

Abstract
The level of RNA polymerase (pol) III transcription is tightly linked to the rate of growth; it is low in resting cells and increases following mitogenic stimulation. When mammalian cells begin to proliferate, maximal pol III activity is reached shortly before the G1/S transition; it then remains high throughout S and G2 phases. Recent data suggest that the retinoblastoma protein RB and its relatives p107 and p130 may be largely responsible for this pattern of expression. During G0 and early G1 phase, RB and p130 bind and repress the pol III-specific factor TFIIIB; shortly before S phase they dissociate from TFIIIB, allowing transcription to increase. At the end of interphase, when cells enter mitosis, pol III transcription is again suppressed; this mitotic repression is achieved through direct phosphorylation of TFIIIB. Thus, pol III transcription levels fluctuate as mammalian cells cycle, being high in S and G2 phases and low during mitosis and early G1. In addition to this cyclic regulation, TFIIIB can be bound and repressed by the tumor suppressor p53. Conversely, it is a target for activation by several viruses, including SV40, HBV, and HTLV-1. Some viruses also increase the activity of a second pol III-specific factor called TFIIIC. A large proportion of transformed and tumor cell types express abnormally high levels of pol III products. This may be explained, at least in part, by the very high frequency with which RB and p53 become inactivated during neoplastic transformation; loss of function of these cardinal tumor suppressors may release TFIIIB from key restraints that operate in normal cells.
Keywords: Cell cycle, p53, RB, Transformation, Transcription, TFIIIB, pol III
RNA polymerase (pol) III is responsible for 5–10% of all nuclear transcription (91). It operates in discrete, localized transcription “factories,” which typically contain about five molecules of active pol III, but no pol I or pol II (91). Pol III synthesizes a variety of essential cellular products, including tRNA, 5S rRNA, the 7SL RNA component of the signal recognition particle, and the U6 small nuclear RNA that is required for splicing mRNA [reviewed in (126,135)]. Common features of these transcripts are that they are short (usually less than 200 bp) and are not translated.
The synthesis of pol III products is clearly an essential component of cellular metabolism and, as such, can be regarded as a housekeeping function. However, although it occurs in all cells with nuclei, pol III transcription is regulated strongly in response to a variety of external stimuli [reviewed in (126)]. For example, it is tightly linked to growth conditions, increasing in response to mitogenic signals and falling when serum factors or nutrients are limiting. It is also subject to cell cycle control in vertebrates, being activated at the G1/S transition and repressed at mitosis. Many different viruses have potent effects upon the rate of pol III transcription. Furthermore, the majority of transformed and tumor cell types display abnormally elevated levels of pol III products. In none of these cases has evidence been found that the polymerase itself is controlled directly. Instead, regulation is mediated through changes in the activities of pol III-specific transcription factors.
THE BASAL POL III TRANSCRIPTION APPARATUS
The nuclear RNA polymerases alone have little specificity for particular DNA sequences. In the case of pol III, recruitment to specific genes requires the presence of TFIIIB, a factor that binds just upstream of the transcription start site (3,49). TFIIIB is a complex comprising a minimum of three subunits, one of which is the TATA binding protein (TBP) [reviewed in (34,94)]. In the yeast Saccharomyces, two TBP-associated factors (TAFs) are also required for TFIIIB activity (50,51,95,99). One of these TAFs has substantial homology to the basal pol II factor TFIIB (9,19,75); for this reason, it is often referred to as TFIIB-related factor or BRF. The other essential TAF in yeast TFIIIB has a SANT domain and is called B″ (1,51,95,99). Mammalian TFIIIB is much less well characterized, with at least one unidentified subunit. Homologues of BRF have been isolated in humans, nematodes, and fruit flies (71,81,117,121), but a gene for B″ has yet to be reported outside of yeast.
A few pol III templates, such as U6 snRNA genes, have TATA sequences located ∼30 bp upstream of the initiation site. This motif is recognized by TBP and therefore allows direct binding by TFIIIB (47,74,78,105). However, most pol III-transcribed genes have no TATA element. In such cases, TFIIIB is recruited to the promoter by protein-protein interactions with the factor TFIIIC. Although TFIIIC is a single entity in yeast, human TFIIIC has been subdivided into two separate components, called TFIIIC1 and TFIIIC2 (138). Little progress has been made in characterizing TFIIIC1, although sedimentation analysis suggested a mass of up to 200 kDa, assuming that it is globular (138). However, human TFIIIC2 has been purified and shown to consist of five poly-peptides, with a cumulative mass approaching 600 kDa (107,122,139). The cloning of cDNAs has now been reported for all of these subunits (41,42,65,66,107). A notable feature of TFIIIC2 is that three of its subunits possess histone acetyltransferase activity, which may help it gain access to promoters that have been incorporated into chromatin (41,63). TFIIIC2 binds directly to DNA sequences that are present within the coding regions of most pol III templates, including tRNA genes (72,138). An exception is provided by the 5S rRNA genes, where the polypeptide TFIIIA provides a platform for TFIIIC (72). Once TFIIIC2 is in position, it interacts with TFIIIB and brings it onto a region of DNA for which it has little or no intrinsic affinity (21,42). TFIIIC1 can join the complex either before or after TFIIIB is recruited (21). Once assembled, TFIIIB binds the pol III enzyme and places it over the initiation site so that transcription can commence (49). Indeed, the assembly factors TFIIIA and TFIIIC are no longer required after they have brought TFIIIB to a promoter, at least in vitro (49). TFIIIB can therefore be regarded as the pivotal initiation factor in the pol III system.
THE TUMOR SUPPRESSOR PROTEINS p53 AND RB
The key role of TFIIIB can explain why it is targeted for tight control by many regulatory proteins (127). One of these is the important tumor suppressor p53, which arrests cell growth or triggers apoptosis in response to stresses such as radiation exposure, on-cogenic stimuli, or hypoxia (28,58). Overexpression of p53 will repress transcription of various pol III templates both in vitro and in transfected cells (10,15). Furthermore, the synthesis of tRNA and 5S rRNA in vivo is elevated four- to sixfold following the targeted disruption of the p53 gene in knockout mice (10). TFIIIB activity is abnormally elevated in fibroblasts derived from such mice (10). Cofractionation, coimmunoprecipitation, and pull-down assays have shown that TFIIIB interacts with both recombinant and endogenous p53 (10,15). It therefore appears that p53 can regulate pol III transcription by binding and repressing TFIIIB. It remains to be determined how this control responds to the various stress conditions that are known to influence p53 activity.
TFIIIB is also bound and regulated by the tumor suppressor RB, the product of the retinoblastoma susceptibility gene (16,69,70,110). This is despite the fact that p53 and RB bear no similarity in sequence or structure. Overexpressing RB in transfected cells produces a significant decrease in pol III activity (16,132). Recombinant RB will also repress transcription that has been reconstituted in vitro using partially purified pol III factors (16,69,70,132). Perhaps most importantly, inactivation of the endogenous Rb gene using knockout technology results in a fivefold increase in the synthesis of tRNA and 5S rRNA in vivo (132). Extracts prepared from Rb-knockout mice display a specific increase in TFIIIB activity (69). Recombinant RB will bind to natural or recombinant TFIIIB subunits (16,69,70,110). Furthermore, the existence of endogenous complexes between cellular RB and TFIIIB can be readily demonstrated by coimmunoprecipitation assays (69,70,110). These data provide a convincing case that TFIIIB is a target for repression by RB in vivo.
RB has two relatives, called p107 and p130, with which it shares 30–35% identity at the amino acid level [reviewed in (32,35,85)]. These three are referred to as the pocket proteins, because most of their homology lies within a bipartite region called the pocket domain. They can each inhibit cell growth and proliferation when overexpressed (18,92,140). A number of common target proteins have been found to interact with the pocket domains of RB, p107, and p130, including members of the E2F family of cellular transcription factors and the oncoprotein products of several DNA tumor viruses [reviewed in (24,35,85,112)]. TFIIIB also falls into this category, because it binds to all three of the pocket proteins, as shown by cofractionation, coimmunoprecipitation, and pulldown assays (110). Like RB, recombinant p107 and p130 will repress transcription of a range of pol III templates both in vitro and in transfected cells (110). Furthermore, fibroblasts derived from p107 −/− p130 −/− double-knockout mice display elevated levels of pol III transcripts (110). The ability to bind and repress TFIIIB is therefore a feature of each of the pocket proteins; this is consistent with deletion and substitution analyses which have shown that regulation of pol III transcription by RB is dependent on the pocket domain (16,132).
