

SUMMARY
Leishmaniasis is a vector-borne neglected tropical disease caused by protozoan parasites of the genus Leishmania for which there is a paucity of effective viable non-toxic drugs. There are 1·3 million new cases each year causing considerable socio-economic hardship, best measured in 2·4 million disability adjusted life years, with greatest impact on the poorest communities, which means that desperately needed new antileishmanial treatments have to be both affordable and accessible. Established medicines with cheaper and faster development times may hold the cure for this neglected tropical disease. This concept of using old drugs for new diseases may not be novel but, with the ambitious target of controlling or eradicating tropical diseases by 2020, this strategy is still an important one. In this review, we will explore the current state-of-the-art of drug repurposing strategies in the search for new treatments for leishmaniasis.
Key words: leishmaniasis, repurposing, repositioning, drug discovery
INTRODUCTION: DRUG REPOSITIONING
The introduction of a new drug from initial concept to public release is a slow expensive process. Developing a new chemical entity (NCE) drug and delivering it to market, de novo drug discovery (Fig. 1), is estimated to take 10–17 years and cost ~$1·8 billion (Paul et al. 2010). Moreover, the probability of success is lower than 10% and the costs of late stage failures in this process are significant and are accounted for in the high cost of patented drugs. Consequently, the discovery of new drugs for leishmaniasis, and other neglected tropical diseases, for which the average patient suffering from visceral leishmaniasis (VL) exists on an income of less than $2 per day (WHO, 2002), is simply not a commercially viable operation for pharmaceutical companies without significant cross-subsidy.
Fig. 1.

Drug discovery and repositioning pathways (adapted from Guha et al. 2015).
Alternative strategies are therefore needed and the process of drug repositioning or repurposing, where new applications for existing drugs or drug candidates are discovered and refined, has become increasingly common (Ashburn and Thor, 2004; Aubé, 2012; Oprea and Mestres, 2012; Andrews et al. 2014; Klug et al. 2016; Jain and Sharma, 2017). When using a drug repositioning strategy several development phases necessary to develop NCE drugs can be bypassed or considerably shortened (Fig. 1) because candidates will have passed various checkpoints and information is available about their pharmacokinetic and safety profiles. Reflecting this, the process of finding new uses for drugs outside their original indication is becoming increasingly successful as more companies are screening their libraries for repositioning candidates. Figure 2 illustrates two examples of approved drugs possessing optimum properties for their new indication. However, it should be noted that these cases are relatively rare and some development work is still commonly required. Moreover, whilst serendipity (phenotypic screening) can play a sizeable role in identifying repurposed drugs, it is important to establish the molecular connection between old drugs and new targets as this information aids the development of the new therapeutics. Despite these caveats drug repositioning can reduce time, cost and risk, and is a particularly attractive approach for neglected tropical diseases where new medications are needed urgently to treat the poorest of people. In this review, we will focus on the neglected tropical disease, leishmaniasis, and how repositioning strategies have shown potential to enable the discovery of new medicines for this disease.
Fig. 2.

Examples of old drugs directly repositioned as treatments for new diseases and dysfunctions.
LEISHMANIASIS
Leishmaniasis is an important vector borne ´Neglected Tropical Disease’ caused by protozoan parasites of the genus Leishmania (Roberts, 2006). With over 350 million people world wide considered at risk, 12 million people currently infected (Kedzierski, 2010), one million new cases reported annually, more than 30 000 deaths each year (WHO, 2016) and an economic cost that can be best estimated in terms of 2·4 million disability adjusted life years, the health challenge is only surpassed amongst parasitic diseases by malaria, schistosomiasis and lymphatic filariasis (Davies et al. 2003; Bern et al. 2008). Consequently, leishmaniasis has been classified by the World Health Organization as Category I: emerging or uncontrolled diseases. In particular, the spread and severity of infection is exacerbated by its status as an important co-infection of AIDS patients and the overlap in prevalence of HIV and Leishmania spp. (Alvar et al. 2008). The disease is endemic in 98 countries across five continents and has strong links with poverty, principally affecting low income regions of Africa, Asia and Latin America (Alvar et al. 2012). However, the impact of the disease is spreading and, reflecting population movements and climate change, leishmaniasis has also been recorded with growing frequency in Southern Europe (Gradoni, 2013).
Leishmaniasis is transmitted through the bite of infected female phlebotomine sandflies. Subsequently, in the mammalian host, flagellated promastigote Leishmania spp. are taken up by macrophages where they transform into the non-flagellated amastigote form, proliferate and cause disease. Infected macrophages can then be taken up by the insect vector during a second blood meal, the parasite then transforms back to the promastigote form and the cycle is perpetuated. Importantly, many Leishmania spp. can infect multiple mammalian species leading to animal reservoirs for human infection, most notably in canines (Solano-Gallego et al. 2011).
Human disease is caused by 20 of the 31 parasite species that infect mammals with the different species leading to different clinical manifestations (Akhoundi et al. 2016). Depending on parasite tropism for skin or visceral macrophages, and host immune status, clinical forms range from localized cutaneous leishmaniasis (CL), diffuse CL, disseminated CL, mucosal CL, to visceral leishmaniasis (VL). The most common form of the disease is CL which, although not fatal, leads to slow growing skin lesions, which leave permanent scars and in some cases cause serious disability. Mucocutaneous leishmaniasis is the most disfiguring form and causes destruction of soft tissue in the nose, mouth and throat. In contrast, VL is the most severe form of the disease as it affects the vital organs of the body and is fatal if left untreated (CDC, 2016).
Treatment of leishmaniasis is challenging and there are currently no vaccines or prophylactic drugs to prevent human infection. For over 70 years, the mainstay of antileishmanial therapy has been pentavalent antimonials (Pentostam 3 and Glucantime 4) which are administered by slow intravenous or intramuscular injections (Kedzierski et al. 2009). The mode of action is not fully understood but one possibility involves the biological reduction of Sb(V) to Sb(III) by the parasite or by infected host cells to create antileishmanial activity (Shaked-Mishan et al. 2001; Krauth-Siegel & Comini 2008). Unfortunately, antimonials are also toxic (Sundar and Chakravarty, 2010b) and reports of resistance are becoming increasingly common. For example, a study of the efficacy of Pentostam 1, in North Bihar, India, revealed that only 36% of patients could be cured when treated with a dose of 20 mg kg−1 for 30 days (Croft et al. 2006).
For the past seven decades, amphotericin B 5 (AmpB) and pentamidine 6 have been used in cases of antimonial failure (Kedzierski et al. 2009). Pentamidine 6 is an antimicrobial medication that was commonly used as a second line, parenteral, treatment for leishmaniasis in the case of antimonial failure. The primary mode of action of pentamidine 6 in kinetoplastids is not well understood but there is some evidence that it inhibits the active transport system and disrupts the mitochondrial membrane potential. However, pentamidine 6 has lower efficacy than amphotericin B 5 (Lockwood and Moore, 2010) and this, coupled with severe toxicity, have resulted in a gradual decline in its use (No, 2016).
The antifungal drug AmpB 5, first isolated in 1955, was reported to have antileishmanial activity in the early 1960s. AmpB 5 binds to ergosterol, forming pores in the membrane, which leads to the death of the parasite (Kamiński, 2014). Whilst severe side effects limited its use, liposomal formulations that circumvent these problems have been developed. However, a course of L-AmpB 5 still requires parenteral administration and, without extensive subsidy is expensive (up to $3000 per course). This makes L-AmpB inaccessible to most patients who live in the poorest, least developed regions. In comparison, an equivalent course of pentavalent antimonials costs between $150 and $198 (de Menezes et al. 2015). Despite this, L-AmpB 5 is now often preferred over Penstostam 3 for VL because it is less toxic and there are fewer reports of parasitic resistance (Croft et al. 2006; Sundar and Singh, 2016).
Along with L-AmpB, paromomycin 7 and miltefosine 8 are now approved drugs for VL (Sundar and Chakravarty, 2015). Paromomycin 7 is an antibiotic and a relatively new therapy for the parenteral treatment of VL in India. It has been shown to be effective and relatively cheap first-line treatment with low toxicity, requiring a course of 15 mg kg−1 for 21 days (Musa et al. 2010; No, 2016). The mechanism of action is not fully understood but it is thought to inhibit protein synthesis (Fernández et al. 2011). As with the other treatments there are drawbacks, the drug is administered by injection (Sundar et al. 2007) and shows poor efficacy in some regions in Africa (Hailu et al. 2010).
As discussed above, these current medications all involve prolonged parenteral administration, which leads to many complications, including poor patient compliance, blood-borne disease from unsanitary conditions and the need for medical facilities. Therefore, in the past two decades there has been a focus on developing new oral therapies. In 2002 miltefosine 8, originally developed as an anticancer agent, was introduced as the first oral antileishmanial agent (Soto and Berman, 2006). The primary mechanism of action is poorly understood, although the drug is thought to involve disruption of ether-phospholipid metabolism, glycosylphosphatidylinositol anchor biosynthesis and signal transduction within the parasite (No, 2016). Miltefosine 8 efficacy against both VL and CL has been reported and, in 2014, the US Food and Drug Administration approved the drug for all forms of leishmaniasis (Dorlo et al. 2012). However, this oral therapy does have limitations, including high costs and teratogenic effects. Moreover, the long-term effectiveness of miltefosine 8 is questionable, with reports of VL relapses in Nepalese patients treated with this drug. In addition, miltefosine 8 resistance is readily induced in Leishmania donovani in vitro (Seifert et al. 2003).
In conclusion, all current treatments (Fig. 3) have serious limitations such as high cost, route of administration, severe toxic side effects and increasing drug resistance. Currently, there are some new therapies under clinical investigation as monotherapies or in combination with existing antileishmanials (Bahamdan et al. 1997; Alrajhi et al. 2002; Momeni et al. 2002; Machado et al. 2007; Sundar et al. 2011; ClinicalTrials.gov, 2016). These include paromomycin 7 topical cream, currently in phase 3 trials as a treatment for CL, and sitamaquine 9 in phase 2 to treat VL.
Fig. 3.

Current antileishmanials.
REPOSITIONING OF DRUGS TARGETING LEISHMANIASIS
Significantly, as noted above in the Introduction, many of the existing therapies for leishmaniasis are licensed or were initially intended for other illnesses. For example, Amphotericin B 5 is an antifungal drug, paromomycin 7 is an aminoglycoside antibiotic used to treat intestinal infections, and miltefosine 8 was originally developed as an anticancer agent. This section will review drugs for different disease states; cancer, microbial infection, depression, allergies and others; that have shown potential to be repurposed as new antileishmanial therapies, and discuss their putative parasitic drug targets.
