walker BR. Elevated plasma cortisol concentrations: a link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–760. doi: 10.1210/jcem.83.3.4634. [DOI] [PubMed] [Google Scholar]
- Picard M. Mitochondrial synapses: intracellular communication and signal integration. Trends Neurosci. 2015;38:468–474. doi: 10.1016/j.tins.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the 'gluc' back in glucocorticoids. Nat Rev Endocrinol. 2014;10:303–310. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]
- Picard M, McEwen BS. Mitochondria impact brain function and cognition. Proc Natl Acad Sci USA. 2014;111:7–8. doi: 10.1073/pnas.1321881111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McEwen BS. Psychological stress and mitochondria: a conceptual framework. Psychosom Med. 2018a;80:126–140. doi: 10.1097/PSY.0000000000000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McEwen BS. Psychological stress and mitochondria: a systematic review. Psychosom Med. 2018b;80:141–153. doi: 10.1097/PSY.0000000000000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McManus MJ. Mitochondrial Signaling and Neurodegeneration. Vol. 1. Springer International Publishing; Cham: 2016. pp. 107–137. [Google Scholar]
- Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304:R393–R406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Turnbull DM. Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes. 2013;62:672–678. doi: 10.2337/db12-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson H, Trounce IA, Procaccio V, Wallace DC. Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA. 2014;111:E4033–E4042. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci USA. 2015;112:E6614–E6623. doi: 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105–116. doi: 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier GL, Imamura N, Zanoletti O, Sandi C. Social deficits induced by peripubertal stress in rats are reversed by resveratrol. J Psychiatr Res. 2014;57:157–164. doi: 10.1016/j.jpsychires.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Prendergast BJ, Onishi KG, Zucker I. Female mice liberated for inclusion in neuroscience and biomedical research. Neurosci Biobehav Rev. 2014;40:1–5. doi: 10.1016/j.neubiorev.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. BBA. 1813;2011:1814–1821. doi: 10.1016/j.bbamcr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Psarra AM, Sekeris CE. Glucocorticoid receptors and other nuclear transcription factors in mitochondria and possible functions. BBA. 2009;1787:431–436. doi: 10.1016/j.bbabio.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ, Vogt GJ, Massung B, Miller AH. Activation of central nervous system inflammatory pathways by interferon-alpha: Relationship to monoamines and depression. Biol Psychiat. 2009;65:296–303. doi: 10.1016/j.biopsych.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Gomez-Valades AG, Schneeberger M, Varela L, Haddad-Tovolli R, Altirriba J, Noguera E, Drougard A, Flores-Martinez A, Imbernon M, Chivite I, Pozo M, Vidal-Itriago A, Garcia A, Cervantes S, Gasa R, Nogueiras R, Gama-Perez P, Garcia-Roves PM, Cano DA, Knauf C, Servitja JM, Horvath TL, Gomis R, Zorzano A, Claret M. Mitochondrial dynamics mediated by mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metab. 2017;25:1390–1399 e6. doi: 10.1016/j.cmet.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Rasgon NL, McEwen BS. Insulin resistance – a missing link no more. Mol Psychiatry. 2016;21:1648–1652. doi: 10.1038/mp.2016.162. [DOI] [PubMed] [Google Scholar]
- Razzoli M, Pearson C, Crow S, Bartolomucci A. Stress, overeating, and obesity: insights from human studies and preclinical models. Neurosci Biobehav Rev. 2017;76:154–162. doi: 10.1016/j.neubiorev.2017.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014;35:8–30. doi: 10.1016/j.yfrne.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Lemieux H, Hoppel CL. Mitochondria in the elderly: is acetylcarnitine a rejuvenator? Adv Drug Deliv Rev. 2009;61:1332–1342. doi: 10.1016/j.addr.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan T, Mlynczak S, Erickson T, Man SF, Man GC. Oxygen consumption during sleep: influence of sleep stage and time of night. Sleep. 1989;12:201–210. [PubMed] [Google Scholar]