THE CELL CYCLE
The function of the pocket proteins is subject to cell cycle control [reviewed in (32,35,82,85,112)]. These proteins are found in an underphosphorylated active state during G0 and early G1 phases; but as cells pass the restriction point in mid to late G1 phase, the pocket proteins are inactivated through hyper-phosphorylation by the cyclin D- and cyclin E-dependent kinases (32,35,82,85,112). For much of the mammalian cell cycle, there is an inverse correlation between the activity of the pocket proteins and the level of pol III transcription. Thus, expression of class III genes is low during G0 and early G1 phases and then increases at the G1/S transition (48,79,128). Recent data suggest that the pocket proteins play a major role in the cell cycle control of pol III transcription.
Whereas TFIIIC activity remains relatively constant in cycling mammalian cells, TFIIIB is regulated strongly (128,129). As a consequence, the limiting component of the pol III machinery changes as cells cycle. During mitosis and early G1 phase, the activity of TFIIIB is severely limiting, but it increases substantially at the G1/S transition and is in relative excess throughout S and G2 (128,129). Detailed time courses show that the rise in pol III transcription occurs in mid to late G1 phase and corresponds well with the time when RB and its relatives are switched off by hyperphosphorylation [(48,79), P. Scott and R. White, unpublished data]. Coimmunoprecipitation experiments confirm that the pocket proteins dissociate from TFIIIB when cells enter S phase (Fig. 1). Indeed, TFIIIB is bound exclusively by the under-phosphorylated active form of RB and not by the hyperphosphorylated forms found after the G1/S transition (J. Sutcliffe and R. White, unpublished data). Consistent with these observations, pol III transcription is stimulated strongly if cells are transfected with vectors encoding cyclin D/cdk4 and cyclin E/cdk2, the kinases responsible for phosphorylating the pocket proteins (C. Cairns and R. White, unpublished data). Having been switched off at the restriction point, RB and its relatives remain inactive until they are de-phosphorylated at the start of the next G1 phase (32,35,82,112).
Figure 1.
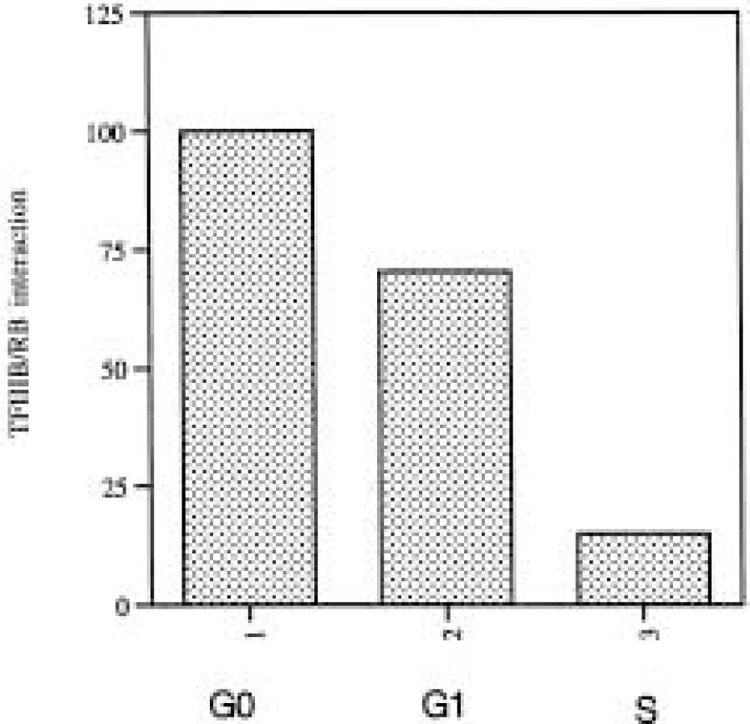
RB binds to TFIIIB during G0 and early G1 phase, but is released as cells pass through the G1/S transition. The graph compares the level of interaction between RB and TFIIIB in Balb/c 3T3 cells harvested in G0, mid-G1, or S phase, as indicated. Arrest in G0 phase was achieved by serum deprivation for 24 h; 20% serum was then added and cells were harvested after 9 h (mid-G1 phase) or 15 h (S phase). Cell cycle phase was confirmed by flow cytometry and by measuring thymidine incorporation. Protein was extracted from the harvested cells and used for immunoprecipitation with the anti-RB antibody C15 (Santa Cruz), as described previously (110). The amounts of RB and associated TFIIIB in the immunoprecipitates were determined by scanning Western blots carried out with C15 and the anti-BRF antibody 128 (10). The amount of BRF in each immunoprecipitate was normalized against the total amount of RB in the same immunoprecipitate. Numbers represent percentages of the interaction seen in G0 phase (designated 100%).
The activity of TFIIIB and the expression of class III genes is maximal during S and G2 phases (128). However, this situation is rapidly reversed once mitosis is reached. Indeed, mitosis in higher eukaryotes is accompanied by a general decrease in transcription by all three nuclear RNA polymerases [reviewed in (29)]. In the case of pol III, this is achieved through the phosphorylation and inactivation of TFIIIB (31,73,129). Thus, TFIIIB isolated from metaphase-arrested frog eggs is unable to support transcription unless it is treated with phosphatase (31). The mitotic kinase cyclin B/cdc2 is at least partially responsible for this effect, because it can inactivate affinity-purified Xenopus TFIIIB (31,136). It is striking that the cyclin-dependent kinases have opposite effects on the class III system at different stages of the cell cycle; pol III activity increases during G1 phase in response to cyclin D/cdk4 and cyclin E/cdk2, but in M phase it is repressed by cyclin B/cdc2. However, there is redundancy in the mechanism of mitotic control and one or more additional kinases can also repress pol III transcription in metaphase-arrested frog eggs (33). A similar situation is found in mitotic HeLa cells, where TFIIIB is also inactivated in a phosphorylation-dependent manner (129). In both the frog and human systems, TBP is found in a hyperphosphorylated state during mitosis (73,129). However, the functional significance of this remains unclear and repression of pol III transcription appears to be due to a specific loss of activity of one or more of the TAF subunits of TFIIIB (73,129).
The hyperphosphorylation of TFIIIB is reversed soon after cycling human cells leave mitosis (128). Despite this, TFIIIB activity remains low during early G1 phase (128). As a consequence, the level of pol III transcription is two- to threefold lower in early G1 than it is in S and G2 phases (128). This repression coincides with the interval during which the pocket proteins are active, following their dephosphorylation at the end of mitosis (82). They bind to TFIIIB and inhibit it until the restriction point is reached in mid to late G1 phase (Fig. 2).
Figure 2.
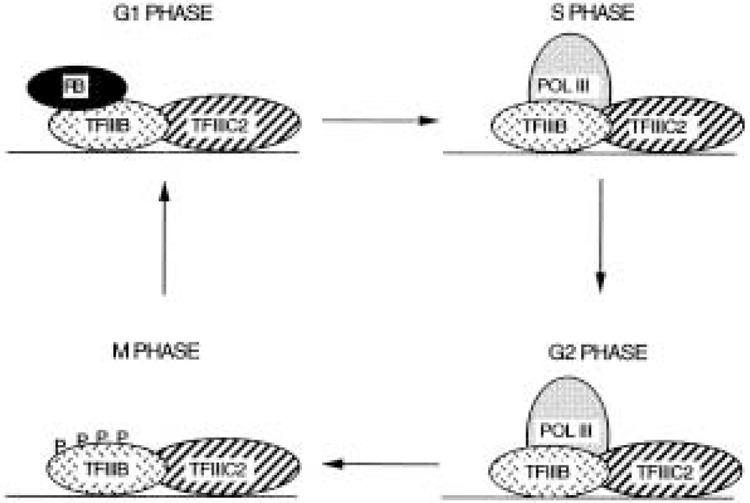
Model illustrating changes in the pol III transcription machinery in cycling mammalian cells. During early G1 phase, TFIIIB is bound and repressed by RB. At the G1/S transition, RB dissociates from TFIIIB, allowing active transcription throughout S and G2 phases. At mitosis, TFIIIB is inactivated through hyperphosphorylation, resulting in a drop in transcription. Although TFIIIB has been depicted as remaining at the promoter throughout the cell cycle, it is possible that it dissociates in response to RB binding and/or mitotic phosphorylation.
In addition to their window of action during early G1, the pocket proteins are also underphosphorylated and active in quiescent cells (32,35). Indeed, the abundance of p130 is especially elevated in serum-starved resting cells (32,35,45,80). It is likely that hypophosphorylated p130 and RB play an important part in suppressing pol III transcription during G0 phase. Thus, TFIIIB is bound by p130 and RB in quiescent fibroblasts (P. Scott and R. White, unpublished data). Furthermore, cells derived from Rb −/− knockout or p107 −/− p130 −/− double knockout mice are compromised in their ability to downregulate pol III activity following serum withdrawal [(110), P. Scott and R. White, unpublished data]. These observations suggest that the pocket proteins make a major contribution towards the serum responsiveness of class III genes.