It should be noted that many natural products often used in traditional medicines have antileishmanial activity, including vast numbers of different alkaloids (Mishra et al. 2009a, b), flavonoids (Mittra et al. 2000; da Silva et al. 2012) chalcones (Chen et al. 1994; Boeck et al. 2006; Aponte et al. 2010; de Mello et al. 2014) and terpenoids (Rosa et al. 2003; Torres-Santos et al. 2004; Arruda et al. 2005; Mazoir et al. 2011). These compounds have been discussed extensively in previous literature (Mishra et al. 2009a, b; Schmidt et al. 2012; Singh et al. 2014a) however, they are structurally complex molecules, often affecting multiple targets and possess properties that make them unlikely to be orally active drugs. As such the greatest potential of natural products is for the identification of novel targets required to underpin target-based drug discovery and not as starting points for a repositioning strategy and therefore they will not be discussed further in this review.
Anticancer drugs
Both cancerous cells and Leishmania spp. have the ability to proliferate in a host organism for prolonged periods of time and some enzymes targeted by anticancer therapies can also be used in the development of antileishmanials (Fig. 4, Table 1) (Klinkert and Heussler, 2006; Uliana and Barcinski, 2009).
Fig. 4.

Treatments for cancer that could be repurposed as antileishmanials.
Table 1.
List of drugs in this review and their activities
| Drug | Species | In vitro form | EC50 (μm) | Selectivity index | Animal study | References |
|---|---|---|---|---|---|---|
| 10 | L. major | Promastigotes | 0·11 | – | Y | Diaz-Gonzalez et al. (2011) |
| L. donovani | Promastigotes | 0·14 | N | |||
| Axenic amastigotes | 0·07 | |||||
| 11 | L. major | Promastigotes | 0·12 | 0·08 | N | Patel et al. (2014) |
| Axenic amastigotes | 2·37 | |||||
| 12 | L. donovani | Intracellular amastigotes | 1·08 | 7·04 | Y | Sanderson et al. (2014) |
| L. mexicana | Intracellular amastigotes | 2·63 | 2·89 | N | ||
| 13 | L. donovani | Intracellular amastigotes | 3·73 | 1·88 | Y | |
| L. amazonensis | Intracellular amastigotes | 6·87 | 1·02 | N | ||
| L. mexicana | Intracellular amastigotes | 4·72 | 1·48 | N | ||
| L. major | Intracellular amastigotes | 3·77 | 1·86 | N | ||
| 14 | L. donovani | Intracellular amastigotes | 2·49 | 2·41 | Y | |
| 15 | L. amazonensis | – | – | – | Y | Wetzel et al. (2012) |
| 16 | L. donovani | Axenic amastigotes | 0·47 | 10 | N | Wang et al. (2015) |
| 17 | L. amazonensis | Promastigotes | 13·51 | – | Y | Trinconi et al. (2016) |
| Intracellular amastigotes | 4·25 | |||||
| 18 | L. amazonensis | Promastigotes | 30·2 | 1·77 | Y | Reimao et al. (2014) |
| Intracellular amastigotes | 16·2 | |||||
| 19 | L. major | Intracellular amastigotes | >105 | – | Y | Berman and Gallalee (1987) |
| 20 | L. major | – | – | – | Y | Zakai and Zimmo (2000) |
| 21 | L. major | Promastigotes | 136 | 1·82 | Y | Shokri et al. (2017) |
| Intracellular amastigotes | 24 | |||||
| 22 | L. amazonensis | Promastigotes | 0·44 | 187·5 | Y | de Macedo-Silva et al. (2013), Zakai and Zimmo (2000) |
| Intracellular amastigotes | 0·08 | |||||
| 23 | L. amazonensis | Promastigotes | 2·74 | 12·2 | Y | de Macedo-Silva et al. (2013) |
| Intracellular amastigotes | 1·63 | |||||
| 24 | L. infantum | Promastigotes | 2–8 | 3·6 | N | Mesquita et al. (2014) |
| Intracellular amastigotes | 11 | |||||
| 25 | L. major | Intracellular amastigotes | 0·015 | >407 | N | Khraiwesh et al. (2016) |
| 26 | 0·061 | >100 | ||||
| 27 | 0·091 | 14·2 | ||||
| 28 | 0·13 | >45·4 | ||||
| 29 | L. infantum | Promastigotes | 139 | 3·09 | N | Mesquita et al. (2013) |
| Intracellular amastigotes | 22 | |||||
| L. donovani | – | – | – | Y | Zhang et al. (2010) | |
| L. amazonensis | Promastigote | 7·2 | 115 | N | Nava-Zuazo et al. (2014) | |
| 30 | L. donovani | Promastigotes | 5·6 | >17·9 | Y | Wyllie et al. (2012) |
| Axenic amastigotes | 2·8 | |||||
| Intracellular amastigotes | Inactive | – | ||||
| 31 | L. amazonensis | – | – | – | Y | Rodrigues et al. (2006) |
| 32 | L. amazonensis | – | – | – | Y | Rodrigues et al. (2006) |
| 33 | L. donovani | Promastigotes | 0·9 | >10 | Y | Patterson et al. (2013) |
| Intracellular amastigotes | 4·92 | |||||
| 34 | L. donovani | Promastigotes | 0·16 | >54 | Y | Patterson et al. (2013) |
| Intracellular amastigotes | 0·93 | |||||
| 35 | L. donovani | Promastigotes | 0·016 | >3125 | Y | Patterson et al. (2016) |
| Axenic amastigotes | 0·005 | >10 000 | ||||
| Intracellular amastigotes | 0·087 | >575 | ||||
| 36 | L. donovani | Intracellular amastigotes | Inactive | – | Y | Smith et al. (2000) |
| 37 | L. donovani | Promastigotes | Inactive | – | N | Buates and Matlashewski (1999) |
| Axenic amastigotes | Inactive | – | ||||
| Intracellular amastigotes | Active | – | ||||
| L. major | Intracellular amastigotes | 21–42 | – | Y | El-On et al. (2007) | |
| 38 | L. infantum | Promastigotes | 34 | 4·14 | Y | Pinto et al. (2014) |
| Intracellular amastigotes | 21 | |||||
| 39 | L. donovani | Intracellular amastigotes | Active | – | N | Clark et al. (2007) |
| 40 | L. mexicana | Promastigotes | 1·24 | – | N | Navin et al. (2017) |
| 41 | L. donovani | Intracellular amastigotes | 4·90 | – | Y | Chan et al. (1998) |
| 42 | L. donovani | Intracellular amastigotes | 8·60 | 0·55 | N | Parveen et al. (2005) |
| 43 | L. donovani | Promastigotes | >50 | 9·68 | N | De Muylder et al. (2011) |
| Axenic amastigotes | >50 | |||||
| Intracellular amastigotes | 3·45 | |||||
| 44 | L. donovani | Promastigotes | 17·30 | 3·02 | N | De Muylder et al. (2011) |
| Axenic amastigotes | >50 | |||||
| Intracellular amastigotes | 5·40 | |||||
| 45 | L. donovani | Promastigotes | 37 | 3·51 | N | Singh et al. (2014b) |
| L. donovani | Intracellular amastigotes | 28 | ||||
| 46 | L. donovani | Promastigotes | 21 | 2·16 | N | Dinesh et al. (2014) |
| L. donovani | Intracellular amastigotes | 46 | ||||
| 47 | L. donovani | Promastigotes | 23·8 | >13 | N | Dinesh et al. (2015) |
| Intracellular amastigotes | 7·5 | |||||
| 48 | L. mexicana | Promastigotes | 0·9 | – | Y | Serrano-Martin et al. (2009b) |
| Intracellular amastigotes | 0·008 | |||||
| 49 | L. mexicana | Promastigotes | 0·115 | – | N | Benaim et al. (2014) |
| Intracellular amastigotes | 0·0007 | |||||
| 50 | L. major | Promastigotes | 0·41 | 1 | N | Firdessa et al. (2015) |
| Intracellular amastigotes | 4 | |||||
| 51 | L. donovani | Promastigotes | 0·25 | – | N | Witschel et al. (2012) |
Kinases have long been recognized as a major target in oncology and this has spawned a number of efforts to repurpose a range of kinase inhibitors for leishmaniasis. For example, miltefosine 8 is the first, and still the only, oral drug in the treatment of VL but was originally developed as a treatment for breast cancer. Like many anticancer drugs, miltefosine 8 induces apoptosis through kinase inhibition. In this case the PI3K/Akt/PkB pathway is targeted and it has been reported that inhibition of this pathway may influence the susceptibility of host cell to Leishmania infection (Dorlo et al. 2012).
The genomes of trypanosomatids encode at least 12 proteins belonging to the PI3K protein superfamily, many of which are unique to the parasites. The Target of Rapamycin (TOR) kinase is a member of the PI3K-related kinase (PKK) subfamily and has a central role in fundamental processes such as growth, cell shape and autophagy. Trypanosomatids possess four distinct genes belonging to the TOR family whereas mammals only possess one mTOR protein. On this basis, a targeted set of eight known mTOR inhibitors with varied potencies and selectivity for mTOR/PI3K were screened against Trypanosoma brucei, Trypanosoma cruzi, cutaneous Leishmania major and visceral L. donovani. From these data, the mTor/PI3K inhibitor, Dactolisib 10, was identified as being the most active against all the species tested and therefore was tested in animal models of T. brucei rhodesiense, T. cruzi and L. major. Unfortunately, no efficacy was observed against either T. cruzi or L. major; this may reflect the need for the drug to cross into the phagolysosomal compartment where the parasite resides. In contrast, and consistent with this hypothesis, Dactolisib significantly reduced the parasite burden and extended the survival of mice infected with T. brucei rhodesiense (Diaz-Gonzalez et al. 2011), a blood stream parasite. Although the exact mechanism of action has yet to be identified this investigation has demonstrated a potential target in the search for antitrypanosomal therapies.
Similar small scale focused screens against specific kinase targets have been conducted. For example, through exploring Aurora kinases, hesperidin 11 has been identified as a putative antileishmanial lead but host cell toxicity was observed when the compound was screened against the hepatic cancer cell line (Patel et al. 2014). Although, further structure-activity relationship (SAR) studies demonstrated that these inhibitors are not general toxins but can be modified to improve selectivity, useful antileishmanial selectivity was not obtained. More significantly, and consistent with other reports (De Muylder et al. 2011), there was no correlation between activity against promastigote and axenic amastigote with in this case the latter being the less sensitive form.
Whilst target-based screening is an attractive strategy, the need for a specific orthologous target protein may not be essential as many kinase inhibitors affect multiple proteins and simply the proven cellular accessibility of these compounds is a sufficient starting point. For example, whilst tyrosine kinases are not expressed in trypanosomatids, protein phosphorylation occurs in parasites and therefore kinase inhibitors may interact with other enzymes (Karaman et al. 2008; Peña et al. 2015). Reflecting this, in a screen of eleven tyrosine kinase inhibitors, sunitib 12, sorafenib 13 and lapatinib 14 showed antileishmanial activity against intracellular L. donovani amastigotes (Sanderson et al. 2014). Control experiments confirmed that the leishmanial targets were unlikely to be the same as those found in mammalian cells and these kinase inhibitors showed a degree of selectivity against amastigotes compared with mammalian cells. In addition, an oral dose of sunitib 12, sorafenib 13 and lapatinib 14 reduced liver parasite burden in mice infected with L. donovani. Whilst these screens identify new leads, the challenge for such studies is in the identification of the actual target to guide further optimization.