- Schinzel R, Dillin A. Endocrine aspects of organelle stress-cell non-autonomous signaling of mitochondria and the ER. Curr Opin Cell Biol. 2015;33:102–110. doi: 10.1016/j.ceb.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastian D, Imbernon M, Castano C, Garcia A, Esteban Y, Gonzalez-Franquesa A, Rodriguez IC, Bortolozzi A, Garcia-Roves PM, Gomis R, Nogueiras R, Horvath TL, Zorzano A, Claret M. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70(6):1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C, Lambertz M, Nelesen RA, Bardwell W, Choi JB, Dimsdale JE. Effects of stress on heart rate complexity – a comparison between short-term and chronic stress. Biol Psychol. 2009;80:325–332. doi: 10.1016/j.biopsycho.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122:1271–1278. doi: 10.1289/ehp.1408418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonkoff JP, Garner AS, C. Committee on Psychosocial Aspects of, H. Family, A. Committee on Early Childhood, C. Dependent, D. Section on, P. Behavioral The lifelong effects of early childhood adversity and toxic stress. Pediatrics. 2012;129:e232–46. doi: 10.1542/peds.2011-2663. [DOI] [PubMed] [Google Scholar]
- Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Fisher S, Reason J, editors. Handbook of Life Stress, Cognition and Health. John Wiley & Sons; New York: 1988. pp. 629–649. [Google Scholar]
- Suga H, Goto Y, Kawaguchi O, Hata K, Takasago T, Saeki A, Taylor TW. Ventricular perspective on efficiency. Basic Res Cardiol. 1993;88(Suppl 2):43–65. [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Reports. 2013:1–7. doi: 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Tang G, Gutierrez Rios P, Kuo SH, Akman HO, Rosoklija G, Tanji K, Dwork A, Schon EA, Dimauro S, Goldman J, Sulzer D. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis. 2013;54:349–361. doi: 10.1016/j.nbd.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tataranni PA, Larson DE, Snitker S, Young JB, Flatt JP, Ravussin E. Effects of glucocorticoids on energy metabolism and food intake in humans. Am J Physiol. 1996;271:E317–E325. doi: 10.1152/ajpendo.1996.271.2.E317. [DOI] [PubMed] [Google Scholar]
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, Sood AK. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- Tome ME, Lee K, Jaramillo MC, Briehl MM. Mitochondria are the primary source of the H(2)O(2) signal for glucocorticoid-induced apoptosis of lymphoma cells. Exp Ther Med. 2012;4:237–242. doi: 10.3892/etm.2012.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooij MA, Hollis F, Lozano L, Zalachoras I, Abad S, Zanoletti O, Grosse J, Guillot de Suduiraut I, Canto C, Sandi C. Diazepam actions in the VTA enhance social dominance and mitochondrial function in the nucleus accumbens by activation of dopamine D1 receptors. Mol Psychiatry. 2017 doi: 10.1038/mp.2017.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Kooi BT, Onuma H, Oeser JK, Svitek CA, Allen SR, Vander Kooi CW, Chazin WJ, O’Brien RM. The glucose-6-phosphatase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Mol Endocrinol. 2005;19:3001–3022. doi: 10.1210/me.2004-0497. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, Garnier A. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 2017;131:803–822. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci USA. 2010;107(Suppl 2):8947–8953. doi: 10.1073/pnas.0914635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA variation in human radiation and disease. Cell. 2015;163:33–38. doi: 10.1016/j.cell.2015.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XL, Herzog B, Waltner-Law M, Hall RK, Shiota M, Granner DK. The synergistic effect of dexamethasone and all-trans-retinoic acid on hepatic phosphoenolpyruvate carboxykinase gene expression involves the coactivator p300. J Biol Chem. 2004;279:34191–34200. doi: 10.1074/jbc.M403455200. [DOI] [PubMed] [Google Scholar]
- Wang J, Lou H, Pedersen CJ, Smith AD, Perez RG. 14-3-3zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J Biol Chem. 2009;284:14011–14019. doi: 10.1074/jbc.M901310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Picard M, Gu Z. Genetic evidence for elevated pathogenicity of mitochondrial DNA heteroplasmy in autism spectrum disorder. PLoS Genet. 2016;12:e1006391. doi: 10.1371/journal.pgen.1006391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner H. Perturbing the Organism: The Biology of Stressful Life Experience. University of Chicago Press; Chicago: 1992. [Google Scholar]
- Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47:3–6. doi: 10.1016/s0026-0495(98)90184-6. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Gralla JD. Multiple ATP-dependent steps in RNA polymerase II promoter melting and initiation. EMBO J. 1997;16:7457–7467. doi: 10.1093/emboj/16.24.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]