As well as using RB and p130 to repress TFIIIB, some mammalian cell types may also inactivate TFIIIC2 when they are deprived of mitogens (11,37,115). In HeLa cells, regulation of TFIIIC2 appears to be achieved by interconversion between two forms: TFIIIC2a, which is active, and TFIIIC2b, which is transcriptionally inactive although still able to bind DNA (37,59,107). The inactive TFIIIC2b form is reported to lack a 110-kDa subunit called TFIIICβ (59,107). Both forms are present in logarithmically growing HeLa cells, but low serum levels trigger a specific decrease in the abundance of TFIIICβ so that the inactive form predominates (107). Thus, both TFIIIB and TFIIIC2 can respond to the availability of mitogens. The relative importance of these two regulatory mechanisms will probably depend upon cell type. We observe no change in TFIIICβ levels when Balb/c 3T3 fibroblasts are deprived of serum (H. Alzuherri, A. Winter, and R. White, unpublished data).
VIRUSES
A variety of viruses have been found to stimulate pol III transcription [reviewed in (126)]. In many cases this seems to reflect a general requirement for increased biosynthetic capacity, but several viral genomes contain class III genes. For example, adenovirus encodes two pol III products, called VAI and VAII, that are synthesized at high levels during the late stages of infection (108,124). These short RNAs are involved in subverting the host cells translational apparatus, in order to ensure the synthesis of viral proteins (114). Multiple mechanisms may contribute to the activation of pol III transcription in response to adenovirus infection. HeLa cells infected with wild-type adenovirus display a marked increase in TFIIIC2 activity (37,38,107,138). This reflects a selective increase in the level of the TFIIICβ subunit, thereby raising the proportion of the active TFIIIC2a form (37,107). The adenoviral transforming protein E1A is required for induction of TFIIICβ (37,107). E1A also binds and inactivates the pocket proteins, allowing it to relieve TFIIIB from repression by RB (132). In addition, E1A can counteract the effects of another cellular repressor called Dr1 (62). Because Dr1 is a potent inhibitor of TFIIIB, both in vitro and in vivo (57,130), this may provide an additional route for adenovirus to ensure high levels of pol III activity.
Rodent fibroblasts display elevated levels of pol III transcripts following transformation by the DNA tumor virus SV40 (12,102,106,131). The mechanisms used to achieve this bear several similarities to those employed by adenovirus. For example, the large transforming (T) antigen of SV40 resembles E1A in being able to bind and inactivate RB (22,26,76,84). This allows large T antigen to release TFIIIB from the repressive influence of RB (70). The proportion of TFIIIB bound by RB is significantly reduced in SV40-transformed fibroblasts and this correlates with an increase in TFIIIB activity (70). Large T antigen has also been reported to bind to TFIIIB, although the functional significance of this interaction has yet to be established (20). In addition to activating TFIIIB, SV40-transformed fibroblasts also overexpress TFIIIC2, which is the limiting factor for pol III transcription in these cells (70,131). Like adenovirus, SV40 induces a substantial increase in the level of TFIIICβ (70). However, SV40-transformed fibro-blasts also overproduce other subunits of TFIIIC2 (70), which is apparently not the case in adenovirus-infected HeLa cells (107). The elevated levels of TFIIIC2 subunits that follow transformation by SV40 reflect an overproduction of the corresponding mRNAs (70). For example, the two SV40-trans-formed lines that were tested express seven- to eightfold more of the mRNA encoding TFIIICβ than the corresponding untransformed parental cells (70). Thus, SV40 resembles adenovirus in deregulating both TFIIIB and TFIIIC2 in order to achieve very high levels of pol III output (Fig. 3).
Figure 3.

Model illustrating some changes in the pol III transcription machinery that occur when Balb/c 3T3 cells are transformed by SV40. Whereas RB binds and represses some of the TFIIIB in untransformed 3T3 cells, SV40 T antigen sequesters RB and releases active TFIIIB. The SV40-transformed cells also overexpress TFIIIC2. As a consequence of these changes, SV3T3 cells have higher pol III transcription levels than their untransformed 3T3 progenitors.
Other viruses may be more selective in their choice of targets within the class III transcription machinery. For example, hepatitis B virus (HBV) induces a specific increase in TFIIIB activity (120). This effect is associated with the viral X gene, which alone is sufficient to stimulate pol III transcription when introduced into a variety of cell lines (2,64,119,120). The X gene provokes a substantial increase in the abundance of TBP, a key component of the TFIIIB complex (119,120). This X-induced overexpression of TBP may be sufficient to account for the higher TFIIIB activity, because TBP is limiting for pol III transcription in the cell lines used in these studies (117,120). The X gene products have been shown to stimulate the protein kinase C(PKC) and Ras-Raf-MEK-MAP kinase signaling cascades (6,53) and both these pathways have been implicated in the effect of X on pol III transcription (119,120). Thus, inhibitors of either PKCor Ras can block the X-mediated increase in TBP abundance and class III gene expression (119,120). This can also be achieved with a dominant negative mutant form of Ras (119). Coexpression of constitutively active Raf-1 overcomes the block imposed by dominant negative Ras (119). It therefore appears that X stimulates pol III transcription via cellular signaling pathways.
A more direct effect on TFIIIB is produced by the Tax protein of human T-cell leukemia virus type 1 (HTLV-1), which can increase the expression of pol III templates both in vitro and in transfected cells (30,89). TFIIIB activity and the synthesis of tRNA and 5S rRNA are elevated in T cells that have been infected with HTLV-1 (30). Furthermore, in reconstituted assays recombinant Tax can raise the effective concentration of active TFIIIB molecules (30). The precise molecular details of this effect have yet to be established.
TRANSFORMATION
A great many studies have observed that the abundance of pol III transcripts is abnormally elevated in transformed and tumor cell lines (7,60,61,67,77,100,102). A broad range of transforming agents can produce this effect. Adenovirus, SV40, HBV, and HTLV-1 provide examples that have already been described above. Both HBV and HTLV-1 are important causative agents in human diseases; HBV is linked strongly with the development of hepatocellular carcinoma (56), while HTLV-1 causes adult T-cell leukemia and a neurodegenerative disease called tropical spastic paraparesis or HTLV-1-associated myelopathy (46,90). In addition to these and other tumor viruses, many chemical carcinogens have also been found to stimulate pol III activity when applied to cells. A tight causal link between pol III activation and transformation is suggested by the fact that two fibroblast lines transformed by temperature-sensitive mutants of the SV40 large T antigen downregulate pol III transcription at the nonpermissive temperature while reverting to normal morphology and phenotype (102). The abundance of pol III transcripts varies substantially between different SV40-transformed lines and the highest levels correlate with progression to a more tumorigenic phenotype (102,131). However, a few rare examples have been reported of transformed lines that do not display the characteristic increase in pol III transcript levels, such as the Rb +/+ p53 +/+ osteosarcoma cell line U2OS (132).
Almost all the work described above was carried out with cultured cells. However, two recent studies have demonstrated clearly that pol III is also consistently deregulated in tumors in vivo. The first of these examined a murine pol III transcript of unknown function called BC1, which is unusual because it is normally only expressed in neurons (23). Northern analysis showed BC1 expression in breast carcinomas, colonic adenocarcinomas, and skin fibro-sarcomas, but not in the corresponding untransformed tissues (14). In situ hybridization studies of these tumors confirmed the presence of BC1 RNA in the neoplastic cells, whereas it was absent from the surrounding tissues (14). Although the fibrosarcomas and adenocarcinomas were induced by local inoculation with cells that had been treated with chemical carcinogens, the breast carcinoma analyzed was a primary tumor caused by ras (14). A more extensive analysis has been carried out with samples from 80 human tumors, representing 19 different types of cancer; it found that BC200 RNA, the primate analogue of BC1, is expressed in many, but not all, primary human tumors (13). Like BC1, BC200 RNA is found exclusively in the malignant cells and not in the adjacent normal tissue (13). An even more striking observation to emerge from this study is that levels of 7SL RNA, an essential pol III product, were elevated in every tumor examined relative to the corresponding normal tissue (13). We have also found that ovarian carcinomas overexpress 7SL RNA, tRNA, and 5S rRNA, relative to healthy ovarian tissue from the same patients (A. Winter, G. Sourvinos, D. Spandidos, and R. White, unpublished data). Thus, abnormal activation of class III gene expression is a very frequent feature of tumors in vivo.