Kinases also proved to be the most common predicted target from a set of 192 antileishmanial compounds identified from a high throughput phenotypic screen of 1·8 million compounds against representative kinetoplastid species, including L. donovani (Peña et al. 2015). However, since most of these compounds have no literature precedent this was not strictly a repurposing study, and further discussion is outside the scope of this review. Finally, an interesting, and somewhat different, study involving the repurposing of kinase inhibitors, was the use of imatinib 15 (Gleevec, another tyrosine kinase inhibitor used in the treatment of multiple cancers) to target the host macrophage AbI and Arg kinases believed to be involved in parasite entry to the cell. Mice infected with L. amazonenesis were treated with oral imatinib, which resulted in smaller lesions that developed later and a reduction in parasite burden relative to controls. (Wetzel et al. 2012). Imatinib is a clinically approved drug with relatively benign side effects when compared with antileishmanials on the market. It would be of great benefit to further understand the cell entry pathway to provide new lines of therapy for leishmaniasis.
The approach of starting with inhibitors with known modes of action, for example against mammalian kinases, and screening for new antileishmanial activities for which orthologous protozoal targets can then be identified is an attractive concept. However, good levels of host parasite selectivity are required before any hits can be repurposed as antiparasitic therapeutics. This was nicely illustrated in a study exploring Lysine Deacetylases (KDAC) as putative antileishmanial drug targets. KDACs are one of the most studied epigenetic drug targets of humans (Wang et al. 2015) and inhibition of their activity is a validated strategy for cancer treatment (Patil et al. 2010). As with the kinases discussed above, Leishmania spp. have multiple genes encoding different HDACs isoenzymes some of which are essential to parasite survival (Patil et al. 2010). Whilst, the homology between kinetoplastid and mammalian KDACs is low (~40%) much higher levels of similarity are found in active site residues (Wang et al. 2015). Reflecting this only one compound, Hydroxamate 16, showed significant selectivity (~10-fold) between host cell and parasite and high (nanomolar) activity against axenic L. donovani amastigotes (Wang et al. 2015).
A final class of anticancer agents that have been shown to have potential as antileishmanials are the selective estrogen-receptor modulators, tamoxifen 17 and raloxifene 18, used to prevent and treat breast cancer. Both have been identified as potential candidates for leishmaniasis treatment with micro-molar potency against intracellular Leishmania amazonensis amastigotes (Reimao et al. 2014; Doroodgar et al. 2016; Trinconi et al. 2016). The mechanism of action of tamoxifen 17 and raloxifene 18 is still unclear as estrogen responses have not been described in Leishmania spp. (Miguel et al. 2007, 2008). However, it has been postulated that tamoxifen 17 causes changes in parasite-membrane properties and disrupts sphingolipid synthesis (Trinconi et al. 2016), whereas raloxifene 18 is thought to damage the cell membrane and the mitochondrion of the parasite (Reimao et al. 2014). Whilst the antileishmanial activity of these compounds has not necessarily been optimized, the proven safety profile of these repurposed drugs allows them to be considered as synergists in combination therapies to help combat growing concerns about the emergence of parasitic resistance. For instance, combination of tamoxifen 17 and miltefosine 8 in vitro and in vivo revealed no interaction between the two drugs and suggested that tamoxifen could slow the emergence of miltefosine 8 resistance (Trinconi et al. 2016).
Antimicrobial drugs
An antimicrobial is any compound that kills or inhibits the growth of a microorganism; this can include bacteria, fungi, viruses and protozoa. Antimicrobial drugs that were originally licensed for other indications have already been repurposed as antileishmanial therapies. For example, amphotericin B 5 and pentamidine 6 are antifungal medications and paromomycin 7 is used to treat the parasitic diseases, cryptosporidiosis and amoebiasis. These examples of successfully repurposed antimicrobials as treatments for leishmaniasis have encouraged further investigation into the leishmanicidal activity of other antimicrobial drugs (Figs 5–8, Table 1).
Fig. 5.

Antifungal drugs that could be repurposed as antileishmanials.
Fig. 6.

Antiparasitic drugs that could be repurposed as antileishmanials.
Fig. 7.

Antibacterial drugs that could be repurposed as antileishmanials.
Fig. 8.

Antiviral drugs that could be repurposed as antileishmanials.
Antifungals
As discussed above, the main use of amphotericin B 5 is in the treatment of a wide range of fungal infections but is now also used to treat VL. Liposomal amphotericin B 5 is the treatment of choice for immunocompetent patients in the Mediterranean region and the preferred drug for HIV/VL co-infection (Sundar and Chakravarty, 2010a). AmpB functions by binding to ergosterol disrupting the plasma membrane. This is the major sterol of the fungal plasma membranes and there are other antifungal azoles, which inhibit C14α-demethylase, an essential enzyme in the biosynthesis of ergosterol,. As the sterol biosynthesis pathway is conserved in Leishmania spp. parasites and is important for their survival (de Souza and Rodrigues, 2009), these antifungal azoles have the potential to be repurposed as antileishmanials. Fluconazole 19 and terbinafine 20, two antifungal azoles, which have similar modes of actions, can be administered orally and possess low levels of toxicity; therefore, the use of these drugs as putative antileishmanials in clinical trials can be approached with some confidence. Oral fluconazole 19 was shown to be a safe and useful drug for the treatment of CL caused by L. major (Alrajhi et al. 2002) and was reported to be more efficacious in higher doses after a phase 2 clinical study (Emad et al. 2011). However, a recent phase 3 clinical trial of the use of high dose fluconazole 19 in the treatment CL caused by L. braziliensis and L. guyanensis was terminated due to a low cure rate, suggesting species dependent activity (ClinicalTrials.gov, 2015a). The potential for terbinafine 20 as a new treatment for CL was first described in 1997. Significantly, at the dosage used, no patients reported any side effects. However, the efficacy appears lower than other azole antifungals notably, ketoconazole 21 and itraconazole 22, and further investigation is required to optimize the efficacy of the drug before it can be considered as an effective solution (Bahamdan et al. 1997). One method being investigated is using terbinafine 20 in combination therapies. For example, oral terbinafine 20 and cryotherapy has completed phase 1 of clinical trials for treatment of CL and results show a similar level of efficacy to glucantime 2 (Farajzadeh et al. 2015).
Positive results, coupled with attractive pharmacokinetic and safety profiles, with these antifungal azoles have encouraged the investigation of similar compounds. For example, itraconazole 22 and posaconazole 23 exhibit in vitro and in vivo activity against L. amazonensis, L. donovani and L. infantum. In addition, in a separate study, econazole 24 showed activity against L. infantum chagasi promastigotes and similar intramacrophage effectiveness to miltefosine 8 (Mesquita et al. 2014).
Antiparasitics
A second source of antimicrobial compounds that have been exploited as antileishmanials are, not surprisingly, other antiparasitics. In one approach, a collection of 400 compounds obtained from Medicines for Malaria Venture (MMV) were screened for antileishmanial activity. Significantly, in contrast to many other such approaches, which use promastigote or axenic amastigotes, this study developed an assay using intracellular amastigotes, thus enabling screening against the most clinically relevant form of the parasite (Freitas-Junior et al. 2012). The results identified 14 antimalarial drugs that have antileishmanial activity. The compound of most interest was the anticancer drug, MMV666080 25, which, with an IC50 of 15 nm, is more potent and also less toxic than amphotericin B 5. In addition, a series of substituted aminobenzimidazoles (26–28) were identified from this screen as active antileishmanials with IC50 values ranging from 61 to 134 nm. A current focus for these compounds is to validate the results using in vivo models (Khraiwesh et al. 2016).
A second set of studies has focused on the use of nitazoxanide 29, an oral drug used in the treatment of infectious diarrhoea caused by the protozoan parasites, Cryptosporidium parvum and Giardia lamblia. The lethal action of nitazoxanide 29 against L. infantum involves upregulation of reactive oxygen species resulting in oxidative stress and parasite death (Mesquita et al. 2013). Nitazoxanide 29 also displays activity against L. donovani and L. amazonensis (Zhang et al. 2010; Nava-Zuazo et al. 2014), with spleen and liver parasite load being reduced by 86% in L. donovani infected mice (Zhang et al. 2010). In addition, nitazoxanide 29 demonstrates low cytotoxicity against mammalian VERO cell line and exhibits activity against Giardia intestinalis and Trichomonas vaginalis; therefore derivatives of nitazoxanide 29 would generally be attractive starting points for new antiparasitic drugs (Nava-Zuazo et al. 2014).
Due to the closer taxonomy lineage, it would be expected that anti-trypanosomal drugs would be more likely repurposed to leishmaniasis than other antiparasitics. However, apart from pentamidine used to treat African trypanosomiasis caused by Trypanosoma gambiense, no other anti-trypanosomal drugs including those used to treat Chagas Disease are presently used against leishmaniasis.
Whilst nitroaromatics, such as nitazoxanide 29, are generally avoided in the pharmaceutical industry due to the known potential mutagenicity and carcinogenicity of the nitro group, recent research has revealed the potential of nitroaromatics to treat neglected parasitic diseases (Patterson and Wyllie, 2014). Reflecting this, another antiparasitic nitroaromatic compound, fexinidazole 30, is being explored as a potential treatment for leishmaniasis. Fexinidazole 30 is in clinical trials for treating African trypanosomiasis (T. brucei) and Chagas disease (T. cruzi) where the mechanism of action is proposed to involve reductive activation of the nitro group by NADH-dependent bacterial-like nitroreductase. A study involving the overexpression of the leishmanial orthologue of the nitroreductase demonstrated that a similar mechanism occurs in Leishmania spp. Building on this fexinidazole 30 and its two predominant in vivo metabolites (fexinidazole sulfoxide and sulfone) displayed excellent activity against L. donovani promastigotes and axenic amastigotes. However, fexinidazole was inactive against intracellular amastigotes whereas fexinidazole sulfoxide and sulfone remained potent against intracellular amastigotes (both EC50 values equal to 5·3 µm). When progressed into a murine model infected with L. donovani a 98·4% suppression of infection was observed with a single daily dose of fexindazole 30 for 5 days (Wyllie et al. 2012). With this positive data, fexinidazole 30 was progressed into clinical trials as a treatment for VL. In 2013, a phase II study tested fexinidazole 30 on 14 patients suffering with VL and all patients showed clinical improvement during treatment. Unfortunately, treatment relapses were observed and thus the study was terminated in 2014 due to lack of efficacy (ClinicalTrials.gov, 2015,b). However, fexinidazole 30 is now being investigated as a combination therapy with miltefosine 8 in Eastern Africa (DNDi, 2016).