Although deregulation of pol III activity has been observed in a broad range of transformed cell types, in most cases the mechanistic basis of this effect has yet to be elucidated. However, a potential explanation is offered by the discovery that RB and p53 play a major role in repressing TFIIIB in untransformed cells. The p53 gene contains missense mutations in approximately half of the major forms of human cancer (39). The effects of such mutations on pol III transcription have yet to be reported, but there is a good chance that many will release TFIIIB from a constraining influence. Furthermore, wild-type p53 can be inactivated by the cellular oncoprotein Mdm2, which is overexpressed in certain tumor types, including 30–40% of human sarcomas (83). Mdm2 might therefore be expected to deregulate TFIIIB. Although highly plausible, it has yet to be established experimentally that p53 inactivation contributes to the elevated pol III activity found in tumors. However, evidence has already been provided that this is the case for RB.
Many human cancers carry mutations in Rb, including retinoblastomas where the gene was first identified (27,123). In some instances the Rb gene is deleted completely; Rb −/− mice provide a model for this situation and have allowed confirmation that pol III transcription is elevated in vivo when RB protein is missing (132). Many other tumors carry mutant forms of RB and in these cases the mutation generally incorporates the pocket domain (43). Deletion and substitution analyses have shown that the pocket domain is essential for RB to regulate pol III activity (16,132). Furthermore, TFIIIB is unable to bind to a mutant form of RB that is found in the osteosarcoma cell line SAOS2 (70), where a C-terminal truncation has removed part of the pocket (104). Several examples have been described in which highly localized mutations inactivate the pocket. For example, in one small-cell lung carcinoma a single base change in a splice acceptor site gave rise to an RB polypeptide that lacked the 35 amino acids encoded by exon 21 (40). In another small-cell lung carcinoma, a point mutation created a stop codon and a novel splice donor site within exon 22, thereby eliminating 38 residues from the pocket domain of the product (40). A third inactivating mutation from a small-cell lung cancer resulted in a single amino acid substitution at codon 706 (52). We tested the ability of each of these three naturally occurring mutants to regulate pol III transcription and found that repression was lost in every case (132). We have also examined the effect of a point mutation at residue 567 of RB, which was isolated from the germ line of a child with bilateral retinoblastoma (103). As shown in Figure 4, this single residue substitution is sufficient to prevent RB from repressing a pol III template following overexpression in transfected cells. This is clearly a limited survey, but it nevertheless demonstrates that mutations that arise in RB in tumors can compromise its ability to regulate pol III transcription.
Figure 4.
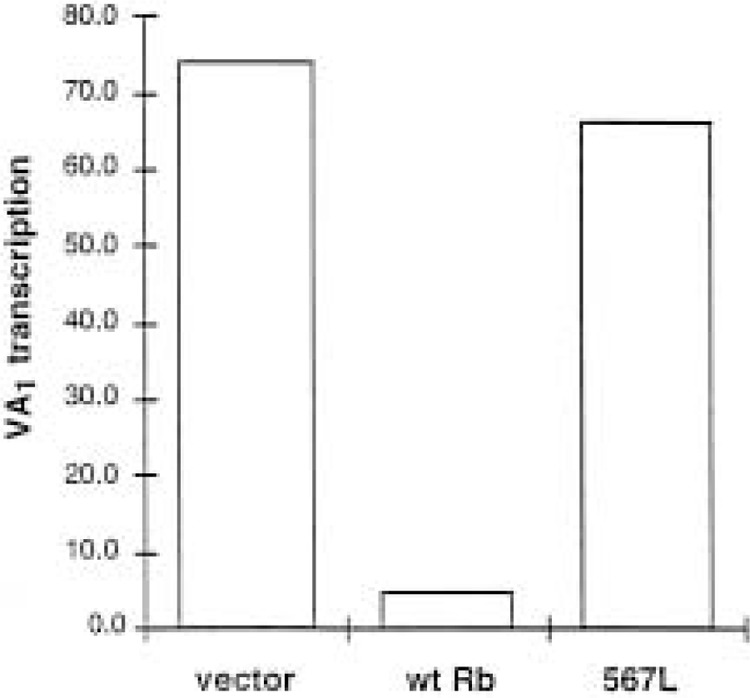
A substitution in RB (567L) that arose in a retinoblastoma patient compromises its ability to repress pol III transcription. The graph shows levels of VA1 transcription by pol III in transfected SAOS2 cells, after normalization to a cotransfected pol II control (pCAT) containing the SV40 promoter. Cells received 4 μg of pVA1, 4 μg of pCAT, and 8 μg of pSG5L vector, pSG5L-HA-RB(wt), or pSG-RB;567L (103), as indicated. RNA was harvested after 48 h and the levels of VA1 and CAT transcripts were determined by primer extension, as previously described (110). A phosphorimager was used for quantitation. Western blotting confirmed that wild-type and 567L mutant RB are expressed at comparable levels, as previously reported (103).
Although most cancers retain wild-type RB, its function is usually found to be compromised. Indeed, it has been suggested that the regulatory pathway involving RB may be disrupted in all human malignancies (123). A survey of seven human cervical carcinoma cell lines found that two had sustained small inactivating mutations in RB (101). Whereas neither of these lines were infected by human papillomavirus (HPV), each of the remaining five lines that expressed wild-type RB also carried HPV DNA (101). HPVs have an etiologic role in most cervical malignancies (118). The viral gene product E7 that is expressed in these tumors binds to the pocket domain of RB and inactivates it (25,86). Thus, RB function may be lost in most if not all cervical cancers, either by gene mutation in the minority of HPV-negative cases or by complex formation with E7 oncoprotein (118). We have shown that pol III transcription can be strongly stimulated in vivo by the E7 product of the highly malignant strain HPV-16 (70,110). This is not an indirect response to cell transformation, because pol III is also activated by a nontransforming mutant version of E7 that has retained its ability to inactivate RB (70). However, deletions or substitutions in the pocket-binding domain of E7 abolish its capacity to stimulate expression of a class III gene (70,110). It is therefore highly likely that E7 deregulates pol III transcription by releasing TFIIIB from repression by RB and its relatives p107 and p130.
The oncoproteins of several other DNA tumor viruses can also bind RB and neutralize its function, including adenoviral E1A (133,134) and SV40 large T antigen (22,84). As already described, both E1A and large T can overcome the repression of pol III activity by RB (70,132). Furthermore, the interaction between RB and TFIIIB is diminished substantially in SV40-transformed cells (70). Release of TFIIIB from the inhibitory effects of the pocket proteins is therefore a feature of cells transformed by various DNA tumor viruses.
As already described, the activity of RB can also be switched off through phosphorylation by the cyclin D- and E-dependent kinases. Cyclin D-dependent kinases are hyperactive in a variety of cancers and this provides another mechanism whereby RB function is lost (4,44,123). For example, cyclin D1 is overexpressed in 30–40% of primary breast tumors (4). In addition to situations in which cyclins are affected directly, many other cancers have lost the function of p16, a specific repressor of the cyclin D-dependent kinases (36,44). For example, the gene for p16 is deleted in many oesophageal, bladder, lung, and pancreatic carcinomas (36). Thus, cyclin D-dependent kinase activity is abnormally elevated in a broad spectrum of cancers, which has the effect of switching off RB. As might have been predicted, pol III transcription is stimulated strongly when various cell types are transfected with vectors encoding cyclin D and cdk4 (C. Cairns and R. White, unpublished data). To mimic the effect of p16 deletion, we have made use of a ribozyme that degrades the p16 mRNA (88); pol III transcription is elevated significantly when this approach is used to deplete p16 from cells (P. Scott and R. White, unpublished data). Thus, aberrant pol III activity can result when RB function is compromised through genetic mutation, hyper-phosphorylation, or binding of viral oncoproteins. Because one or another of these mechanisms is thought to apply to the vast majority of tumors (123), these observations are likely to go a long way towards explaining the very high incidence of pol III deregulation in human cancers.