Levamisole 31 is a heterocyclic compound that is immunoregulatory and an effective antihelminthic agent (Goldstein, 1978). Since the 1970s it has been studied as a therapy for CL (Butler, 1978) and a more recent study compared the effects of levamisole 31 and imidocarb 32, veterinary medicine used to treat parasite infection, in murine models infected with L. amazonensis. The results from a number of parameters (IgG levels, vacuolar area, megakaryocyte count in spleen and parasite burden) demonstrate that imidocarb 32 has the most potential as a therapy for CL (Rodrigues et al. 2006).
Antibacterials
The activity and safety profile of fexinidazole 30 sparked the search for other nitroimidazoles, which may have potential as oral treatments for VL. This identified the antituberculosis drugs PA-824 (33–34) and delamanid 35, which can also undergo bioactivation by a nitroreductase, as potential leads (Patterson et al. 2013; Gupta et al. 2015; Patterson et al. 2016; Thompson et al. 2016). As with fexinidazole 30 the presence of the nitro group has been shown to be essential for antileishmanial activity (Patterson et al. 2013, 2016). However the nitroreductase found in Mycobacterium tuberculosis [deazaflavin (F420)-dependent nitroreductase (Ddn)] (Manjunatha et al. 2009), is absent in Leishmania and over-expression of the leishmanial nitroreductase did not provide enhanced sensitivity to the drugs, suggesting a different mode of action. This suggests the potential for future combination therapies involving fexinidazole 30 and these antituberculosis drugs. These differences are reinforced by the observation that whilst the (S)-enantiomer of PA-824 33 is currently in phase II clinical trials for tuberculosis it is the (R)-enantiomer of PA-824 34, which shows superior activity against L. donovani parasites, both in vitro and in vivo, with a twice daily dose of (R)-PA-824 34 at 100 mg kg−1 effectively curing a murine model of infection, suppressing infection by 99·9% (Patterson et al. 2013). More recently, the structurally similar compound delamanid 35 (R-enantiomer) has been demonstrated to display high activity in vitro against intracellular L. donovani amastigotes. In addition, a twice-daily oral dose of this nitroimidazole at 30 mg kg−1 for 5 days cured the mice infected with L. donovani (Patterson et al. 2016). Whilst these compounds appear to show considerable potential, a cautionary note arises from the history of the chimeric nitroimidazole, DNDI-VL-2098 36. This compound, following promising early results, was at the final stages of preclinical development for the treatment of VL. However, results from animal models showed a link between dose, length of treatment and testicular toxicity leading to a decision to halt progression (DNDi, 2015). Whether this is a compound specific effect or a class problem remains to be established.
Antivirals
Tucaresol 36 is an investigational drug that has completed phase 2 clinical trials as a treatment for HIV (Gori et al. 2004; ClinicalTrials.gov, 2008) and, like the antiparasitic drug Levamisole 31, is an immunomodulator that demonstrates antileishmanial activity in vivo. This novel immunomodulator was inactive in L. donovani intracellular amastigote assays but showed a reduction in parasite burden in mice infected with L. donovani. Tucaresol 36 could be delivered orally at an optimum dose of 5 mg kg−1 to produce a 44 to 62% suppression of liver amastigotes (Smith et al. 2000).
Another drug with immunomodulatory effects being studied as a therapy for leishmaniasis is imiquimod 37. This immune-response modifier is a treatment for gentil warts caused by human papillomaviruses and works through activation of immune cells, including macrophages. Macrophages are the host cells of Leishmania and therefore in 1999 a study was conducted to determine if imiquimod 37 could be used as potential therapy for leishmania. In vitro results determined that imiquimod 37 could induce leishmanicidal properties in infected macrophages and a possible mode of action is through stimulated signal transduction increasing the synthesis of nitric oxide, which are toxic to the intracellular parasites. After the positive in vitro results it was important to test the capabilities of imiquimod 37 in vivo. L. major infected mice were treated with 5% imiquimod 37 cream and a significant reduction in the severity of the lesions was observed (Buates and Matlashewski, 1999) but the progression of this treatment to human trials demonstrated that topical application of imiquimod 37 alone was ineffective at treating CL (Seeberger et al. 2003). In 2005, a study focused on new approaches such as combination therapies and conducted a clinical trial of parental antimonial plus topical imiquimod 37 to treat patients with CL. Results from this study showed that 72% of patients treated with the combination containing 37 achieved a cure at 3 months vs 35% treated with the vehicle cream. In addition, accelerated healing and less prominent scarring was observed in the group treated with antimonial and imiquimod 37 compared with therapy with antimonial alone demonstrating that the next clinically available treatment for leishmaniasis might not rely on one but a combination of drugs (Miranda-Verástegui et al. 2005).
Antihistamines
Antiallergic compounds, such as quercetin found in Kalanchoe plant extracts, display potent antileishmanial effects. This reflects a capacity to restore the Th1/Th2T cell balance and also to inhibit mast cell histamine release (Cruz et al. 2008, 2012; Muzitano et al. 2008; Gomes et al. 2009; Mlcek et al. 2016). Consistent with these observations, a set of seven H1-antagonists were reported to display activity against L. infantum promastigotes with IC50 values in the range 13–84 µm (Pinto et al. 2014). The activity recorded for cinnarizine 38 (Fig. 9, Table 1) was particularly noteworthy as, unlike the other compounds tested, it also demonstrated potency against intracellular amastigotes (EC50 = 21 µm and NCTC cells EC50 = 87 µm). Whilst, initial in vivo experiments using hamsters infected with L. infantum revealed a lack of efficacy, a liposomal formulation circumvented this problem and enabled effective treatment of infection in the liver but not the spleen, which could be ascribed to the higher parasite burden (amastigotes/gram of organ) in this latter organ. Cinnarizine 38 has also been described as a calcium channel blocker, which is significant because Reimão et al. (2010) demonstrated the activity of eight calcium channel blockers against intracellular L. infantum amastigotes (EC50 = 5–176 µm) (Reimão et al. 2010). Further development of cinnarizine 38 will require the synthesis of more active analogues, an approach challenged by the fact that no target has yet been identified (Pinto et al. 2014).
Fig. 9.
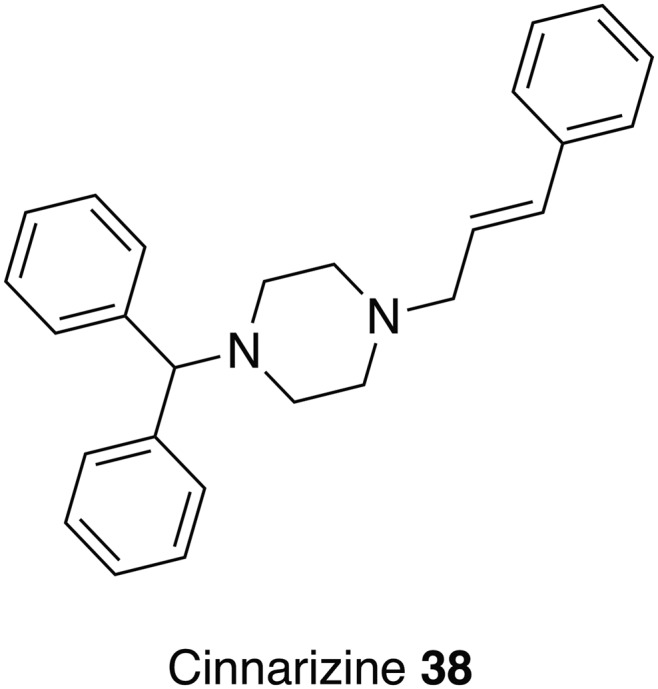
An antihistamine that could be repurposed as an antileishmanial.
In a separate study we have successfully developed an enzyme assay to explore the potential of the Leishmania sphingolipid synthase as a new antileishmanial drug target (Mina et al. 2010, 2011). This assay was employed to screen a set of 1040 pharmacologically active compounds selected from the National Institute of Neurological Disorders and Stroke (NINDS). Many of the most active and selective hits have reported antihistamine activity. As these maintained activity in infected cell models they have considerable potential for repurposing as target specific antileishmanials (Brown et al. in preparation).
Central nervous system (CNS) active drugs
Benzodiazepines are sedatives used to treat anxiety, insomnia and seizures and various analogues have been reported to show activity against visceral and CL (Fig. 10). Compound 39, structurally related to paullone, demonstrated efficacy against L. donovani amastigotes with no observed toxicity (Clark et al. 2007). In a separate study, benzodiazepine 40 was effective against L. mexicana promastigotes with a better IC50 value than that observed for miltefosine 8 (Navin et al. 2017). However, metabolic studies using rat hepatocytes and microsomal preparation showed that the potential of these compounds is limited by extensive and rapid metabolism. Therefore, further rounds of medicinal chemistry optimisation will be required (Thi et al. 2009).
Fig. 10.

CNS drugs that could be repurposed as antileishmanials.
Tricycylic heterocycles are part of numerous therapeutic agents and therefore important structures in medicinal chemistry (Marcu et al. 2016). Chlorpromazine 41 is perhaps the best known of the phenothiazine drugs (tricycylic heterocycles) for treatment of neurological disorders. In 1984 this compound was identified as having activity against L. donovani in vitro and in vivo (Pearson et al. 1984) and in a separate study also showed it could kill L. mexicana, L. aethiopica and L.major promastigotes at a concentration of 7·5 µg mL−1 (El-On et al. 1986). The same study determined the in vivo effect for chlorpromazine 41 in a murine model infected with L. major and L. mexicana and demonstrated leishmanicidal effect in the spleen but not in the skin lesion (El-On et al. 1986). Therefore, like pentamidine, this compound was shown to be effective against VL but with only partial effect against the cutaneous form. Chlorpromazine 41 will probably not be useful in the treatment of VL due to toxicity concerns but these results have led to the investigation of structurally similar compounds (Chan et al. 1998; Mukherjee et al. 2012; Marcu et al. 2016) to explore the SAR. For example, chlorpromazine analogues with a quaternary nitrogen atom 42 have demonstrated antileishmanial, antitrypanosomal and antimalarial activity (Khan et al. 2000; Parveen et al. 2005). These analogues are trypanothione reductase inhibitors, which are likely to affect the redox defense of the parasite and hence increase the parasites sensitivity to redox-damaged-based drugs. In addition, the enzyme, trypanothione reductase, is present in Leishmania and Trypanosoma but not the mammalian hosts providing the opportunity to design a selective inhibitor (Khan et al. 2000; Parveen et al. 2005).
HTS against the L. major inositol phosphorylceramide synthase (IPCS) formatted into a cell-based yeast assay identified a class of benzazepanes as potent inhibitors of the enzyme with good activity against both L. major and intramacrophage L. donovani with good levels of selectivity vs Human THP-1 cells; on target effects were shown by comparison with a mutant L. major strain with redundant sphingolipid synthase activity. However, again further progression requires improvements to the pharmacokinetic profile of these compounds (Norcliffe et al., in prep).