RAPID GROWTH REQUIRES HIGH RATES OF POL III TRANSCRIPTION
That TFIIIB is targeted by cellular tumor suppressors and a range of viral oncoproteins can be explained by the fact that pol III transcriptional activity is tightly linked to the rate of growth. To maintain a constant size, cells must double their mass prior to division. Because protein constitutes the bulk of a cell’s dry mass, a high rate of translation is a prerequisite of rapid growth. Indeed, growth rate is directly proportional to the rate of accumulation of protein (5). A 50% decrease in the level of protein synthesis causes cells to withdraw from cycle and quiesce (8,96). An important determinant of the rate of translation is the availability of rRNA and tRNA. High pol III activity is therefore necessary to sustain rapid growth. Thus, mitogenic stimulation provokes a rapid and coordinate induction of rRNA, tRNA, ribosomal proteins, and translation factors, such that the rate of protein synthesis increases substantially before cells reach S phase (17,48,54,79,93,97,98,109,111). Indeed, cells are unable to enter S phase and duplicate their chromosomes until they have accumulated an adequate level of protein (55,113). These observations can readily explain why pol III transcription is targeted by so many viruses that need the host cell’s DNA synthesis machinery to replicate their genomes. It may also account, at least in part, for the frequent activation of pol III transcription in transformed and tumor cells.
CONCLUSION
TFIIIB is a target for many regulatory proteins, including several viral oncoproteins and the tumor suppressors p53 and RB. It is also subject to mitotic repression through direct phosphorylation. These observations suggest strongly that TFIIIB is a crucial cellular control point. The activity of TFIIIB is clearly a major determinant of biosynthetic capacity, dictating the rate of production of tRNA, 5S rRNA, and several other essential small RNA molecules. It has been suggested that the repression of pol III transcription may provide a mechanism for restraining cell growth (68,87,125). The fact that this process is targeted by two unrelated tumor suppressors and a range of viral oncoproteins provides support for this contention. It seems very likely that the loss of function of p53 and/or RB will deregulate pol III transcription in a large proportion of malignancies. This may constitute an important step towards tumor development.
ACKNOWLEDGMENTS
The authors thank William Kaelin, Jr., for the pSG5L, pSG5L-HA-RB(wt), and pSG-RB;567L plasmids. Our research is supported by project grants CO5766 and 17/C10311 from the Biotechnology and Biological Sciences Research Council, SP2314/0102 from the Cancer Research Campaign, and 98-46 from the Association for International Cancer Research. P.H.S. is a Wellcome Trust Research Career Development Fellow and R.J.W. is a Jenner Research Fellow of the Lister Institute of Preventive Medicine.
REFERENCES
- 1. Aasland R.; Stewart A. F.; Gibson T. The SANT domain: A putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional core-pressor N-CoR and TFIIIB. Trends Biochem. Sci. 21: 87–88; 1996. [PubMed] [Google Scholar]
- 2. Aufiero B.; Schneider R. J. The hepatitis B virus X-gene product transactivates both RNA polymerase II and III promoters. EMBO J. 9:497–504; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartholomew B.; Kassavetis G. A.; Geiduschek E. P. Two components of Saccharomyces cerevisiae transcription factor IIIB (TFIIIB) are stereospecifically located upstream of a tRNA gene and interact with the second-largest subunit of TFIIIC. Mol. Cell. Biol. 11:5181–5189; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bates S.; Peters G. Cyclin D1 as a cellular proto-oncogene. Semin. Cancer Biol. 6:73–82; 1995. [DOI] [PubMed] [Google Scholar]
- 5. Baxter G. C.; Stanners C. P. The effect of protein degradation on cellular growth characteristics. J. Cell Physiol. 96:139–146; 1978. [DOI] [PubMed] [Google Scholar]
- 6. Benn J.; Schneider R. J. Hepatitis B virus HBx protein activates Ras–GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl. Acad. Sci. USA 91:10350–10354; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brickell P. M.; Latchman D. S.; Murphy D.; Willison K.; Rigby P. W. J. Activation of a Qa/Tla class I major histocompatibility antigen gene is a general feature of oncogenesis in the mouse. Nature 306:756–760; 1983. [DOI] [PubMed] [Google Scholar]
- 8. Brooks R. F. Continuous protein synthesis is required to maintain the probability of entry into S phase. Cell 12:311–317; 1977. [DOI] [PubMed] [Google Scholar]
- 9. Buratowski S.; Zhou H. A suppressor of TBP mutations encodes an RNA polymerase III transcription factor with homology to TFIIB. Cell 71:221–230; 1992. [DOI] [PubMed] [Google Scholar]
- 10. Cairns C. A.; White R. J. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 17: 3112–3123; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carey M. F.; Singh K. Enhanced B2 transcription in simian virus 40-transformed cells is mediated through the formation of RNA polymerase III transcription complexes on previously inactive genes. Proc. Natl. Acad. Sci. USA 85:7059–7063; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carey M. F.; Singh K.; Botchan M.; Cozzarelli N. R. Induction of specific transcription by RNA polymerase III in transformed cells. Mol. Cell. Biol. 6: 3068–3076; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen W.; Bocker W.; Brosius J.; Tiedge H. Expression of neural BC200 RNA in human tumours. J. Pathol. 183:345–351; 1997. [DOI] [PubMed] [Google Scholar]
- 14. Chen W.; Heierhorst J.; Brosius J.; Tiedge H. Expression of neural BC1 RNA: Induction in murine tumours. Eur. J. Cancer 33:288–292; 1997. [DOI] [PubMed] [Google Scholar]
- 15. Chesnokov I.; Chu W.-M.; Botchan M. R.; Schmid C. W. p53 inhibits RNA polymerase III-directed transcription in a promoter-dependent manner. Mol. Cell. Biol. 16:7084–7088; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chu W.-M.; Wang Z.; Roeder R. G.; Schmid C. W. RNA polymerase III transcription repressed by Rb through its interactions with TFIIIB and TFIIIC2. J. Biol. Chem. 272:14755–14761; 1997. [DOI] [PubMed] [Google Scholar]
- 17. Clarke E. M.; Peterson C. L.; Brainard A. V.; Riggs D. L. Regulation of the RNA polymerase I and III transcription systems in response to growth conditions. J. Biol. Chem. 271:22189–22195; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Claudio P. P.; Howard C. M.; Baldi A.; De Luca A.; Fu Y.; Condorelli G.; Sun Y.; Colburn N.; Calabretta B.; Giordano A. p130/Rb2 has growth sup-pressive properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 54:5556–5560; 1994. [PubMed] [Google Scholar]
- 19. Colbert T.; Hahn S. A yeast TFIIB-related factor involved in RNA polymerase III transcription. Genes Dev. 6:1940–1949; 1992. [DOI] [PubMed] [Google Scholar]
- 20. Damania B.; Mital R.; Alwine J. C. Simian virus 40 large T antigen interacts with human TFIIB-related factor and small nuclear RNA-activating protein complex for transcriptional activation of TATA-containing polymerase III promoters. Mol. Cell. Biol. 18: 1331–1338; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dean N.; Berk A. J. Ordering promoter binding of class III transcription factors TFIIIC1 and TFIIIC2. Mol. Cell. Biol. 8:3017–3025; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DeCaprio J. A.; Ludlow J. W.; Figge J.; Shew J.-Y.; Huang C.-M.; Lee W.-H.; Marsilio E.; Pau-cha E.; Livingston D. M. SV40 large tumour antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54:275–283; 1988. [DOI] [PubMed] [Google Scholar]
- 23. DeChiara T. M.; Brosius J. Neural BC1 RNA: cDNA clones reveal nonrepetitive sequence content. Proc. Natl. Acad. Sci. USA 84:2624–2628; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245–2262; 1998. [DOI] [PubMed] [Google Scholar]
- 25. Dyson N.; Howley P. M.; Munger K; Harlow E. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937; 1989. [DOI] [PubMed] [Google Scholar]
- 26. Ewen M. E.; Ludlow J. W.; Marsilio E.; DeCaprio J. A.; Millikan R. C.; Cheng S. H.; Paucha E.; Livingston D. M. An N-terminal transformation-governing sequence of SV40 large T antigen contributes to the binding of both p110Rb and a second cellular protein, p120. Cell 58:257–267; 1989. [DOI] [PubMed] [Google Scholar]