The opioid receptor antagonist naloxonazine 43 was identified in a screen comparing promastigote and amastigote activity in which it was uniquely the only compound identified that showed activity against the intracellular amastigote and not the promastigote. Curiously, the structurally related opioid naloxone (Narcan®) was completely inactive against both parasitic forms. Since homologs of opioid receptors have not been identified in the Leishmania genome it is possible that opioids are involved in modulation of host resistance to parasite infection. In support of this, Loperamide 44, a μ-opioid receptor agonist, was also identified in this study as inhibiting parasite growth with greater activity against the intracellular stage of the parasite (De Muylder et al. 2011).
Finally, antileishmanial activity was observed when the broad spectrum antidepressants, ketanserin 45 and mianserin 46, were screened against L. donovani promastigotes and intramacrophage amastigotes. Amongst other enzymes these compounds are known to target 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR). This is the rate-limiting enzyme of the sterol biosynthesis, which is also the putative pathway targeted by amphotericin B 5. In support of this proposal both compounds inhibited recombinant L. donovani HMGR, although given the diverse reported activities, other modes of action cannot be discounted (Dinesh et al. 2014; Singh et al. 2014b).
Other drugs
Similar to ketanserin 45 and mianserin 46 discussed above, statins target HMGR. Statins are currently on the market to reduce cardiovascular disease by lowering the level of low-density lipoprotein cholesterol (‘bad cholesterol’) in the blood. Studies have shown that statins also have the potential to be repurposed as antileishmanials (Kumar et al. 2016; Parihar et al. 2016). For example, mevastatin 47 also inhibits recombinant L. donovani HMGR enzyme with an IC50 value of 42·2 ± 3·0 µm. It also shows correspondingly good activity against both L. donovani promastigotes and intracellular amastigotes with no toxicity exhibited towards the host cell line (Dinesh et al. 2015).
Amiodarone 48 and dronedarone 49, (Fig. 11) used to treat cardiac arrhythmias, have antileishmanial activity against L. mexicana promastigotes and intramacrophage amastigotes, and inhibit oxidosqualene cyclase (OSC), another enzyme essential for ergosterol biosynthesis. However, as with the antihistamines discussed above, these drugs are also known to disrupt Ca2+ homeostasis in Saccharomyces cerevisiae and Trypanosoma cruzi, so may have multiple modes of action (Serrano-Martin et al. 2009a). The in vivo activity of amiodarone 48 was investigated in a murine model infected with L. mexicana. amiodarone 48 was given as an oral therapy at 50 mg kg−1 day−1 and although preventing the development of lesions during the course of treatment, reoccurrences of the infection were observed once treatment had ended. Amiodarone 48 was then tested in combination with miltefosine 8 at a dose of 50 and 20 mg kg−1 day−1 respectively, and showed permanent control of lesion size. Hence, using this drug in combination with currently available antiparasitics could lower the dosage and reduce the known side effects of amiodarone (e.g. cardiotoxicity, thyroid dysfunction and pulmonary fibrosis) (Serrano-Martín et al. 2009,b). Alternatively, dronedarone 49 appears to have great potential to be repurposed as it has greater antiparasitic activity coupled with lower mammalian toxicity than amiodarone 48 (Benaim et al. 2014).
Fig. 11.

Other drugs that could be repurposed as antileishmanials.
This review has focused on repurposing synthetic small molecules, though there are studies that have investigated polymers and natural products. For instance, a recent study confirmed that polyhexanide 50, a wound antiseptic and disinfectant, has antileishmanial activity with IC50 values of 0·41 µm against L. major promastigotes, 69-fold more potent than miltefosine 8, and 4 µm against intracellular L. major amastigotes. Polyhexanide 50 is thought to kill the parasite by disruption to membrane structure and selective chromosome condensation and damage. Interestingly, it was also discovered that this cationic polymer can be used as a vehicle to transport cargoes to the macrophage and thus has the ability to deliver immunomodulatory agents (Firdessa et al. 2015).
Whilst the focus of this review has been on repurposing human therapeutics, there is the potential for investigating other groups of bioactive chemicals, including both agrochemicals and veterinary medicines for human use as treatments for neglected diseases. For example, more than 600 commercial agrochemicals were screened against parasitic protozoans and zoxamide 51 was found to be the most effective against L. donovani promastigotes. Zoxamide 51 is an oomyceticidal compound used in fruit and vegetables, and acts through the inhibition of microtubule formation. Microtubule inhibitors may be a class of potential antileishmanials since tubulin is one of the most abundant leishmanial proteins (Kapoor et al. 1999). Zoxamide 51 killed the promastigotes with an IC50 value of 250 nm (Witschel et al. 2012). Although explored in other contexts (Andrews et al. 2014), there has been little reported effort to repurpose veterinary medicines for leishmanaisis, possibly due to the potential human toxicity issues which could arise and negate the benefits of the repurposing approach.
Concluding remarks
The repositioning of old drugs for new uses is not a new concept and is a particularly attractive approach for neglected tropical diseases. Here the use of approved drugs is particularly beneficial as clinical trials have already successfully been conducted and often their patents have expired leading to shorter, cheaper discovery pipelines. As discussed in the various sections above, a wide range of structures have been explored for antileishmanial activity. Significantly, most of the medicines used to treat leishmaniasis, both on the market or under development in the drug discovery process, were initially intended for other indications. Furthermore, more novel approaches show drug repositioning is extending beyond the scope of human medication as investigation into the human use of agrochemicals and veterinary therapeutics is underway. However, drug repurposing is limited by the number of approved drugs and compounds with established pharmacokinetic data, in addition the repositioning discovery process tends to mean that there is less chance of finding treatments that act through a new mechanism of action. Overall, whilst not the sole solution, drug repositioning represents a valuable, relatively fast and cost-effective, strategy for developing essential new therapies, particularly for neglected tropical diseases such as leishmaniasis.
FINANCIAL SUPPORT
We thank CNPq (PVE Grant: 400894/2014-9 to BRB and PGS); The Royal Society (The Royal Society International Collaboration Awards for Research Professors 2016: IC160044 to BRB and PGS); and BBSRC (BB/M024156/1 to PWD) for financial support.
REFERENCES
- Akhoundi M., Kuhls K., Cannet A., Votýpka J., Marty P., Delaunay P. and Sereno D. (2016). A historical overview of the classification, evolution, and dispersion of leishmania parasites and sandflies. PLoS Neglected Tropical Diseases 10, e0004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrajhi A. A., Ibrahim E. A., De Vol E. B., Khairat M., Faris R. M. and Maguire J. H. (2002). Fluconazole for the treatment of cutaneous leishmaniasis caused by leishmania major. New England Journal of Medicine 346, 891–895. [DOI] [PubMed] [Google Scholar]
- Alvar J., Aparicio P., Aseffa A., Den Boer M., Cañavate C., Dedet J.-P., Gradoni L., Ter Horst R., López-Vélez R. and Moreno J. (2008). The relationship between leishmaniasis and AIDS: the second 10 years. Clinical Microbiology Reviews 21, 334–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J., Vélez I. D., Bern C., Herrero M., Desjeux P., Cano J., Jannin J. and den Boer M. (2012). Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 7, e35671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K. T., Fisher G. and Skinner-Adams T. S. (2014). Drug repurposing and human parasitic protozoan diseases. International Journal for Parasitology: Drugs and Drug Resistance 4, 95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte J. C., Castillo D., Estevez Y., Gonzalez G., Arevalo J., Hammond G. B. and Sauvain M. (2010). In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorganic & Medicinal Chemistry Letters 20, 100–103. [DOI] [PubMed] [Google Scholar]
- Arruda D. C., D'Alexandri F. L., Katzin A. M. and Uliana S. R. B. (2005). Antileishmanial activity of the terpene nerolidol. Antimicrobial Agents and Chemotherapy 49, 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn T. T. and Thor K. B. (2004). Drug repositioning: identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery 3, 673–683. [DOI] [PubMed] [Google Scholar]
- Aubé J. (2012). Drug repurposing and the medicinal chemist. ACS Medicinal Chemistry Letters 3, 442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahamdan K. A., Tallab T. M., Johargi H., Nourad M. M., Ibrahim K., el Sherbini A. H., Karkashan E., Khare A. K. and Nauri M. M. (1997). Terbinafine in the treatment of cutaneous leishmaniasis: a pilot study. International Journal of Dermatology 36, 59–60. [DOI] [PubMed] [Google Scholar]
- Benaim G., Casanova P., Hernandez-Rodriguez V., Mujica-Gonzalez S., Parra-Gimenez N., Plaza-Rojas L., Concepcion J. L., Liu Y.-L., Oldfield E., Paniz-Mondolfi A. and Suarez A. I. (2014). Dronedarone, an amiodarone analog with improved anti-leishmania mexicana efficacy. Antimicrobial Agents and Chemotherapy 58, 2295–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman J. D. and Gallalee J. V. (1987). In vitro antileishmanial activity of inhibitors of steroid biosynthesis and combinations of antileishmanial agents. Journal of Parasitology 73, 671–673. [PubMed] [Google Scholar]
- Bern C., Maguire J. H. and Alvar J. (2008). Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Neglected Tropical Diseases 2, e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeck P., Bandeira Falcão C. A., Leal P. C., Yunes R. A., Filho V. C., Torres-Santos E. C. and Rossi-Bergmann B. (2006). Synthesis of chalcone analogues with increased antileishmanial activity. Bioorganic & Medicinal Chemistry 14, 1538–1545. [DOI] [PubMed] [Google Scholar]
- Buates S. and Matlashewski G. (1999). Treatment of experimental leishmaniasis with the immunomodulators imiquimod and S-28463: efficacy and mode of action. Journal of Infectious Diseases 179, 1485–1494. [DOI] [PubMed] [Google Scholar]
- Butler P. G. (1978). Levamisole therapy of chronic Leishmania tropica. Journal of Tropical Medicine and Hygiene 81, 221–224. [PubMed] [Google Scholar]
- CDC (2016). Leishmaniasis – resources for health professionals. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/.
- Chan C., Yin H., Garforth J., McKie J. H., Jaouhari R., Speers P., Douglas K. T., Rock P. J., Yardley V., Croft S. L. and Fairlamb A. H. (1998). Phenothiazine inhibitors of trypanothione reductase as potential antitrypanosomal and antileishmanial drugs. Journal of Medicinal Chemistry 41, 148–156. [DOI] [PubMed] [Google Scholar]
- Chen M., Christensen S. B., Theander T. G. and Kharazmi A. (1994). Antileishmanial activity of licochalcone A in mice infected with Leishmania major and in hamsters infected with Leishmania donovani. Antimicrobial Agents and Chemotherapy 38, 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. L., Carter K. C., Mullen A. B., Coxon G. D., Owusu-Dapaah G., McFarlane E., Duong Thi M. D., Grant M. H., Tettey J. N. A. and Mackay S. P. (2007). Identification of the benzodiazepines as a new class of antileishmanial agent. Bioorganic & Medicinal Chemistry Letters 17, 624–627. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov (2008). Tucaresol as Add-On to HAART (Highly Active Antiretroviral Therapy) In Chronic HIV-1 Infected Adults. https://clinicaltrials.gov/ct2/show/NCT00343941.
- ClinicalTrials.gov (2015a). High Dose Fluconazole in Cutaneous Leishmaniasis in Bahia and Manaus. https://clinicaltrials.gov/ct2/show/NCT01953744.
- ClinicalTrials.gov (2015b). Trial to Determine Efficacy of Fexinidazole in Visceral Leihmaniasis Patients in Sudan. https://clinicaltrials.gov/ct2/show/NCT01980199.
- ClinicalTrials.gov (2016). Phase 3 Study of Walter Reed (WR) 279,396 and Paromomycin Alone for the Treatment of Cutaneous Leishmaniasis in Panama. https://clinicaltrials.gov/ct2/show/NCT01790659.
- Croft S. L., Sundar S. and Fairlamb A. H. (2006). Drug resistance in leishmaniasis. Clinical Microbiology Reviews 19, 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz E. A., Da-Silva S. A. G., Muzitano M. F., Silva P. M. R., Costa S. S. and Rossi Bergmann B. (2008). Immunomodulatory pretreatment with Kalanchoe pinnata extract and its quercitrin flavonoid effectively protects mice against fatal anaphylactic shock. International Immunopharmacology 8, 1616–1621. [DOI] [PubMed] [Google Scholar]
- Cruz E. A., Reuter S., Martin H., Muzitano M. F., Costa S. S., Rossi-Bergmann B. and Taube C. (2012). Kalanchoe pinnata inhibits mast cell activation and prevents allergic airway disease. Phytomedicine 19, 115–121. [DOI] [PubMed] [Google Scholar]
- da Silva E. R., do Maquiaveli C. C. and Magalhães P. P. (2012). The leishmanicidal flavonols quercetin and quercitrin target Leishmania (Leishmania) amazonensis arginase. Experimental Parasitology 130, 183–188. [DOI] [PubMed] [Google Scholar]
- Davies C. R., Kaye P., Croft S. L. and Sundar S. (2003). Leishmaniasis: new approaches to disease control. BMJ: British Medical Journal 326, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Macedo-Silva S. T., Urbina J. A., de Souza W. and Rodrigues J. C. F. (2013). In vitro activity of the antifungal azoles itraconazole and posaconazole against leishmania amazonensis. PLOS ONE 8, e83247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mello T. F. P., Bitencourt H. R., Pedroso R. B., Aristides S. M. A., Lonardoni M. V. C. and Silveira T. G. V. (2014). Leishmanicidal activity of synthetic chalcones in Leishmania (Viannia) braziliensis. Experimental Parasitology 136, 27–34. [DOI] [PubMed] [Google Scholar]
- de Menezes J. P. B., Guedes C. E. S., Petersen A., Almeida nio L. de O., Fraga D. B. M., Veras P., Tavares cia S., de Menezes J. P. B., Guedes C. E. S., Petersen A., Almeida nio L. de O., Fraga D. B. M., Veras P. and Tavares cia S. (2015). Advances in development of new treatment for leishmaniasis, advances in development of new treatment for leishmaniasis. BioMed Research International, BioMed Research International 2015, e815023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muylder G., Ang K. K. H., Chen S., Arkin M. R., Engel J. C. and McKerrow J. H. (2011). A Screen against leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Neglected Tropical Diseases 5, e1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza W. and Rodrigues J. C. F. (2009). Sterol biosynthesis pathway as target for anti-trypanosomatid drugs. Interdisciplinary Perspectives on Infectious Diseases 2009, e642502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Gonzalez R., Kuhlmann F. M., Galan-Rodriguez C., da Silva L. M., Saldivia M., Karver C. E., Rodriguez A., Beverley S. M., Navarro M. and Pollastri M. P. (2011). The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLOS Neglected Tropical Diseases 5, e1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinesh N., Kaur P. K., Swamy K. K. and Singh S. (2014). Mianserin, an antidepressant kills Leishmania donovani by depleting ergosterol levels. Experimental Parasitology 144, 84–90. [DOI] [PubMed] [Google Scholar]
- Dinesh N., Soumya N. and Singh S. (2015). Antileishmanial effect of mevastatin is due to interference with sterol metabolism. Parasitology Research 114, 3873–3883. [DOI] [PubMed] [Google Scholar]
- DNDi (2015). VL-2098. http://www.dndi.org/diseases-projects/portfolio/completed-projects/vl-2098/.
- DNDi (2016). Fexinidazole/miltefosine combination (VL). http://www.dndi.org/diseases-projects/portfolio/fexinidazole-vl/.
- Dorlo T. P. C., Balasegaram M., Beijnen J. H. and de Vries P. J. (2012). Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. Journal of Antimicrobial Chemotherapy 67, 2576–2597. [DOI] [PubMed] [Google Scholar]
- Doroodgar M., Delavari M., Doroodgar M., Abbasi A., Taherian A. A. and Doroodgar A. (2016). Tamoxifen induces apoptosis of leishmania major promastigotes in vitro. Korean Journal of Parasitology 54, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-On J., Rubinstein N., Kernbaum S. and Schnur L. F. (1986). In vitro and in vivo anti-leishmanial activity of chlorpromazine alone and combined with N-meglumine antimonate. Annals of Tropical Medicine & Parasitology 80, 509–517. [DOI] [PubMed] [Google Scholar]
- El-On J., Bazarsky E. and Sneir R. (2007). Leishmania major: in vitro and in vivo anti-leishmanial activity of paromomycin ointment (Leshcutan) combined with the immunomodulator Imiquimod. Experimental Parasitology 116, 156–162. [DOI] [PubMed] [Google Scholar]
- Emad M., Hayati F., Fallahzadeh M. K. and Namazi M. R. (2011). Superior efficacy of oral fluconazole 400 mg daily versus oral fluconazole 200 mg daily in the treatment of cutaneous leishmania major infection: a randomized clinical trial. Journal of the American Academy of Dermatology 64, 606–608. [DOI] [PubMed] [Google Scholar]
- Farajzadeh S., Esfandiarpour I., Haghdoost A. A., Mohammadi S., Mohebbi A., Mohebbi E. and Mostafavi M. (2015). Comparison between combination therapy of oral terbinafine and cryotherapy versus systemic meglumine antimoniate and cryotherapy in cutaneous leishmaniasis: a randomized clinical trial. Iranian Journal of Parasitology 10, 1–8. [PMC free article] [PubMed] [Google Scholar]
- Fernández M. M., Malchiodi E. L. and Algranati I. D. (2011). Differential effects of paromomycin on ribosomes of leishmania mexicana and mammalian cells. Antimicrobial Agents and Chemotherapy 55, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firdessa R., Good L., Amstalden M. C., Chindera K., Kamaruzzaman N. F., Schultheis M., Roeger B., Hecht N., Oelschlaeger T. A., Meinel L., Luehmann T. and Moll H. (2015). Pathogen- and host-directed antileishmanial effects mediated by polyhexanide (PHMB). Plos Neglected Tropical Diseases 9, e0004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas-Junior L. H., Chatelain E., Kim H. A. and Siqueira-Neto J. L. (2012). Visceral leishmaniasis treatment: what do we have, what do we need and how to deliver it? International Journal for Parasitology: Drugs and Drug Resistance 2, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein G. (1978). Mode of action of levamisole. Journal of Rheumatology. Supplement 4, 143–148. [PubMed] [Google Scholar]
- Gomes D. C. O., Muzitano M. F., Costa S. S. and Rossi-Bergmann B. (2009). Effectiveness of the immunomodulatory extract of Kalanchoe pinnata against murine visceral leishmaniasis. Parasitology 137, 613–618. [DOI] [PubMed] [Google Scholar]
- Gori A., Trabattoni D., Bandera A., Saresella M., Marchetti G., Gazzola L., Biasin M., Rhodes J., McDade H., Panebianco R., Galli M., Moroni M., Ferrante P., Thomas N., Franzetti F., Bray D. and Clerici M. (2004). Immunomodulation induced by tucaresol in HIV infection: results of a 16 week pilot Phase I/II trial. Antiviral Therapy 9, 603–614. [PubMed] [Google Scholar]
- Gradoni L. (2013). Epidemiological surveillance of leishmaniasis in the European Union: operational and research challenges. Eurosurveillance 18, 20539. [DOI] [PubMed] [Google Scholar]
- Guha M. (2015). Repositioning existing drugs for cancer treatment. The Pharmaceutical Journal 294, 7867. [Google Scholar]
- Gupta S., Yardley V., Vishwakarma P., Shivahare R., Sharma B., Launay D., Martin D. and Puri S. K. (2015). Nitroimidazo-oxazole compound DNDI-VL-2098: an orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. Journal of Antimicrobial Chemotherapy 70, 518–527. [DOI] [PubMed] [Google Scholar]
- Hailu A., Musa A., Wasunna M., Balasegaram M., Yifru S., Mengistu G., Hurissa Z., Hailu W., Weldegebreal T., Tesfaye S., Makonnen E., Khalil E., Ahmed O., Fadlalla A., El-Hassan A., Raheem M., Mueller M., Koummuki Y., Rashid J., Mbui J., Mucee G., Njoroge S., Manduku V., Musibi A., Mutuma G., Kirui F., Lodenyo H., Mutea D., Kirigi G., Edwards T., et al. and Group, for the L. E. A. P. (LEAP) (2010). Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: a multicentre, open-label, randomized trial. PLOS Neglected Tropical Diseases 4, e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V. and Sharma A. (2017). Repurposing of potent drug candidates for multiparasite targeting. Trends in Parasitology 33, 158–161. [DOI] [PubMed] [Google Scholar]
- Kamiński D. M. (2014). Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. European biophysics journal: EBJ 43, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P., Sachdeva M. and Madhubala R. (1999). Effect of the microtubule stabilising agent taxol on leishmanial protozoan parasites in vitro. FEMS Microbiology Letters 176, 429–435. [DOI] [PubMed] [Google Scholar]
- Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M. and Zarrinkar P. P. (2008). A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnology 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Kedzierski L. (2010). Leishmaniasis vaccine: where are we today? Journal of Global Infectious Diseases 2, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski L., Sakthianandeswaren A., Curtis J., Andrews P., Junk P. and Kedzierska K. (2009). Leishmaniasis: current treatment and prospects for new drugs and vaccines. Current Medicinal Chemistry 16, 599–614. [DOI] [PubMed] [Google Scholar]
- Khan M. O. F., Austin S. E., Chan C., Yin H., Marks D., Vaghjiani S. N., Kendrick H., Yardley V., Croft S. L. and Douglas K. T. (2000). Use of an additional hydrophobic binding site, the Z site, in the rational drug design of a new class of stronger trypanothione reductase inhibitor, quaternary alkylammonium phenothiazines. Journal of Medicinal Chemistry 43, 3148–3156. [DOI] [PubMed] [Google Scholar]