- 27. Friend S. H.; Bernards R.; Rogelj S.; Weinberg R. A.; Rapaport J. M.; Alberts D. M.; Dryja T. P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323:643–646; 1986. [DOI] [PubMed] [Google Scholar]
- 28. Giaccia A. J.; Kastan M. B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 12:2973–2983; 1998. [DOI] [PubMed] [Google Scholar]
- 29. Gottesfeld J. M.; Forbes D. J. Mitotic repression of the transcriptional machinery. Trends Biochem. Sci. 22:197–202; 1997. [DOI] [PubMed] [Google Scholar]
- 30. Gottesfeld J. M.; Johnson D. L.; Nyborg J. K. Transcriptional activation of RNA polymerase III-dependent genes by the human T-cell leukaemia virus type 1 Tax protein. Mol. Cell. Biol. 16:1777–1785; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gottesfeld J. M.; Wolf V. J.; Dang T.; Forbes D. J.; Hartl P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science 263:81–84; 1994. [DOI] [PubMed] [Google Scholar]
- 32. Grana X.; Garriga J.; Mayol X. Role of the retino-blastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 17:3365–3383; 1998. [DOI] [PubMed] [Google Scholar]
- 33. Hartl P.; Gottesfeld J.; Forbes D. J. Mitotic repression of transcription in vitro. J. Cell Biol. 120:613–624; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hernandez N. TBP, a universal eukaryotic transcription factor? Genes Dev. 7:1291–1308; 1993. [DOI] [PubMed] [Google Scholar]
- 35. Herwig S.; Strauss M. The retinoblastoma protein: A master regulator of cell cycle, differentiation and apoptosis. Eur. J. Biochem. 246:581–601; 1997. [DOI] [PubMed] [Google Scholar]
- 36. Hirama T.; Koeffler H. P. Role of the cyclin-dependent kinase inhibitors in the development of cancer. Blood 86:841–854; 1995. [PubMed] [Google Scholar]
- 37. Hoeffler W. K.; Kovelman R.; Roeder R. G. Activation of transcription factor IIIC by the adenovirus E1A protein. Cell 53:907–920; 1988. [DOI] [PubMed] [Google Scholar]
- 38. Hoeffler W. K.; Roeder R. G. Enhancement of RNA polymerase III transcription by the E1A gene product of adenovirus. Cell 41:955–963; 1985. [DOI] [PubMed] [Google Scholar]
- 39. Hollstein M.; Rice K.; Greenblatt M. S.; Soussi T.; Fucks R.; Sorlie T.; Hovig E.; Smith-Sorensen B.; Montesano R.; Harris C. C. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 22:3551–3555; 1994. [PMC free article] [PubMed] [Google Scholar]
- 40. Horowitz J. M.; Park S.-H.; Bogenmann E.; Cheng J.-C.; Yandell D. W.; Kaye F. J.; Minna J. D.; Dryja T. P.; Weinberg R. A. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumour cells. Proc. Natl. Acad. Sci. USA 87:2775–2779; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsieh Y.-J.; Kundu T. K.; Wang Z.; Kovelman R.; Roeder R. G. The TFIIIC90 subunit of TFIIIC interacts with multiple components of the RNA polymerase III machinery and contains a histone-specific acetyltransferase activity. Mol. Cell. Biol. 19:7697–7704; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hsieh Y.-J.; Wang Z.; Kovelman R.; Roeder R. G. Cloning and characterization of two evolutionarily conserved subunits (TFIIIC102 and TFIIIC63) of human TFIIIC and their involvement in functional interactions with TFIIIB and RNA polymerase III. Mol. Cell. Biol. 19:4944–4952; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu Q.; Dyson N.; Harlow E. The regions of the retinoblastoma protein needed for binding to adenovirus E1A or SV40 large T antigen are common sites for mutations. EMBO J. 9:1147–1155; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hunter T.; Pines J. Cyclins and cancer II: Cyclin D and CDK inhibitors come of age. Cell 79:573–582; 1994. [DOI] [PubMed] [Google Scholar]
- 45. Hurford R. K.; Cobrinik D.; Lee M.-H.; Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 11:1447–1463; 1997. [DOI] [PubMed] [Google Scholar]
- 46. Jacobson S.; Raine C. S.; Mingioli E. S.; McFarlin D. E. Isolation of an HTLV-I-like retrovirus from patients with tropical spastic paraparesis. Nature 331: 540–543; 1988. [DOI] [PubMed] [Google Scholar]
- 47. Joazeiro C. A. P.; Kassavetis G. A.; Geiduschek E. P. Identical components of yeast transcription factor IIIB are required and sufficient for transcription of TATA box-containing and TATA-less genes. Mol. Cell. Biol. 14:2798–2808; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson L. F.; Abelson H. T.; Green H.; Penman S. Changes in RNA in relation to growth of the fibro-blast. I. Amounts of mRNA, rRNA, and tRNA in resting and growing cells. Cell 1:95–100; 1974. [DOI] [PubMed] [Google Scholar]
- 49. Kassavetis G. A.; Braun B. R.; Nguyen L. H.; Geiduschek E. P. S. cerevisiae TFIIIB is the transcription initiation factor proper of RNA polymerase III, while TFIIIA and TFIIIC are assembly factors. Cell 60:235–245; 1990. [DOI] [PubMed] [Google Scholar]
- 50. Kassavetis G. A.; Joazeiro C. A. P.; Pisano M.; Geiduschek E. P.; Colbert T.; Hahn S.; Blanco J. A. The role of the TATA-binding protein in the assembly and function of the multisubunit yeast RNA polymerase III transcription factor, TFIIIB. Cell 71:1055–1064; 1992. [DOI] [PubMed] [Google Scholar]
- 51. Kassavetis G. A.; Nguyen S. T.; Kobayashi R.; Kumar A.; Geiduschek E. P.; Pisano M. Cloning, expression, and function of TFC5, the gene encoding the B″ component of the Saccharomyces cerevisiae RNA polymerase III transcription factor TFIIIB. Proc. Natl. Acad. Sci. USA 92:9786–9790; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaye F. J.; Kratzke R. A.; Gerster J. L.; Horowitz J. M. A single amino acid substitution results in a retinoblastoma protein defective in phosphorylation and oncoprotein binding. Proc. Natl. Acad. Sci. USA 87:6922–6926; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kekule A. S.; Lauer U.; Weiss L.; Luber B.; Hofschneider P. H. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature 361:742–745; 1993. [DOI] [PubMed] [Google Scholar]
- 54. Kief D. R.; Warner J. R. Coordinate control of syntheses of ribosomal ribonucleic acid and ribosomal proteins during nutritional shift-up in Saccharomyces cerevisiae . Mol. Cell. Biol. 1:1007–1015; 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Killander D.; Zetterberg A. A quantitative cyto-chemical investigation of the relationship between cell mass and initiation of DNA synthesis in mouse fibroblasts in vitro . Exp. Cell Res. 40:12–20; 1965. [DOI] [PubMed] [Google Scholar]
- 56. Kim C.-M.; Koike K.; Saito I.; Miyamura T.; Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351:317–320; 1991. [DOI] [PubMed] [Google Scholar]
- 57. Kim S.; Na J. G.; Hampsey M.; Reinberg D. The Dr1/DRAP1 heterodimer is a global repressor of transcription in vivo. Proc. Natl. Acad. Sci. USA 94:820–825; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ko L. J.; Prives C. p53: Puzzle and paradigm. Genes Dev. 10:1054–1072; 1996. [DOI] [PubMed] [Google Scholar]
- 59. Kovelman R.; Roeder R. G. Purification and characterization of two forms of human transcription factor IIIC. J. Biol. Chem. 267:24446–24456; 1992. [PubMed] [Google Scholar]
- 60. Kramerov D. A.; Lekakh I. V.; Samarina O. P.; Ryskov A. P. The sequences homologous to major interspersed repeats B1 and B2 of mouse genome are present in mRNA and cytoplasmic poly(A)+ RNA. Nucleic Acids Res. 10:7477–7491; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kramerov D. A.; Tillib S. V.; Shumyatsky G. P.; Georgiev G. P. The most abundant nascent poly(A)+ RNAs are transcribed by RNA polymerase III in murine tumor cells. Nucleic Acids Res. 18:4499–4506; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kraus V. B.; Inostroza J. A.; Yeung K.; Reinberg D.; Nevins J. R. Interaction of the Dr1 inhibitory factor with the TATA binding protein is disrupted by adenovirus E1A. Proc. Natl. Acad. Sci. USA 91: 6279–6282; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kundu T. K.; Wang Z.; Roeder R. G. Human TFIIIC relieves chromatin-mediated repression of RNA polymerase III transcription and contains an intrinsic histone acetyltransferase activity. Mol. Cell. Biol. 19:1605–1615; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kwee L.; Lucito R.; Aufiero B.; Schneider R. J. Alternate translation initiation on hepatitis B virus X mRNA produces multiple polypeptides that differentially transactivate class II and III promoters. J. Virol. 66:4382–4389; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. L’Etoile N. D.; Fahnestock M. L.; Shen Y.; Aebersold R.; Berk A. J. Human transcription factor IIIC box B binding subunit. Proc. Natl. Acad. Sci. USA 91:1652–1656; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lagna G.; Kovelman R.; Sukegawa J.; Roeder R. G. Cloning and characterization of an evolutionarily divergent DNA-binding subunit of mammalian TFIIIC. Mol. Cell. Biol. 14:3053–3064; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lania L.; Pannuti A.; La Mantia G.; Basilico C. The transcription of B2 repeated sequences is regulated during the transition from quiescent to proliferative state in cultured rodent cells. FEBS Lett. 219: 400–404; 1987. [DOI] [PubMed] [Google Scholar]