- Khraiwesh M., Leed S., Roncal N., Johnson J., Sciotti R., Smith P., Read L., Paris R., Hudson T., Hickman M. and Grogl M. (2016). Antileishmanial activity of compounds derived from the medicines for malaria venture open access box against intracellular leishmania major amastigotes. American Journal of Tropical Medicine and Hygiene 94, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert M.- and Heussler V. (2006). The use of anticancer drugs in antiparasitic chemotherapy. Mini-Reviews in Medicinal Chemistry 6, 131–143. [DOI] [PubMed] [Google Scholar]
- Klug D. M., Gelb M. H. and Pollastri M. P. (2016). Repurposing strategies for tropical disease drug discovery. Bioorganic & Medicinal Chemistry Letters 26, 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauth-Siegel R. L., Comini M. A. (2008). Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochimica et Biophysica Acta 1780, 1236–1248. [DOI] [PubMed] [Google Scholar]
- Kumar G. A., Roy S., Jafurulla M., Mandal C. and Chattopadhyay A. (2016). Statin-induced chronic cholesterol depletion inhibits Leishmania donovani infection: relevance of optimum host membrane cholesterol. Biochimica et Biophysica Acta (BBA) – Biomembranes 1858, 2088–2096. [DOI] [PubMed] [Google Scholar]
- Lockwood D. and Moore E. (2010). Treatment of visceral leishmaniasis. Journal of Global Infectious Diseases 2, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado P. R. L., Lessa H., Lessa M., Guimarães L. H., Bang H., Ho J. L. and Carvalho E. M. (2007). Oral pentoxifylline combined with pentavalent antimony: a randomized trial for mucosal leishmaniasis. Clinical Infectious Diseases 44, 788–793. [DOI] [PubMed] [Google Scholar]
- Manjunatha U., Boshoff H. I. M. and Barry C. E. (2009). The mechanism of action of PA-824. Communicative & Integrative Biology 2, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu A., Schurigt U., Müller K., Moll H., Krauth-Siegel R. L. and Prinz H. (2016). Inhibitory effect of phenothiazine- and phenoxazine-derived chloroacetamides on Leishmania major growth and Trypanosoma brucei trypanothione reductase. European Journal of Medicinal Chemistry 108, 436–443. [DOI] [PubMed] [Google Scholar]
- Mazoir N., Benharref A., Bailén M., Reina M., González-Coloma A. and Martínez-Díaz R. A. (2011). Antileishmanial and antitrypanosomal activity of triterpene derivatives from latex of two Euphorbia species. Zeitschrift Fur Naturforschung. C, Journal of Biosciences 66, 360–366. [DOI] [PubMed] [Google Scholar]
- Mesquita J. T., Pinto E. G., Taniwaki N. N., Galisteo A. J. Jr. and Tempone A. G. (2013). Lethal action of the nitrothiazolyl-salicylamide derivative nitazoxanide via induction of oxidative stress in Leishmania (L.) infantum. Acta Tropica 128, 666–673. [DOI] [PubMed] [Google Scholar]
- Mesquita J. T., da Costa-Silva T. A., Borborema S. E. T. and Tempone A. G. (2014). Activity of imidazole compounds on Leishmania (L.) infantum chagasi: reactive oxygen species induced by econazole. Molecular and Cellular Biochemistry 389, 293–300. [DOI] [PubMed] [Google Scholar]
- Miguel D. C., Yokoyama-Yasunaka J. K. U., Andreoli W. K., Mortara R. A. and Uliana S. R. B. (2007). Tamoxifen is effective against Leishmania and induces a rapid alkalinization of parasitophorous vacuoles harbouring Leishmania (Leishmania) amazonensis amastigotes. Journal of Antimicrobial Chemotherapy 60, 526–534. [DOI] [PubMed] [Google Scholar]
- Miguel D. C., Yokoyama-Yasunaka J. K. U. and Uliana S. R. B. (2008). Tamoxifen is effective in the treatment of Leishmania amazonensis infections in mice. PLoS Neglected Tropical Diseases 2, e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina J. G., Mosely J. A., Ali H. Z., Shams-Eldin H., Schwarz R. T., Steel P. G. and Denny P. W. (2010). A plate-based assay system for analyses and screening of the Leishmania major inositol phosphorylceramide synthase. International Journal of Biochemistry & Cell Biology 42, 1553–1561. [DOI] [PubMed] [Google Scholar]
- Mina J. G., Mosely J. A., Ali H. Z., Denny P. W. and Steel P. G. (2011). Exploring leishmania majorInositol phosphorylceramide synthase (LmjIPCS): insights into the ceramide binding domain. Organic & Biomolecular Chemistry 9, 1823–1830. [DOI] [PubMed] [Google Scholar]
- Miranda-Verástegui C., Llanos-Cuentas A., Arévalo I., Ward B. J. and Matlashewski G. (2005). Randomized, double-blind clinical trial of topical imiquimod 5% with parenteral meglumine antimoniate in the treatment of cutaneous leishmaniasis in Peru. Clinical Infectious Diseases 40, 1395–1403. [DOI] [PubMed] [Google Scholar]
- Mishra B. B., Singh R. K., Srivastava A., Tripathi V. J. and Tiwari V. K. (2009a). Fighting against leishmaniasis: search of alkaloids as future true potential anti-leishmanial agents. Mini Reviews in Medicinal Chemistry 9, 107–123. [DOI] [PubMed] [Google Scholar]
- Mishra B. B., Kale R. R., Singh R. K. and Tiwari V. K. (2009b). Alkaloids: future prospective to combat leishmaniasis. Fitoterapia 80, 81–90. [DOI] [PubMed] [Google Scholar]
- Mittra B., Saha A., Chowdhury A. R., Pal C., Mandal S., Mukhopadhyay S., Bandyopadhyay S. and Majumder H. K. (2000). Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Molecular Medicine 6, 527–541. [PMC free article] [PubMed] [Google Scholar]
- Mlcek J., Jurikova T., Skrovankova S. and Sochor J. (2016). Quercetin and its anti-allergic immune response. Molecules 21, 623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni A. Z., Reiszadae M. R. and Aminjavaheri M. (2002). Treatment of cutaneous leishmaniasis with a combination of allopurinol and low-dose meglumine antimoniate. International Journal of Dermatology 41, 441–443. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Mukherjee B., Mukhopadhyay R., Naskar K., Sundar S., Dujardin J. C., Das A. K. and Roy S. (2012). Imipramine is an orally active drug against both antimony sensitive and resistant Leishmania donovani clinical isolates in experimental infection. PLoS Neglected Tropical Diseases 6, e1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa A. M., Younis B., Fadlalla A., Royce C., Balasegaram M., Wasunna M., Hailu A., Edwards T., Omollo R., Mudawi M., Kokwaro G., El-Hassan A. and Khalil E. (2010). Paromomycin for the treatment of visceral leishmaniasis in Sudan: a randomized, open-label, dose-finding study. PLoS Neglected Tropical Diseases 4, e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzitano M. F., Falcão C. A. B., Cruz E. A., Bergonzi M. C., Bilia A. R., Vincieri F. F., Rossi-Bergmann B. and Costa S. S. (2008). Oral metabolism and efficacy of Kalanchoe pinnata flavonoids in a murine model of cutaneous leishmaniasis. Planta Medica 75, 307–311. [DOI] [PubMed] [Google Scholar]
- Nava-Zuazo C., Chavez-Silva F., Moo-Puc R., Jesus Chan-Bacab M., Otto Ortega-Morales B., Moreno-Diaz H., Diaz-Coutino D., Hernandez-Nunez E. and Navarrete-Vazquez G. (2014). 2-acylamino-5-nitro-1,3-thiazoles: preparation and in vitro bioevaluation against four neglected protozoan parasites. Bioorganic & Medicinal Chemistry 22, 1626–1633. [DOI] [PubMed] [Google Scholar]
- Navin P., Sarvil P., Amit P., Divyesh P., Dhansukh R., Moo-Puc R. and Rivera G. (2017). Synthesis and biological evaluation of newer 1,3,4-oxadiazoles incorporated with benzothiazepine and benzodiazepine moieties. Zeitschrift für Naturforschung C 72, 133–146. [DOI] [PubMed] [Google Scholar]
- No J. H. (2016). Visceral leishmaniasis: revisiting current treatments and approaches for future discoveries. Acta Tropica 155, 113–123. [DOI] [PubMed] [Google Scholar]
- Oprea T. I. and Mestres J. (2012). Drug repurposing: far beyond new targets for old drugs. AAPS Journal 14, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar S. P., Hartley M.-A., Hurdayal R., Guler R. and Brombacher F. (2016). Topical simvastatin as host-directed therapy against severity of cutaneous leishmaniasis in mice. Scientific Reports 6, 33458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen S., Khan M. O. F., Austin S. E., Croft S. L., Yardley V., Rock P. and Douglas K. T. (2005). Antitrypanosomal, antileishmanial, and antimalarial activities of quaternary arylalkylammonium 2-amino-4-chlorophenyl phenyl sulfides, a new class of trypanothione reductase inhibitor, and of N-acyl derivatives of 2-amino-4-chlorophenyl phenyl sulfide. Journal of Medicinal Chemistry 48, 8087–8097. [DOI] [PubMed] [Google Scholar]
- Patel G., Roncal N. E., Lee P. J., Leed S. E., Erath J., Rodriguez A., Sciotti R. J. and Pollastri M. P. (2014). Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. MedChemComm 5, 655–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil V., Guerrant W., Chen P. C., Gryder B., Benicewicz D. B., Khan S. I., Tekwani B. L. and Oyelere A. K. (2010). Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorganic & Medicinal Chemistry 18, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S. and Wyllie S. (2014). Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends in Parasitology 30, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S., Wyllie S., Stojanovski L., Perry M. R., Simeons F. R. C., Norval S., Osuna-Cabello M., Rycker M. D., Read K. D. and Fairlamb A. H. (2013). The R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for visceral leishmaniasis. Antimicrobial Agents and Chemotherapy 57, 4699–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S., Wyllie S., Norval S., Stojanovski L., Simeons F. R. C., Auer J. L., Osuna-Cabello M., Read K. D. and Fairlamb A. H. (2016). The anti-tubercular drug delamanid as a potential oral treatment for visceral leishmaniasis. Elife 5, e09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S. M., Mytelka D. S., Dunwiddie C. T., Persinger C. C., Munos B. H., Lindborg S. R. and Schacht A. L. (2010). How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature Reviews Drug Discovery 9, 203–214. [DOI] [PubMed] [Google Scholar]
- Pearson R. D., Manian A. A., Hall D., Harcus J. L. and Hewlett E. L. (1984). Antileishmanial activity of chlorpromazine. Antimicrobial Agents and Chemotherapy 25, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña I., Pilar Manzano M., Cantizani J., Kessler A., Alonso-Padilla J., Bardera A. I., Alvarez E., Colmenarejo G., Cotillo I., Roquero I., de Dios-Anton F., Barroso V., Rodriguez A., Gray D. W., Navarro M., Kumar V., Sherstnev A., Drewry D. H., Brown J. R., Fiandor J. M. and Julio Martin J. (2015). New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Scientific Reports 5, 8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto E. G., da Costa-Silva T. A. and Tempone A. G. (2014). Histamine H1-receptor antagonists against Leishmania (L.) infantum: an in vitro and in vivo evaluation using phosphatidylserine-liposomes. Acta Tropica 137, 206–210. [DOI] [PubMed] [Google Scholar]
- Reimão J. Q., Scotti M. T. and Tempone A. G. (2010). Anti-leishmanial and anti-trypanosomal activities of 1,4-dihydropyridines: in vitro evaluation and structure–activity relationship study. Bioorganic & Medicinal Chemistry 18, 8044–8053. [DOI] [PubMed] [Google Scholar]
- Reimao J. Q., Miguel D. C., Taniwaki N. N., Trinconi C. T., Yokoyama-Yasunaka J. K. U. and Uliana S. R. B. (2014). Antileishmanial activity of the estrogen receptor modulator raloxifene. Plos Neglected Tropical Diseases 8, e2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M. T. M. (2006). Current understandings on the immunology of leishmaniasis and recent developments in prevention and treatment. British Medical Bulletin 75–76, 115–130. [DOI] [PubMed] [Google Scholar]
- Rodrigues F. H., Afonso-Cardoso S. R., Gomes M. A. B., Beletti M. E., Rocha A., Guimarães A. H. B., Candeloro I. and de Souza M. A. (2006). Effect of imidocarb and levamisole on the experimental infection of BALB/c mice by Leishmania (Leishmania) amazonensis. Veterinary Parasitology 139, 37–46. [DOI] [PubMed] [Google Scholar]
- Rosa M. d. S. S., Mendonca-Filho R. R., Bizzo H. R., Rodrigues I. d. A., Soares R. M. A., Souto-Padron T., Alviano C. S. and Lopes A. H. C. S. (2003). Antileishmanial Activity of a Linalool-Rich Essential Oil from Croton cajucara. Antimicrobial Agents and Chemotherapy 47, 1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson L., Yardley V. and Croft S. L. (2014). Activity of anti-cancer protein kinase inhibitors against Leishmania spp. Journal of Antimicrobial Chemotherapy 69, 1888–1891. [DOI] [PubMed] [Google Scholar]
- Schmidt T. J., Khalid S. A., Romanha A. J., Alves T. M., Biavatti M. W., Brun R., Da Costa F. B., de Castro S. L., Ferreira V. F., de Lacerda M. V. G., Lago J. H. G., Leon L. L., Lopes N. P., das Neves Amorim R. C., Niehues M., Ogungbe I. V., Pohlit A. M., Scotti M. T., Setzer W. N., de N C Soeiro M., Steindel M. and Tempone A. G. (2012). The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases – part II. Current Medicinal Chemistry 19, 2176–2228. [PubMed] [Google Scholar]
- Seeberger J., Daoud S. and Pammer J. (2003). Transient effect of topical treatment of cutaneous leishmaniasis with imiquimod. International Journal of Dermatology 42, 576–579. [DOI] [PubMed] [Google Scholar]
- Seifert K., Matu S., Javier Pérez-Victoria F., Castanys S., Gamarro F. and Croft S. L. (2003). Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine). International Journal of Antimicrobial Agents 22, 380–387. [DOI] [PubMed] [Google Scholar]
- Serrano-Martin X., Garcia-Marchan Y., Fernandez A., Rodriguez N., Rojas H., Visbal G. and Benaim G. (2009a). Amiodarone destabilizes intracellular Ca2+ homeostasis and biosynthesis of sterols in Leishmania mexicana. Antimicrobial Agents and Chemotherapy 53, 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Martín X., Payares G., De Lucca M., Martinez J. C., Mendoza-León A. and Benaim G. (2009b). Amiodarone and miltefosine act synergistically against Leishmania mexicana and can induce parasitological cure in a murine model of cutaneous leishmaniasis. Antimicrobial Agents and Chemotherapy 53, 5108–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked-Mishan P., Ulrich N., Ephros M. and Zilberstein D. (2001). Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. Journal of Biological Chemistry 276, 3971–3976. [DOI] [PubMed] [Google Scholar]
- Shokri A., Emami S., Fakhar M., Teshnizi S. H. and Keighobadi M. (2017). In vitro antileishmanial activity of novel azoles (3-imidazolylflavanones) against promastigote and amastigote stages of Leishmania major. Acta Tropica 167, 73–78. [DOI] [PubMed] [Google Scholar]
- Singh N., Mishra B. B., Bajpai S., Singh R. K. and Tiwari V. K. (2014a). Natural product based leads to fight against leishmaniasis. Bioorganic & Medicinal Chemistry 22, 18–45. [DOI] [PubMed] [Google Scholar]
- Singh S., Dinesh N., Kaur P. K. and Shamiulla B. (2014b). Ketanserin, an antidepressant, exerts its antileishmanial action via inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) enzyme of Leishmania donovani. Parasitology Research 113, 2161–2168. [DOI] [PubMed] [Google Scholar]
- Smith A. C., Yardley V., Rhodes J. and Croft S. L. (2000). Activity of the novel immunomodulatory compound tucaresol against experimental visceral leishmaniasis. Antimicrobial Agents and Chemotherapy 44, 1494–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano-Gallego L., Miró G., Koutinas A., Cardoso L., Pennisi M., Ferrer L., Bourdeau P., Oliva G. and Baneth G. (2011). LeishVet guidelines for the practical management of canine leishmaniosis. Parasites & Vectors 4, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto J. and Berman J. (2006). Treatment of new world cutaneous leishmaniasis with miltefosine. Transactions of The Royal Society of Tropical Medicine and Hygiene 100, S34–S40. [DOI] [PubMed] [Google Scholar]
- Sundar S. and Chakravarty J. (2010a). Liposomal amphotericin B and leishmaniasis: dose and response. Journal of Global Infectious Diseases 2, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S. and Chakravarty J. (2010b). Antimony toxicity. International Journal of Environmental Research and Public Health 7, 4267–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S. and Chakravarty J. (2015). An update on pharmacotherapy for leishmaniasis. Expert Opinion on Pharmacotherapy 16, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S., Jha T. K., Thakur C. P., Sinha P. K. and Bhattacharya S. K. (2007). Injectable paromomycin for visceral leishmaniasis in India. New England Journal of Medicine 356, 2571–2581. [DOI] [PubMed] [Google Scholar]
- Sundar S. and Singh A. (2016). Recent developments and future prospects in the treatment of visceral leishmaniasis Therapeutic Advances in Infectious Diseases 3, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S., Sinha P. K., Dixon S. A., Buckley R., Miller A. K., Mohamed K. and Al-Banna M. (2011). Pharmacokinetics of oral sitamaquine taken with or without food and safety and efficacy for treatment of visceral leishmaniais: a randomized study in Bihar, India. American Journal of Tropical Medicine and Hygiene 84, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi M. D. D., Grant M. H., Mullen A. B., Tettey J. N. A., MacKay S. P. and Clark R. L. (2009). Metabolism of two new benzodiazepine-type anti-leishmanial agents in rat hepatocytes and hepatic microsomes and their interaction with glutathione in macrophages. Journal of Pharmacy and Pharmacology 61, 399–406. [DOI] [PubMed] [Google Scholar]
- Thompson A. M., O'Connor P. D., Blaser A., Yardley V., Maes L., Gupta S., Launay D., Martin D., Franzblau S. G., Wan B., Wang Y., Ma Z. and Denny W. A. (2016). Repositioning antitubercular 6-nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: structure-activity studies on a preclinical candidate for visceral leishmaniasis. Journal of Medicinal Chemistry 59, 2530–2550. [DOI] [PubMed] [Google Scholar]
- Torres-Santos E. C., Lopes D., Oliveira R. R., Carauta J. P. P., Falcão C. A. B., Kaplan M. A. C. and Rossi-Bergmann B. (2004). Structure - activity relationship of triterpenoids from Pourouma guyanensis with antileishmanial activity. Phytomedicine 11, 114–120. [DOI] [PubMed] [Google Scholar]
- Trinconi C. T., Reimão J. Q., Coelho A. C. and Uliana S. R. B. (2016). Efficacy of tamoxifen and miltefosine combined therapy for cutaneous leishmaniasis in the murine model of infection with Leishmania amazonensis. Journal of Antimicrobial Chemotherapy 71, 5. [DOI] [PubMed] [Google Scholar]
- Uliana S. R. B and Barcinski M. A. (2009). Repurposing for neglected diseases. Science 326, 935–935. [DOI] [PubMed] [Google Scholar]
- Wang Q., Rosa B. A., Nare B., Powell K., Valente S., Rotili D., Mai A., Marshall G. R. and Mitreva M. (2015). Targeting lysine deacetylases (KDACs) in parasites. Plos Neglected Tropical Diseases 9, e0004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel D. M., McMahon-Pratt D. and Koleske A. J. (2012). The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal leishmania infection. Molecular and Cellular Biology 32, 3176–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2002). New treatment for leishmaniasis is 95% effective. Bulletin of the World Health Organisation 80, 688. [PMC free article] [PubMed] [Google Scholar]
- WHO (2016). Leishmaniasis fact sheet. http://www.who.int/mediacentre/factsheets/fs375/en/.
- Witschel M., Rottmann M., Kaiser M. and Brun R. (2012). Agrochemicals against malaria, sleeping sickness, leishmaniasis and Chagas disease. PLOS Negl Trop Dis 6, e1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S., Patterson S., Stojanovski L., Simeons F. R. C., Norval S., Kime R., Read K. D. and Fairlamb A. H. (2012). The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Science Translational Medicine 4, 119re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakai H. A. and Zimmo S. K. (2000). Effects of itraconazole and terbinafine on Leishmania major lesions in BALB/c mice. Annals of Tropical Medicine & Parasitology 94, 787–791. [DOI] [PubMed] [Google Scholar]
- Zhang R., Shang L., Jin H., Ma C., Wu Y., Liu Q., Xia Z., Wei F., Zhu X.-Q. and Gao H. (2010). In vitro and in vivo antileishmanial efficacy of nitazoxanide against Leishmania donovani. Parasitology Research 107, 475–479. [DOI] [PubMed] [Google Scholar]