- 68. Larminie C. G. C.; Alzuherri H. M.; Cairns C. A.; McLees A.; White R. J. Transcription by RNA polymerases I and III: A potential link between cell growth, protein synthesis and the retinoblastoma protein. J. Mol. Med. 76:94–103; 1998. [DOI] [PubMed] [Google Scholar]
- 69. Larminie C. G. C.; Cairns C. A.; Mital R.; Martin K.; Kouzarides T.; Jackson S. P.; White R. J. Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J. 16:2061–2071; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Larminie C. G. C.; Sutcliffe J. E.; Tosh K.; Winter A. G.; Felton- Edkins Z. A.; White R. J. Activation of RNA polymerase III transcription in cells transformed by simian virus 40. Mol. Cell. Biol. 19:4927–4934; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Larminie C. G. C.; White R. J. Identification of a putative BRF homologue in the genome of Caenorhabditis elegans . DNA Sequence 9:49–58; 1998. [DOI] [PubMed] [Google Scholar]
- 72. Lassar A. B.; Martin P. L.; Roeder R. G. Transcription of class III genes: Formation of preinitiation complexes. Science 222:740–748; 1983. [DOI] [PubMed] [Google Scholar]
- 73. Leresche A.; Wolf V. J.; Gottesfeld J. M. Repression of RNA polymerase II and III transcription during M phase of the cell cycle. Exp. Cell Res. 229: 282–288; 1996. [DOI] [PubMed] [Google Scholar]
- 74. Lobo S. M.; Lister J.; Sullivan M. L.; Hernandez N. The cloned RNA polymerase II transcription factor IID selects RNA polymerase III to transcribe the human U6 gene in vitro. Genes Dev. 5:1477–1489; 1991. [DOI] [PubMed] [Google Scholar]
- 75. Lopez-de-Leon A.; Librizzi M.; Tuglia K.; Willis I. PCF4 encodes an RNA polymerase III transcription factor with homology to TFIIB. Cell 71:211–220; 1992. [DOI] [PubMed] [Google Scholar]
- 76. Ludlow J. W.; DeCaprio J. A.; Huang C.-M.; Lee W.-H.; Paucha E.; Livingston D. M. SV40 large T antigen binds preferentially to an underphosphorylated member of the retinoblastoma susceptibility gene product family. Cell 56:57–65; 1989. [DOI] [PubMed] [Google Scholar]
- 77. Majello B.; La Mantia G.; Simeone A.; Boncinelli E.; Lania L. Activation of major histocompatibility complex class I mRNA containing an Alu-like repeat in polyoma virus-transformed rat cells. Nature 314: 457–459; 1985. [DOI] [PubMed] [Google Scholar]
- 78. Margottin F.; Dujardin G.; Gerard M.; Egly J. M.; Huet J.; Sentenac A. Participation of the TATA factor in transcription of the yeast U6 gene by RNA polymerase C. Science 251:424–426; 1991. [DOI] [PubMed] [Google Scholar]
- 79. Mauck J. C.; Green H. Regulation of pre-transfer RNA synthesis during transition from resting to growing state. Cell 3:171–177; 1974. [DOI] [PubMed] [Google Scholar]
- 80. Mayol X.; Garriga J.; Grana X. The transition from G1 to G0 involves G1 cyclin/cdk-independent phosphorylation of p130 and its association with E2F-4. Oncogene 13:237–246; 1996. [PubMed] [Google Scholar]
- 81. Mital R.; Kobayashi R.; Hernandez N. RNA polymerase III transcription from the human U6 and adenovirus type 2 VAI promoters has different requirements for human BRF, a subunit of human TFIIIB. Mol. Cell. Biol. 16:7031–7042; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mittnacht S. Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 8:21–27; 1998. [DOI] [PubMed] [Google Scholar]
- 83. Momand J.; Jung D.; Wilczynski S.; Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 26:3453–3459; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moran E. A region of SV40 large T antigen can substitute for a transforming domain of the adenovirus E1A products. Nature 334:168–170; 1988. [DOI] [PubMed] [Google Scholar]
- 85. Mulligan G.; Jacks T. The retinoblastoma gene family: Cousins with overlapping interests. Trends Genet. 14:223–229; 1998. [DOI] [PubMed] [Google Scholar]
- 86. Munger K.; Werness B. A.; Dyson N.; Phelps W. C.; Harlow E.; Howley P. M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumour suppressor gene product. EMBO J. 8:4099–4105; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nasmyth K. Another role rolls in. Nature 382:28–29; 1996. [DOI] [PubMed] [Google Scholar]
- 88. Nylandsted J.; Rohde M.; Bartek J.; Strauss M. Expression of a p16INK4a-specific ribozyme downmodulates p16INK4a abundance and accelerates cell proliferation. FEBS Lett. 436:41–45; 1998. [DOI] [PubMed] [Google Scholar]
- 89. Piras G.; Dittmer J.; Radonovich M. F.; Brady J. N. Human T-cell leukaemia virus type I Tax protein transactivates RNA polymerase III promoter in vitro and in vivo . J. Biol. Chem. 271:20501–20506; 1996. [DOI] [PubMed] [Google Scholar]
- 90. Poiesz B. J.; Ruscetti F. W.; Gazdar A. F.; Bunn P. A.; Minna J. D.; Gallo R. C. Detection and isolation of type Cretrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 77:7415–7419; 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pombo A.; Jackson D. A.; Hollinshead M.; Wang Z.; Roeder R. G.; Cook P. R. Regional specialization in human nuclei: Visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 18: 2241–2253; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Qin X.; Chittenden T.; Livingston D. M.; Kaelin W. G. Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 6:953–964; 1992. [DOI] [PubMed] [Google Scholar]
- 93. Redpath N. T.; Proud C. G. Molecular mechanisms in the control of translation by hormones and growth factors. Biochim. Biophys. Acta 1220:147–162; 1994. [DOI] [PubMed] [Google Scholar]
- 94. Rigby P. W. J. Three in one and one in three: It all depends on TBP. Cell 72:7–10; 1993. [DOI] [PubMed] [Google Scholar]
- 95. Roberts S.; Miller S. J.; Lane W. S.; Lee S.; Hahn S. Cloning and functional characterization of the gene encoding the TFIIIB90 subunit of RNA polymerase III transcription factor TFIIIB. J. Biol. Chem. 271: 14903–14909; 1996. [DOI] [PubMed] [Google Scholar]
- 96. Ronning O. W.; Lindmo T.; Pettersen E. O.; Seglen P. O. The role of protein accumulation in the cell cycle control of human NHIK 3035 cells. J. Cell. Physiol. 109:411–418; 1981. [DOI] [PubMed] [Google Scholar]
- 97. Rosenwald I. B. Deregulation of protein synthesis as a mechanism of neoplastic transformation. Bioessays 18:243–250; 1996. [DOI] [PubMed] [Google Scholar]
- 98. Rosenwald I. B. Upregulated expression of the genes encoding translation initiation factors eIF-4E and eIF-2α in transformed cells. Cancer Lett. 102:113–123; 1996. [DOI] [PubMed] [Google Scholar]
- 99. Ruth J.; Conesa C; Dieci G.; Lefebvre O.; Duster-hoft A.; Ottonello S.; Sentenac A. A suppressor of mutations in the class III transcription system encodes a component of yeast TFIIIB. EMBO J. 15:1941–1949; 1996. [PMC free article] [PubMed] [Google Scholar]
- 100. Ryskov A. P.; Ivanov P. L.; Tokarskaya O. N.; Kramerov D. A.; Grigoryan M. S.; Georgiev G. P. Major transcripts containing B1 and B2 repetitive se-quences in cytoplasmic poly(A)+ RNA from mouse tissues. FEBS Lett. 182:73–76; 1985. [DOI] [PubMed] [Google Scholar]
- 101. Scheffner M.; Munger K.; Byrne J. C.; Howley P. M. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 88:5523–5527; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Scott M. R. D.; Westphal K.-H.; Rigby P. W. J. Activation of mouse genes in transformed cells. Cell 34:557–567; 1983. [DOI] [PubMed] [Google Scholar]
- 103. Sellers W. R.; Novitch B. G.; Miyake S.; Heith A.; Otterson G. A.; Kaye F. J.; Lassar A. B.; Kaelin W. G. Jr. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 12:95–106; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shew J.-Y.; Lin B. T.-Y.; Chen P.-L.; Tseng B. Y.; Yang-Feng T. L.; Lee W.-H. C-terminal truncation of the retinoblastoma gene product leads to functional inactivation. Proc. Natl. Acad. Sci. USA 87:6–10; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Simmen K. A.; Bernues J.; Parry H. D.; Stunnen-berg H. G.; Berkenstam A.; Cavallini B.; Egly J. M.; Mattaj I. W. TFIID is required for in vitro transcription of the human U6 gene by RNA polymerase III. EMBO J. 10:1853–1862; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Singh K.; Carey M.; Saragosti S.; Botchan M. Expression of enhanced levels of small RNA polymerase III transcripts encoded by the B2 repeats in simian virus 40-transformed mouse cells. Nature 314:553–556; 1985. [DOI] [PubMed] [Google Scholar]
- 107. Sinn E.; Wang Z.; Kovelman R.; Roeder R. G. Cloning and characterization of a TFIIIC2 subunit (TFIIICP) whose presence correlates with activation of RNA polymerase III-mediated transcription by adenovirus E1A expression and serum factors. Genes Dev. 9:675–685; 1995. [DOI] [PubMed] [Google Scholar]
- 108. Soderlund H.; Pettersson U.; Vennstom B.; Philipson L.; Mathews M. B. A new species of virus-coded low molecular weight RNA from cells infected with adenovirus type 2. Cell 7:585–593; 1976. [DOI] [PubMed] [Google Scholar]
- 109. Stanners C. P.; Adams M. E.; Harkins J. L.; Pollard J. W. Transformed cells have lost control of ribosome number through their growth cycle. J. Cell. Physiol. 100:127–138; 1979. [DOI] [PubMed] [Google Scholar]
- 110. Sutcliffe J. E.; Cairns C. A.; McLees A.; Allison S. J.; Tosh K.; White R. J. RNA polymerase III transcription factor IIIB is a target for repression by pocket proteins p107 and p130. Mol. Cell. Biol. 19: 4255–4261; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tatsuka M.; Mitsui H.; Wada M.; Nagata A.; No-jima H.; Okayama H. Elongation factor-1α gene determines susceptibility to transformation. Nature 359: 333–335; 1992. [DOI] [PubMed] [Google Scholar]
- 112. Taya Y. RB kinases and RB-binding proteins: New points of view. Trends Biochem. Sci. 22:14–17; 1997. [DOI] [PubMed] [Google Scholar]
- 113. Terasima T.; Yasukawa M. Synthesis of G1 protein preceding DNA synthesis in cultured mammalian cells. Exp. Cell. Res. 44:669; 1966. [DOI] [PubMed] [Google Scholar]
- 114. Thimmappaya B.; Weinberger C.; Schneider R. J.; Shenk T. Adenovirus VAI RNA is required for efficient translation of viral mRNA at late times after infection. Cell 31:543–551; 1982. [DOI] [PubMed] [Google Scholar]
- 115. Tower J.; Sollner-Webb B. Polymerase III transcription factor B activity is reduced in extracts of growth-restricted cells. Mol. Cell. Biol. 8:1001–1005; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Trivedi A.; Vilalta A.; Gopalan S.; Johnson D. L. TATA-binding protein is limiting for both TATA-containing and TATA-lacking RNA polymerase III promoters in Drosophila cells. Mol. Cell. Biol. 16: 6909–6916; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vilalta A.; Trivedi A.; Wang Z.; Roeder R. G.; Johnson D. L. An RNA polymerase III-defective mutation in TATA-binding protein disrupts its interaction with a transcription factor IIIB subunit in Drosophila cells. J. Biol. Chem. 272:18087–18092; 1997. [DOI] [PubMed] [Google Scholar]
- 118. Vousden K. H. Regulation of the cell cycle by viral oncoproteins. Semin. Cancer Biol. 6:109–116; 1995. [DOI] [PubMed] [Google Scholar]
- 119. Wang H.-D.; Trivedi A.; Johnson D. L. Hepatitis B virus X protein induces RNA polymerase III-dependent gene transcription and increases cellular TATA-binding protein by activating the Ras signalling pathway. Mol. Cell. Biol. 17:6838–6846; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang H.-D.; Yuh C.-H.; Dang C. V.; Johnson D. L. The hepatitis B virus X protein increases the cellular level of TATA-binding protein, which mediates transactivation of RNA polymerase III genes. Mol. Cell. Biol. 15:6720–6728; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang Z.; Roeder R. G. Structure and function of a human transcription factor TFIIIB subunit that is evolutionarily conserved and contains both TFIIB-and high-mobility-group protein 2-related domains. Proc. Natl. Acad. Sci. USA 92:7026–7030; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang Z.; Roeder R. G. TFIIIC1 acts through a downstream region to stabilize TFIIIC2 binding to RNA polymerase III promoters. Mol. Cell. Biol. 16: 6841–6850; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Weinberg R. A. The retinoblastoma protein and cell cycle control. Cell 81:323–330; 1995. [DOI] [PubMed] [Google Scholar]
- 124. Weinmann R.; Raskas H. J.; Roeder R. G. Role of DNA-dependent RNA polymerases II and III in transcription of the adenovirus genome late in productive infection. Proc. Natl. Acad. Sci. USA 71:3426–3430; 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. White R. J. Regulation of RNA polymerases I and III by the retinoblastoma protein: A mechanism for growth control? Trends Biochem. Sci. 22:77–80; 1997. [DOI] [PubMed] [Google Scholar]
- 126. White R. J. RNA polymerase III transcription. Berlin: Springer-Verlag; 1998. [Google Scholar]
- 127. White R. J. Transcription factor IIIB: An important determinant of biosynthetic capacity that is targeted by tumour suppressors and transforming proteins. Int. J. Oncol. 12:741–748; 1998. [PubMed] [Google Scholar]
- 128. White R. J.; Gottlieb T. M.; Downes C. S.; Jackson S. P. Cell cycle regulation of RNA polymerase III transcription. Mol. Cell. Biol. 15:6653–6662; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. White R. J.; Gottlieb T. M.; Downes C. S.; Jackson S. P. Mitotic regulation of a TATA-binding-protein-containing complex. Mol. Cell. Biol. 15:1983–1992; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. White R. J.; Khoo B. C.-E.; Inostroza J. A.; Rein-berg D.; Jackson S. P. The TBP-binding repressor Dr1 differentially regulates RNA polymerases I, II and III. Science 266:448–450; 1994. [DOI] [PubMed] [Google Scholar]
- 131. White R. J.; Stott D.; Rigby P. W. J. Regulation of RNA polymerase III transcription in response to Simian virus 40 transformation. EMBO J. 9:3713–3721; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. White R. J.; Trouche D.; Martin K.; Jackson S. P.; Kouzarides T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 382:88–90; 1996. [DOI] [PubMed] [Google Scholar]
- 133. Whyte P.; Buchkovich K. J.; Horowitz J. M.; Friend S. H.; Raybuck M.; Weinberg R. A.; Harlow E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124–129; 1988. [DOI] [PubMed] [Google Scholar]
- 134. Whyte P., Williamson N. M., and Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell 56:67–75; 1989. [DOI] [PubMed] [Google Scholar]
- 135. Willis I. M. RNA polymerase III. Genes, factors and transcriptional specificity. Eur. J. Biochem. 212:1–11; 1993. [DOI] [PubMed] [Google Scholar]
- 136. Wolf V. J.; Dang T.; Hartl P.; Gottesfeld J. M. Role of maturation-promoting factor (p34cdc2-cyclin B) in differential expression of the Xenopus oocyte and somatic-type 5S RNA genes. Mol. Cell. Biol. 14: 4704–4711; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yoshinaga S.; Dean N.; Han M.; Berk J. A. Adeno-virus stimulation of transcription by RNA polymerase III: Evidence for an E1A-dependent increase in transcription factor IIIC concentration. EMBO J. 5:343–354; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yoshinaga S. K.; Boulanger P. A.; Berk A. J. Resolution of human transcription factor TFIIIC into two functional components. Proc. Natl. Acad. Sci. USA 84:3585–3589; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yoshinaga S. K.; L’Etoile N. D.; Berk A. J. Purification and characterization of transcription factor IIIC2. J. Biol. Chem. 264:10726–10731; 1989. [PubMed] [Google Scholar]
- 140. Zhu L.; van den Heuvel S.; Helin K.; Fattaey A.; Ewen M.; Livingston D.; Dyson N.; Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 7:1111–1125; 1993. [DOI] [PubMed] [Google Scholar]